
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-based non-isocyanate poly(hydroxy urethane)s (PHU) derived from vanillin and CO2†
Noé
Fanjul-Mosteirín
 abc,
Lucas Polo
Fonseca
c,
Andrew P.
Dove
*b and
Haritz
Sardon
*c
abc,
Lucas Polo
Fonseca
c,
Andrew P.
Dove
*b and
Haritz
Sardon
*c
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: A.Dove@bham.ac.uk
cPOLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, Avda Tolosa 72, 20018 Donostia-San Sebastian, Spain. E-mail: haritz.sardon@ehu.es
First published on 16th May 2023
Abstract
As an alternative to the use of hazardous phosgene-based isocyanates for polyurethane preparation, non-isocyanate poly(hydroxy urethanes) (PHU) based on 5-membered cyclic carbonates have been developed. However, to date, most aromatic PHUs are oil-based or based on toxic precursors such as bisphenol-A. In this work, bio-based non-isocyanate poly(hydroxy urethanes) (PHUs) prepared from vanillin are reported for the first time. First, three different vanillin-derived bis-cyclic carbonates were synthesized. Subsequently, each monomer was reacted with two different bis-amines to yield six different PHUs, which were characterized in depth by 1H and 13C NMR spectroscopy, FTIR, SEC and DSC. PHUs based on vanillic acid were found to exhibit thermal properties superior to bisphenol A-based PHUs, with a Tg of around 66 °C. It is envisioned that vanillin-based PHUs could potentially be a safer alternative to harmful bisphenol A-based PHUs and provide a useful strategy for CO2 revalorization, especially considering that vanillin is an abundant byproduct of Kraft lignin production.
Introduction
Polyurethanes represent a highly versatile class of materials as a consequence of their good physical and mechanical properties, which are the result of the high density of intermolecular hydrogen bonds.1 For this reason, coupled with the wide variety of available monomers, polyurethanes are commonly used in a broad range of applications, such as rigid and soft foams, coatings, adhesives, sealants, elastomers and biomedical applications.2–8 In industry, polyurethanes are typically prepared by the reaction between diisocyanates and polyols,9 catalyzed by toxic organometallic compounds.1,10,11 Although this strategy is highly efficient, it also poses significant health and environmental risks.11–13 The major issue is the use of intrinsically toxic diisocyanate monomers. This is particularly important as the industrial production of isocyanates uses phosgene as a precursor. Therefore, non-isocyanate polyurethanes (NIPUs) have attracted great interest in both academia and industry as an alternative to the classical synthesis of polyurethanes.14One of the most promising strategies to bypass the use of toxic isocyanates for the synthesis of polyurethanes is the synthesis of NIPUs by reacting a bis-cyclic carbonate with a bis-amine, which generates poly(hydroxy urethanes) (PHU).14–19 Indeed, this reaction not only has 100% atom economy but also benefits from the availability of a large portfolio of poly(cyclic carbonate)s, which can be synthesized by a facile chemical [3+2] CO2 insertion into the corresponding epoxy precursor. This reaction has been extensively studied and a plethora of metal-based and organocatalytic methodologies have been reported.20–25 While there is no doubt about the reduced carbon footprint in comparison with isocyanate chemistry, oil still is the main source for the production of cyclic carbonates, especially those based on aromatic precursors. This fact, combined with the growing concern over global warming, represents a worrying challenge in terms of energy and sustainability.26–28 In this sense the development of monomers with potential bio-based origin and good performance is attracting a lot of attention as an alternative towards a carbon-neutral and circular polymer industry.29–37
Among all the renewable resources available, lignin represents one of the most abundant biopolymers as it is present in all vascular plants along with cellulose and hemicellulose.38–40 These three biopolymers are present in different distributions depending on the plant. In the case of lignin, this percentage range is from 25 to 35%.41 Lignin is a cross-linked polymer formed by copolymerization of three different monolignols and, despite its heterogeneity, lignin can be envisaged as the largest bio-based aromatic feedstock in nature. Depending on the depolymerization process, a large variety of different added-value chemicals can be isolated from the depolymerization of lignin.41 One of the compounds that can be isolated from lignin depolymerization in the so-called Kraft process is vanillin.42 This aromatic compound has been extensively used in many applications, such as in the food industry as a flavoring, the fragrance industry in perfumes and as intermediates in the production of fine chemicals. Vanillin, with its aldehyde and phenol functionality, represents a very versatile platform for the synthesis of a plethora of monomers for polymer synthesis. Boutevin and co-workers reported the synthesis of a total of 18 compounds with different classes of functionalities,43 some of which have been reported for the synthesis of epoxy resins44–49 and polymers obtained by acyclic diene metathesis (ADMET) polymerization.50,51
The aromaticity of lignin and its derivatives can be highly valuable for non-isocyanate polyurethane production, as it can potentially increase the cohesion of polymer materials due to strong π-interactions, which limit chain mobility.52,53 This can lead to materials with properties analogous to those of oil-based PUs, but with a “green” feedstock that has reduced environmental impact.54,55 The use of bio-based aromatic precursors, such as vanillin, is even more interesting, due to their reduced toxicity when compared to bisphenol-A, and due to their natural occurrence, thus minimizing potential negative impacts associated with plastic production.
Herein, following our interest in CO2 revalorization56 along with our experience in the synthesis of NIPUs,57,58 we present the metal-free synthesis of three different bis-carbonates derived from vanillin and the subsequent synthesis of novel bio-based PHUs via step growth polymerization using 1,4-butanediamine (BDA) and 1,6-hexamethylenediamine (HDMA). The effect of the vanillin-based bis-carbonate structure and the length of the diamine on the thermal properties of the PHUs is evaluated.
Experimental section
Material and methods
All commercially available starting materials were purchased from Sigma-Aldrich, Alfa Aesar or Acros Organics and used as received unless otherwise indicated. CO2 was purchased from Air Liquide (99.995%). NMR spectra were recorded on a Bruker Avance 300 MHz spectrometer or Bruker Avance III HD 300 or 400 MHz spectrometer. Chemical shifts are reported in parts per million (ppm) and referenced to the residual solvent signal (CDCl3: 1H, δ = 7.26 ppm, 13C, δ = 77.2 ppm, TMS: 1H, δ = 0.00 ppm). Multiplicities are reported as s = singlet, brs = broad singlet, d = doublet, dd = doublet of doublets, dt = doublet of triplets, ddd = doublet of doublet of doublets, ddt = doublet of doublet of triplets, t = triplet, tt = triplet of triplets q = quartet, quint = quintet, m = multiplet. Multiplicity is followed by coupling constant (J) in Hz and integration. Size exclusion chromatography (SEC) was used to determine the number average molar mass (Mn) and dispersities (ĐM) of the PHUs. SEC analysis in DMF was conducted on a system composed of a Varian 390-LC-Multi detector suite fitted with differential refractive index, light scattering and viscosimeter detectors equipped with a guard column (Varian Polymer Laboratories PLGel 5 μM, 50 × 7.5 mm) and two mixed D columns (Varian Polymer Laboratories PLGel 5 μM, 300 × 7.5 mm). The mobile phase was DMF containing 2% TEA + 0.01% of salt at a steady flow rate of 1.0 mL min−1. FT-IR spectra of PHUs were obtained using a PerkinElmer Spectrum 100 FT-IR spectrometer. Thermal characterization by differential scanning calorimetry (DSC) was performed using Mettler Toledo DSC1 Star.Synthesis of [HDBU]I
Ammonium iodide (970 mg, 6.69 mmol) was dissolved in MeOH (6.7 mL, 1 M), then DBU (6 mL, 6.69 mmol) was added. A reflux condenser was attached to the flask and the mixture was heated up to reflux overnight. Solvent was evaporated and the crude was recrystallized from CH2Cl2/hexane. A solid was obtained (1.818 g, 6.49 mmol, 97%). Characterization data in agreement with those reported previously1. 1H NMR (300 MHz, CDCl3) δ 8.46 (bs, 1H, N–H), 3.57–3.53 (m, 4H, NH–CH2–CH2–CH2–N, C–(CH2)5–CH2–N), 3.40–3.35 (broad quint, 2H, CH2–NH), 2.93–2.90 (m, 2H, CH2–C), 2.06–1.99 (quint, 2H, 3JH–H = 5.9 Hz, CH2–CH2–NH), 1.79–1.73 (m, 6H, CH2–CH2–CH2). 13C APT NMR (75 MHz, CDCl3) δ 166.0 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 54.8 (C–(CH2)5–CH2–N), 48.9 (NH–CH2–CH2–CH2–N), 37.7 (CH2–NH), 32.7 (CH2–C), 28.7 (C–(CH2)3–CH2–CH2–N), 26.5 (CH2–CH2–C), 23.7 (C–(CH2)2–CH2–CH2–CH2–N), 19.2 (CH2–CH2–NH).
N), 54.8 (C–(CH2)5–CH2–N), 48.9 (NH–CH2–CH2–CH2–N), 37.7 (CH2–NH), 32.7 (CH2–C), 28.7 (C–(CH2)3–CH2–CH2–N), 26.5 (CH2–CH2–C), 23.7 (C–(CH2)2–CH2–CH2–CH2–N), 19.2 (CH2–CH2–NH).
Synthesis of methoxyhydroquinone (2)
A two-necked RBF was charged with a solution of vanillin (15.00 g, 98.6 mmol) in THF (395 mL). Deionized water (158 mL) was then added. The mixture was degassed with N2. Sodium percarbonate (Na2CO3·1.5H2O2, 17.03 g, 108.4 mmol) was then added in portions under N2 and agitation. The reaction was stirred for 3 h at room temperature. Portions of a HCl solution (0.1 mol L−1) were added to the mixture under vigorous stirring until pH = 3 was reached to quench the reaction. The volatiles were evaporated and the aqueous phase was extracted with EtOAc. The organic phases were collected, washed with brine, dried over anhydrous Na2SO4 before the EtOAc was removed under reduced pressure. A white solid was obtained (12.16 g, 86.77 mmol, 88%). Characterization data in agreement with those reported previously.21H NMR (300 MHz, DMSO) δ 8.69 (s, 1H, CH3O–CC–OH), 8.10 (s, 1H, CH3O–C–CHC–OH), 6.54 (d, 1H, 3JH–H = 8.4 Hz, CH3O–CC–CH), 6.35 (d, 1H, 4JH–H = 2.5 Hz, CH3O–C–CH), 6.14 (dd, 1H, 4JH–H = 2.5 Hz, 3JH–H = 8.4 Hz, CH3O–CC–CHCH), 3.69 (s, 3H, OCH3). 13C APT NMR (75 MHz, DMSO) δ 150.3 (CH3O–C–CHC–OH), 148.1 (CH3O–C), 138.7 (CH3O–CC–OH), 115.7 (CH3O–CC–CH), 106.2 (CH3O–CC–CHCH), 100.7 (CH3O–C–CH), 55.4 (CH3).
Synthesis of vanillyl alcohol (4)
Vanillin (15.00 g, 98.6 mmol) was dissolved in MeOH (300 mL) and the mixture was cooled down with an ice-bath. NaBH4 (7.46 g, 197.15 mmol) was added to the previous mixture in small portions. Once the addition was finished, the mixture was diluted with EtOAc and extracted 3 times before organic layers were combined and washed with NH4Cl, brine and dried over Na2SO4. The EtOAc was removed by evaporation and the afforded solid was dried under high vacuum. A white solid was obtained (13.83 g, 89.73 mmol, 91%). Characterization data was in agreement with those reported previously.31H NMR (300 MHz, DMSO) δ 8.78 (s, 1H, CH3O–CC–OH), 6.88 (s, 1H, CH3O–CC–CHCH), 6.71–6.70 (m, 2H, CH3O–CC–CH, CH3O–C–CH) 5.00 (t, 1H, 3JH–H = 5.7 Hz, CH2-OH), 4.38 (d, 2H, 3JH–H = 5.7 Hz, HO-CH2), 3.75 (s, 3H, OCH3). 13C NMR (75 MHz, DMSO) δ 147.4 (CH3O–C), 145.3 (CH3O–CC–OH), 133.5 (CH3O–C–CHC–CH2OH), 119.1 (HO–CH2–C–CHCH), 115.1 (HO–CH2–C–CHCH), 111.0 (CH3O–C–CH), 63.0 (HO–CH2), 55.4 (CH3).
General method for the synthesis of bis-epoxide (5)
A RBF was charged with vanillyl alcohol (5 g, 32.43 mmol) and TEBAC (739 mg, 3.24 mmol). Epichlorohydrin (25.6 mL, 324.32 mmol) was added and the mixture was stirred for 4 h until a slightly pink solution was obtained. This solution was cooled down to 0 °C with an ice bath. A NaOH solution (19.5 g, 486 mmol) in deionized water (39 mL) was poured into the cold mixture under vigorous stirring. The reaction was conducted overnight at room temperature (ice bath was left to melt over time). Deionized water was added to the mixture to dilute 4 times the NaOH solution. An equal volume of EtOAc was added. The mixture was stirred and the aqueous solution was extracted 2 more times with EtOAc. The organic phases were combined, rinsed with brine and dried over anhydrous Na2SO4. EtOAc and excess epichlorohydrin were removed under high vacuum. Further purification was achieved by recrystallization in EtOAc. A white solid was obtained (5.82 g, 21.73 mmol, 67%). Characterization data was in agreement with those reported previously.21H NMR (300 MHz, CDCl3) δ 6.91–6.83 (m, 3H, CH3O–C–CH, CH3O–CC–CH, CH3O–CC–CHCH), 4.51 (q, 2H, 3JH–H = 11.6 Hz, CH3O–C–CHC–CH2), 4.25–4.20 (m, 1H, CH3O–CC–O–CHb) 4.05–4.00 (m, 1H, CH3O–C–CHC–CH2–O–CHb), 3.87 (s, 3H, CH3), 3.78–3.73 (m, 1H, CH3O–CC–O–CHa), 3.41–3.37 (m, 2H, CH3O–C–CHC–CH2–O–CH2–CH, CH3O–CC–O–CH2–CH) 3.18–3.16 (m, 1H, CH3O–C–CHC–CH2–O–CHa), 2.90–2.78 (m, 2H, CH3O–CC–O–CH2–CH–CHb, CH3O–C–CHC–CH2–O–CH2–CH–CHb), 2.74–2.59 (m, 2H, CH3O–CC–O–CH2–CH–CHa, CH3O–C–CHC–CH2–O–CH2–CH–CHa). 13C APT NMR (75 MHz, CDCl3) δ 149.8 (CH3O–C), 147.8 (CH3O–CC), 131.7 (CH3O–C–CHC), 120.5 (CH3O–CC–CHCH), 114.1 (CH3O–C–CH), 111.8 (CH3O–CC–CH), 73.3 (CH3O–C–CHC–CH2), 70.8 (CH3O–CC–O–CH2), 70.4 (CH3O–C–CHC–CH2–O–CH2) 56.0 (CH3), 51.0 (CH3O–CC–O–CH2–CH), 50.3 (CH3O–C–CHC–CH2–O–CH2–CH), 45.1 (CH3O–CC–O–CH2–CH–CH2), 44.4 (CH3O–C–CHC–CH2–O–CH2–CH–CH2).
General method for the synthesis of bis-epoxides (6) and (7)
A RBF was charged with methoxyhydroquinone (8.33 g, 59.47 mmol) and TEBAC (1.36 g, 5.95 mmol). Epichlorohydrin (46.5 mL, 594 mmol) was added and the mixture was heated to 80 °C and stirred for 1 h. The solution was cooled down to room temperature. An aqueous solution of TEBAC (1.36 g, 5.95 mmol) and NaOH (9.50 g, 237.9 mmol) in deionized water (48 mL) was added before the mixture was stirred for 30 minutes at room temperature. EtOAc (50 mL) and deionized water (50 mL) were then added. The mixture was stirred, and the aqueous phase was extracted with EtOAc. The organic phases were combined, rinsed with brine and dried over Na2SO4. Ethyl acetate and excess epichlorohydrin were removed under high vacuum. Further purification was achieved by recrystallization from EtOAc. A white solid was obtained (10.80 g, 42.82 mmol, 72%). Bis-epoxide (7) was prepared following the same procedure depicted above. A white solid was obtained (12.22 g, 50.55 mmol, 85%). Characterization data was in agreement with those reported previously.21H NMR (300 MHz, CDCl3) δ 6.86 (d, 1H, 3JH–H = 8.7 Hz, CH3O–CC–CH), 6.56 (d, 1H, 4JH–H = 2.6 Hz, CH3O–C–CH), 6.38 (dd, 1H, 4JH–H = 2.7 Hz, 3JH–H = 8.7 Hz, CH3O–CC–CHCH), 4.20–4.15 (m, 2H, CH3O–CC–O–CHb, CH3O–C–CHC–O–CHb), 3.99–3.91 (m, 2H, CH3O–CC–O–CHa, CH3O–C–CHC–O–CHa), 3.84 (s, 3H, CH3), 3.33 (m, 2H, CH3O–CC–O–CH2–CH, CH3O–C–CHC–O–CH2–CH), 2.91–2.85 (m, 2H, CH3O–CC–O–CH2–CH–CHb, CH3O–C–CHC–O–CH2–CH–CHb), 2.75–2.70 (m, 2H, CH3O–CC–O–CH2–CH–CHa, CH3O–C–CHC–O–CH2–CH–CHa). 13C APT NMR (75 MHz, CDCl3) δ 154.2 (CH3O–C–CHC), 151.0 (CH3O–C), 142.8 (CH3O–CC), 116.0 (C5 CH3O–CC–CH), 104.3 (CH3O–CC–CHCH), 101.4 (CH3O–C–CH), 71.5 (CH3O–CC–O–CH2), 69.4 (CH3O–C–CHC–O–CH2), 56.0 (CH3), 50.5 (CH3O–CC–O–CH2–CH), 50.3 (CH3O–C–CHC–O–CH2–CH), 45.0 (CH3O–CC–O–CH2–CH–CH2), 44.8 (CH3O–C–CHC–O–CH2–CH–CH2).
General method of the synthesis of bis-cyclic carbonates (8–10)
The corresponding bis-epoxide (24.97 mmol) was dissolved in DMF (25 mL) in a cylindrical glass vessel. To this mixture [HDBU]I (699 mg, 2.50 mmol) was added before it was placed in a stainless steel autoclave. The autoclave was subsequently sealed and filled to a pressure of 3 MPa of CO2. The autoclave was heated to 110 °C using an oil bath for 48 h. After 48 h the autoclave was allowed to cool down in an ice bath and the CO2 was vented. The crude reaction mixture was diluted in EtOAc (150 mL) and transferred to a separation funnel. Distilled water (150 mL) was added and aqueous phase was extracted three times. The organic layers were combined and washed with brine, then dried over Na2SO4, filtered and concentrated under vacuum. Compounds 9 and 10 were further purified by recrystallization in EtOAc and compound 8 was further purified by flash column chromatography using a mixture of ethyl acetate/hexane.4-((2-methoxy-4-(((2-oxo-1,3-dioxolan-4-yl)methoxy)methyl)phenoxy)methyl)-1,3-dioxolan-2-one (8)
A slightly yellow oil was obtained and further purified by flash column chromatography (6.97 g, 20.48 mmol, 82%). Characterization data was in agreement with those reported in the literature.21H NMR (300 MHz, DMSO) δ 6.98 (d, 2H, 3JH–H = 8.2 Hz, CH3O–C–CH, CH3O–CC–CH), 6.85 (d, 1H, 3JH–H = 8.1 Hz, CH3O–CC–CHCH), 5.13–5.12 (m, 1H, CH3O–CC–O–CH2–CH), 4.96–4.94 (m, 1H, CH3O–C–CHC–CH2–O–CH2–CH), 4.62 (t, 1H, 3JH–H = 8.5 Hz, CH3O–C–CHC–O–CH2–CH–CHb), 4.55–4.48 (m, 3H, CH3O–C–CHC–CH2, CH3O–C–CHC–O–CH2–CH–CHa), 4.42–4.37 (m, 1H, CH3O–C–CHC–CH2–O–CH2–CH–CHb), 4.31–4.14 (m, 3H, CH3O–CC–O–CH2, CH3O–C–CHC–CH2–O–CH2–CH–CHa), 3.87 (s, 3H, CH3), 3.70–3.56 (m, 2H, CH3O–C–CHC–CH2–O–CH2). 13C APT NMR (75 MHz, DMSO) δ 155.0 (CH3O–C–CHC–O–CH2–CH–OCO2, CH3O–CC–CH2–O–CH2–CH–OCO2), 149.2 (CH3O–C), 147.0 (CH3O–C–CHC–CH2), 131.7 (CH3O–C–CHC), 120.0 (CH3O–CC–CHCH), 114.4 (CH3O–CC–CH), 112.0 (CH3O–C–CH), 75.5 (CH3O–C–CHC–CH2–O–CH2–CH), 75.0 (CH3O–CC–O–CH2–CH), 72.1 (CH3O–C–CHC–CH2), 68.9 (CH3O–CC–O–CH2) 68.8 (CH3O–C–CHC–CH2–O–CH2), 66.2 (CH3O–C–CHC–CH2–O–CH2–CH–CH2), 66.0 (CH3O–CC–O–CH2–CH–CH2), 55.6 (CH3).
4,4′-(((3-methoxy-1,4-phenylene)bis(oxy))bis(methylene))bis(1,3-dioxolan-2-one) (9)
A white solid was obtained (6.60 g, 20.23 mmol, 81%). Characterization data was in agreement with those reported in the literature.21H NMR (300 MHz, DMSO) δ 6.93 (d, 1H, 3JH–H = 8.8 Hz, CH3O–CC–CH), 6.63 (d, 1H, 4JH–H = 2.6 Hz, CH3O–C–CH), 6.47 (dd, 1H, 4JH–H = 2.7 Hz, 3JH–H = 8.7 Hz, CH3O–CC–CHCH), 5.14–5.07 (m, 2H, CH3O–C–CHC–O–CH2–CH, CH3O–CC–O–CH2–CH), 4.65–4.58 (m, 2H, CH3O–CC–O–CH2–CH–CHb, CH3O–C–CHC–O–CH2–CH–CHb), 4.42–4.35 (m, 2H, CH3O–CC–O–CH2–CH–CHa, CH3O–C–CHC–O–CH2–CH–CHa), 4.27–4.06 (m, 4H, CH3O–CC–O–CH2, CH3O–C–CHC–O–CH2) 3.76 (s, 3H, CH3). 13C APT NMR (75 MHz, DMSO) δ 161.9 (CH3O–C–CHC–O–CH2–CH–OCO2, CH3O–CC–O–CH2–CH–OCO2), 153.6 (CH3O–C–CHC), 150.5 (CH3O–C), 142.1 (CH3O–C–CHC–CH2), 116.3 (CH3O–CC–CH), 104.8 (CH3O–CC–CHCH), 101.3 (CH3O–C–CH), 75.1 (CH3O–CC–O–CH2–CH), 74.9 (CH3O–C–CHC–O–CH2–CH), 69.7 (CH3O–CC–O–CH2), 67.9 (CH3O–C–CHC–O–CH2), 66.0 (CH3O–CC–O–CH2–CH–CH2), 65.9 (CH3O–C–CHC–O–CH2–CH–CH2), 55.7 (CH3).
(2-oxo-1,3-dioxolan-4-yl)methyl 3-methoxy-4-((2-oxo-1,3-dioxolan-4-yl)methoxy)benzoate (10)
A white solid was obtained (7.61 g, 20.48 mmol, 82%). Characterization data was in agreement with those reported in the literature.21H NMR (300 MHz, DMSO) δ 7.57 (dd, 1H, 4JH–H = 1.7 Hz, 3JH–H = 8.4 Hz, CH3O–CC–CHCH, 7.47 (d, 1H, 4JH–H = 1.7 Hz, CH3O–C–CH), 7.15 (d, 1H, 3JH–H = 8.5 Hz, CH3O–CC–CH), 5.19–5.17 (m, 2H, CH3O–CC–O–CH2–CH, CH3O–C–CHC–CO2–CH2–CH), 4.67–4.61 (m, 2H, CH3O–C–CHC–O–CH2–CH–CHb, CH3O–C–CHC–CO2–CH2–CH–CHb), 4.52–4.32 (m, 6H, CH3O–C–CHC–O–CH2–CH–CHa, CH3O–CC–O–CH2, CH3O–CC–O–CH2, CH3O–C–CHC–CO2–CH2–CH–CHa), 3.82 (s, 3H, CH3). 13C APT NMR (75 MHz, DMSO) δ 164.4 (CH3O–C–CHC–CO2), 154.9 (CH3O–C–CHC–O–CH2–CH–OCO2), 154.7 (CH3O–CC–CO2–CH2–CH–OCO2), 151.9 (CH3O–C–CHC–CH2), 148.8 (CH3O–C), 123.1 (CH3O–CC–CHCH), 122.1 (CH3O–C–CHC), 113.0 (CH3O–C–CH), 112.4 (CH3O–CC–CH), 74.7 (CH3O–CC–O–CH2–CH), 74.4 (CH3O–C–CHC–CO2–CH2–CH), 68.4 (CH3O–CC–O–CH2), 66.4 (CH3O–CC–O–CH2–CH–CH2), 65.9 (CH3O–C–CHC–CO2–CH2–CH–CH2), 64.3 (CH3O–C–CHC–CO2–CH2), 55.7 (CH3).
General method of the synthesis of PHUs
In a vial, equimolecular amounts of bis-amine and bis-carbonate were dissolved in DMSO (1 mL). The mixture was heated up to 80 °C for 24 h in an oil bath. Then, the mixture was allowed to cool down before being precipitated into CH2Cl2 in a falcon tube. The suspension was centrifuged and CH2Cl2 was decanted. The polymer obtained was dried under high vacuum affording a solid foam. Detailed procedures are available in the ESI.†The detailed synthetic procedures of all the compounds together with their 1H and 13C NMR analysis results are available in the ESI.†
NIPU 11
1H NMR (300 MHz, DMSO) δ 7.20–7.08 (m, 2H, NH–CO2), 6,93–6.90 (m, 2H, CH3O–C–CHC–CHCH), 6.83–6.80 (s, 1H, CH3O–CC–CHCH), 5.19 (s, 1H, CH3O–C–CHC–CH2–O–CH2–CH–OH), 4.96 (s, 1H, CH3O–CC–O–CH2–CH–CH2–OH), 4.89–4.71 (m, 1H, CH3O–CC–O–CH2–CH), 4.39 (m, 2H, CH3O–CC–CH2), 4.00–3.89 (m, 7H, CH3O–C–CHC–CH2–O–CH2–CH–CHanti–O–CONH, CH3O–CC–O–CH2–CH–CH2–OH), 3.74 (m, 3H, CH3O), 3.57, (s, 1H, CH3O–C–CHC–CH2–O–CH2–CH–OH), 2.95–2.93 (m, 4H), 1.36–1.22 (m, 8H, CH2–CH2–NH–CO2). 13C APT NMR (75 MHz, DMSO) δ 156.2 (CH3O–CC–O–CH2–CH–O–CONH), 149.0 (CH3O–CC), 147.4 (CH3O–C), 131.3 (CH3O–C–CHC), 120.0 (CH3O–CC–CHCH), 113.3 (CH3O–CC–CH), 111.9 (CH3O–C–CH), 72.2 (CH3O–CC–O–CH2), 71.3 (CH3O–C–CHC–CH2), 70.3 (CH3O–C–CHC–CH2–O–CH2–CH–CH2), 67.8 (CH3O–CC–O–CH2–CH2), 67.3 (CH3O–C–CHC–CH2–O–CH2–CH), 65.5 (CH3O–CC–O–CH2–CH2–CH–CH2), 65.1 (CH3O–C–CHC–CH2–O–CH2), 55.5 (CH3O), 40.2 (CH2–NH–CO2), 29.4 (CH2–CH2–NH–CO2), 26.0 (CH2–CH2–CH2–NH–CO2). Mn = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, ĐM = 1.50 (determined by SEC analysis in DMF against poly(styrene) (PS) standards using RI detection).
000 g mol−1, ĐM = 1.50 (determined by SEC analysis in DMF against poly(styrene) (PS) standards using RI detection).
NIPU 12
1H NMR (300 MHz, DMSO) δ 7.15–7.13 (m, 2H, NH–CO2), 6,92–6.90 (m, 2H, CH3O–C–CHC–CHCH), 6.83–6.81 (s, 1H, CH3O–CC–CHCH), 5.20 (s, 1H, CH3O–C–CHC–CH2–O–CH2–CH–OH), 4.96 (s, 1H, CH3O–CC–O–CH2–CH–CH2–OH), 4.85–4.73 (m, 1H, CH3O–C–CHC–CH2–O–CH2–CH–CHsyn–O–CONH), 4.39 (m, 2H, CH3O–CC–CH2) 4.00–3.89 (m, 7H, CH3O–C–CHC–CH2–O–CH2–CH–CHanti–O–CONH, CH3O–CC–O–CH2–CH–CH2–OH), 3.75 (m, 3H, CH3O), 3.57, (s, 1H, CH3O–C–CHC–CH2–O–CH2–CH–OH), 2.94 (m, 4H), 1.36 (m, 4H, CH2–CH2–NH–CO2). 13C APT NMR (75 MHz, DMSO) δ 156.2 (CH3O–CC–O–CH2–CH–O–CONH), 149.0 (CH3O–CC), 147.4 (CH3O–C), 131.3 (CH3O–C–CHC), 120.0 (CH3O–CC–CHCH), 113.2 (CH3O–CC–CH), 111.8 (CH3O–C–CH), 72.2 (CH3O–CC–O–CH2), 71.3 (CH3O–C–CHC–CH2), 70.3 (CH3O–C–CHC–CH2–O–CH2–CH–CH2), 67.9 (CH3O–CC–O–CH2–CH2), 67.3 (CH3O–C–CHC–CH2–O–CH2–CH), 65.5 (CH3O–CC–O–CH2–CH2–CH–CH2), 65.2 (CH3O–C–CHC–CH2–O–CH2), 55.5 (CH3O), 40.2 (CH2–NH–CO2), 28.7 (CH2–CH2–NH–CO2). Mn = 7,000 g mol−1, ĐM = 1.71 (determined by SEC analysis in DMF against poly(styrene) (PS) standards using RI detection).
NIPU 13
1H NMR (300 MHz, DMSO) δ 7.20–7.10 (m, 2H, NH–CO2), 6,85 (d, 1H, 3JH–H = 8.8 Hz, CH3O–CC–CH), 6.56 (s, 1H, CH3O–C–CH), 6.42–6.38 (m, 1H, CH3O–CC–CHCH), 5.19 (s, 1H, CH3O–CC–O–CH2–CH–CH2–OH), 5.13 (s, 1H, CH3O–C–CHC–O–CH2–CH–OH), 4.91–4.81 (m, 1H, CH3O–CC–O–CH2–CH–CH2–OH), 4.06–3.78 (m, 8H, CH3O–C–CHC–O–CH2–CH–CH2–O–CONH, CH3O–CC–O–CH2–CH–CH2–OH), 3.74 (m, 3H, CH3O), 3.57, (s, 1H, CH3O–C–CHC–O–CH2–CH–OH), 2.94 (d, 4H, 3JH–H = 6.4 Hz, CH2–NH–CO2), 1.36–1.22 (m, 8H, CH2–CH2–NH–CO2). 13C APT NMR (75 MHz, DMSO) δ 156.2 (CH3O–CC–O–CH2–CH–O–CONH), 153.6 (CH3O–C–CHC), 150.2 (CH3O–C), 142.3 (CH3O–CC), 115.2 (CH3O–CC–CH), 104.3 (CH3O–CC–CHCH), 101.1 (CH3O–C–CH), 71.2 (CH3O–C–CHC–O–CH2–CH–CH2), 69.7 (CH3O–CC–O–CH2), 67.4 (CH3O–C–CHC–O–CH2–CH), 65.1 (CH3O–CC–O–CH2–CH), 55.6 (CH3O), 40.2 (CH2–NH–CO2), 29.4 (CH2–CH2–NH–CO2), 26.0 (CH2–CH2–CH2–NH–CO2). Mn = 17000 g mol−1, ĐM = 1.76 (determined by SEC analysis in DMF against poly(styrene) (PS) standards using RI detection).
NIPU 14
1H NMR (300 MHz, DMSO) δ 7.22–7.16 (m, 2H, NH–CO2), 6,85 (d, 1H, 3JH–H = 8.7 Hz, CH3O–CC–CH), 6.56 (s, 1H, CH3O–C–CH), 6.42–6.39 (m, 1H, CH3O–CC–CHCH), 5.21 (s, 1H, CH3O–CC–O–CH2–CH–CH2–OH), 5.16 (s, 1H, CH3O–C–CHC–O–CH2–CH–OH), 4.93–4.84 (m, 1H, CH3O–CC–O–CH2–CH–CH2–OH), 4.02–3.82 (m, 8H, CH3O–C–CHC–O–CH2–CH–CH2–O–CONH, CH3O–CC–O–CH2–CH–CH2–OH), 3.74 (m, 3H, CH3O), 3.56, (s, 1H, CH3O–C–CHC–O–CH2–CH–OH), 2.95 (d, 4H, 3JH–H = 6.4 Hz, CH2–NH–CO2), 1.36 (m, 4H, CH2–CH2–NH–CO2). 13C APT NMR (75 MHz, DMSO) δ 156.2 (CH3O–CC–O–CH2–CH–O–CONH), 153.6 (CH3O–C–CHC), 150.2 (CH3O–C), 142.3 (CH3O–CC), 115.2 (CH3O–CC–CH), 104.3 (CH3O–CC–CHCH), 101.1 (CH3O–C–CH), 71.2 (CH3O–C–CHC–O–CH2–CH–CH2), 69.7 (CH3O–CC–O–CH2), 67.4 (CH3O–C–CHC–O–CH2–CH), 65.2 (CH3O–CC–O–CH2–CH), 55.6 (CH3O), 40.0 (CH2–NH–CO2), 26.8 (CH2–CH2–NH–CO2). Mn = 14000 g mol−1, ĐM = 1.86 (determined by SEC analysis in DMF against poly(styrene) (PS) standards using RI detection).
NIPU 15
1H NMR (300 MHz, DMSO) δ 7.61–7.49 (m, 2H, CH3O–C–CHC–CHCH), 7.13–7.06 (m, 3H, CH3O–C–CHC–CH, NH–CO2), 5.30 (s, 1H, CH3O–C–CHC–CO2–CH2–CH–OH), 4.94 (s, 1H, CH3O–CC–O–CH2–CH–CH2–OH), 4.19 (s, 2H, CH3O–C–CHC–CO2–CH2), 4.00 (s, 6H, CH3O–C–CHC–CO2–CH2–CH–CH2, CH3O–CC–O–CH2–CH–CH2), 3.82 (m, 3H, CH3O), 3.59, (s, 1H, CH3O–C–CHC–CO2–CH2–CH), 2.93 (s, 4H, CH2–NH–CO2), 1.35–1.21 (m, 8H, CH2–CH2–CH2–NH–CO2). 13C APT NMR (75 MHz, DMSO) δ 165.4 (CH3O–C–CHC–CO2), 156.1 (CH3O–CC–O–CH2–CH–O–CONH), 152.3 (CH3O–CC), 148.6 (CH3O–C), 122.0 (CH3O–C–CHC), 112.2 (CH3O–C–CH, CH3O–C–CHC–CHCH), 70.1 (CH3O–C–CHC–CO2–CH2–CH–CH2), 70.0 (CH3O–CC–O–CH2), 67.1 (CH3O–C–CHC–O–CH2–CH), 66.7 (CH3O–C–CHC–CO2–CH2–CH), 65.0 (CH3O–C–CHC–CO2–CH2–CH–CH2), 64.9 (CH3O–CC–O–CH2–CH–CH2), 55.7 (CH3O), 40.2 (CH2–NH–CO2), 29.4 (CH2–CH2–NH–CO2), 26.0 (CH2–CH2–CH2–NH–CO2). Mn = 13000 g mol−1, ĐM = 2.00 (determined by SEC analysis in DMF against poly(styrene) (PS) standards using RI detection).
NIPU 16
1H NMR (300 MHz, DMSO) δ 7.61–7.44 (m, 2H, CH3O–C–CHC–CHCH), 7.25–7.06 (m, 3H, CH3O–C–CHC–CH, NH–CO2), 5.33–5.29 (m, 1H, CH3O–C–CHC–CO2–CH2–CH–OH), 4.99–4.94 (s, 1H, CH3O–CC–O–CH2–CH–CH2–OH), 4.21–4.17 (s, 2H, CH3O–C–CHC–CO2–CH2), 4.00 (s, 6H, CH3O–C–CHC–CO2–CH2–CH–CH2, CH3O–CC–O–CH2–CH–CH2), 3.82 (m, 3H, CH3O), 3.59, (s, 1H, CH3O–C–CHC–CO2–CH2–CH), 2.93 (s, 4H, CH2–NH–CO2), 1.36 (s, 4H, CH2–CH2–NH–CO2). 13C APT NMR (75 MHz, DMSO) δ 165.4 (CH3O–C–CHC–CO2), 156.2 (CH3O–CC–O–CH2–CH–O–CONH), 152.3 (CH3O–CC), 148.5 (CH3O–C), 123.3 (CH3O–C–CHC–CH), 122.0 (CH3O–C–CHC), 112.1 (CH3O–C–CHC–CHCH), 70.2 (CH3O–C–CHC–CO2–CH2–CH–CH2), 70.1 (CH3O–CC–O–CH2), 67.1 (CH3O–C–CHC–CO2–CH2–CH), 66.7 (CH3O–C–CHC–O–CH2–CH), 65.7 (CH3O–C–CHC–CO2–CH2–CH–CH2), 65.0 (CH3O–CC–O–CH2–CH–CH2), 55.6 (CH3O), 40.0 (CH2–NH–CO2), 26.7 (CH2–CH2–NH–CO2). Mn = 12000 g mol−1, ĐM = 1.92 (determined by SEC analysis in DMF against poly(styrene) (PS) standards using RI detection).
Characterization
NMR spectra were recorded on a Bruker Avance 300 MHz spectrometer or Bruker Avance III HD 300 or 400 MHz spectrometer on DMSO-d6. Size exclusion chromatography (SEC) was conducted on systems composed of a Varian 390-LC-Multi detector suite fitted with differential refractive index (RI), light scattering, and ultraviolet detectors, equipped with a guard column (Varian Polymer Laboratories PLGel 5 μM, 50 × 7.5 mm) and two mixed D columns (Varian Polymer Laboratories PLGel 5 μM, 300 × 7.5 mm). The mobile phase used was dimethylformamide (DMF) with a flow rate of 1.0 mL min−1. SEC samples were calibrated against Varian Polymer Laboratories Easi-Vials linear poly(styrene) standards (103–106 g mol−1). Differential scanning calorimetry (DSC) analysis was performed using a Mettler Toledo DSC1 star system. DSC heating and cooling curves were run in duplicate from −100 °C to 200 °C in series under a nitrogen atmosphere at a heating rate of ±10 °C min−1 in a 40 μL aluminum crucible. FTIR spectra were obtained using a PerkinElmer Spectrum 100 FT-IR spectrometer. Spectra were acquired with 16 scans, spectral window of 600 to 4000 cm−1, and a resolution of 2 cm−1.Results and discussion
The aromatic aldehyde functional group of vanillin, 1, can undergo different reduction and oxidation reactions to render three different platform functionalities (Scheme 1): (a) 2-methoxyhydroquinone, 2, via a Dakin reaction,59 (b) aldehyde reduction with NaBH4 to render a vanillyl alcohol, 4, (c) aldehyde oxidation to obtain vanillic acid, 3. As vanillic acid is commercially available it was used directly. Compounds 2 and 4 were obtained with high yields (88 and 91% respectively) following procedures previously reported in the literature.43,60 Sodium percarbonate is a more sustainable oxidizing agent as compared to other analogous reagents such as peroxyacetic acid, peroxybenzoic acid or mCPBA. Glycidyl ether intermediates 5–7 were obtained with moderate to high yields (67–85%) following a previously reported procedure (Scheme 1).59 Finally, bis-cyclic carbonates 8–10 derived from vanillin were efficiently prepared (81–82% yield) from the corresponding bis-glycidyl compounds through a metal-free CO2 insertion methodology inspired by previous work from our group.56 Overall, bis-carbonate monomers 8–10 derived from vanillin were successfully prepared with overall yields ranging between 50–70% and characterized by 1H and 13C NMR spectroscopy (Scheme 1). | ||
| Scheme 1 Synthetic route for the synthesis of functional bis-carbonates derived from vanillin: (a) vanillin alcohol-based bis-carbonate, (b) hydroquinone-based bis-carbonate, and (c) vanillin acid-based bis-carbonate. | ||
After successful synthesis of three different bis-carbonate vanillin derivatives 8–10, six different PHU formulations were subsequently prepared by reacting each bis-carbonate monomer with two different bis-amines, 1,4-butanediamine (BDA) and 1,6-hexamethylenediamine (HDMA). Polymerizations were carried out using DMSO as solvent, at 80 °C, for 24 h. As compound 10 has an ester functionality, control over the reaction temperature is important in order to avoid ester aminolysis.61 Therefore, polymerizations were conducted at 80 °C to ensure the chemoselective reaction between bis-amines and bis-carbonates, leading to satisfactory formation of PHUs (Scheme 2).
 | ||
| Scheme 2 Synthesis of vanillin-based non-isocyanate poly(hydroxyurethanes): (a) vanillin alcohol-based PHU, (b) hydroquinone-based PHU, and (c) vanillin acid-based PHU. | ||
The obtained PHUs were characterized by FT-IR spectroscopy along with 1H and 13C NMR spectroscopy and SEC analysis. The thermal properties were studied using DSC analysis. 1H NMR spectroscopy confirmed the formation of the desired PHUs (Fig. 1). On the one hand, the disappearance of the α protons (δ = 5.12, 4.61 and 4.39 ppm) with respect to the carbonate functionalities was observed. On the other hand, three new characteristic signals appeared. Between δ = 7.20–7.10 ppm a peak corresponding to the proton of the new carbamate functionality appeared. In the range of δ = 5.21–5.16 ppm signals corresponding to the formation of primary and secondary alcohols were also observed. Finally, a signal around δ = 2.95 ppm appeared which was attributed to the methylene protons in the α position with respect to the –NH– moiety of the newly formed urethane functionality. In this paper the PHUs 11, 13 and 15 are based on the HDMA, while the PHUs 12, 14 and 16 are based on BDA.
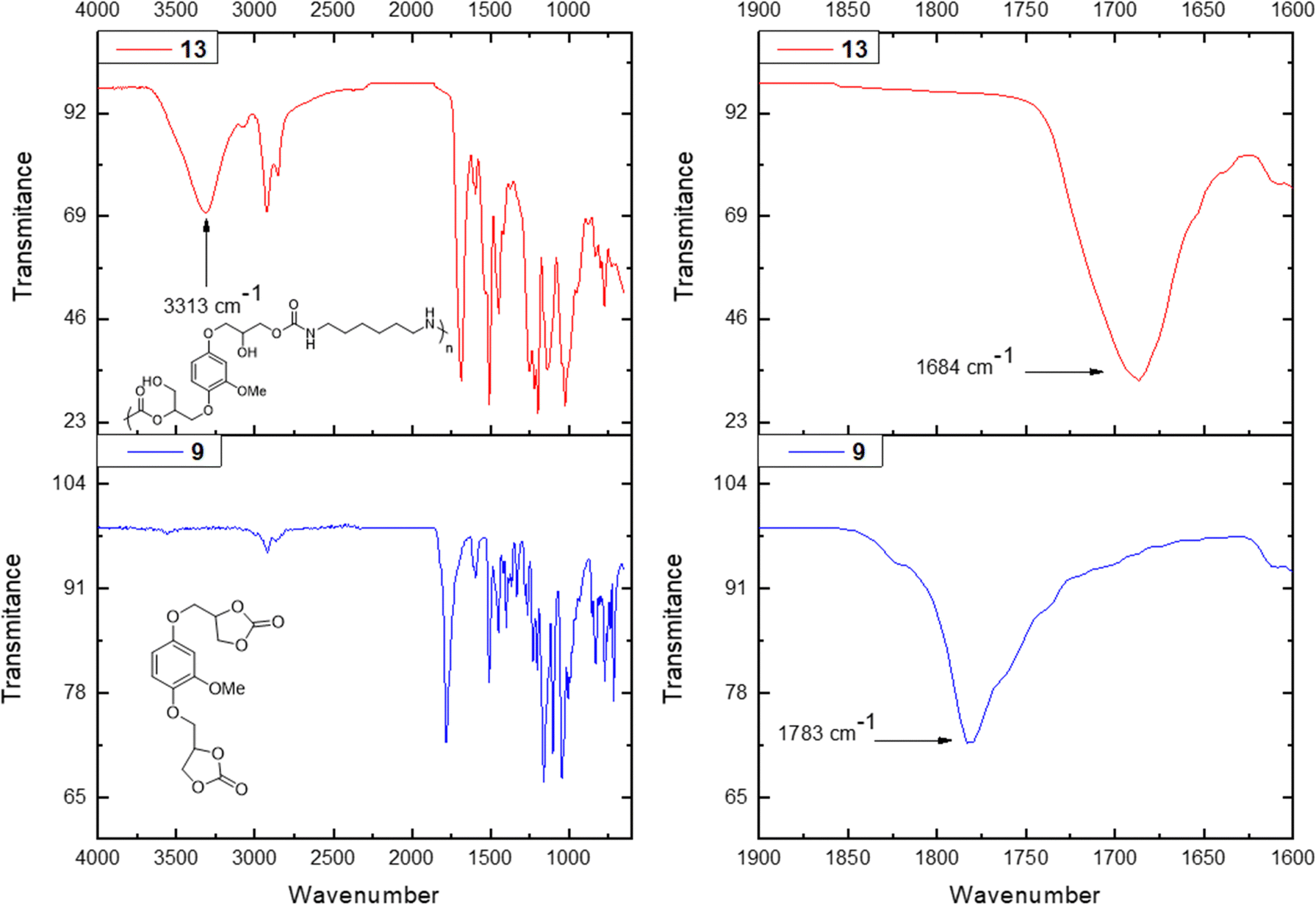 | ||
| Fig. 1 FT-IR spectrum of hydroquinone-based PHUs (13) prepared from hydroquinone-based bis-carbonate (9) (left), zoom of carbonyl stretching region of hydroquinone-based PHUs (13) prepared from hydroquinone-based bis-carbonate (9) (right). | ||
The analysis of the obtained material by FT-IR spectroscopy further confirmed the success of the polymerization (Fig. 1). In all examples the same trend was observed, the peak corresponding to the carbonate functionality of each monomer (i.e. based on vanillyl alcohol, 1779 cm−1, hydroquinone, 1783 cm−1, vanillic acid, 1779 cm−1 and 1798 cm−1) disappeared and two new signals appeared. Broad signals around 3300 cm−1 were observed, which were attributed to the newly formed primary and secondary alcohol functionalities in the PHU and the –NH– stretching vibration in the carbamate moiety. A second signal appeared around 1684–1689 cm−1 depending on the starting material, which corresponds to the CO deformation vibration in the formed carbamate moiety.
Molecular weight distributions were determined by SEC using DMF as solvent (Table 1). Molecular weight (Mn) values obtained were between 7.0–20.0 kDa and dispersities (ĐM) were between 1.50–2.00, as expected for step growth polymerizations (Fig. 2). Thermal characterization performed by DSC showed a glass transition temperature (Tg) in all formulations in a range between 41–66 °C (Fig. 3). The fact that the PHUs present similar values of Tg when comparing those obtained with butylamine and hexylamine, supports the idea that they have molar mass within the same order of magnitude, although SEC results obtained by calibration relative to PS standards show moderately lower molar mass for PHU12 (vanillyl alcohol/butylamine-based). This may be due to a distinct lower hydrodynamic volume-conformation that PHU12 adopts when in DMF solution when compared to the other PHU or to a small stoichiometry imbalance. Further studies are required to confirm the impact of the structure in the SEC results.
| Vanillin precursor | Entry | PHU | M w (kg mol−1)a | M n (kg mol−1)a | Đ M (Mw/Mn)a | T g (°C)b |
|---|---|---|---|---|---|---|
| a Determined by SEC analysis in DMF against poly(styrene) (PS) standards. b Thermal transition calculated from the second heating run of the DSC analysis. | ||||||
| Vanillyl alcohol | 1 | 11 | 30 | 20 | 1.50 | 41 |
| 2 | 12 | 12 | 7 | 1.71 | 41 | |
| Hydroquinone | 3 | 13 | 30 | 17 | 1.76 | 50 |
| 4 | 14 | 26 | 14 | 1.86 | 52 | |
| Vanillic acid | 5 | 15 | 26 | 13 | 2.00 | 60 |
| 6 | 16 | 23 | 12 | 1.92 | 66 | |
 | ||
| Fig. 2 GPC traces of the different PHUs obtained from functionalized vanillin bis-carbonates. | ||
 | ||
| Fig. 3 DSC curves (2nd heating scan) of the different PHUs obtained from functionalized vanillin bis-carbonates. | ||
The PHUs derived from hydroquinone showed a lower Tg compared with those derived from vanillic acid (Table 1, entries 4 and 6), which presented the highest Tg between 60–66 °C. On the other hand, PHUs based on vanillyl alcohol bis-carbonate exhibited the lowest Tg values (Table 1, entries 1 and 2), of 41 °C. The presence of the ester functionality in the vanillic acid PHUs confers a rotational hindrance not found on the hydroquinone and vanillyl alcohol PHUs.62–65 As a possible resonance structure the electron rich aromatic ring can donate electron density to the ester bond, thus favoring its planarity with respect to the aromatic ring. This favors π-interactions and therefore increases the Tg. The same effect is not found in the other PHUs, in which the aromatic ring of the hydroquinone (13, 14) and vanillyl alcohol (11, 12) possess a higher degree of rotational freedom. The higher Tg of the hydroquinone-based PHUs (13, 14) with respect to the vanillin alcohol-based PHUs (11, 12) is probably due to the methanolic group attached to the aromatic ring of the vanillin alcohol, which increases the mobility of the polymer chain.
Vanillin based PHUs can represent a safer alternative to the widely used bisphenol A-based polyurethanes. Although both compounds possess a substituted aromatic ring and therefore their thermal properties are expected to be similar to some extent, bisphenol A is a known endocrine disruptor and it is suspected to produce harmful effects on the reproductive system.61 To look into the potential for replacement of bisphenol A with vanillin derivatives, the thermal properties reported above were compared to those of a comparative bisphenol A-based PHU. In the case of the bisphenol A material, the observed Tg was 56 °C. This is lower than the Tg values of the vanillin-based PHUs synthesized using vanillic acid as precursor (Table 1, entries 5 and 6), which suggests that these bio-based PHUs can be used in similar application areas.
Conclusions
To the best of our knowledge, this work provides the first example of a vanillin-based bis-cyclic carbonate for the synthesis of bio-based non-isocyanate poly(hydroxy urethane)s (PHUs). Bis-cyclic carbonates were prepared using a metal-free methodology, which also represents an efficient way to revalorize CO2. The thermal properties of the PHUs were studied by DSC and subsequently compared to an analogous bisphenol A-based PHU. The PHUs prepared using bis-cyclic carbonates derived from vanillic acid as monomer exhibited superior thermal properties in comparison to the bisphenol A-based PHU. This result demonstrates that vanillin-based PHUs may be used to provide a safer alternative to bisphenol A-based polyurethanes.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge financial support from the European Commission through SUSPOL-EJD 642671 project. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 101034379. The authors acknowledge grant TED2021-129852B-C22 funded by MCIN/AEI/10.13039/501100011033 and by the European Union NextGenerationEU/PRTR.References
- N. Polyurethane, E. Delebecq, J. P. Pascault, B. Boutevin, U. De Lyon and F. Ganachaud, On the Versatility of Urethane/Urea Bonds: Reversibility, Blocked Isocyanate, and Non-Isocyanate Polyurethane, Chem. Rev., 2013, 113(1), 80–118, DOI:10.1021/cr300195n.
- Z. S. Petrovíc and J. Ferguson, Polyurethane Elastomers, Prog. Polym. Sci., 1991, 16, 695–836, DOI:10.1007/978-94-009-1531-2_60.
- H. W. Engels, H. G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Polyurethanes: Versatile Materials and Sustainable Problem Solvers for Today's Challenges, Angew. Chem., Int. Ed., 2013, 52(36), 9422–9441, DOI:10.1002/anie.201302766.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, Polyurethane Types, Synthesis and Applications-a Review, RSC Adv., 2016, 6(115), 114453–114482, 10.1039/c6ra14525f.
- M. B. Lowinger, S. E. Barrett, F. Zhang and R. O. Williams, Sustained Release Drug Delivery Applications of Polyurethanes, Pharmaceutics, 2018, 10(2), 1–19, DOI:10.3390/pharmaceutics10020055.
- F. E. Golling, R. Pires, A. Hecking, J. Weikard, F. Richter, K. Danielmeier and D. Dijkstra, Polyurethanes for Coatings and Adhesives – Chemistry and Applications, Polym. Int., 2019, 68(5), 848–855, DOI:10.1002/pi.5665.
- L. Polo Fonseca and M. I. Felisberti, Thermo- and UV-Responsive Amphiphilic Nanogels via Reversible [4 + 4] Photocycloaddition of PEG/PCL-Based Polyurethane Dispersions, Eur. Polym. J., 2021, 160(September), 110800, DOI:10.1016/j.eurpolymj.2021.110800.
- L. Polo Fonseca, R. B. Trinca and M. I. Felisberti, Amphiphilic Polyurethane Hydrogels as Smart Carriers for Acidic Hydrophobic Drugs, Int. J. Pharm., 2018, 546(1–2), 106–114, DOI:10.1016/j.ijpharm.2018.05.034.
- B. Otto, Das Di-Lsocganat-Poluadditionsverfahren (Polyurethane), Angew. Chemie, 1947, 59(9), 257–288 CrossRef.
- A. J. Bloodworth and A. G. Davies, 975. Organometallic Reactions. Part I. The Addition of Tin Alkoxides to Isocyanates, J. Chem. Soc., 1965, 433(5238), 5238–5244, 10.1039/JR9650005238.
- V. R. Dhara and R. Dhara, The Union Carbide Disaster in Bhopal: A Review of Health Effects, Arch. Environ. Health, 2002, 57(5), 391–404, DOI:10.1080/00039890209601427.
- N. Fanjul-Mosteirín, R. Aguirresarobe, N. Sadaba, A. Larrañaga, E. Marin, J. Martin, N. Ramos-Gomez, M. C. Arno, H. Sardon and A. P. Dove, Crystallization-Induced Gelling as a Method to 4D Print Low-Water-Content Non-Isocyanate Polyurethane Hydrogels, Chem. Mater., 2021, 33(18), 7194–7202, DOI:10.1021/acs.chemmater.1c00913.
- A. Hernández, H. A. Houck, F. Elizalde, M. Guerre, H. Sardon and F. E. Du Prez, Internal Catalysis on the Opposite Side of the Fence in Non-Isocyanate Polyurethane Covalent Adaptable Networks, Eur. Polym. J., 2022, 168, 111100, DOI:10.1016/j.eurpolymj.2022.111100.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Non-Isocyanate Polyurethanes: Synthesis, Properties, and Applications, Polym. Adv. Technol., 2015, 26(7), 707–761, DOI:10.1002/pat.3522.
- B. Bizet, B. Bizet, É. Grau, H. Cramail and J. M. Asua, Water-Based Non-Isocyanate Polyurethane-Ureas (NIPUUs), Polym. Chem., 2020, 11(23), 3786–3799, 10.1039/d0py00427h.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, Non-Isocyanate Polyurethanes: From Chemistry to Applications, RSC Adv., 2013, 3(13), 4110–4129, 10.1039/c2ra21938g.
- H. Blattmann, M. Fleischer, M. Bähr and R. Mülhaupt, Isocyanate- and Phosgene-Free Routes to Polyfunctional Cyclic Carbonates and Green Polyurethanes by Fixation of Carbon Dioxide, Macromol. Rapid Commun., 2014, 35(14), 1238–1254, DOI:10.1002/marc.201400209.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Isocyanate-Free Routes to Polyurethanes and Poly(Hydroxy Urethane)S, Chem. Rev., 2015, 115(22), 12407–12439, DOI:10.1021/acs.chemrev.5b00355.
- Y. Ma, Y. Xiao, Y. Zhao, Y. Bei, L. Hu, Y. Zhou and P. Jia, Biomass Based Polyols and Biomass Based Polyurethane Materials as a Route towards Sustainability, React. Funct. Polym., 2022, 175, 105285, DOI:10.1016/j.reactfunctpolym.2022.105285.
- G. Fiorani, W. Guo and A. W. Kleij, Sustainable Conversion of Carbon Dioxide: The Advent of Organocatalysis, Green Chem., 2015, 17(3), 1375–1389, 10.1039/c4gc01959h.
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Organocatalyzed Coupling of Carbon Dioxide with Epoxides for the Synthesis of Cyclic Carbonates: Catalyst Design and Mechanistic Studies, Catal. Sci. Technol., 2017, 7(13), 2651–2684, 10.1039/c7cy00438a.
- R. R. Shaikh, S. Pornpraprom and V. D’Elia, Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions, ACS Catal., 2018, 8(1), 419–450, DOI:10.1021/acscatal.7b03580.
- R. Dalpozzo, N. D. Ca, B. Gabriele and R. Mancuso, Recent Advances in the Chemical Fixation of Carbon Dioxide: A Green Route to Carbonylated Heterocycle Synthesis, Catalysts, 2019, 9(6), 11, DOI:10.3390/catal9060511.
- T. Weidlich and B. Kamenická, Utilization of CO2 Available Organocatalysts for Reactions with Industrially Important Epoxides, Catalysts, 2022, 12(3), 298, DOI:10.3390/catal12030298.
- T. Yan, H. Liu, Z. X. Zeng and W. G. Pan, Recent Progress of Catalysts for Synthesis of Cyclic Carbonates from CO2and Epoxides, J. CO2 Util., 2023, 68, 102355, DOI:10.1016/j.jcou.2022.102355.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, The Path Forward for Biofuels and Biomaterials, Renew. Energy Four Vol. Set, 2018, 3–4, 271–283 Search PubMed.
- A. Demirbas, Progress and Recent Trends in Biofuels, Prog. Energy Combust. Sci., 2007, 33(1), 1–18, DOI:10.1016/j.pecs.2006.06.001.
- M. Höök and X. Tang, Depletion of Fossil Fuels and Anthropogenic Climate Change-A Review, Energy Policy, 2013, 52, 797–809, DOI:10.1016/j.enpol.2012.10.046.
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Biobased Amines: From Synthesis to Polymers; Present and Future, Chem. Rev., 2016, 116(22), 14181–14224, DOI:10.1021/acs.chemrev.6b00486.
- M. Janvier, P. H. Ducrot and F. Allais, Isocyanate-Free Synthesis and Characterization of Renewable Poly(Hydroxy)Urethanes from Syringaresinol, ACS Sustain. Chem. Eng., 2017, 5(10), 8648–8656, DOI:10.1021/acssuschemeng.7b01271.
- A. Agrawal, R. Kaur and R. S. Walia, PU Foam Derived from Renewable Sources: Perspective on Properties Enhancement: An Overview, Eur. Polym. J., 2017, 95, 255–274, DOI:10.1016/j.eurpolymj.2017.08.022.
- L. Zhang, X. Luo, Y. Qin and Y. Li, A Novel 2,5-Furandicarboxylic Acid-Based Bis(Cyclic Carbonate) for the Synthesis of Biobased Non-Isocyanate Polyurethanes, RSC Adv., 2017, 7(1), 37–46, 10.1039/c6ra25045a.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, From the Synthesis of Biobased Cyclic Carbonate to Polyhydroxyurethanes: A Promising Route towards Renewable Non-Isocyanate Polyurethanes, ChemSusChem, 2019, 12(15), 3410–3430, DOI:10.1002/cssc.201900737.
- F. Magliozzi, G. Chollet, E. Grau and H. Cramail, Benefit of the Reactive Extrusion in the Course of Polyhydroxyurethanes Synthesis by Aminolysis of Cyclic Carbonates, ACS Sustain. Chem. Eng., 2019, 7(20), 17282–17292, DOI:10.1021/acssuschemeng.9b04098.
- M. Ghasemlou, F. Daver, E. P. Ivanova and B. Adhikari, Bio-Based Routes to Synthesize Cyclic Carbonates and Polyamines Precursors of Non-Isocyanate Polyurethanes: A Review, Eur. Polym. J., 2019, 118, 668–684, DOI:10.1016/j.eurpolymj.2019.06.032.
- J. Brzeska and A. Piotrowska-Kirschling, A Brief Introduction to the Polyurethanes According to the Principles of Green Chemistry, Processes, 2021, 9(11), 1929, DOI:10.3390/pr9111929.
- A. Gandini and T. M. Lacerda, Monomers and Macromolecular Materials from Renewable Resources: State of the Art and Perspectives, Molecules, 2022, 27(1), 1–26, DOI:10.3390/molecules27010159.
- L. Polo Fonseca, W. R. Waldman and M. A. De Paoli, ABS Composites with Cellulose Fibers: Towards Fiber-Matrix Adhesion without Surface Modification, Compos. Part C Open Access, 2021, 5, 100142, DOI:10.1016/j.jcomc.2021.100142.
- S. Luo, J. Cao and W. Sun, Evaluation of Kraft Lignin as Natural Compatibilizer in Wood Flour/Polypropylene Composites, Polym. Compos., 2017, 38(11), 2387–2394, DOI:10.1002/pc.23821.
- H. Ludmila, J. Michal, Š. Andrea and H. Aleš, Lignin, Potential Products and Their Market Value, Wood Res., 2015, 60(6), 973–986 Search PubMed.
- S. Laurichesse and L. Avérous, Chemical Modification of Lignins: Towards Biobased Polymers, Prog. Polym. Sci., 2014, 39(7), 1266–1290, DOI:10.1016/j.progpolymsci.2013.11.004.
- P. Varanasi, P. Singh, M. Auer, P. D. Adams, B. A. Simmons and S. Singh, Survey of Renewable Chemicals Produced from Lignocellulosic Biomass during Ionic Liquid Pretreatment, Biotechnol. Biofuels, 2013, 6, 16, DOI:10.1186/1754-6834-6-14.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Vanillin, a Promising Biobased Building-Block for Monomer Synthesis, Green Chem., 2014, 16(4), 1987–1998, 10.1039/c3gc42613k.
- A. Salanti, L. Zoia and M. Orlandi, Chemical Modifications of Lignin for the Preparation of Macromers Containing Cyclic Carbonates, Green Chem., 2016, 18(14), 4063–4072, 10.1039/c6gc01028h.
- M. Fache, R. Auvergne, B. Boutevin and S. Caillol, New Vanillin-Derived Diepoxy Monomers for the Synthesis of Biobased Thermosets, Eur. Polym. J., 2015, 67, 527–538, DOI:10.1016/j.eurpolymj.2014.10.011.
- Z. Fang, M. C. Weisenberger and M. S. Meier, Utilization of Lignin-Derived Small Molecules: Epoxy Polymers from Lignin Oxidation Products, ACS Appl. Bio Mater., 2020, 3(2), 881–890, DOI:10.1021/acsabm.9b00954.
- Z. Wang, P. Gnanasekar, S. Sudhakaran Nair, R. Farnood, S. Yi and N. Yan, Biobased Epoxy Synthesized from a Vanillin Derivative and Its Reinforcement Using Lignin-Containing Cellulose Nanofibrils, ACS Sustain. Chem. Eng., 2020, 8(30), 11215–11223, DOI:10.1021/acssuschemeng.0c02559.
- A. S. Mora, R. Tayouo, B. Boutevin, G. David and S. Caillol, Vanillin-Derived Amines for Bio-Based Thermosets, Green Chem., 2018, 20(17), 4075–4084, 10.1039/c8gc02006j.
- E. D. Hernandez, A. W. Bassett, J. M. Sadler, J. J. La Scala and J. F. Stanzione, Synthesis and Characterization of Bio-Based Epoxy Resins Derived from Vanillyl Alcohol, ACS Sustain. Chem. Eng., 2016, 4(8), 4328–4339, DOI:10.1021/acssuschemeng.6b00835.
- L. Vlaminck, S. Lingier, A. Hufendiek and F. E. Du Prez, Lignin Inspired Phenolic Polyethers Synthesized via ADMET: Systematic Structure-Property Investigation, Eur. Polym. J., 2017, 95, 503–513, DOI:10.1016/j.eurpolymj.2017.08.042.
- E. A. Baroncini, D. M. Rousseau, C. A. Strekis and J. F. Stanzione, Optimizing Conductivity and Cationic Transport in Crosslinked Solid Polymer Electrolytes, Solid State Ionics, 2020, 345, 115161, DOI:10.1016/j.ssi.2019.115161.
- R. B. Trinca and M. I. Felisberti, Effect of Diisocyanates and Chain Extenders on the Physicochemical Properties and Morphology of Multicomponent Segmented Polyurethanes Based on Poly(l-Lactide), Poly(Ethylene Glycol) and Poly(Trimethylene Carbonate), Polym. Int., 2015, 64(10), 1326–1335, DOI:10.1002/pi.4920.
- R. B. Trinca and M. I. Felisberti, Segmented Polyurethanes Based on Poly(l-Lactide), Poly(Ethylene Glycol) and Poly(Trimethylene Carbonate): Physico-Chemical Properties and Morphology, Eur. Polym. J., 2015, 62, 77–86, DOI:10.1016/j.eurpolymj.2014.11.008.
- J. Ke, X. Li, S. Jiang, C. Liang, J. Wang, M. Kang, Q. Li and Y. Zhao, Promising Approaches to Improve the Performances of Hybrid Non-Isocyanate Polyurethane, Polym. Int., 2019, 68(4), 651–660, DOI:10.1002/pi.5746.
- Y. Ecochard and S. Caillol, Hybrid Polyhydroxyurethanes: How to Overcome Limitations and Reach Cutting Edge Properties, Eur. Polym. J., 2020, 137, 109915, DOI:10.1016/j.eurpolymj.2020.109915.
- N. Fanjul-Mosteirín, C. Jehanno, F. Ruipérez, H. Sardon and A. P. Dove, Rational Study of DBU Salts for the CO2 Insertion into Epoxides for the Synthesis of Cyclic Carbonates. ACS Sustain, Chem. Eng., 2019, 7(12), 10633–10640, DOI:10.1021/acssuschemeng.9b01300.
- A. Yuen, A. Bossion, E. Gómez-bengoa, F. Ruipérez, M. Isik, J. L. Hedrick, D. Mecerreyes, Y. Y. Yang and H. Sardon, Polymer Chemistry Polyurethanes (NIPUs) Using Highly Reactive, Polym. Chem., 2016, 7, 2105–2111, 10.1039/c6py00264a.
- A. Bossion, G. O. Jones, D. Taton, D. Mecerreyes, J. L. Hedrick, Z. Y. Ong, Y. Y. Yang and H. Sardon, Non-Isocyanate Polyurethane Soft Nanoparticles Obtained by Surfactant-Assisted Interfacial Polymerization, Langmuir, 2017, 33(8), 1959–1968, DOI:10.1021/acs.langmuir.6b04242.
- G. W. Kabalka, N. K. Reddy and C. Narayana, Sodium Percarbonate: A Convenient Reagent for the Dakin Reaction, Tetrahedron Lett., 1992, 33(7), 865–866, DOI:10.1016/S0040-4039(00)91561-2.
- Z. Lu, C. Hu, J. Guo, J. Li, Y. Cui and Y. Jia, Water-Controlled Regioselectivity of Pd-Catalyzed Domino Reaction Involving a C-H Activation Process: Rapid Synthesis of Diverse Carbo- And Heterocyclic Skeletons, Org. Lett., 2010, 12(3), 480–483, DOI:10.1021/ol902672a.
- F. Camara, S. Benyahya, V. Besse, G. Boutevin, R. Auvergne, B. Boutevin and S. Caillol, Reactivity of Secondary Amines for the Synthesis of Non-Isocyanate Polyurethanes, Eur. Polym. J., 2014, 55(1), 17–26, DOI:10.1016/j.eurpolymj.2014.03.011.
- E. Bisz, A. Piontek, B. Dziuk, R. Szostak and M. Szostak, Barriers to Rotation in Ortho-Substituted Tertiary Aromatic Amides: Effect of Chloro-Substitution on Resonance and Distortion, J. Org. Chem., 2018, 83(6), 3159–3163, DOI:10.1021/acs.joc.8b00019.
- X. Yu, J. Jia, S. Xu, K. U. Lao, M. J. Sanford, R. K. Ramakrishnan, S. I. Nazarenko, T. R. Hoye, G. W. Coates and R. A. DiStasio, Unraveling Substituent Effects on the Glass Transition Temperatures of Biorenewable Polyesters, Nat. Commun., 2018, 9(1), 2880, DOI:10.1038/s41467-018-05269-3.
- T. Drakenberg, J. Sommer and R. Jost, The Torsional Barrier in Aromatic Carbonyl Compounds, J. Chem. Soc. Perkin Trans. 2, 1980, 11(2), 363–369, 10.1039/p29800000363.
- R. Mattes and E. G. Rochow, Internal Motion in Some Aromatic Polyesters and Linear Aromatic Chains, J. Polym. Sci. Part A-2 Polym. Phys., 1966, 4(3), 375–384, DOI:10.1002/pol.1966.160040308.
Footnote |
| † Electronic supplementary information (ESI) available: Complementary procedures and 1H and 13C NMR spectra (PDF). See DOI: https://doi.org/10.1039/d3ma00111c |
| This journal is © The Royal Society of Chemistry 2023 |