DOI:
10.1039/D3MA00121K
(Paper)
Mater. Adv., 2023,
4, 4436-4443
Degradation suppression effect of amorphous-hard-carbon-bundled Si-based negative electrode†
Received
14th March 2023
, Accepted 25th August 2023
First published on 30th August 2023
Abstract
To improve the electrode calendar life of high-capacity Si, a non-core–shell type Si negative electrode (hard-carbon-bundled Si, HCS) was investigated for preventing volume changes and particle breakage of nano-sized Si particles. This modified electrode, which consists of hard-carbon (HC)-bundled multiple nano-sized Si particles, prevents direct contact between Si and a liquid electrolyte and enables suppression of physical changes to the core HC. In this study, HCS samples were prepared at various carbonization temperatures and their physical and electrochemical properties were also evaluated. The charge–discharge properties of electrochemical cells with a HCS/C6 mixed negative electrode gave adequate cycling performances and coulombic efficiencies, i.e. almost flat capacity retention and more than 99% coulombic efficiency over 100 cycles. The effects of the HCS carbonization temperature on the charge–discharge properties were investigated by determining the reaction electrode potential of the HCS. The physical and electrochemical properties of the HC phase in the HCS particle surface contributed to the charge–discharge capacity and good cycling properties. The reactivity of the nano-Si in HC effectively suppressed electrochemical cell degradation. The developed HCS is an appropriate material for protecting negative electrodes against significant volume changes in the Si and improving their electrochemical performances.
1. Introduction
Lithium-ion batteries (LIBs), which are energy-storage devices with high voltages and high energy densities, are widely used in portable electronic devices and power tools. The development of innovative batteries with performances that exceed those of LIBs for widespread use in areas such as renewable energy and electric vehicles is important.1,2 The use of alloys based on materials such as Si, Ge, and Sn as the negative electrodes in high-performance secondary batteries is being actively investigated because the capacities of such materials are expected to be considerably higher than those of currently used graphite-based materials.3 A Si negative electrode has a low electrode potential, and one Si atom is estimated to absorb and release up to 4.4 Li ions, with a theoretical capacity of 4200 mA h g−1.4–6 The capacity of elemental Si is approximately 10 times higher than that of C6, which is the carbon-based negative electrode material conventionally used in LIBs, and its use is expected to improve the cell capacity. Even use of a small amount of Si can give huge capacity differences between Si and other electrodes.7–10 Furthermore, Si is produced by reducing and purifying SiO2, which is abundant in the Earth's crust. Si has the second-highest Clarke number (existence ratio: 25.8) and is considered to be a promising negative electrode material for storage batteries, which are expected to be produced in increasing quantities in the future. However, the change in the volume of a Si negative electrode (approximately 300%), which arises from alloying-dealloying of Li ions, causes pulverization. This leads to blockage of the conducting paths and an increase in the reaction area. Consequently, the cycling performance is significantly decreased by unstable interface formation and continuous growth of an interphase layer.5,11–13 The addition of vinylene carbonate (VC) or fluoroethylene carbonate (FEC) to the electrolyte solution, which leads to formation of a stable surface film and improves the cell life, has been used to deal with these problems.14–17 However, these chemically formed films do not have sufficient uniformity and physical strength, and the additive becomes depleted during repeated charge–discharge cycling. The use of additives therefore promotes film formation but the battery capacity decreases in the medium to long term because of structural degradation such as unstable film formation caused by a significant change in the Si volume. Use of nanostructured Si materials significantly suppresses volume changes, and satisfactory long-term performances have been reported. This is attributable to charge–discharge operation over short Li-ion diffusion distances and suppression of structural degradation.18–22 However, a disadvantage of nanostructured Si materials is that their increased surface area promotes side reactions with the electrolyte, which leads to unstable film formation and high interparticle resistance.23–25 In this study, we investigated the use of a shell structure consisting of fine Si particles bundled with a carbon material and void control of the morphological structure to suppress changes in the Si volume and ensure stable electrical conductivity. Carbon structures are resistant to volume changes during charging and discharging, and are therefore important in this proposed method. The design of structures (core–shell-type electrode particle) with high physical stability is desirable.26–34 We developed a non-core–shell Si-based negative electrode material, i.e., hard-carbon-bundled Si (HCS), with large particles; it consists of multiple Si nanoparticles restrained by hard carbon (HC; Fig. 1). The use of HCS avoids direct contact between Si particles and the electrolyte, therefore side reactions originating at the electrode/electrolyte interface are minimized. The use of HC derived from a phenolic resin precursor to physically suppress the volume change of the entire electrode and provide high electrical conductivity was investigated. Moreover, HCS particles can be designed to target sizes such as the submicron to micrometer level, as well as for the case of simple HC negative electrodes. HCS preparation is simple and cheap because it does not involve a complex, multi-step process. We report the physical properties of the HCS and the results of electrochemical investigation of the effects of HCS through charge–discharge measurements for [Li metal|HCS] cells.
 |
| | Fig. 1 Schematic diagram of charge–discharge reactions of Si in HCS particles. | |
2. Experimental
2.1 Preparation of Si-based electrode materials
A kneader (Labo Plastomill R30, Toyo Seiki Seisaku-sho, Ltd) was preheated to 343 K. Resol phenolic resin (20 g, PR-X13437, Sumitomo Bakelite Co., Ltd) was added under stirring at 50 rpm to obtain a uniform melt. Crystalline Si powder (2.8 g, average particle size <50 nm, laser synthesized from the vapor phase, Alfa Aesar) was added, and stirring was continued until a gel was formed. The obtained composite material was calcined at 787 K for 1.5 h at a heating rate of 100 K h−1 in a N2 atmosphere, and then pulverized to obtain the carbon precursor. The precursor was heated at a rate of 10 K h−1 from 298 K in a N2 atmosphere to two target temperatures (1273 or 1373 K) and held at these temperatures for 6 h to obtain the HCS samples. The samples obtained by carbonization at 1273 and 1373 K are denoted by HCS-1 and HCS-2, respectively. Simple HC materials without Si powder, denoted by HC-1 and HC-2, were prepared under the same temperature conditions.
2.2 Physical properties of Si-based electrode materials
The physical properties of the prepared hybrid carbon–Si electrode materials (HCS) were investigated by using scanning electron microscopy/energy-dispersive X-ray spectroscopy (SEM-EDX), field-emission transmission electron microscopy (FE-TEM), thermogravimetry/differential thermal analysis (TG-DTA), X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET) specific surface area measurements, and Raman spectroscopy. The surface morphologies and elemental mappings of the HCS samples were investigated by using SEM-EDX (JCM-6000/EX-37001, JEOL). The dispersion of Si nanoparticles in the HCS samples was observed by FE-TEM (HF-2200, Hitachi High-Tech Science) after thin-film processing with a focused ion beam fabrication and observation system (FB-2200, Hitachi High-Tech Science). The Si contents of the HCS samples were determined by TG-DTA (STA7200RV, Hitachi High-Tech Science); experiments were performed from 303 to 1073 K at a heating rate of 10 K min−1. The crystal structures of the HCS samples were identified by XRD (MiniFlex600, Rigaku) with Cu Kα radiation (λ = 0.15418 nm) in the scanning range 10°–90°, at a scanning rate of 3° min−1, applied voltage of 40 kV, and current of 15 mA. The surface chemical states of the HCS samples were also investigated by Raman spectroscopy (NRS-4500, Jasco) at 3.1 mW and 532 nm; the optical resolution was approximately 2.3 cm−1. The BET specific surface areas of the HCS samples were evaluated by using a specific surface area measuring device (BELSORP-MAX, MicrotracBEL) at 77 K and by the Barrett–Joyner–Halenda method after pretreatment of the HCS samples for 6 h under reduced pressure, i.e., 2 kPa.
2.3 Preparation and performance evaluation of [Li metal|HCS] cells
Electrode sheets were prepared by mixing the active material [HCS or HC (C6, SCMG-BH, Showa Denko), acetylene black (Denka Black Li-100, Denka), and poly(vinylidene difluoride) (PVDF: Kureha). Table 1 lists the mass ratios of the electrode materials. The electrode materials were dissolved in N-methylpyrrolidone and mixed in a paste mixer to form a homogeneous slurry. The obtained slurries were coated on Cu current collectors and dried in an oven at 353 K for 12 h. The electrode sheets were compressed to increase the packing density and improve the electrical conductivity (approximate loading masses 30 wt% HCS, 50 wt% HCS, 70 wt% HCS, 100 wt% HCS, and 100 wt% HC: 1.29–1.31, 0.33–0.44, 0.62–0.66, 0.28–1.47, and 0.49–0.66 mg cm−2, respectively). [Li|electrolyte|HCS or HC electrode sheet] cells were prepared by placing the electrode sheet, a glass separator (thickness 0.21 mm, GA-55, Advantec), ethylene carbonate (EC)/diethyl carbonate (DEC) = 3/7 by mass, 1.0 M LiN(SO2F)2 (LiFSI) electrolyte solution, and Li metal (Honjo Metal) in a 2032-type coin cell. All cell-preparation processes were performed in an Ar-filled glovebox ([O2] < 10 ppm, dewpoint < 193 K; Miwa Mfg Co., Ltd) Constant current charge–discharge measurements were performed in galvanostatic mode using a charge–discharge measurement system (HJ1020mSD8, Hokuto Denko) at 0–2.5 V and a current density of approximately 50 μA cm−2 at 303 K. At least three cells were prepared for each electrochemical measurement; their properties were evaluated using the average values obtained, with their error bars.
Table 1 Relationship between HCS mixing ratio and weight ratio (χ) for electrode slurries in HCS/C6 electrodes
| Mixing ratio of HCS χ [wt%] |
30 |
50 |
70 |
100 |
| HCS |
21.75 |
31.25 |
43.75 |
62.5 |
| C6 |
50.75 |
31.25 |
18.75 |
— |
| Acetylene black |
12.8 |
17.5 |
17.5 |
17.5 |
| PVDF |
14.7 |
20 |
20 |
20 |
3. Results and discussion
3.1 Characterization of Si-based electrode materials
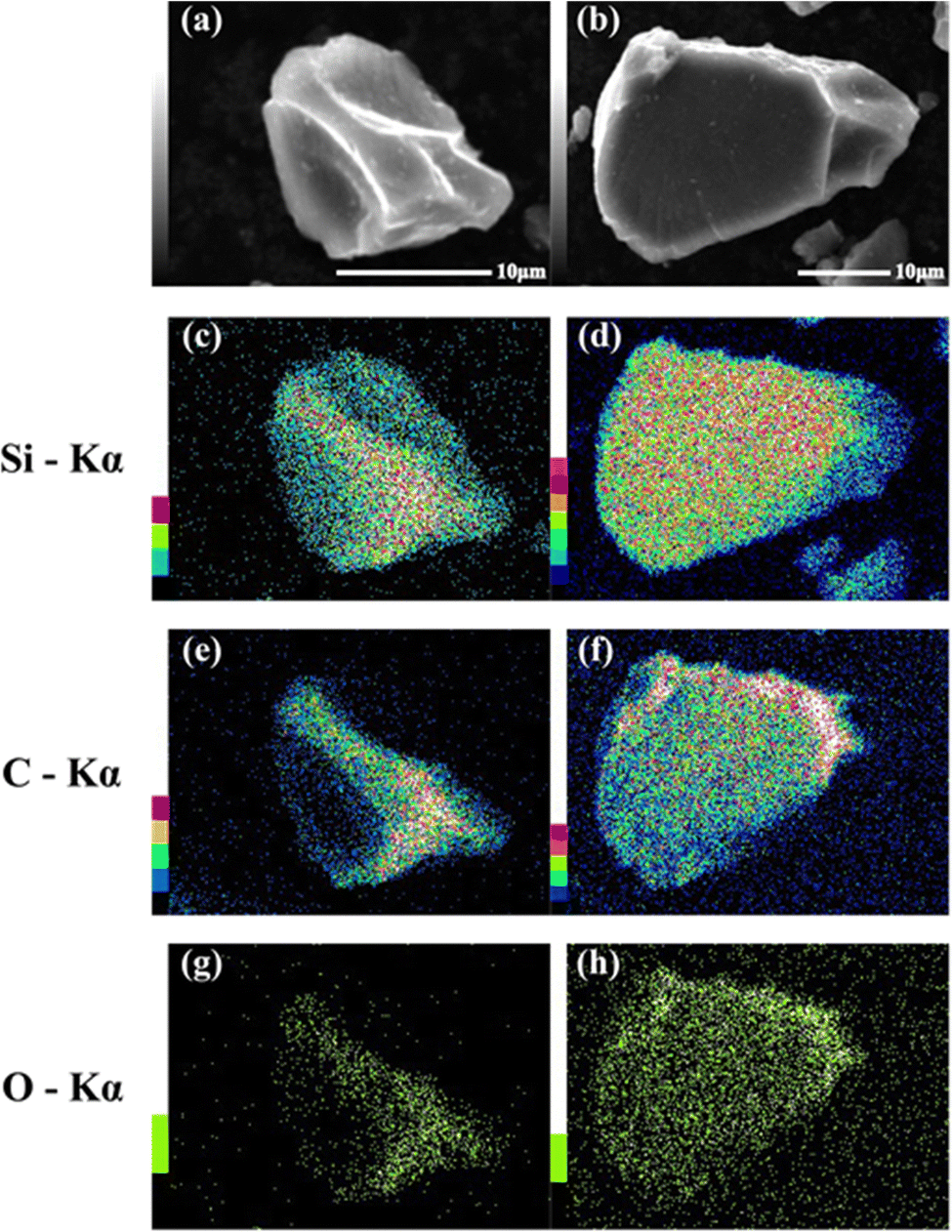
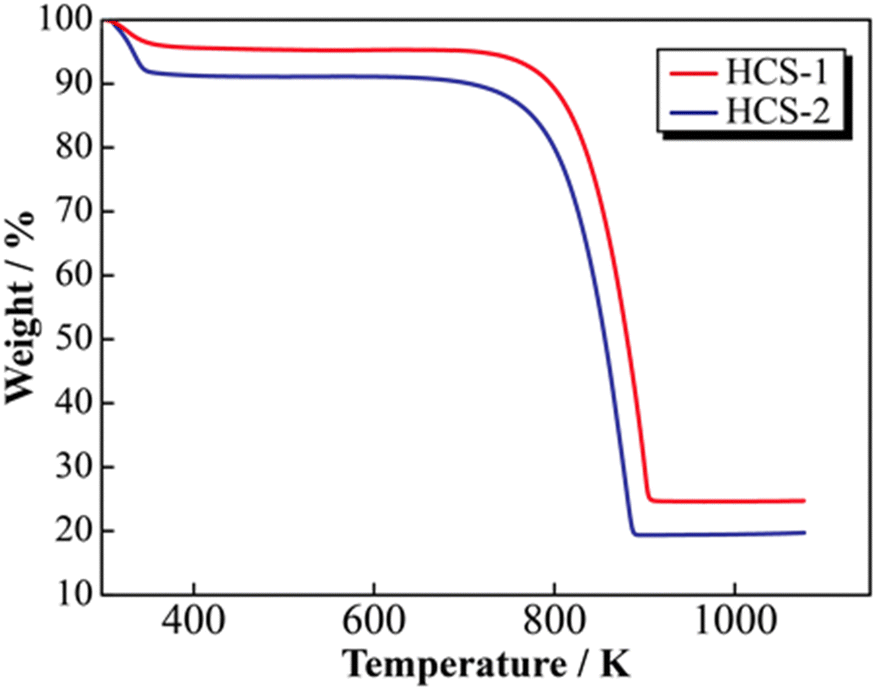
SEM images of the prepared HCS samples are shown in Fig. 2 (a: HCS-1, b: HCS-2). The particle sizes of both HCS samples were 9–17 μm, and there were no significant differences between the HCS-1 and HCS-2 particle sizes (Fig. S1, ESI†). EDX mappings of Si, C, and O for each sample showed nanometer-sized Si dispersed across the entire particle surface [Fig. 2(c)–(h)]. Cross-sectional FE-TEM images of HCS-2 (Fig. 3) showed Si aggregates of size approximately 1 μm; this is 20 times the size of the primary Si nanoparticles (average particle size: <50 nm). The presence of Si particles on the entire cross-section of the HCS particles confirmed that Si dispersion was satisfactory. These morphological observations confirm that the HCS particles are secondary particles consisting of multiple Si aggregates dispersed homogeneously in HC. Fig. 4 shows the TG curves of the prepared HCS samples. Weight losses of 8% and 3% attributable to desorption of adsorbed water were observed in the range 310–350 K, and weight losses of 71% and 65% attributable to carbon combustion were observed in the range 700–900 K, for HCS-1 and HCS-2, respectively. The weights at 473 and 1073 K were therefore designated as the absolute dry weight and residual weight, respectively, and the Si content was calculated from the difference between them. The calculated amounts of Si in HCS-1 and HCS-2 were 21.6 and 25.9 wt%, respectively. The calculated theoretical capacities of HCS-1 and HCS-2 were 1189 and 1355 mA h g−1, respectively, based on their theoretical capacity ratios (HC and C6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :360 mA h g−1, Si: 4200 mA h g−1).
:360 mA h g−1, Si: 4200 mA h g−1).
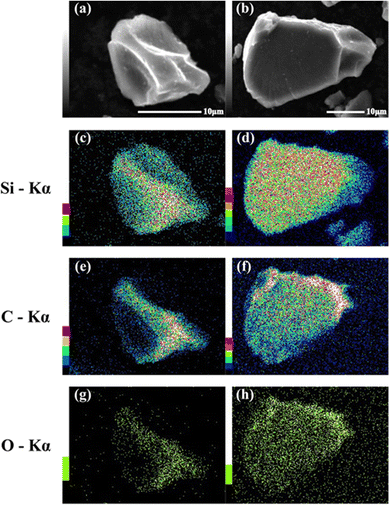 |
| | Fig. 2 SEM images of HCS-1 (a) and HCS-2 (b), and EDX mapping images for (c), (d) Si, (e), (f) O, and (g), (h) C in HCS. | |
 |
| | Fig. 3 Cross-sectional TEM images of HCS-2. | |
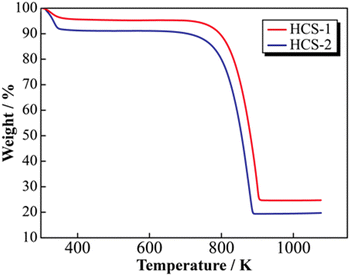 |
| | Fig. 4 Thermogravimetric profiles for HCS. | |
3.2 Si-based electrode material structure
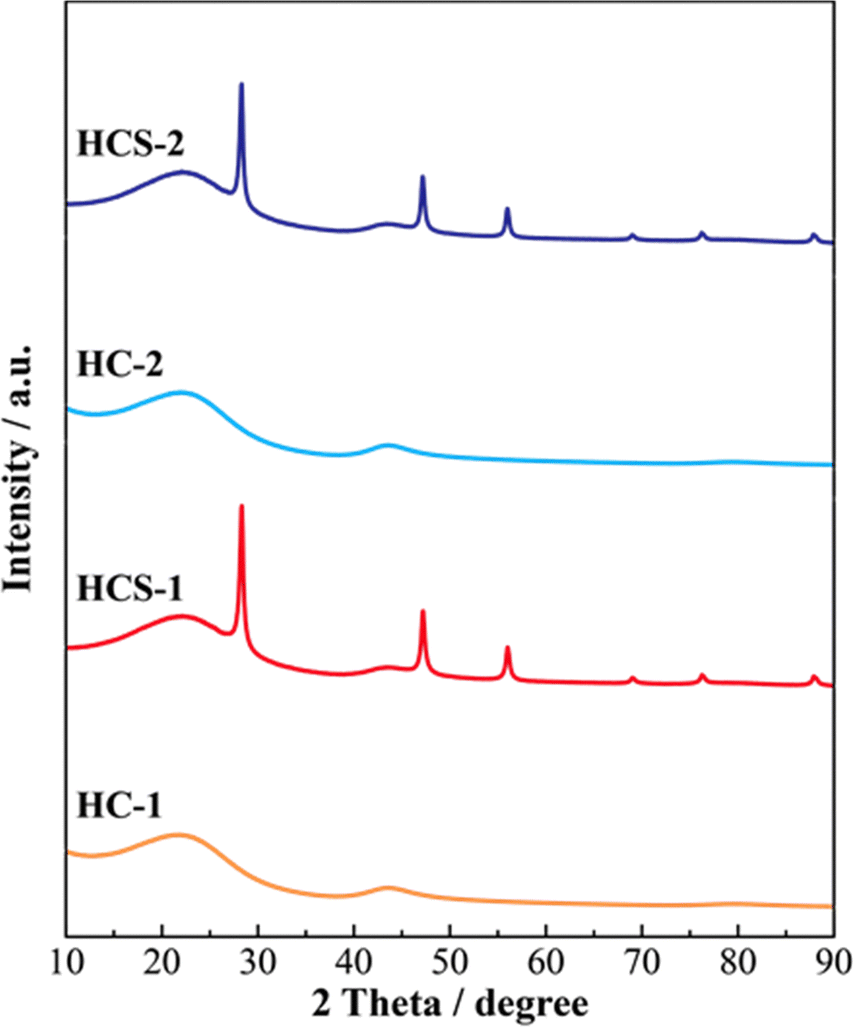
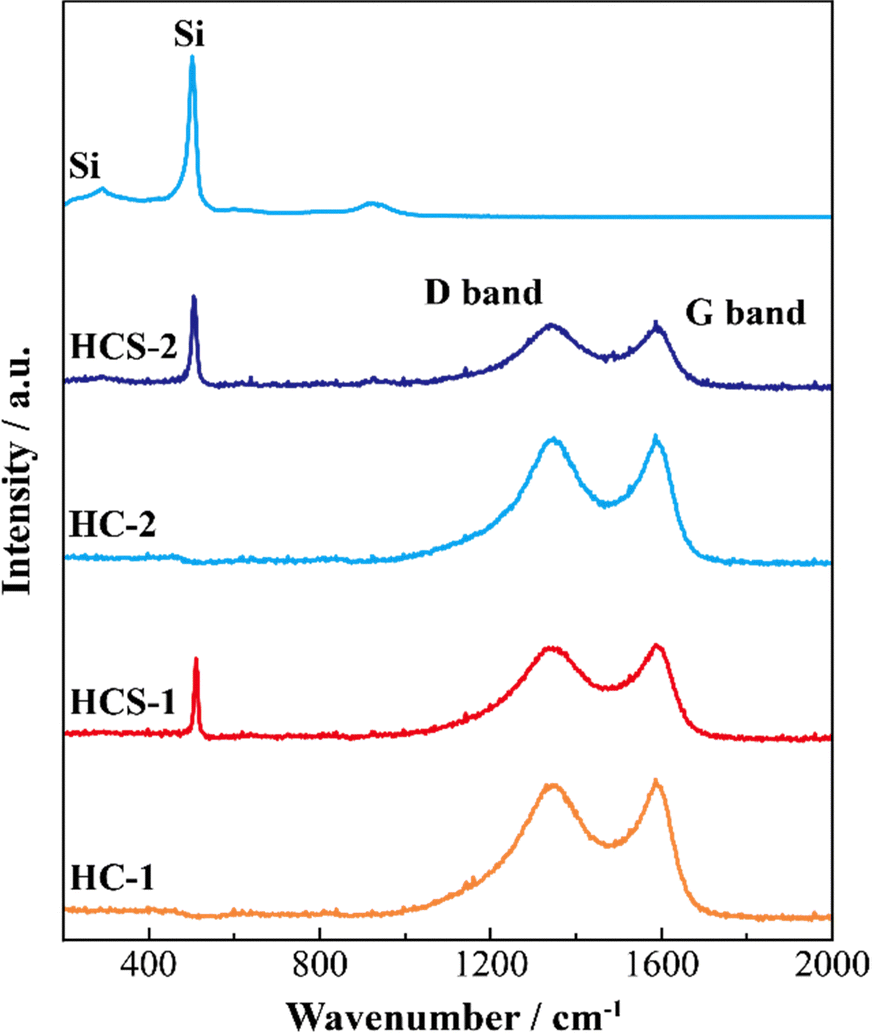
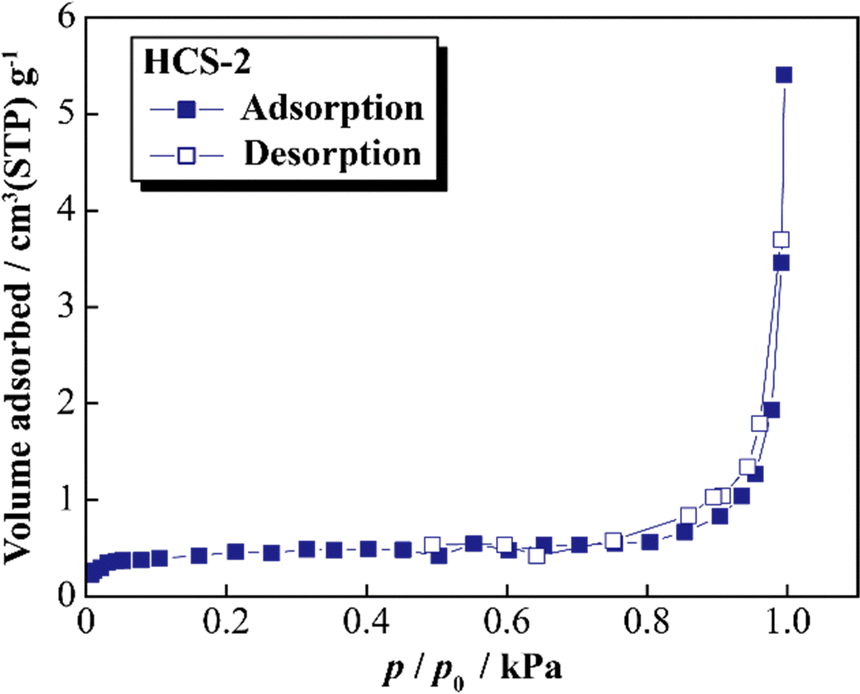
Fig. 5 shows the XRD patterns of Si, HCS-1, HC-1, HCS-2, and HC-2 powders. The patterns for the prepared HCS samples all show sharp peaks at approximately 2θ = 28°, 47°, and 56°, which correspond to crystalline Si (PDF#35-0412). These peaks do not differ from those of the original Si, therefore the crystal structure of Si did not change during HCS preparation. The broad peaks centered at approximately 2θ = 23° indicate the (002) plane corresponding to the graphene layer spacing. This indicates that all the HC samples are highly amorphous and confirms that both HCS samples consist mainly of crystalline Si nanoparticles and amorphous HC. The average lattice spacing values (d002) calculated from the 2θ = 23° (002) diffraction peak are 0.426 and 0.421 nm for HC-1 and HC-2, respectively. The crystallinity values of HC-1 and HC-2 are 11.4% and 11.5%, respectively. The crystallinities of HC-1 and HC-2 therefore did not change with changing carbonization temperature. Fig. 6 shows the Raman spectra of Si, HCS-1, HC-1, HCS-2, and HC-2 powders. The spectrum of each prepared HCS shows a sharp peak at approximately 520 cm−1, which is attributed to crystalline Si,35 and the carbon-derived D and G bands appear at approximately 1350 and 1600 cm−1, respectively.36 These results indicate that the HCS surface consists of well-dispersed Si particles and carbon layers, which is in agreement with the XRD results in Fig. 5. The structure of the prepared HCS therefore consists of small crystalline Si particles dispersed in an amorphous carbon framework. This provides a suitable material for electric and ionic conductors, and electrode materials. Fig. 7 shows the adsorption–desorption isotherms of the HCS-2 samples. Precise evaluation of the prepared HCS-1 sample to obtain stable adsorption–desorption data was difficult because of its high porosity. The HCS-2 sample gave a type II adsorption–desorption isotherm at P/P0 = 0.02–0.10.37 The increased adsorption at P/P0 values greater than 0.9 suggests the absence of fine pores or the presence of macropores and aggregation of fine particles. The HCS surface area was calculated by the BET method to be 1.93 m2 g−1. The HCS-2 sample therefore has a dense structure and is expected to have a high mechanical strength.
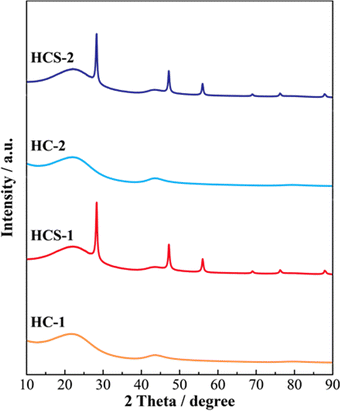 |
| | Fig. 5 XRD patterns of Si-based electrode materials. | |
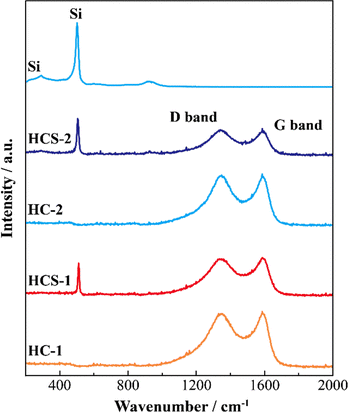 |
| | Fig. 6 Raman spectra of Si-based electrode materials. | |
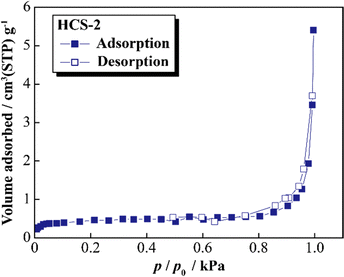 |
| | Fig. 7 Nitrogen adsorption–desorption isotherms for HCS. | |
3.3 Charge–discharge characteristics of [Li metal|HCS] cells
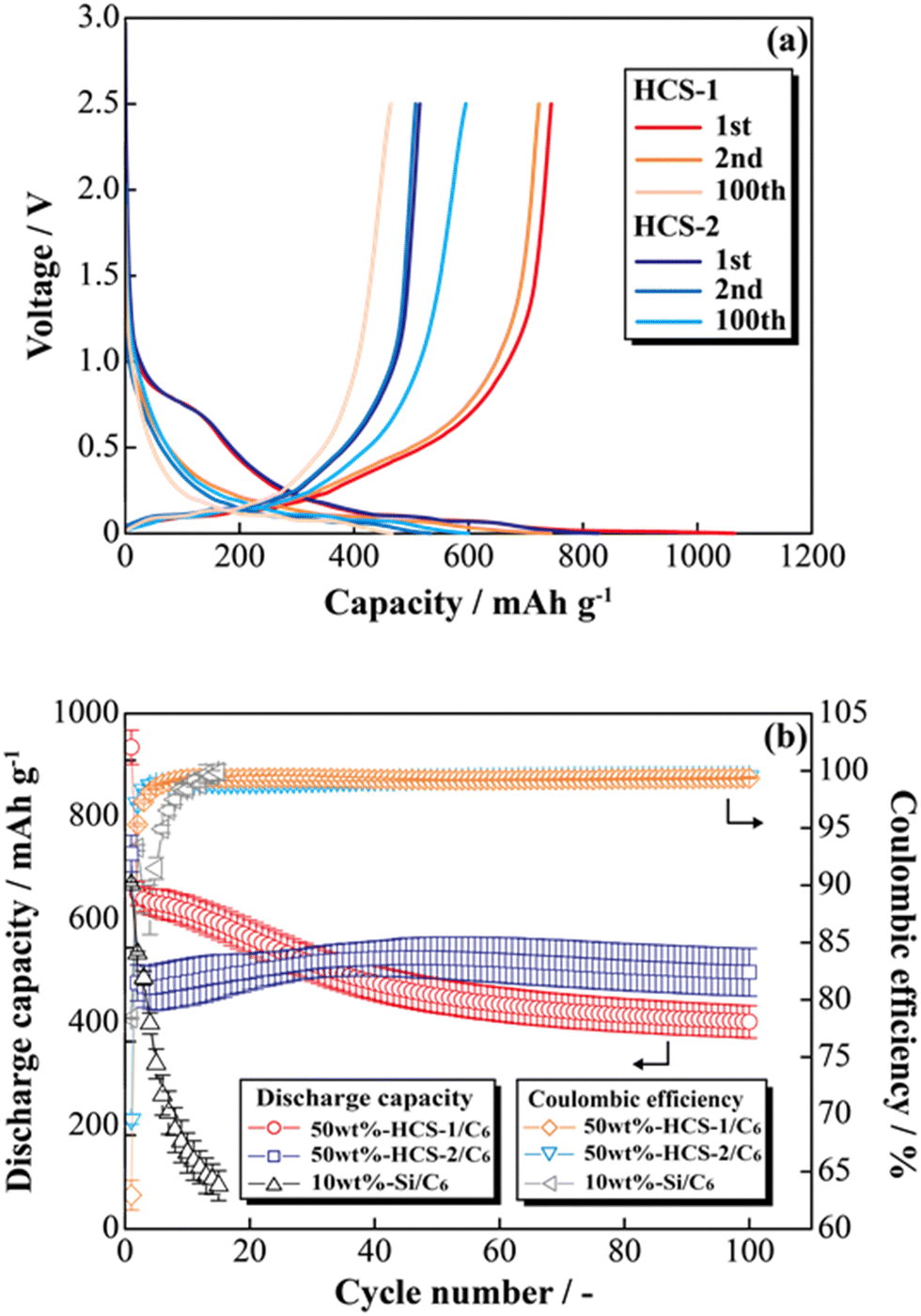
Fig. 8(a) shows the charge–discharge profiles of a prepared [Li|50 wt%-HCS/C6] cell at 303 K. The initial discharge capacities of HCS-1 and HCS-2 were approximately 1064 and 831 mA h g−1, respectively. These values are more than twice those of the high charge–discharge capacities of existing C6 negative electrodes. The cells using HCS-1 and HCS-2 both had large initial irreversible capacities, and showed stable charge–discharge capacities over 100 cycles, without the significant capacity decreases observed with conventional Si negative electrodes. The charge–discharge cycling characteristics of the HCS electrodes are better than that of a Si electrode, and modification of the Si surface with HC suppresses degradation. Fig. 8(b) shows the cycle-number dependence of the discharge capacities and the coulombic efficiencies calculated from the charge–discharge capacities of the prepared [Li|50 wt%-HCS/C6] cell and a simple mixed [Li|10 wt%-untreated Si/C6] cell. A significant decrease in the discharge capacity of the [Li|10 wt%-Si/C6] cell was observed after the first few cycles. This suggests that miniaturization and isolation of the active material occurred as a result of a change in the Si volume during the charge–discharge process, and oxidation–reduction cannot be stably performed inside the electrode. Decomposition of the electrolyte on the surfaces of the Si particles as a result of miniaturization would also decrease the coulombic efficiency in the first few cycles, which would result in a continuous irreversible reaction after the second cycle. In contrast, the discharge capacity of a [Li|50 wt%-HCS/C6] cell with HCS-2 increased with increasing cycle number. This increasing capacity with increasing cycle number is attributed to adequate soaking of the electrolyte into the electrodes and progressive conductive path formation. The capacity retention rates in the 100th cycle, except for the initial cycle, were 62% (HCS-1) and 104% (HCS-2). This indicates excellent cycling characteristics and a high capacity. Coulombic efficiencies greater than 99% were obtained for each HCS, except in the initial cycle. These results show that HCS electrodes are potentially effective negative electrodes because side reactions between Si and the electrolyte are minimized by compositing with amorphous carbon.
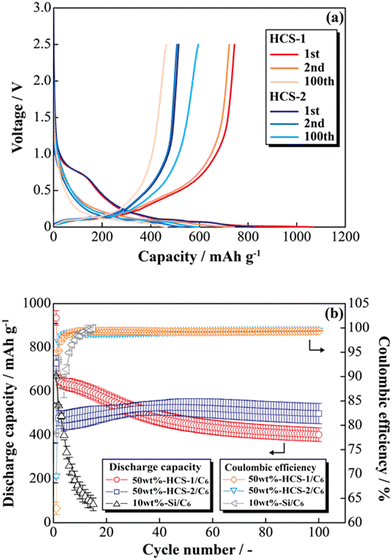 |
| | Fig. 8 Charge–discharge profiles for [Li|1.0 M-LiFSI EC/DEC|50 wt%-HCS/C6] cells (a) and relationships between cycle number and discharge capacity (b). | |
3.4 Investigation of factors for improving capacity retention of HCS cells
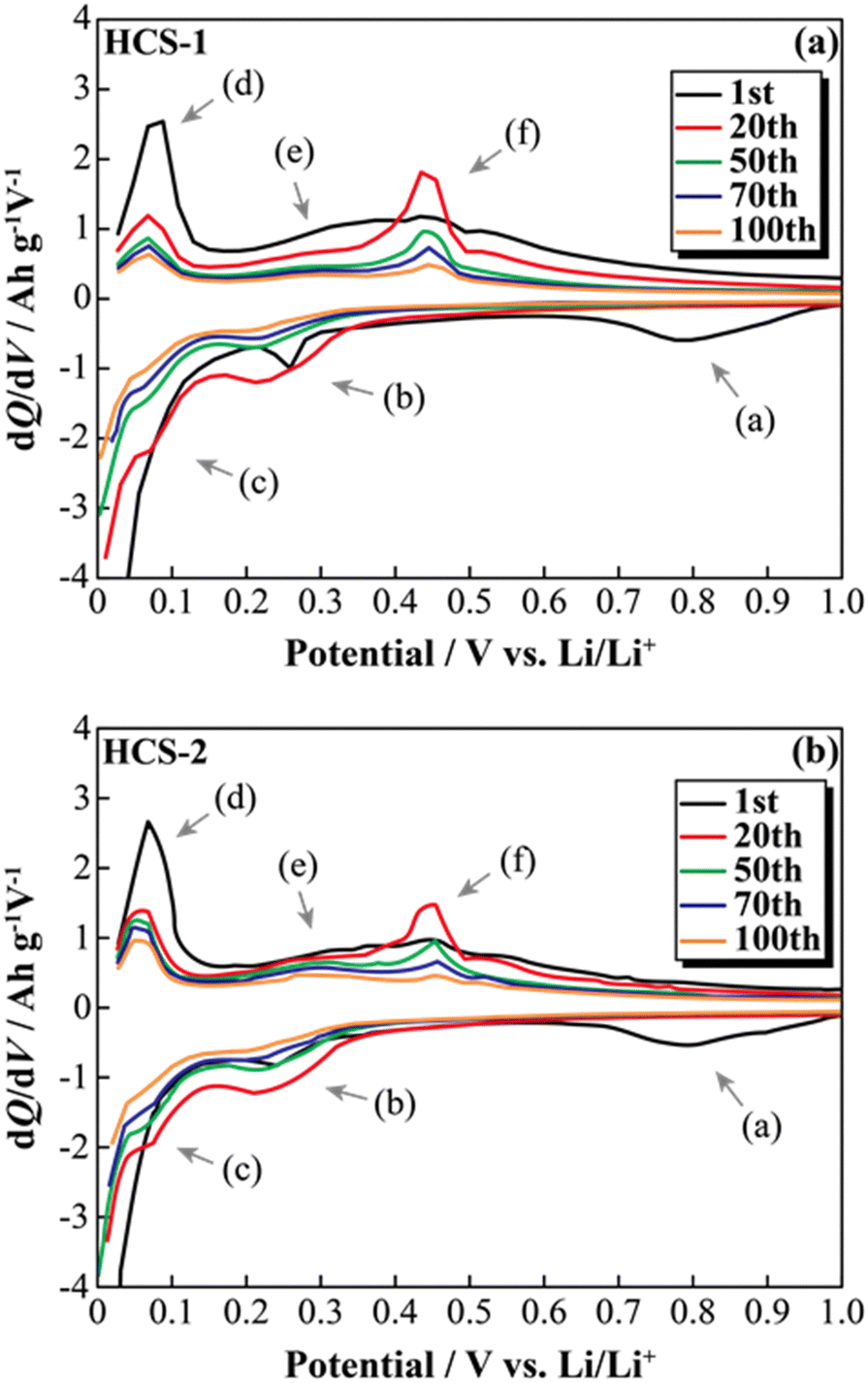
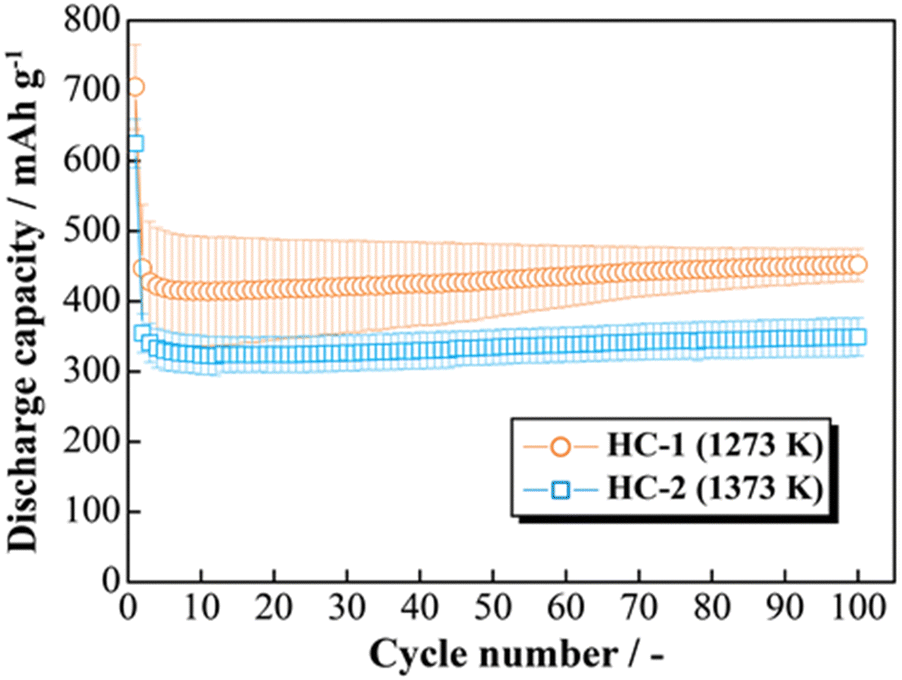
The effects of increasing the HCS capacity were investigated in terms of the reaction potential. The presence/absence and magnitude of the effects caused by different materials on the electrochemical activity of Si were investigated by differentiating the charge–discharge curves shown in Fig. S2 (ESI†). Fig. 9 shows the dQ/dV curves for two [Li|100 wt%-HCS] cells (a: HCS-1, b: HCS-2). For each HCS, clear peaks were observed at approximately (a) 0.7–1.0 V, (b) 0.2–0.3 V, and (c) 0.09–0.1 V during the discharge process, and (d) 0.05–0.1 V, (e) 0.27 V, and (f) 0.45 V during the charge process. Peak identification was achieved by reference to, and analysis of, the dQ/dV curves of HC (Fig. S3, ESI†). The peaks at (a) and (d) were confirmed to be derived from HC. A peak at approximately 0.25 V, which was observed for HC-2 in only the first cycle, is attributed to differences between the structures of HC materials obtained at different carbonization temperatures, and reflects the differences between the charge–discharge characteristics of HCS-1 and HCS-2. In contrast, the peaks at (b), (c), (e), and (f) correspond to the alloying-dealloying of Li into Si.38,39 For the 100 wt%-HCS electrodes, alloying and dealloying of Li into Si were confirmed, and there was no difference between the reaction potentials of Si in HCS-1 and HCS-2. When the discharge voltage is limited, the sharp peak corresponding to Li desorption into Si broadens and remains amorphous throughout the cycle.33 However, both sharp and broad peaks were observed during the charge processes in HCS-1 and HCS-2. The Si in the HCS obtained in this study is a mixture of crystalline and amorphous Si. The ratio of the intensities of the HC peak (d) and Si peak (f) during the charge process was higher in the case of HCS-1. This indicates that the proportion of crystalline Si was higher in HCS-1 than in HCS-2. The main cause of the capacity decrease corresponding to crystalline Si was assumed to be isolation of the active material, which is prone to miniaturization, and an inability to perform stable redox reactions in the electrode. The HCS capacity reduction observed in this study is therefore mainly caused by crystalline Si during charge–discharge cycling. Cracks in the HC layer, which are caused by significant changes in the Si volume, can potentially be associated with a degradation mode during charge–discharge reactions. The difference between the physical strengths of HCS-1 and HCS-2 is also considered to be related to the long-term charge–discharge properties. The role of HC in improving the cycling characteristics when HCS is used was investigated by preparing electrodes with only HC and performing charge–discharge tests. Fig. 10(a) shows the cycle-number dependence of the discharge capacities of the prepared [Li|HC] cells. For both HC-1 and HC-2, the trend in the capacity decrease did not differ from those for HCS [Fig. 7(b) and Fig. S2(b), ESI†], and the discharge capacities were stable over 100 cycles. A capacity decrease was not observed in the case of the HC only electrode, therefore the capacity decrease observed for HCS can be attributed to the significant change in the Si volume; this is in agreement with the results obtained from the dQ/dV curves in Fig. 9. In addition, a difference between the absolute charge–discharge capacities of HC-1 and HC-2 was observed. This is in agreement with the results in Fig. 8(b), and the observed difference in the charge–discharge capacity of HCS can be attributed to the charge–discharge characteristics of HC. The reaction mechanism with HC is considered to involve a modified graphite interlayer, the voids between these units, and the heteroatoms on the surface of the carbon structure.40 Similarly, it has been reported40 that the amount of Li-storage sites, which dominate the capacity, decreased with increasing carbonization temperature, and the charge–discharge capacity also decreased. In the case of hard carbon, carbonization generally increases with increasing carbonization temperature, resulting in a decrease in voids between units. Therefore, in the case of HCS-1, although there are many mechanical/structural stresses and a large initial capacity, the expansion and contraction of Si is likely to occur and cycle degradation is observed, while in the case of HCS-2, there are few mechanical/structural stresses, which suppresses the expansion and contraction of Si and results in good cycling characteristics. In the charge–discharge curve for HC only (Fig. S4, ESI†), the charge–discharge capacities close to a low potential of 0–0.12 V, which corresponds to the interlayer of graphene and the void, decreased with increasing carbonization temperature. The charge–discharge capacity derived from the graphene interlayer also decreased, therefore HC-1 is considered to have a smoother structure for Li alloying-dealloying. These results show that the HCS developed in this study should be able to control the Si reactivity by introducing a HC layer, and consequently suppress degradation of electrochemical cells.
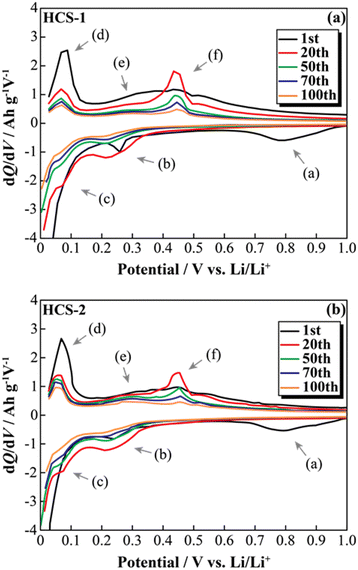 |
| | Fig. 9 Differential capacity curves (dQ/dV) for various cycles between 0 and 1.0 V for [Li|1.0 M-LiFSI EC/DEC|100 wt%-HCS-1/C6] cell (a) and [Li|1.0 M-LiFSI EC/DEC|100 wt%-HCS-2/C6] cell (b). | |
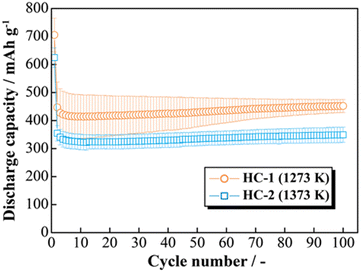 |
| | Fig. 10 Relationship between cycle number and discharge capacity for [Li|1.0 M-LiFSI EC/DEC|100 wt%-HC-1 or HC-2] cells. | |
3.5 Electrochemical and structural characteristics of HCS-2
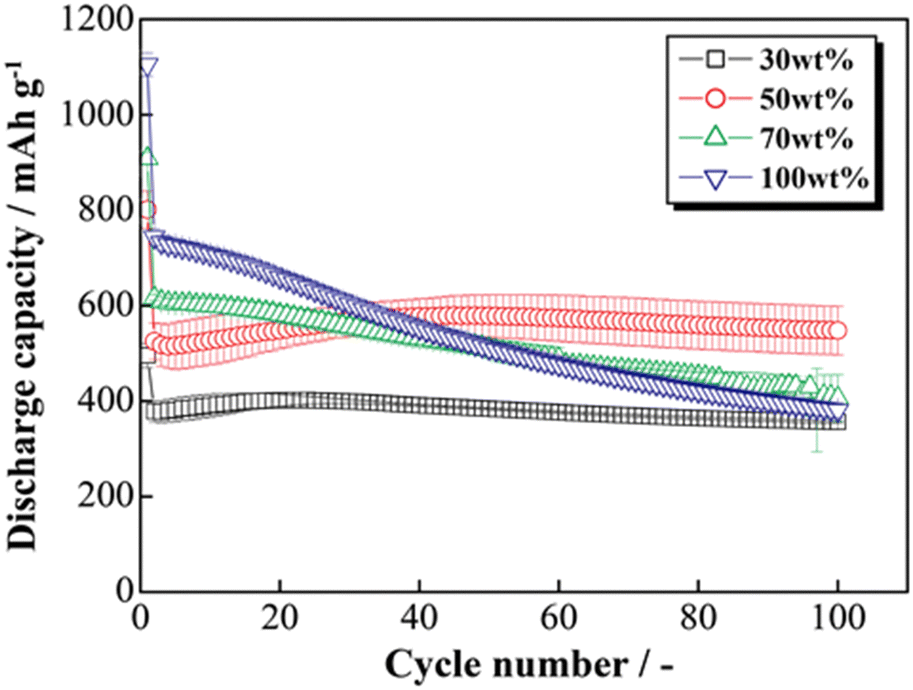
The HCS application range was investigated by performing charge–discharge tests using [Li|HCS/C6] cells with various HCS contents; HCS-2, which has satisfactory cycling characteristics, was used. Fig. 11 shows the cycle-number dependences of the discharge capacities of the prepared [Li|x wt%-HCS/C6] (x = 30, 50, 70, and 100) cells. Fig. S5 (ESI†) shows the charge–discharge curves of [Li|x wt%-HCS/C6] (x = 30, 50, 70, 100) cells at 303 K. Initially, the charge–discharge capacity increased with increasing HCS-2 content, and a discharge capacity of 1105 mA h g−1 was obtained in the first cycle with the [Li|100 wt%-HCS-2] cell. This is more than three times the charge–discharge capacities of conventional C6 negative electrode materials. However, the capacity retention decreased with increasing HCS-2 content. Table 2 shows the relationship between the weight percentage of the active material/C6 and the expansion rate of the electrode for the initial cycle and after 100 cycles. Fig. 12 shows the appearances of the HCS and Si/C6 electrode sheets obtained by disassembling the prepared coin cells. The expansion rates of the 10 wt%-Si/C6 and 25 wt%-Si/C6 electrodes were large, namely +74% and +68%, respectively. In contrast, low expansion rates of less than +50% were observed for HCS-1 and HCS-2. This is clear evidence for suppression of the volume change by HCS.
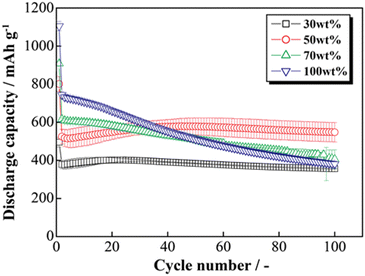 |
| | Fig. 11 Relationship between cycle number and discharge capacity for [Li|1.0 M-LiFSI EC/DEC|x wt%-HCS/C6] cells (x = 30, 50, 70, and 100). | |
Table 2 Relationship between weight percentage of AM/C6 and expansion rate of electrode (initial state and after 100 cycles, AM = HCS or Si)
| AM/C6 |
10 wt% |
25 wt% |
30 wt% |
50 wt% |
100 wt% |
| HCS-1 |
— |
— |
+21% |
+23% |
+43% |
| HCS-2 |
— |
— |
+47% |
+32% |
— |
| Si |
+74% |
+68% |
— |
— |
— |
 |
| | Fig. 12 Appearances of HCS and Si/C6 electrodes after 100 charge–discharge cycles. | |
Delamination of the electrode materials was observed for the 25 wt%-Si/C6 electrode because the adhesive strength of the electrode decreased during charge–discharge operations. The 100 wt%-HCS electrode also showed delamination, which led to a decrease in the charge–discharge capacity, as shown in Fig. 11. The electrochemical performance at the Si/HC interface layer formed via carbonization of the phenolic resin into the electrode material. A significant decrease in the charge–discharge capacity was observed for the 100 wt%-HCS-2 electrode. Suppression of the physical expansion and contraction of Si is therefore important for suppressing capacity degradation of HCS.
4. Conclusions
Micrometer-sized non-core–shell Si negative electrodes were developed by using HC-bundled multiple-nanometer-sized Si powders. HCS samples were prepared at different carbonization temperatures and their physical and electrochemical properties were investigated. The prepared HCS samples consisted of secondary particles with multiple aggregated Si uniformly dispersed in HC. The average lattice spacing values (d002) and crystallinity degrees of the HC-1 and HC-2 samples were similar, which suggests that the carbonization temperature did not affect the crystallinity. HCS electrodes with satisfactory charge–discharge properties, without the significant capacity degradation shown by conventional Si electrodes, were obtained. The expansion rates of the various electrodes after charge–discharge cycling were compared, and the HCS electrode showed clear suppression of volume changes. Physical control of the expansion/contraction of Si affected the HCS capacity degradation. Our proposed HCS will be useful for designing electrodes with improved volume changes and good cycling performances. These techniques will enable the use of low-cost large-particle Si powders in practical applications.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the Precise Measurement Technology Promotion Foundation, the Murata Science Foundation, and the Iketani Science and Technology Foundation, Japan.
References
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- C. M. Park, J. H. Kim, H. Kim and H. J. Sohn, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC.
- B. A. Boukamp, G. C. Lesh, R. A. Huggins and J. E. Soc, J. Electrochem. Soc., 1981, 128, 725–729 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- S. Chae, S. Choi, N. Kim, J. Sung and J. Cho, Angew. Chem., Int. Ed., 2020, 59, 110–135 CrossRef CAS PubMed.
- M. Ge, C. Cao, G. Biesold, C. D. Sewell, S. Hao, J. Huang, W. Zhang, Y. Lai and Z. Lin, Adv. Mater., 2021, 33, 2004577 CrossRef CAS PubMed.
- P. Li, H. Kim, S. Myung and Y. Sun, Energy Storage Mater., 2021, 35, 550–576 CrossRef.
- M. Vila, E. Bernardez, W. Li, C. Stackhouse, C. Kern, A. Head, X. Tong, S. Yan, L. Wang, D. Bock, K. Takeuchi, L. Housel, A. Marschilok and E. Takeuchi, ACS Appl. Mater. Interfaces, 2022, 14, 20404–20417 CrossRef CAS PubMed.
- H. Li, Z. Wang, L. Chen and X. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4985 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- L. Chen, K. Wang, X. Xie and J. Xie, J. Power Sources, 2007, 174, 538–543 CrossRef CAS.
- C. C. Nguyen and B. L. Lucht, J. Electrochem. Soc., 2014, 161, A1933–A1938 CrossRef.
- V. Etacheri, O. Haik, Y. Goffer, G. A. Roberts, I. C. Stefan, R. Fasching and D. Aurbach, Langmuir, 2012, 28, 965–976 CrossRef CAS PubMed.
- N. S. Choi, K. H. Yew, K. Y. Lee, M. Sung, H. Kim and S. S. Kim, J. Power Sources, 2006, 161, 1254–1259 CrossRef CAS.
- M. Ashuri, Q. He and L. L. Shaw, Nanoscale, 2016, 8, 74–103 RSC.
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed.
- T. H. Hwang, Y. M. Lee, B. S. Kong, J. S. Seo and J. W. Choi, Nano Lett., 2012, 12, 802–807 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- C. K. Chan, R. Ruffo, S. S. Hong and Y. Cui, J. Power Sources, 2009, 189, 1132–1140 CrossRef CAS.
- N. Liu, Z. Lu, J. Zhao, M. T. Mcdowell, H. W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- S. H. Ng, J. Wang, D. Wexler, K. Konstantinov, Z. P. Guo and H. K. Liu, Angew. Chem., Int. Ed., 2006, 45, 6896–6899 CrossRef CAS PubMed.
- X. Li, P. Meduri, X. Chen, W. Qi, M. H. Engelhard, W. Xu, F. Ding, J. Xiao, W. Wang, C. Wang, J. G. Zhang and J. Liu, J. Mater. Chem., 2012, 22, 11014–11017 RSC.
- S. Chen, M. L. Gordin, R. Yi, G. Howlett, H. Sohn and D. Wang, Phys. Chem. Chem. Phys., 2012, 14, 12741–12745 RSC.
- L. F. Cui, R. Ruffo, C. K. Chan, H. Peng and Y. Cui, Nano Lett., 2009, 9, 491–495 CrossRef CAS PubMed.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS PubMed.
- C. Shen, X. Fang, M. Ge, A. Zhang, Y. Liu, Y. Ma, M. Mecklenburg, X. Nie and C. Zhou, ACS Nano, 2018, 12, 6280–6291 CrossRef CAS PubMed.
- C. Liu, Y. Zhao, R. Yi, Y. Sun, Y. Li, L. Yang, I. Mitrovic, S. Taylor, P. Chalker and C. Zhao, Electrochim. Acta, 2019, 306, 45–53 CrossRef CAS.
- Z. Zhang, X. Han, L. Li, P. Su, W. Huang, J. Wang, J. Xu, C. Li, S. Chen and Y. Yang, J. Power Sources, 2020, 450, 227593 CrossRef CAS.
- Q. He, M. Ashuri, Y. Liu, B. Liu and L. Shaw, ACS Appl. Energy Mater., 2021, 4, 4744–4757 CrossRef CAS.
- S. A. Lyon and R. J. Nemanich, Phys. B + C, 1983, 117–118, 871–873 CrossRef CAS.
- D. S. Knight and W. B. White, J. Mater. Res., 1989, 4, 385–393 CrossRef CAS.
- R. Haul, Z. Phys. Chem., 1969, 63, 220–221 CrossRef.
- M. N. Obrovac and L. Christensen, Electrochem. Solid-State Lett., 2004, 7, 93–96 CrossRef.
- M. J. Loveridge, M. J. Lain, I. D. Johnson, A. Roberts, S. D. Beattie, R. Dashwood, J. A. Darr and R. Bhagat, Sci. Rep., 2016, 6, 1–11 CrossRef PubMed.
- C. Wan Park, S. H. Yoon, S. I. Lee and S. M. Oh, Carbon, 2000, 38, 995–1001 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Yoshikazu
Kobayashi
b,
Takao
Kunimi
b and
Shiro
Seki
a,
Yoshikazu
Kobayashi
b,
Takao
Kunimi
b and
Shiro
Seki