
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploration of a NiFeV multi-metal compositional space for the oxygen evolution reaction†
Anagha Usha
Vijayakumar
a,
Jael George
Mathew
b,
Anya
Muzikansky
a,
Hannah-Noa
Barad
*a and
David
Zitoun
 *a
*a
aDepartment of Chemistry and Bar-Ilan Institute for Technology and Advanced Materials (BINA), Bar-Ilan University, Ramat Gan 5290002, Israel. E-mail: David.Zitoun@biu.ac.il; hannah-noa.barad@biu.ac.il
bMax Planck Institute for Intelligent Systems (MPI-IS), Heisenbergstr. 3, 70569 Stuttgart, Germany
First published on 30th August 2023
Abstract
A sustainable future based on hydrogen fuel rests on the rapid advancement of non-precious metal catalysts for the oxygen evolution reaction (OER). We demonstrate the efficient utilisation in the analysis of a large compositional space of binary NiFe and ternary NiFeV alloys for OER using a combinatorial method. We fabricated a gradient library of these multi-metal alloys using physical vapor deposition and characterised them using high-throughput techniques. The electrocatalytic OER activity was studied using an automated electrochemical scanning droplet cell (SDC) set-up designed in our lab. From the overpotential (@10 mA cm−2) heatmaps of the libraries, the compositional regime of interest is funnelled down to 10–15% of Fe and 85–90% of Ni for the NiFe alloy and 1–3% V, 10–15% Fe, and 80–90% Ni for the NiFeV alloy with their overpotential values falling between 300–320 mV. Due to its oxidation state versatility, vanadium, is capable of inducing drastic changes in the electronic environment of the host element, boosting the OER activity. The effects of doping in the form of lattice distortions and electronic structure modifications have been investigated with XRD and XPS techniques. Our work shows the reliability and efficiency of the combinatorial approach in material discovery and applications, whilst also providing insights into the design of high performing OER catalysts.
Introduction
Hydrogen as a clean and high energy density fuel, is regarded as one of the countermeasures to the energy crisis. Electrochemical water splitting is a promising approach for the generation of high purity hydrogen at large scale.1–3 However, the anodic half-cell reaction in a water electrolyser, the oxygen evolution reaction (OER), is constrained by the kinetically sluggish four-electron transfer process, limiting the overall efficiency of hydrogen production.4,5 Although noble metals such as Ir or Ru and their alloys are used to catalyse the OER, they suffer from high cost and scarcity.6–8 To this end, development of active, inexpensive, and durable catalysts with low OER overpotentials is crucial.Among the efforts at designing non-noble metal-based materials as OER catalysts in alkaline media, Ni and its multi-metal composites are the highly promising.9–18 After much research, Fe incorporation into Ni has been deemed integral for high catalytic activity, and NiFe hydroxides/oxyhydroxides are undoubtedly some of the best performing binary systems reported so far for OER.19–28 When combining elements, the synergy between them amounts to enhancing conductivity, optimising the electronic structure, and decreasing the reaction activation energy barrier, thereby leading to improved activity. In our previous work, we demonstrated the incorporation of Fe and Co into a Ni matrix as an effective strategy to improve the catalytic performance.29 Although cobalt improves the OER performance, cobalt is a critical raw material and its mining raises environmental, social, and governance issues. Herein, we chose vanadium as a suitable candidate to drive down the overpotential of the NiFe based catalysts relying on vanadium's multivalency, relative abundance,30 and stability.31–40 There are previous reports studying the effect of vanadium using spectroscopic techniques such as XAS and theoretical DFT calculations.41–44 By delving into the charge transfer in the d orbitals and the binding energy of intermediates, it was found that vanadium with its high valence has the potential of driving these traits to their optimum and improving the overall catalytic performance for OER. The addition of vanadium is also expected to have an impact on the structural stabilisation of the active phase during OER.45–47 The inhibiting effect on phase transition induced by vanadium prevents the degradation of the catalyst over time and provides overall enhancement in activity.
Varying the relative ratios of the constituent elements in multi-metallic systems affects the intrinsic activity and chemical properties. Compositional variation can result in tuning the material's electronic environment and enhancing the active sites, but beyond an optimal level, deteriorating impacts start gradually coming into view. The identification of this balance by a systematic analysis that effectively spans the entire compositional space of multi-metal catalysts, surpasses the scope of the traditional approach, where each composition is individually synthesised and studied. We thus devised a combinatorial approach to take over this overwhelming task. This approach thrives on the principal of parallel processing, which makes it much more efficient than the traditional approach by accelerating material discovery and an increased access to a broader range of materials. The combinatorial approach is put into practice by depositing a library of materials (a sample with a systematic variation of a particular material property, for instance a composition gradient), which are subsequently processed, and then analysed rapidly in a high-throughput manner using a suitable analytical technique. Thin film synthesis techniques are often used for the fabrication of combinatorial libraries with compositional spreads.48–56 Such thin film gradient libraries ensure the same processing and measurement conditions for all the samples in question.
In this work, we fabricate a thin film materials library with compositional gradients of bimetallic NiFe and trimetallic NiFeV using physical vapour deposition (PVD). We then use these material libraries to draw a composition-activity relationship of the metal components for OER catalytic activity. A custom-made electrochemical scanning droplet cell (SDC) set-up, completely automated using LABVIEW, was used to measure the OER activity of the material libraries. The overpotential heatmaps at 10 mA cm−2 indicate an optimal compositional window with the best catalytic activity of 10–15% Fe and 85–90% Ni for the NiFe alloy, and 1–3% V, 10–15% Fe, and 80–90% Ni for the NiFeV alloy. Using various characterisation tools such as X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS), we unravel the synergistic effect of the dopants by examining the lattice distortions and electronic structure modifications. We believe that our results successfully demonstrate the impact of high-throughput techniques in guiding materials discovery and the compositional mapping of catalytic activity in multi-metal systems, leading to the development of high performing materials for electrocatalytic water splitting.
Results and discussion
Compositional spread in the material libraries
The compositional gradient in the material libraries was measured using the X-ray fluorescence (XRF) and have been reported in at% for all samples. We made the XRF measurements at 49 points on the gradient library. The bimetallic library spanned a compositional space of 5–30 at% Fe and 70–100 at% Ni, and the trimetallic NiFeV library 0–8 at% V, 5–60 at% Fe, 30–95 at% Ni, as shown in Fig. 1. We have obtained linear gradients by co-deposition of materials on an ITO substrate, using our PVD technique. This was achieved by adjusting the coverage of the barrier placed above the material source to control the vapor flux arriving at the substrate. The XRF measured composition gradients were in agreement to the trend expected during deposition. We have also conducted EDAX measurements on our NiFeV trimetallic sample to further confirm the composition trend observed from XRF (Fig. S5, ESI†).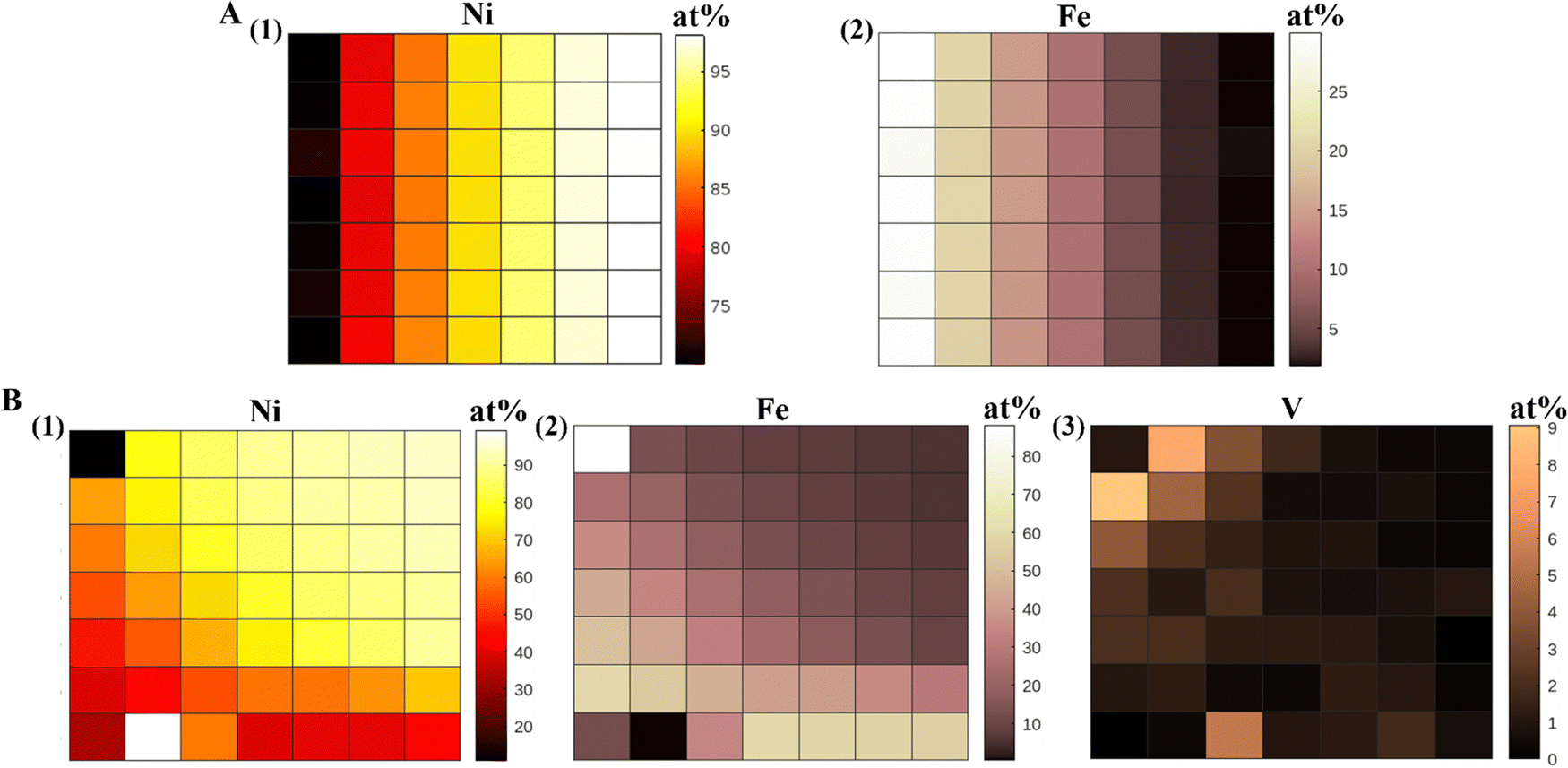 | ||
| Fig. 1 XRF heatmaps (at%) of (A) NiFe alloy, (1) Ni and (2) Fe, (B) NiFeV alloy, (1) Ni, (2) Fe, and (3) V. | ||
Structural properties of the material libraries
The grazing incidence X-ray diffraction (GIXRD) patterns of all the libraries show that they are crystallise with a single phase in a cubic crystal geometry (Fig. 2(A) shows the GIXRD pattern of a representative point from the library and Fig. S1, ESI†). We performed a slow scan measurement at selected points (selected to give a reasonable representation of the compositional space and corresponding overpotential values) on the NiFeV sample to distinguish the crystal structure peaks more clearly (Fig. 2(B)). The values of the d-spacing and the unit cell dimensions (a) of a few selected compositions derived from these measurements are given for comparison in Table S1 (ESI†). The a values for all the doped alloys are higher than that of pristine nickel (a = 3.45 A, taken from literature) due to the lattice distortions induced when additional elements are added into the crystal lattice. It can be observed that the 2 theta value of the diffraction peak shifts systematically to lower Bragg angles with increased level of compositional changes. Correspondingly, there is an increase in the d-spacing values, the magnitude of increase being 0.056 Å for the composition with highest doping of V (Ni79Fe13.5V7.5).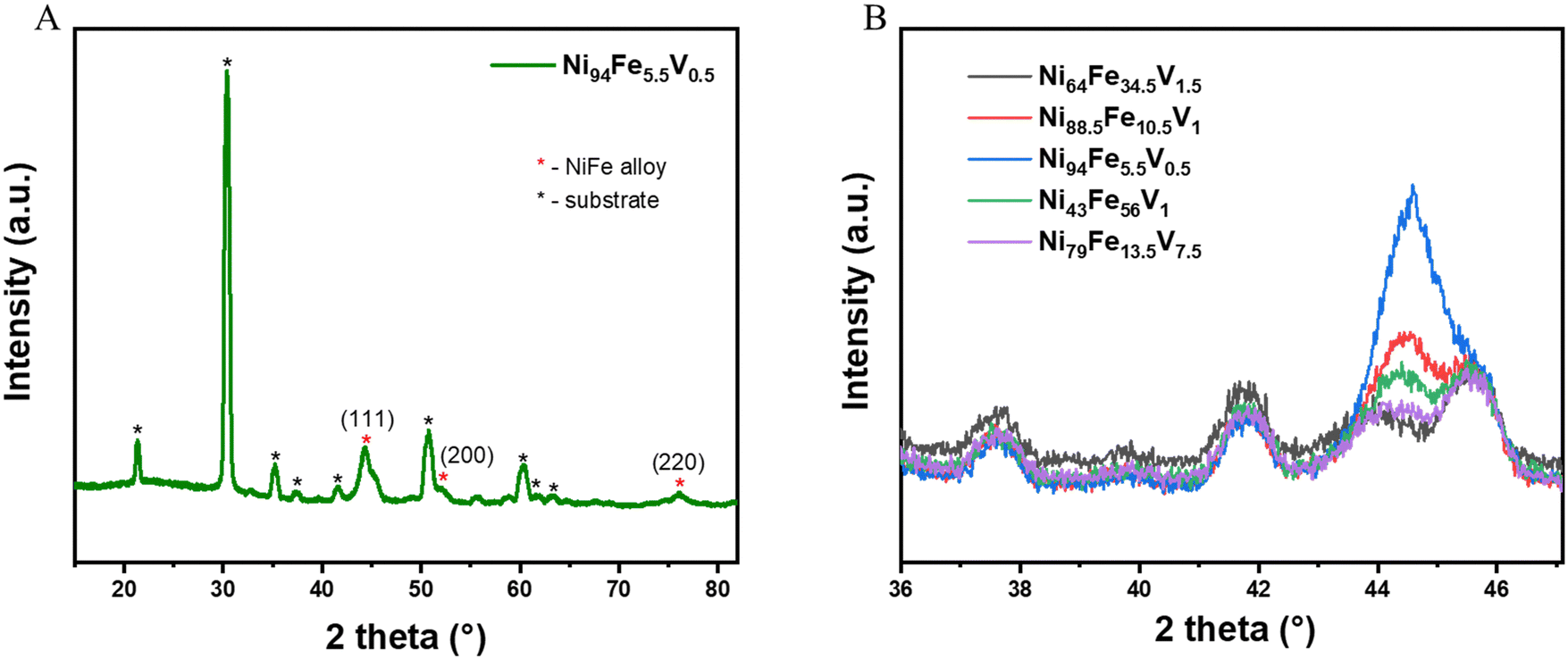 | ||
| Fig. 2 (A) GIXRD of Ni94Fe5.5V0.5 showing the sample and substrate peaks (B) slow scan measurements at selected compositions on NiFeV alloy, depicting shifting in peak position with relation to composition. | ||
The morphology of the thin film NiFeV sample was studied using FESEM (Fig. S6, ESI†). Images were taken at various points across the sample after OER. A dense compact structure with continuous distribution of particles could be observed throughout the sample. We also performed AFM measurements to analyse the surface roughness of the film. Fig. S7 (ESI†) shows the AFM images (30 × 30 μm) taken at various points on the NiFeV thin film after OER measurements. The surface roughness (ra) of the thin film was calculated to be in the range of 1.14–2.03 nm (Table S4, ESI†). The morphology of the thin film can therefore be considered uniform throughout the sample. We measured the thickness of the film at various points using the XRR technique (Fig. S8, ESI†). The reflectivity oscillations were fitted and analyzed using FFT to calculate the film thickness. The results proved that the thickness throughout the film is uniform (96–99 nm).
Electrochemical OER measurements
The electrochemical OER measurements were carried out using a custom made automated SDC set up. The SDC sequentially scans 36 predetermined points on the materials library. The SDC setup is described in detail in our previous work.29Overpotential vs. composition maps of the binary NiFe, and ternary NiFeV libraries are shown in two different representations in Fig. 3(A). Fig. 3(A)(1) and Fig. 3(A)(2) is a 3D plot of overpotential vs. composition shown at different angles for all the points of measurement on the binary and ternary samples. The x and y axes depict the composition of Fe and V, while the z axis depicts the overpotential at 10 mA cm−2. The red spots correspond to the actual measurement points of NiFeV sample and the green spots to that of NiFe sample. In order to visualize the trend in activity with variation in composition, we have plotted a contour heatmap of overpotential vs. compostion (Fig. 3(A)(3)). In addition, we have also given the point-by-point heatmap of the overpotential of the NiFe and the NiFeV library individually in Fig. S2 (ESI†). From these maps, we can deduce a composition-activity relationship. For the bimetallic NiFe sample, we observe that the lowest overpotentials fall within 10–15% of Fe and 85–90% of Ni. In a volcano type behaviour, we see that initially the overpotential decreases with doping, reaches an optimum and then deteriorates further as the iron content exceeds 25%. Further, the trend in the trimetallic NiFeV library points to an optimal regime of 1–3% V, 10–15% Fe, and 80–90% Ni, with the overpotentials reaching as low as 300 mV vs. RHE. The effect of V can be confirmed by comparing the overpotentials in the regions with nearly 0% V doping to that with 1–3% V doping. Fig. 3(B) shows the Tafel slopes for representative compositions from the NiFe and the NiFeV library. Within the NiFeV library, the Tafel slope values follow the trend expected from the overpotential-composition map, with the lowest Tafel slope for Ni80.5Fe18V1.5 (86.67 mV dec−1). The lowest Tafel slope of 55.85 mV dec−1 for Ni85.51Fe14.49 could be due to a different reaction mechanism (with a variation in the rate limiting step) in the NiFe library as opposed to that with the addition of vanadium.
 | ||
| Fig. 3 (A)(1) and (2) Overpotential vs. composition of Fe and V (the green spots correspond to the actual measurement points of NiFe sample and the red points to that of NiFeV sample) at different angles (3) Contour heatmap of overpotential vs. composition of Fe and V (B) The Tafel slopes of selected compositions. | ||
We also show the evolution of CV curves and LSV with compositional variation in Fig. 4. The mechanism of OER proceeds by the formation of M–OOH from the M–O surface. All catalysts must undergo this surface transformation to the M–OOH to start catalyzing OER.21 The redox peak appearing in the LSVs between 1.35 V to 1.45 V correspond to this conversion, featuring an oxidation state change of nickel from +2 to +3. In order to ensure activation of our catalysts prior to OER measurements, we perform 50 CV cycles (until saturation of the redox peak currents). The oxidation potential of the Ni2+/3+ couple shifts to higher potentials with increasing Fe content (Fig. 4(A)(1)). This effect in NiFe alloys has been previously reported and can be attributed to the partial charge transfer effect exerted from nickel to iron. After the addition of vanadium, the Ni oxidation potentials shift to lower potentials in comparison to their corresponding NiFe compositions confirming the “galvanic” effect of vanadium (Fig. 4(B)(1)). The LSVs of both NiFe and NiFeV (Fig. 4(A)(2) and (B)(2)) verify the composition-activity trend observed in the overpotential heatmaps.
 | ||
| Fig. 4 Electrochemical cycling data of few selected compositions of (A) NiFe, (1) CV, (2) LSV, (B) NiFeV, (1) CV, (2) LSV. | ||
In order to estimate the electrochemical surface areas, we calculated the capacitances of the double layer at the solid/liquid interface. CVs were collected on the NiFeV sample in the region of 0.92–1.12 V vs. RHE by varying the scan rate from 10 mV s−1 to 60 mV s−1, where the current response should only be due to the charging of the double layer. We performed these measurements at five different points on our NiFeV sample (Fig. S9, ESI†). The capacitance values for the five points measured are 0.575 mF, 0.467 mF, 0.615 mF, 0.425 mF and 0.342 mF. We further calculated the ECSA for these five points from the ratio Cdl/Cs, where, Cs is the specific capacitance of the sample or the capacitance of an atomically smooth planar surface of each catalyst. We used Cs = 0.39 mF cm−2 based on experiments performed on a flat nickel foil. The values of ECSA for the five points measured on the sample are as follows 0.185, 0.150, 0.198, 0.136, 0.110 cm2. The ECSA values do not show much deviation as would be expected for a uniform thin film with uniform thickness.
To understand the role of V doping on the electrocatalysis, we performed EIS measurements. The Nyquist plots are shown in Fig. S3(A) (ESI†). The EIS fitting for the best performing catalyst is shown in Fig. S3(B) (ESI†) and the inset shows the equivalent circuit used for fitting. This corresponds to a very thin film catalyst. In the equivalent circuit shown, Rs is the uncompensated solution resistance, Rct and Cct are the charge transfer resistance and double layer capacitance at the catalyst – electrolyte interface during OER, and Rc and Cc are the resistance and capacitance across the substrate – catalyst interface.57 The lowest charge transfer resistance (Rct = 25.2 Ohm) was obtained for the composition regime of 10–15% Fe and 1–3%, indicating optimal interfacial electron transfer at this composition.
Chemical states before and after OER
In order to study the surface chemical states of the elements, XPS was performed. The deconvoluted spectra of Ni 2p3/2 for NiFe and NiFeV before OER, show peaks corresponding to metallic nickel as well as oxidised Ni2+/3+ species (Fig. 5(A)). It can be observed on comparison that the Ni2p shifts to lower binding energies after V doping, validating the charge transfer effect of vanadium. As shown in Fig. 5(B) and (C), after OER, the oxyhydroxide formation on the surface is clearly evidenced by the suppression of the metallic peak and rise of the peak at 855.8 eV. Also, a slight shift of the spectra to higher binding energies can be observed, indicative of the conversion to hydroxide. On extracting the ratio of the metal:hydroxide peaks for different points on the NiFeV sample, it can be inferred that surface hydroxides are higher for those compositions delivering lower overpotentials for OER (metal: hydroxide ratios for compositions in order of decreasing activity: 0.07![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.09:1, 0.12:1). The three peaks in the deconvoluted O1s spectra of NiFe and NiFeV pristine samples, can be designated to M–O, M–OH and water adsorbates (Fig. 5(F)). After OER, the O1s spectra evolves to emphasise the surface hydroxides with the surface oxides becoming less prominent (Fig. 5(F) and Fig. S4(D), ESI†). Also, the hydroxide:oxide ratio is higher in the pristine NiFeV sample (1:0.28) compared to pristine NiFe sample (1:0.4) which implies an ease of formation of the surface hydroxide during OER with the addition of vanadium. In addition, the hydroxide:oxide ratio within the NiFeV sample is higher for the points with better overpotential, in agreement with a higher conversion for these compositions (hydroxide:oxide ratios for compositions in order of decreasing activity: 1:0.07, 1:0.08, 1:0.1). From the analysis of the Fe 2p spectra a case can be made that Fe primarily exists in its metallic state before electroctalytic activation, whereas, after catalysis the spectra shifts to a higher binding energy and Fe3+ is the predominant species (Fig. 5(E) and Fig. S4(B), ESI†). There was a decrease in the binding energy of iron by ∼0.2 eV after V doping, indicating a modification in its chemical environment. The V 2p spectra centred around 516.5 eV confirms the presence of vanadium on the sample (Fig. 5(E)), however, deconvolution of these peaks was not attempted due to the low quality of the spectrum. This could be due to the low amounts of V in our sample. In order to understand the effect of vanadium on OER, we compared the V 2p XPS spectra before and after OER (Fig. S10, ESI†). We observed that the vanadium spectrum after OER is more noisy and difficult to distinguish. This could be due to the hydroxide layer formation over vanadium on the surface, during OER. Another possible reason could be the leaching of V out of the surface. The vanadium peak is also slightly shifted to lower binding energies after OER, which however cannot be clearly examined due to noise. Vanadium exhibits a galvanic effect by influencing the electronic states of the active metals and not directly taking part in catalysis. Therefore, it is possible that vanadium gets buried under the nickel-iron hydroxide layer formed during catalysis, making it less distinguishable in the post mortem analysis.
:1, 0.09:1, 0.12:1). The three peaks in the deconvoluted O1s spectra of NiFe and NiFeV pristine samples, can be designated to M–O, M–OH and water adsorbates (Fig. 5(F)). After OER, the O1s spectra evolves to emphasise the surface hydroxides with the surface oxides becoming less prominent (Fig. 5(F) and Fig. S4(D), ESI†). Also, the hydroxide:oxide ratio is higher in the pristine NiFeV sample (1:0.28) compared to pristine NiFe sample (1:0.4) which implies an ease of formation of the surface hydroxide during OER with the addition of vanadium. In addition, the hydroxide:oxide ratio within the NiFeV sample is higher for the points with better overpotential, in agreement with a higher conversion for these compositions (hydroxide:oxide ratios for compositions in order of decreasing activity: 1:0.07, 1:0.08, 1:0.1). From the analysis of the Fe 2p spectra a case can be made that Fe primarily exists in its metallic state before electroctalytic activation, whereas, after catalysis the spectra shifts to a higher binding energy and Fe3+ is the predominant species (Fig. 5(E) and Fig. S4(B), ESI†). There was a decrease in the binding energy of iron by ∼0.2 eV after V doping, indicating a modification in its chemical environment. The V 2p spectra centred around 516.5 eV confirms the presence of vanadium on the sample (Fig. 5(E)), however, deconvolution of these peaks was not attempted due to the low quality of the spectrum. This could be due to the low amounts of V in our sample. In order to understand the effect of vanadium on OER, we compared the V 2p XPS spectra before and after OER (Fig. S10, ESI†). We observed that the vanadium spectrum after OER is more noisy and difficult to distinguish. This could be due to the hydroxide layer formation over vanadium on the surface, during OER. Another possible reason could be the leaching of V out of the surface. The vanadium peak is also slightly shifted to lower binding energies after OER, which however cannot be clearly examined due to noise. Vanadium exhibits a galvanic effect by influencing the electronic states of the active metals and not directly taking part in catalysis. Therefore, it is possible that vanadium gets buried under the nickel-iron hydroxide layer formed during catalysis, making it less distinguishable in the post mortem analysis.
 | ||
| Fig. 5 XPS measurements of: (A) Deconvoluted Ni 2p spectra comparison of NiFe and NiFeV before OER. (B) Deconvoluted Ni 2p spectra comparison of NiFeV before and after OER. (C) Deconvoluted Ni 2p spectra of various points on NiFeV after OER. (D) Fe 2p spectra comparison of NiFe and NiFeV before and after OER. (E) V 2p spectra at two different points on NiFeV. (F) Deconvoluted O 1s spectra comparison of NiFeV before and after OER. | ||
Discussion
In alkaline media, NiOOH has long been identified as the active site for OER. Trotochaud et al. observed that even trace amounts of Fe can drastically improve the OER performance of Ni due to a partial charge transfer activation effect.21 By conducting in situ Raman spectroscopy, Bell and co-workers demonstrated that Fe efficiently boosts the catalytic activity of NiOOH.58 Friebel et al. proved this effect of Fe using operando X-ray absorption spectroscopy and density functional theory (DFT) calculations.59 Concerted efforts by various groups have been dedicated to the study of NiFe based OER catalysts.10,60–62 In our previous work, we have shown that the addition of modest amounts of cobalt to the NiFe system can have a significant impact on the OER activity.29 Various such transition elements (Cr, Ce, V, Mn) owing to their tuneable d-band structure, have been incorporated into the standard NiFe system to improve its catalytic properties.63–67 In the study of multimetal alloys, obtaining the same volume of data as we obtained and effectively spanning the same compositional space as achieved in our work would require a substantially higher number of experiments, time, and cost. In this context, a combinatorial approach is an appealing practice to derive the formulations that will produce the best catalyst and generate a composition – activity map.Among the transition metals studied as dopants to the NiFe system, of particular interest, is the effect of multivalent vanadium. It has been established that the addition of vanadium alters the local structural environment, which reduces the Gibbs free energy of O binding (ΔGO*).41 Detailed electrochemical analysis and DFT calculations were done on a trimetallic NiFeV layered double hydroxide (LDH) catalyst, which revealed that V incorporation resulted in modulating the electronic structure, enhancing electric conductivity, and favouring fast electron transfer.44 V doping was also demonstrated to have the potential to decrease the barrier of the rate determining step.34 The synergistic interaction among Ni, Fe, and V was proposed to originate from the different 3d valence electronic configuration of these elements. When carefully probing these interactions using X-ray absorption spectroscopy (XAS), it turned out that in the trimetallic NiFeV catalysts, V has a highly distorted local coordination structure and short V–O bond lengths, which leads to near optimal binding energies of oxygen intermediates.43 Our choice of vanadium stems from such insightful examination of the effects V doping in the literature.36,38,39,42,68
Another significant impact of alloying Ni with V and Fe that is worth mentioning, is the α-Ni(OH)2 phase stabilisation, which leads to inhibition of long term degradation. The Ni(OH)2 structures, which are precursors of the OER active NiOOH phases, were previously shown to have a prominent effect on the OER activity. Iron was first reported in 1908 by Edison to influence Ni(OH)2 behaviour during charge discharge in alkaline storage batteries.69 Later, in 1987, Corrigan examined its influence on the OER activity.70 According to the Bode diagram, α-Ni(OH)2 transforms to γ-NiOOH while the β phase of Ni(OH)2 converts to β-NiOOH.71 Ni in β-NiOOH has a valency of +3, in contrast to the +3.6–3.7 in γ-NiOOH, which leads to a higher electron transfer number of ∼1.7 in the α → γ phase transition.72–74 In addition, the γ-NiOOH is also more stable due to a higher interlayer spacing (6.9A in comparison to 4.8 A for β-NiOOH), which enables water molecules and ions to penetrate the layers. We thus desire an electrochemical cycling from α → γ, however, the disordered α phase usually transforms to the more crystalline β phase under the applied potential and pH conditions of the reaction. Such an irreversible phase transformation results in the degradation of the OER activity over time. The incorporation of elements such as iron, vanadium, aluminium, cobalt, etc. induce an inhibition of the β-Ni(OH)2 crystallisation and ordering by chemical aging.45–47,75,76 These elements although not directly involved in catalysis, evidently influence the OER activity by altering the structure and chemical environment of the OER active NiOOH. It must also be noted that we are effectively reducing the active metal loading (Ni(OH)2), taking into consideration the supporting elements incorporated and the water molecules associated with the α phase. As the active site of catalysis is the NiOOH and the secondary and tertiary elements indirectly contribute to the stabilisation and improvement of the active material, beyond a particular level of addition, we compromise on the performance. The realisation of this optimum level of incorporation was made possible by our combinatorial approach in a time and cost-effective way. We concluded that more than 3% V and 15% Fe conversely affects the OER activity by compromising on the active metal Ni.
In order to analyse how our catalyst stands in comparison to others from literature, we have added two tables for comparison in the ESI† (Table S2 and S3). It can be observed that in general, the overpotential values are higher in thin films studied using high throughput screening techniques like SDC than in powder catalysts studied in a rotating disk electrode. This can be attributed to the different measurement conditions existing in the two scenarios. In the case of NiFeV system, the literature reports values of lamellar double hydroxide (as a powder film) with huge surface areas compared to our dense PVD thin films.
However, in a high throughput experiment on dense thin films like ours, we can assure that all the catalysts experience the same process and measurement conditions making comparisons among them very reliable. Compared to other real SDC combinatorial works, the overpotential measured for a flat surface are among the lowest @ 10 mA cm−2. Moreover, as far as our knowledge, we have systematically investigated a NiFeV trimetallic system using a combinatorial approach for the first time.
Conclusion
Various transition metal dopants have been explored for the enhancement of Ni based catalysts for OER. However, a comprehensive comparison over a large range of element ratios is very rare and incomplete. We have carried out a deliberate analysis on a series of bimetallic (NiFe) and trimetallic (NiFeV) alloys over a considerable span of compositional space in order to validate the synergistic modulation of activity by compositional tuning. We have used a combinatorial tool for obtaining fast, reliable, and reproducible results regarding the OER activity of the multimetallic material libraries. Thereby, we could achieve a higher level of data acquisition and comprehensive exploration of the compositional space compared to other reports, which would require a significantly larger number of experiments, time, and resources. A PVD technique was optimised to grow thin film libraries with a compositional gradient and a custom-made SDC set up was used for the electrocatalytic OER analysis. A substantial “galvanic” effect of vanadium doping could be inferred from the overpotential heatmaps plotted across the library. The lattice distortions induced by the incorporation of V could be derived from the increase of the lattice parameter after doping addition, calculated from the GIXRD patterns. XPS analysis revealed considerable modification of the chemical environment inducive to better catalytic performance. Overall, our study directs the rapid design and development of new high performing catalysts.Materials and methods
Combinatorial thin film synthesis using PVD
The thin films that form the material libraries were fabricated on ITO substrates, which were 50 × 50 × 1 mm in size. The substrates were cleaned by washing in lens cleaning soap, followed by sonication for 5 minutes in the lens cleaning soap solution. The samples were then sonicated in acetone for 5 minutes, sonicated in MilliQ water for 5 minutes and dried under N2 stream and stored in MilliQ water. Prior to deposition the samples were plasma cleaned for 5 minutes (Diener Electronic Plasma Surface Technology) at maximum power with an Ar plasma. The substrates were then placed in an e-beam deposition system (Thermionics Inc.) for film fabrication. The materials, Ni 99.995%, Fe 99.95%, and V 99.7% (Kurt J. Lesker Co.) were placed in the deposition chamber in separate crucibles, and then co-deposited on the substrate to form the libraries. To control the gradient formation, barriers were placed inside the chamber at specific positions between the crucible and the substrates to control the vapor flux arriving on the sample as previously described.55 The base pressure was 1.2 × 10−6 torr, and during the deposition the pressure was 5 × 10−6 torr. The temperature was kept at 25 °C and the sample remained static to achieve the gradients within the libraries. For the Ni–Fe–V libraries, co-deposition of half the amount of the Ni took place together with the Fe and half of the Ni together with the V, sequentially. After the thin films were formed, the samples were removed from the deposition chamber and measured directly.Our samples are coated on a single 50 × 50 mm2 FTO plate, forming a gradient of compositions. This was considered as a 6 × 6 array of 36 discrete samples for sake of measurement. All measurements were made in a 1 M KOH solution with Hg/HgO as reference and Pt as counter using a SP-150 Biologic Workstation. Oxygen evolution activity of each of these samples was studied by serial scanning. The scanning procedure involved the following steps: (i) SDC moved to the point of interest, (ii) electrolyte was pumped in, (iii) electrochemical measurements was performed, (iv) electrolyte was pumped out.
We have converted all our measurements to the Reversible Hydrogen Electrode (RHE) scale for easy comparison. The potential used for conversion from Hg/HgO to RHE was decided by performing the HOR/HER reaction on a Pt disk electrode (potential window: −0.9 V to −0.2 V) in hydrogen saturated 1 M KOH. We performed CV measurements after obtaining a stable OCV. The average potential of crossover at zero current was calculated and this was used as the conversion factor.
We used the geometrical surface area (0.1256 cm2) for current normalization due to uniform roughness over the entire thin film sample.
The LSV plots shown in Fig. 4 are smoothed versions of the original data due to the limited number of data points measured. We are forced to put this limit, due to the large amount of data being measured which can overload and slow down the working of the system.
We performed CV measurements in the non-faradaic region for the calculation of Cdl at different scan rates ranging from 10 to 60 mV s−1.
GIXRD measurements were conducted with a Rigaku Smartlab workstation with a θ–2θ range of 10–90°. Slow scan rate measurements were performed within a range of 30–80° at few relevant points for better understanding. X-ray reflectivity (XRR) measurements and analysis by FFT were also performed using the Rigaku Smartlab workstation.
XPS was conducted with a Nexsa X-ray photoelectron spectrometer system (Thermo Scientific) with a monochromated Al Kα X-ray source. Measurements were taken at room temperature, under high vacuum (< 3.0 10−9 torr). A 400 μm spot size was used, with a pass energy of 40 eV, and a flood gun was operational for all measurements. The samples were placed whole on the sample holder, with the points measured from the edges using the Avantage (Thermo Scientific) software. Survey scans were performed before higher resolution scans for the respective regions for each element. The spectra were corrected to the adventitious carbon peak at 284.8 eV, before fitting with a Smart background (Avantage, a Shirley background with an added condition that background must be less than the spectrum energy).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Vineesh Thazhe Veettil for the fruitful scientific discussions and Manoj Shanmugasundaram for the XPS measurements.References
- J. Chow, R. J. Kopp and P. R. Portney, Science, 2003, 302, 1528–1531 CrossRef PubMed.
- S. Venkata Mohan and A. Pandey, Biomass, Biofuels, Biochemicals: Biohydrogen, 2nd edn, 2019, vol. 305, pp. 1–23 Search PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- D. G. Nocera and M. P. Nash, Proc. Natl. Acad. Sci. U. S. A., 2007, 104 Search PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- H. Over, Chem. Rev., 2012, 112, 3356–3426 CrossRef CAS PubMed.
- X. Yu, M. Zhang, W. Yuan and G. Shi, J. Mater. Chem. A, 2015, 3, 6921–6928 RSC.
- M. A. Oliver-Tolentino, J. Vázquez-Samperio, A. Manzo-Robledo, R. D. G. González-Huerta, J. L. Flores-Moreno, D. Ramírez-Rosales and A. Guzmán-Vargas, J. Phys. Chem. C, 2014, 118, 22432–22438 CrossRef CAS.
- Y. Pan, Y. Liu, J. Zhao, K. Yang, J. Liang, D. Liu, W. Hu, D. Liu, Y. Liu and C. Liu, J. Mater. Chem. A, 2015, 3, 1656–1665 RSC.
- S. Anantharaj, J. Kennedy and S. Kundu, ACS Appl. Mater. Interfaces, 2017, 9, 8714–8728 CrossRef CAS PubMed.
- L. Yan, L. Cao, P. Dai, X. Gu, D. Liu, L. Li, Y. Wang and X. Zhao, Adv. Funct. Mater., 2017, 27, 1–10 Search PubMed.
- F. Yan, Y. Wang, K. Li, C. Zhu, P. Gao, C. Li, X. Zhang and Y. Chen, Chem. – Eur. J., 2017, 23, 10187–10194 CrossRef CAS PubMed.
- Z. Yin, Y. Sun, C. Zhu, C. Li, X. Zhang and Y. Chen, J. Mater. Chem. A, 2017, 5, 13648–13658 RSC.
- Y. Wang, B. Zhang, W. Pan, H. Ma and J. Zhang, ChemSusChem, 2017, 10, 4170–4177 CrossRef CAS PubMed.
- R. Gao and D. Yan, Nano Res., 2018, 11, 1883–1894 CrossRef CAS.
- R. Gao and D. Yan, Adv. Energy Mater., 2020, 10, 1900954 CrossRef CAS.
- M. Gong and H. Dai, Nano Res., 2014, 8, 23–39 CrossRef.
- M. Gorlin, J. F. De Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6(1), 1–7 Search PubMed.
- J. R. Swierk, S. Klaus, L. Trotochaud, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2015, 119, 19022–19029 CrossRef CAS.
- F. Song and X. Hu, Nat. Commun., 2014, 5(1), 4477 CrossRef CAS PubMed.
- W. Ma, R. Ma, C. Wang, J. Liang, X. Liu, K. Zhou and T. Sasaki, ACS Nano, 2015, 9, 1977–1984 CrossRef CAS PubMed.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377–384 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- J. Landon, E. Demeter, N. Inoǧlu, C. Keturakis, I. E. Wachs, R. Vasić, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef CAS.
- A. U. Vijayakumar, N. Aloni, V. T. Veettil, G. Rahamim, S. S. Hardisty, M. Zysler, S. Tirosh and D. Zitoun, ACS Appl. Energy Mater., 2022, 5, 4017–4024 CrossRef CAS.
- T. Lam Pham, H. Kino, K. Terakura, T. Miyake, K. Tsuda, I. Takigawa and H. Chi Dam, Sci. Technol. Adv. Mater., 2017, 18, 756–765 CrossRef PubMed.
- Z. Zand, P. Salimi, M. R. Mohammadi, R. Bagheri, P. Chernev, Z. Song, H. Dau, M. Görlin and M. M. Najafpour, ACS Sustainable Chem. Eng., 2019, 7, 17252–17262 CrossRef CAS.
- Y. Yang, Y. Ou, Y. Yang, X. Wei, D. Gao, L. Yang, Y. Xiong, H. Dong, P. Xiao and Y. Zhang, Nanoscale, 2019, 11, 23296–23303 RSC.
- Q. Li, X. Liu, Z. Zheng, G. Chen, R. Ma and H. Wan, Dalton Trans., 2022, 51, 11098–11107 RSC.
- R. Wei, X. Bu, W. Gao, R. A. B. Villaos, G. MacAm, Z. Q. Huang, C. Lan, F. C. Chuang, Y. Qu and J. C. Ho, ACS Appl. Mater. Interfaces, 2019, 11, 33012–33021 CrossRef CAS PubMed.
- C. T. Moi, S. Bhowmick, T. K. Sahu and M. Qureshi, Electrochim. Acta, 2021, 370, 137726 CrossRef CAS.
- Z. Wang, W. Liu, Y. Hu, L. Xu, M. Guan, J. Qiu, Y. Huang, J. Bao and H. Li, Inorg. Chem. Front., 2019, 6, 1890–1896 RSC.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- K. N. Dinh, P. Zheng, Z. Dai, Y. Zhang, R. Dangol, Y. Zheng, B. Li, Y. Zong and Q. Yan, Small, 2018, 14, 1–9 CrossRef PubMed.
- P. Zhao, L. Ma and J. Guo, J. Phys. Chem. Solids, 2022, 164, 110634 CrossRef CAS.
- K. Bera, A. Karmakar, S. Kumaravel, S. Sam Sankar, R. Madhu, H. N. Dhandapani, S. Nagappan and S. Kundu, Inorg. Chem., 2022, 61, 4502–4512 CrossRef CAS PubMed.
- W. Wan, H. Wu, Z. Wang, G. Cai, D. Li, H. Zhong, T. Jiang, C. Jiang and F. Ren, Appl. Surf. Sci., 2023, 611, 155732 CrossRef CAS.
- H. S. Chavan, C. H. Lee, A. I. Inamdar, J. Han, S. Park, S. Cho, N. K. Shreshta, S. U. Lee, B. Hou, H. Im and H. Kim, ACS Catal., 2022, 12, 3821–3831 CrossRef CAS.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9, 1–12 CrossRef PubMed.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 8(15), 1703341 CrossRef.
- Q. Cao, Y. Yuan, K. Wang, W. Huang, Y. Zhao, X. Sun, R. Ding, W. Lin, E. Liu and P. Gao, J. Colloid Interface Sci., 2022, 618, 411–418 CrossRef CAS PubMed.
- D. Böhm, M. Beetz, C. Kutz, S. Zhang, C. Scheu, T. Bein and D. Fattakhova-Rohlfing, Chem. Mater., 2020, 32, 10394–10406 CrossRef.
- M. S. Ansari and H. Kim, Fuel, 2021, 304, 121309 CrossRef CAS.
- S. Guerin, B. E. Hayden, C. E. Lee, C. Mormiche and A. E. Russell, J. Phys. Chem. B, 2006, 110, 14355–14362 CrossRef CAS PubMed.
- S. Guerin, B. E. Hayden, D. Pletcher, M. E. Rendall, J. P. Suchsland and L. J. Williams, J. Comb. Chem., 2006, 8, 791–798 CrossRef CAS PubMed.
- C. Zhang, X. Zhang, K. Daly, C. P. Berlinguette and S. Trudel, ACS Catal., 2017, 7, 6385–6391 CrossRef CAS.
- C. Zhang, C. P. Berlinguette and S. Trudel, Chem. Commun., 2016, 52, 1513–1516 RSC.
- L. Zhou, A. Shinde, D. Guevarra, M. H. Richter, H. S. Stein, Y. Wang, P. F. Newhouse, K. A. Persson and J. M. Gregoire, J. Mater. Chem. A, 2020, 8, 4239–4243 RSC.
- A. Shinde, R. J. R. Jones, D. Guevarra, S. Mitrovic, N. Becerra-Stasiewicz, J. A. Haber, J. Jin and J. M. Gregoire, Electrocatalysis, 2015, 6, 229–236 CrossRef CAS.
- K. C. Neyerlin, G. Bugosh, R. Forgie, Z. Liu and P. Strasser, J. Electrochem. Soc., 2009, 156, B363 CrossRef CAS.
- H. N. Barad, M. Alarcón-Correa, G. Salinas, E. Oren, F. Peter, A. Kuhn and P. Fischer, Mater. Today, 2021, 50, 89–99 CrossRef CAS.
- X. Cao, E. Johnson and M. Nath, ACS Sustainable Chem. Eng., 2019, 7, 9588–9600 CrossRef CAS.
- C. G. Morales-Guio, L. Liardet and X. Hu, J. Am. Chem. Soc., 2016, 138, 8946–8957 CrossRef CAS PubMed.
- A. T. Bell, Chapter 3: Understanding the Effects of Composition and Structure on the Oxygen Evolution Reaction (OER) Occurring on NiFeOx Catalysts, 2019.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- M. Görlin, P. Chernev, J. F. De Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- A. Mu, C. Scheu, A. Pokharel, S. Bo, T. Bein and D. Fattakhova-rohlfing, ACS Nano, 2015, 9, 5180–5188 CrossRef PubMed.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380–5387 CrossRef CAS.
- P. Chakthranont, J. Kibsgaard, A. Gallo, J. Park, M. Mitani, D. Sokaras, T. Kroll, R. Sinclair, M. B. Mogensen and T. F. Jaramillo, ACS Catal., 2017, 7, 5399–5409 CrossRef CAS.
- Y. He, T. Yu, H. Wen and R. Guo, Dalton Trans., 2021, 50, 9746–9753 RSC.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- T. Tian, H. Gao, X. Zhou, L. Zheng, J. Wu, K. Li and Y. Ding, ACS Energy Lett., 2018, 3, 2150–2158 CrossRef CAS.
- Y. Deng, Y. Lu, R. Dai, M. Xiang, Z. Zhang, X. Zhang, Q. Zhou, H. Gu and J. Bai, J. Colloid Interface Sci., 2022, 627, 215–223 CrossRef CAS PubMed.
- T. A. Edison, Electrolyte for alkaline storage batteries, U.S. Pat., US876445A, 1908 Search PubMed.
- D. A. Corrigan and S. P. Maheswari, Electrochem. Soc. Ext. Abstr., 1985, 85–1, 934–935 Search PubMed.
- A. Van der Ven, D. Morgan, Y. S. Meng and G. Ceder, J. Electrochem. Soc., 2006, 153, A210 CrossRef CAS.
- S. Y. Lee, I. S. Kim, H. S. Cho, C. H. Kim and Y. K. Lee, Appl. Catal., B, 2021, 284, 119729 CrossRef CAS.
- L. Wang, V. A. Saveleva, M. J. Eslamibidgoli, D. Antipin, C. Bouillet, I. Biswas, A. S. Gago, S. S. Hosseiny, P. Gazdzicki, M. H. Eikerling, E. R. Savinova and K. A. Friedrich, ACS Appl. Energy Mater., 2022, 5, 2221–2230 CrossRef CAS.
- S. Chakrabarty, I. Offen-Polak, T. Y. Burshtein, E. M. Farber, L. Kornblum and D. Eisenberg, J. Solid State Electrochem., 2021, 25, 159–171 CrossRef CAS.
- R. Li, P. Ren, P. Yang, Y. Li, H. Zhang, A. Liu, S. Wen, J. Zhang and M. An, J. Colloid Interface Sci., 2023, 631, 173–181 CrossRef CAS PubMed.
- J. G. Baker, J. R. Schneider, J. A. Garrido Torres, J. A. Singh, A. J. M. MacKus, M. Bajdich and S. F. Bent, ACS Appl. Energy Mater., 2019, 2, 3488–3499 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00368j |
| This journal is © The Royal Society of Chemistry 2023 |