
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing crystal structures of dicarbamate phase change materials to inform structural design†
Samantha L.
Piper
 a,
Craig M.
Forsyth
a,
Mega
Kar
b,
Luke A.
O’Dell
b,
Jisheng
Ma
c,
Jennifer M.
Pringle
b,
Douglas R.
MacFarlane
a and
Karolina
Matuszek
*a
a,
Craig M.
Forsyth
a,
Mega
Kar
b,
Luke A.
O’Dell
b,
Jisheng
Ma
c,
Jennifer M.
Pringle
b,
Douglas R.
MacFarlane
a and
Karolina
Matuszek
*a
aSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: Karolina.Matuszek@monash.edu
bInstitute for Frontier Materials, Deakin University, Burwood Campus, Burwood, Victoria 3125, Australia
cMonash X-ray Platform & Department of Materials and Engineering, Monash University, Clayton, Victoria 3800, Australia
First published on 1st September 2023
Abstract
The isothermal nature of latent energy storage makes phase change materials (PCMs) uniquely suited to facilitate highly efficient “Thermal Batteries” for the storage of renewable energy. Nevertheless, suitable PCMs for this application are scarce, which is largely due to the challenges of targeting a high enthalpy of fusion among the various other property requirements of this application, including a Tm between 100 and 220 °C and the excellent thermal stability required for long-term operation at these temperatures. Key to targeting the diverse suite of properties needed is a comprehensive understanding of the structure–property relationships that govern them. Here, we probe the molecular origins of the thermal properties of five aliphatic dicarbamate PCMs that melt in this intermediate temperature range, for two of which we have previously demonstrated excellent thermal stability indicating long lifetimes in this application. With the use of single-crystal X-ray crystallography, in situ powder X-ray diffraction and static solid-state nuclear magnetic resonance spectroscopy, for the first time we study the solid-state structures of the dicarbamate PCMs and correlate structural features with packing arrangements and intermolecular interactions with thermal properties. The results elucidate structural features that can be incorporated into new PCMs to achieve favourable thermal properties for renewable energy applications, as well as some features which may encourage solid-state transitions in these compounds.
Introduction
Since its discovery by Joseph Black while observing a pot of water boiling isothermally in 1762,1 the property of latent heat has been recognised as a ubiquitous feature of all first order phase transitions. Materials utilising this property, named phase change materials (PCMs), have found various energy applications including in electronic devices,2 cold-chain transportation,3 and space4 and water5 heating. The suitability of a PCM for a given application is determined by the nature of the transition (solid–solid, solid–liquid, liquid–gas, etc.), the temperature of the transition, Tt, and its associated enthalpy change, ΔHt.Despite their diverse application in a range of contexts, PCMs have untapped potential for facilitating an increased share of renewables in the global energy system.6 In recent years, the ability of intermediate temperature solid–liquid PCMs (i.e. Tm = 100–220 °C) to accommodate the storage of solar PV and excess wind energy in “Thermal Batteries” has been recognised,7 with temperatures within this range optimising the performance of the heat to power conversion (discharge) step.8 PCMs are favourable over conventional sensible heat storage materials in these technologies due to the isothermal nature of the energy storage process, which results in narrow operating temperature ranges that can benefit efficiency.9 This temperature range has hence become a target for new materials.
While this emerging application of PCMs offers exciting potential for meaningful global impact, targeting this temperature range creates an additional design challenge. Proposed PCMs, which are typically organic, require exceptional thermal stability to facilitate a long lifetime in application. This is no minor task, given the accelerated rates of any decomposition reactions when operating at these elevated temperatures. Proposed materials must also exhibit all other properties necessary for PCM applications, including an appropriate Tm, good thermal conductivity, low-flammability and toxicity, etc. Crucially, to facilitate an efficient thermal energy storage system, a given PCM should also exhibit a high enthalpy of fusion, ΔHf.
To meet these various design requirements, it is crucial that the structure–property relationships governing the target properties are well-understood. While the molecular mechanisms underpinning properties such as thermal conductivity10 and heat capacity11 are well-known, and the effect of different structural features on Tm are relatively well-established12 (with some computational methods even offering accurate predictions13), approaches to predicting ΔHf remain under-developed. This is largely due to the complexity of the phenomenon, as ΔHf is determined by the difference in free energy between the solid and liquid states, and is therefore highly sensitive to the intricacies of molecular packing and crystal symmetry; the absence of an a priori approach to predicting crystal structures hinders reliable estimations of this property. As is common for supramolecular systems,14 the development of high ΔHf materials is therefore often a result of the synergistic effects of serendipity and rational design.
Nevertheless, various correlations between chemical structures, intermolecular interactions and ΔHf have been identified that can aid the design of new PCMs. For example, hydrogen bonds have been recognised as dominant interactions in many high ΔHf materials, including glycerol (Tm = 18 °C, ΔHf = 200 J g−1),15 sugar alcohols (e.g. erythritol, Tm = 117 °C, ΔHf = 380 J g−1)16 and protic organic salts (e.g. guanidinium methanesulfonate, Tm = 208 °C, ΔHf = 190 J g−1).17 Notably, the latter two material types display appropriate melting temperatures for intermediate PCM applications and have been studied in this context. Unfortunately, despite the possibility of very high energy densities,18 supercooling tendencies that inhibit the energy release mechanism in sugar alcohols has prohibited their practical application thus far. Guanidinium methanesulfonate, on the other hand, evades the supercooling issue often associated with extensively hydrogen bonded systems, possibly indicating the importance of complementary interactions that guide crystal packing in these materials (in this case, Coulombic). Similarly to hydrogen bonds, coordination bonds between protons and metals cations can be favourable for achieving high ΔHf materials with intermediate temperature melting points (e.g. in metal-salt hydrates19 and metal–organic coordination complexes20). However, phase instability hinders cyclability of metal-salt hydrates21 while metal–organic coordination complexes utilise less abundant, or rare, transition metals.
Van der Waals interactions may also contribute to high ΔHf through their cumulative contributions to crystal packing, despite being inherently weaker in nature. In fact, van der Waals interactions dominate in paraffins (e.g. eicosane: Tm = 36.3 °C, ΔHf = 247 J g−1),22 which are the most common PCMs used currently. Nevertheless, this highlights the need for new material discovery.
A combination of these various interactions can also result in high ΔHf materials – fatty acids, for example, interact via intermolecular hydrogen bonds and van der Waals interactions and typically exhibit high ΔHf (e.g. stearic acid, Tm = 71 °C, ΔHf = 208 J g−1).23 However, this requires a synergism between interactions that can be challenging to achieve by design; competing interactions may result in geometrical frustration and high energy crystal lattices,24 resulting in low values of ΔHf. Further demonstrating the complexities of this property, the distribution of functional groups about a molecule can have remarkable effects on ΔHf, for example repulsive interactions between functional groups (e.g. oxygen atoms in hydroxyl groups) can also destabilise a crystal lattice. The significance of this is demonstrated by comparing the ΔHf of sugar alcohol isomers galactitol (ΔHf = 273 J g−1) and iditol (ΔHf = 127 J g−1), which differ only by the distribution of the hydroxyl groups about the carbon chain.25 In some cases, the variation in ΔHf for a set of isomers can be much greater than the variation in Tm for the same compounds.26
In a previous paper,27 we reported five aliphatic dicarbamate PCMs (Fig. 1), and determined the practical applicability of the two highest ΔHf materials (C18-hx and C18-MDP) for intermediate temperature PCM applications through thermal stability studies and sustainability assessments. Here, we use synchrotron single-crystal X-ray crystallography to elucidate the relationships between molecular structure, crystal packing and ΔHf in these materials. We also probe the nature of the solid–solid transitions observed in two of the materials, C18-hx and C18-MDP, with the use of in situ PXRD and variable temperature 1H NMR experiments, to understand the impact of these additional transitions on ΔHf and the suitability of the materials as PCMs. Notably, materials exhibiting such solid-state transitions are often targeted in other energy storage applications, such as solid-state battery electrolytes. In the final section, we consider the effects of structural variations on thermal properties from a thermodynamic perspective, elucidating structural features that can be used to tailor ΔHf and Tm alongside other design requirements in the development of next-generation PCMs.
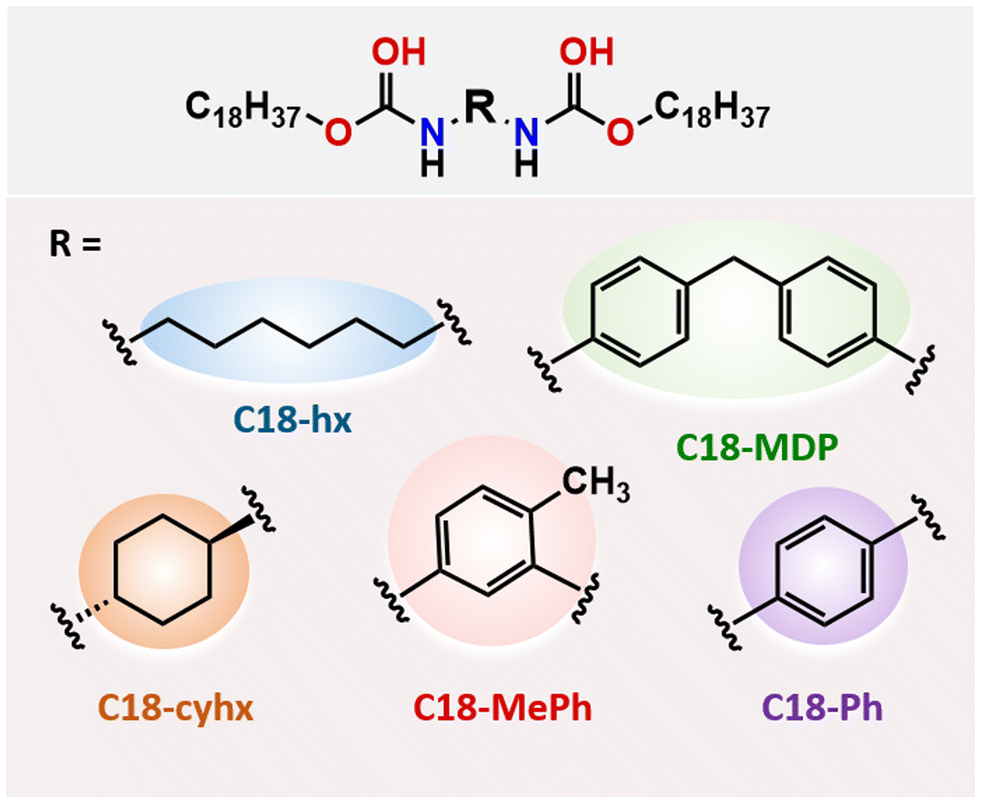 | ||
| Fig. 1 Chemical structures of the five dicarbamate PCMs studied here. | ||
Results and discussion
1. Thermal properties
The thermal properties of the five materials studied here are summarised in Table 1. The DSC traces are shown in Fig. 2.| Compound | T m °C ± 2 °C | T s–s °C ± 2 °C | ΔHf J g−1 ± 5% | ΔHf kJ mol−1 ± 5% | ΔHs–s kJ mol−1 ± 5% | ΔSf J mol−1 K−1 ± 5% | ΔSs–s J mol−1 K−1 ± 5% | ΔStotal J mol−1 K−1 ± 5% | N (melting) |
|---|---|---|---|---|---|---|---|---|---|
| a As the thermal properties of C18-MePh are dependent on the ramp rate, properties determined at heating rates of 1 °C min−1, 10 °C min−1 and 20 °C min−1 are reported for this compound. Due to the breadth of the endothermic transitions for some compounds, the transition temperatures, Tt, are determined by the temperature at peak maximum instead of peak onset. The value N is derived from the Boltzmann equation and describes the ratio of the number of molecular conformations in the liquid state over the number of conformations in the solid state, calculated from the entropy of melting ΔSf, as discussed in Section 5. | |||||||||
| C18-hx | 119 | — | 200 | 142 | — | 326 | — | 362 | 8 × 1018 |
| C18-cyhx | 156 | 134 | 68 | 48 | 21 | 112 | 52 | 164 | 7 × 105 |
| C18-MDP | 129 | — | 147 | 116 | — | 289 | — | 289 | 1 × 1015 |
| C18-Ph | 149 | — | 135 | 95 | — | 224 | — | 224 | 5 × 1011 |
| C18-MePha | |||||||||
| 1 °C min−1 | 105 | — | 144 | 100 | — | 265 | — | 265 | 7 × 1013 |
| 10 °C min−1 | 104 | 85, 92 | 113 | 81 | 1, 29 | 214 | 2, 80 | 296 | 1 × 1011 |
| 20 °C min−1 | 104 | 67, 80, 92 | 115 | 82 | 4, 4, 20 | 218 | 11, 10, 55 | 294 | 2 × 1011 |
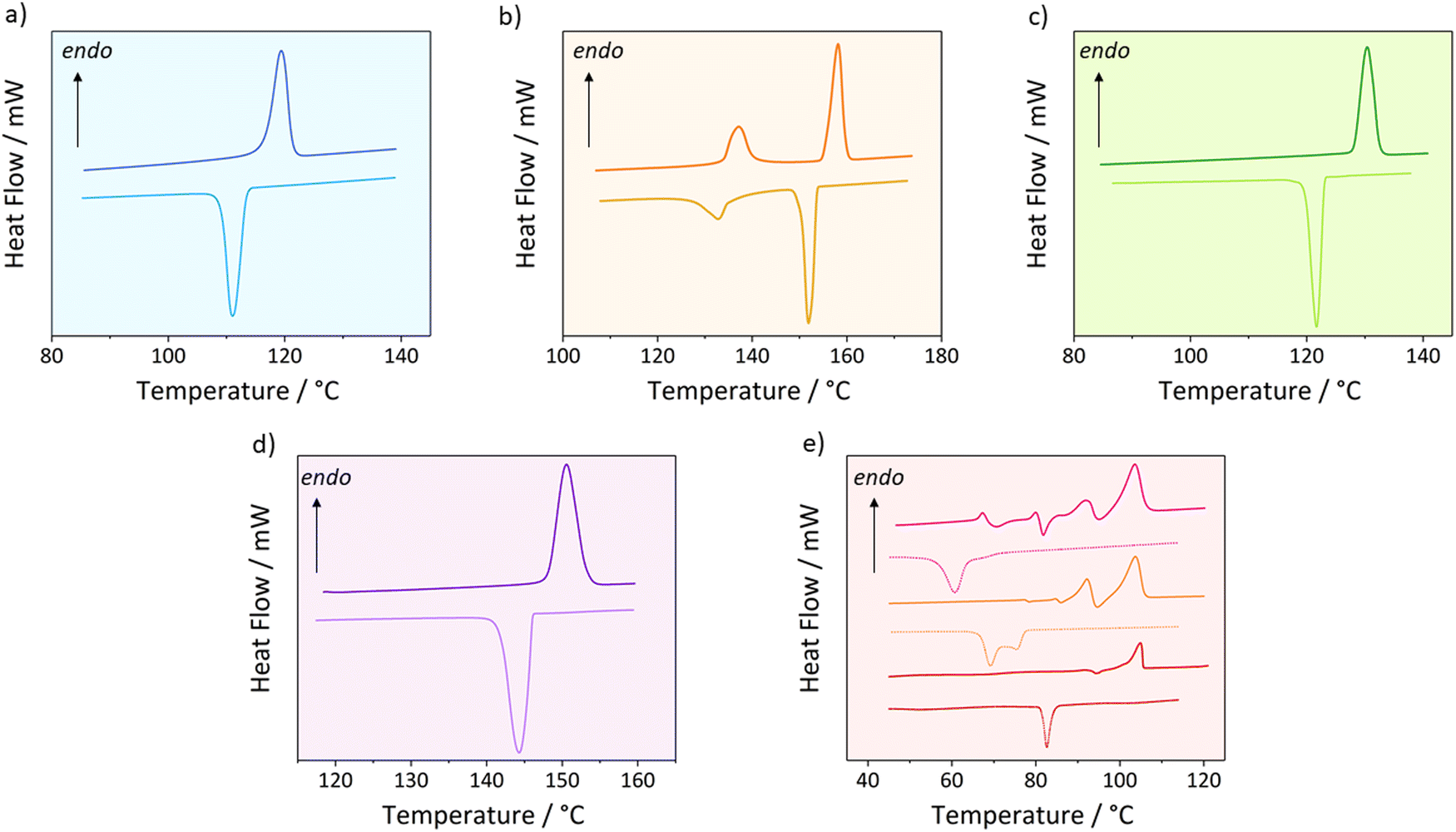 | ||
| Fig. 2 DSC traces showing the second heating cycle for C18-hx (a), C18-cyhx (b), C18-MDP (c), C18-Ph (d) and C18-MePh (e). (e) shows the DSC curves of the second heating cycle of C18-MePh at ramp rates of 1 °C min−1, 10 °C min−1, and 20 °C min−1 (bottom to top), as the features of the traces vary depending on the ramp rate used. | ||
Since all of the studied compounds have very similar molecular weights, we focus our discussion here on the chemically meaningful kJ mol−1 quantity, rather than the J g−1 that is often reported. When comparing compounds of vastly different molecular weights, this quantity may obscure the differences, since important interactions may not scale linearly with molecular weight. In that case the more practically important J g−1 is used.
The DSC traces of C18-hx, C18-MDP and C18-Ph demonstrate simple melting/crystallisation behaviour, with single endo- and exothermic peaks on the heating and cooling cycles respectively. ΔHf is highest for C18-hx (142 kJ mol−1), followed by C18-MDP (116 kJ mol−1) and C18-Ph (95 kJ mol−1). Interestingly, Tm follows the reverse order for these three materials, with values of 119 °C for C18-hx, 129 °C for C18-MDP and 149 °C for C18-Ph.
C18-cyhx also adheres to the above trend, having the lowest ΔHf and highest Tm of the five compounds (ΔHf = 48 kJ mol−1, Tm = 156 °C). However, the DSC trace of C18-cyhx shows an additional endothermic solid–solid transition on heating prior to its melting transition, with a ΔHs–s of 21 kJ mol−1 (Ts–s = 136 °C). An exothermic peak attributed to the reverse transition is observed on the subsequent cooling cycle (Fig. 2b).
The thermal behaviour of C18-MePh is more complex than that described for the previous four compounds, with varying transition behaviour observed by DSC when the sample is subjected to different heating and cooling rates (Fig. 2e). When cycled at a rate of 10 °C min−1 (which is consistent with the rate used for property determination of the other compounds), an exotherm is seen on the heating cycle at 92 °C (24 kJ mol−1), followed immediately by an exotherm at 95 °C (21 kJ mol−1), which transitions immediately into an endotherm attributed to melting at 103 °C (75 kJ mol−1). When cycled at an increased rate of 20 °C min−1, similar exo- and endotherms appear on the heating cycle, with additional lower temperature transitions (detailed in Table 1, additional information in Table S5, ESI†). Conversely, when the ramp rate is reduced to 1 °C min−1, only one endotherm is observed on the heating cycle, attributed to the melting transition (Tm = 104 °C, ΔHf = 100 kJ mol−1). Notably, a small exotherm of −15 kJ mol−1 can be seen on the heating cycle immediately before melting, at 94 °C (Fig. 2e). The dependence of these solid–solid transitions on the ramp rate is unlike the solid–solid transition behaviour observed in C18-cyhx, indicating the nature of the transitions is unique between the two compounds, as further discussed in Section 4.
In the next section we analyse the single-crystal structures of these compounds in order to correlate packing arrangements with the observed thermal properties.
2. Single-crystal X-ray crystallography
2.1. Molecular conformations
C18-hx, C18-MDP, C18-cyhx and C18-Ph all crystallise in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. C18-hx, C18-cyhx and C18-Ph all have half of one molecule in the asymmetric unit (ASU), with trans orientation of the urethane functionalities (Fig. 3). The urethane functionalities are tilted relative to the plane of the core groups in C18-cyhx and C18-Ph, which are almost isostructural, whereas the urethane group is almost in plane with the C6 core group in C18-hx (at a tilt of 10.1°). As a result, the molecules of C18-cyhx and C18-Ph are near linear (Fig. 3b and d), while C18-hx exists in a Z conformation (Fig. 3a).
space group. C18-hx, C18-cyhx and C18-Ph all have half of one molecule in the asymmetric unit (ASU), with trans orientation of the urethane functionalities (Fig. 3). The urethane functionalities are tilted relative to the plane of the core groups in C18-cyhx and C18-Ph, which are almost isostructural, whereas the urethane group is almost in plane with the C6 core group in C18-hx (at a tilt of 10.1°). As a result, the molecules of C18-cyhx and C18-Ph are near linear (Fig. 3b and d), while C18-hx exists in a Z conformation (Fig. 3a).
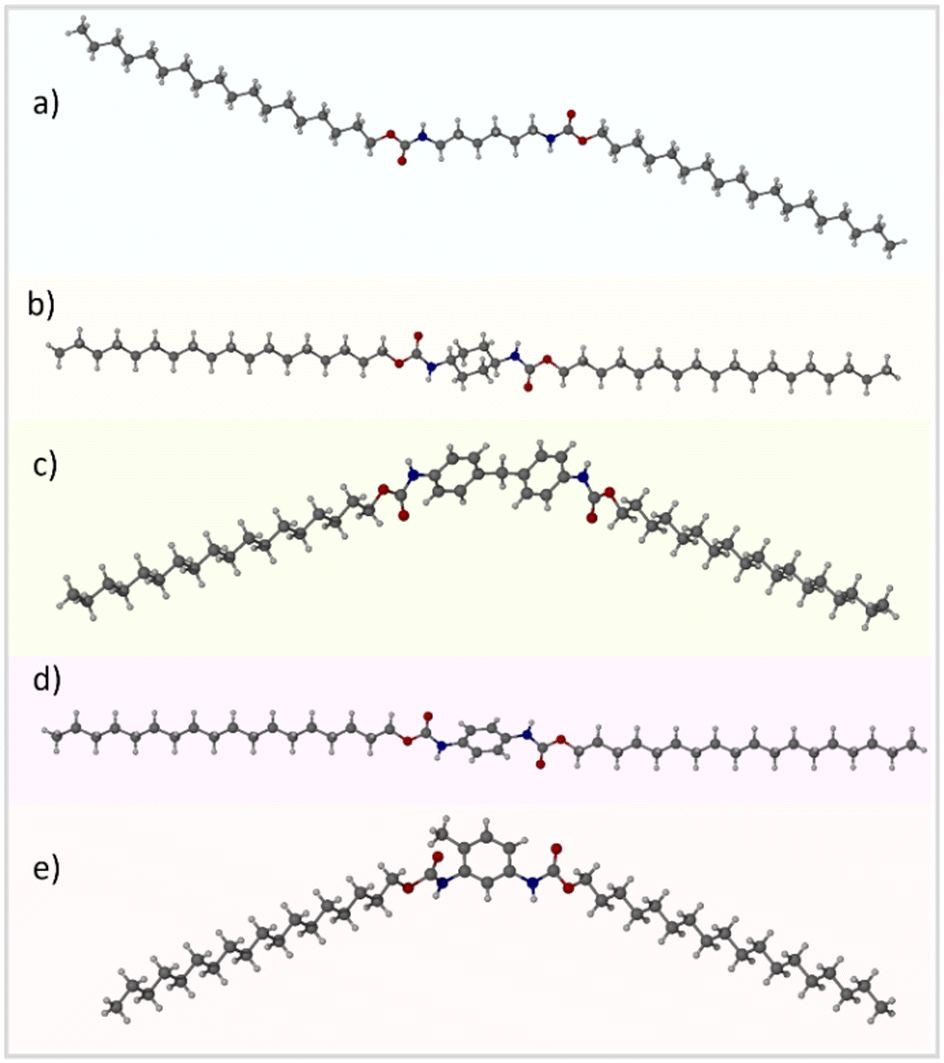 | ||
| Fig. 3 Crystallographically determined structures of C18-hx (a), C18-cyhx (b), C18-MDP (c), C18-Ph (d) and C18-MePh (e). | ||
C18-MDP, which has one molecule per ASU, exists in bent geometry as a result of the tetrahedral methylene carbon at the center of the MDP core group. In this structure, the urethane groups are in cis orientation, i.e. both carbonyl groups point in the same direction (Fig. 3c). The two phenylene rings of the MDP core are modelled with half occupancy over two perpendicular positions, indicating that the orientation of the rings alternate throughout the structure.
Disorder is also present in the structure of C18-MePh, which solves in the Pmc21 space group with half of two different molecules in ASU. In this structure, the methyl group of the toluene core and the urethane carbonyl groups are modelled over two positions, each with half occupancy. This indicates that the orientation of the toluene group alternates throughout the structure, and that the orientation of the urethane groups alternates to minimise steric hinderance. As observed in the structure of C18-MDP, the urethane groups of the C18-MePh molecule are in cis orientation relative to one another (Fig. 3e).
2.2. Hydrogen bonding interactions
In all five structures, each molecule participates in four intermolecular hydrogen bonding interactions between the urethane functionalities, where each N–H group donates a proton to a carbonyl group from a neighbouring molecule. These hydrogen bonds form a ladder motif, resulting in the formation of hydrogen-bonded sheets that propagate down the plane of the urethane functionalities (Fig. 4). The hydrogen bonds are of similar length in all five structures (d(N⋯O) = 2.8–3.0 Å, values shown in Fig. 4 and tabulated in Table S2, ESI†), but differ substantially in their angles. Hydrogen bonds are the most linear in C18-hx (∠(N–H⋯O) = 175°), and decrease in the following order: C18-MePh (av. 171°) > C18-cyhx (166°) > C18-MDP (161°) > C18-Ph (152°). Notably, the disorder of the urethane groups in C18-MePh leads to challenges identifying the hydrogen bonds; here it is assumed that the disordered components occupy alternating sites that yield hydrogen bonds with the most favourable geometries.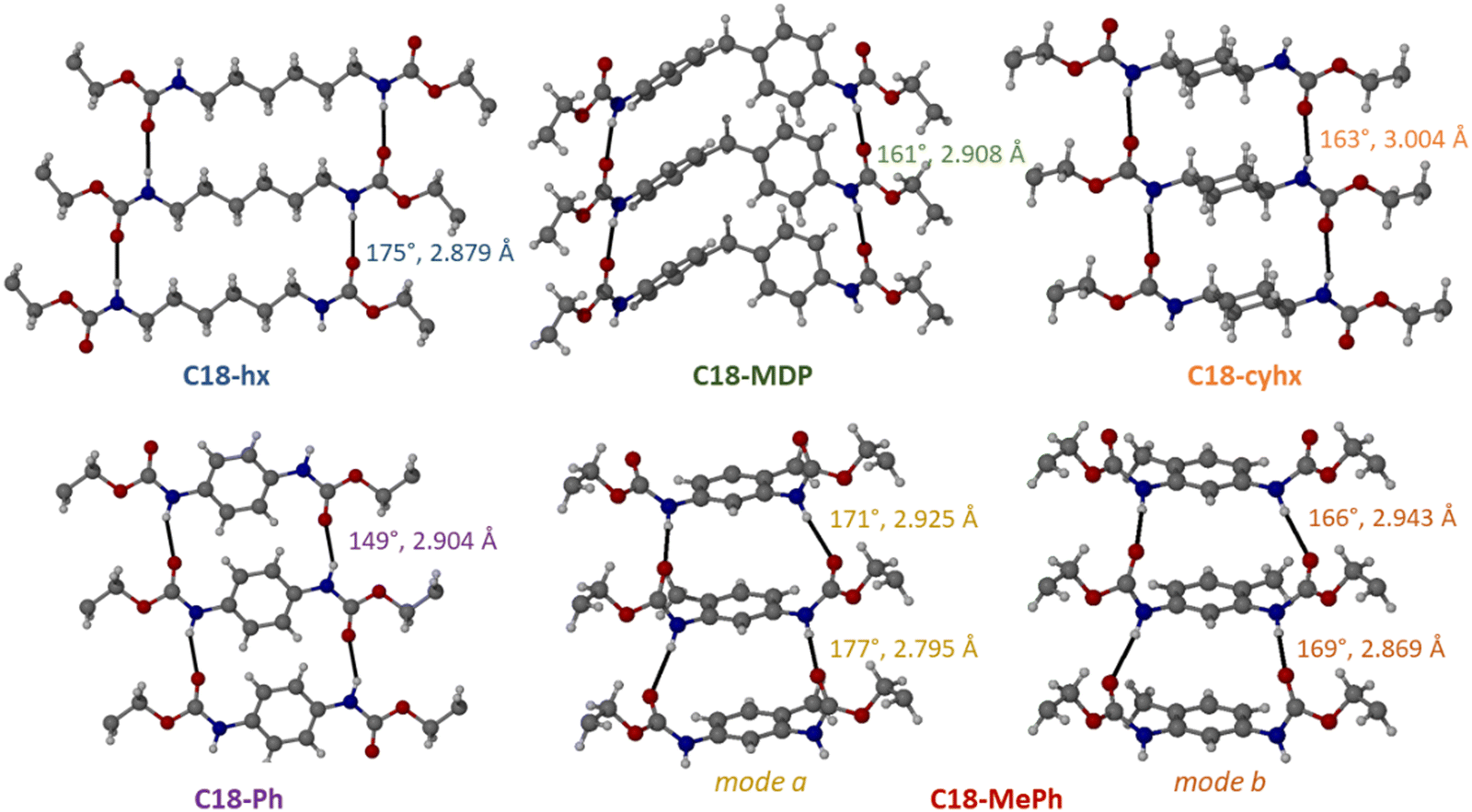 | ||
| Fig. 4 Hydrogen bonding and core group interactions in C18-hx, C18-MDP, C18-cyhx, C18-Ph and C18-MePh. There are four unique H-bonding interactions in C18-MePh as there are two half molecules in the asymmetric unit for this structure. | ||
These values suggest a broad trend that increasing the rigidity of the core group results in less favourable hydrogen bonding geometries (with the exception of C18-MePh).
2.3. Core group interactions
The hexane groups in C18-hx have edge-on van der Waals interactions with other hexane groups within the same hydrogen-bonded sheet, with a slight offset down the long-axis of the molecules as required to achieve the intermolecular hydrogen bonds (Fig. 4). Neighbouring C6 chains from adjacent sheets are offset face-on, with an interplanar distance of 3.11 Å. These groups are also offset down the long-axis of the molecules, as adjacent H-bonded sheets are offset by two carbons. Therefore, only four of the six carbons from the hexane core groups are aligned to participate in these inter-sheet interactions.In the structure of C18-cyhx, molecules from adjacent sheets are offset by four carbons, which hinders any inter-sheet interactions between core groups in this structure. Cyclohexane moieties from neighbouring molecules within the same H-bonded sheet pack in a parallel displaced chair conformation, with distances between the mean planes of cyclohexane groups of 3.94 Å.
C18-MDP, C18-Ph and C18-MePh all pack with π–π stacking as the dominant interaction between core groups of molecules within the same H-bonded sheets. In all three structures, phenylene groups within the same H-bonded sheet are parallel and offset, with centroid–centroid distances (Cg⋯Cg) of 5.075 Å in C18-Ph, 5.077 Å in C18-MDP, and av. 4.634 Å in C18-MePh (an average is taken for C18-MePh as there are two different packing modes as a result of the two half molecules in the ASU).
In the structure of C18-Ph, neighbouring H-bonded sheets are offset by four carbons and therefore phenylene groups do not align for any inter-sheet core group interactions in this structure, as was observed in the structure of C18-cyhx. Conversely, while H-bonded sheets in C18-MDP and C18-MePh are also offset (by two and three carbons respectively), the bent geometry of the molecules result in no net offset down the c axis of the molecules, and core groups therefore align for inter-sheet interactions. Phenylene groups from adjacent sheets in C18-MDP are parallel (Cg⋯Cg = 5.649 Å), while those in C18-MePh are almost perpendicular (av. 75°), resulting in a tilted T-shaped geometry with a centroid–centroid distance of 5.785 Å.
Notably, the two compounds without any inter-sheet core group interactions – C18-cyhx and C18-Ph – display the lowest values of ΔHf. This suggests the importance of strong intermolecular interactions, both within and between sheets, in achieving high ΔHf materials.
2.4. Hydrocarbon chain interactions and packing
All five structures show 3D packing of C18 chains, irrespective of the different molecular geometries. In the structures of C18-hx, C18-MDP, C18-cyhx and C18-Ph, the interacting hydrocarbon chains of the molecules are parallel and pack with triclinic symmetry of the methylene subcells (which is the smallest spatial unit of repetition down the long axis of the chains, illustrated in Fig. 5). Each chain therefore participates in four face-on interactions with neighbouring chains (two of which are significantly offset) and two (approximately) edge-on interactions with neighbouring chains. Due to the urethane group lying in a plane close to the plane of the C6 core group in C18-hx, interactions between chains from the same H-bonded sheet are edge-on in this structure, while those in C18-Ph, C18-cyhx and C18-MDP are offset face-on (Fig. 5). The volume of the methylene subcells can be used as an indication of the packing efficiency in these structures. This volume is smallest for C18-hx, at 8.4 Å3, followed by C18-MDP (8.7 Å3), C18-Ph (9.2 Å3) and C18-cyhx (11.55 Å3). A key difference in packing in these structures, which contributes to these different volumes via its impact on the b axis of the subcell – illustrated in Fig. 5 – is the extent of offsetting between neighbouring H-bonded sheets. The offset of two carbons between sheets in C18-hx and C18-MDP results in an only slightly elongated b axis; therefore, 16 of the 18 alkyl chain carbons align to interact with adjacent chains from neighbouring sheets. There is an increased offset between sheets in C18-cyhx and C18-Ph, of four carbons, which is reflected in the increased length of the b axes of their methylene subcells (relative to those of C18-hx and C18-MDP – Fig. 5). Therefore, only 14 of the 18 alkyl carbons are aligned to interact with alkyl chains from neighbouring sheets. There is therefore a greater number of attractive van der Waals forces between hydrocarbon chains in the structures of C18-hx and C18-MDP; this presumably contributes to the increased energy required to disrupt packing in these structures, which is reflected in their higher values of ΔHf.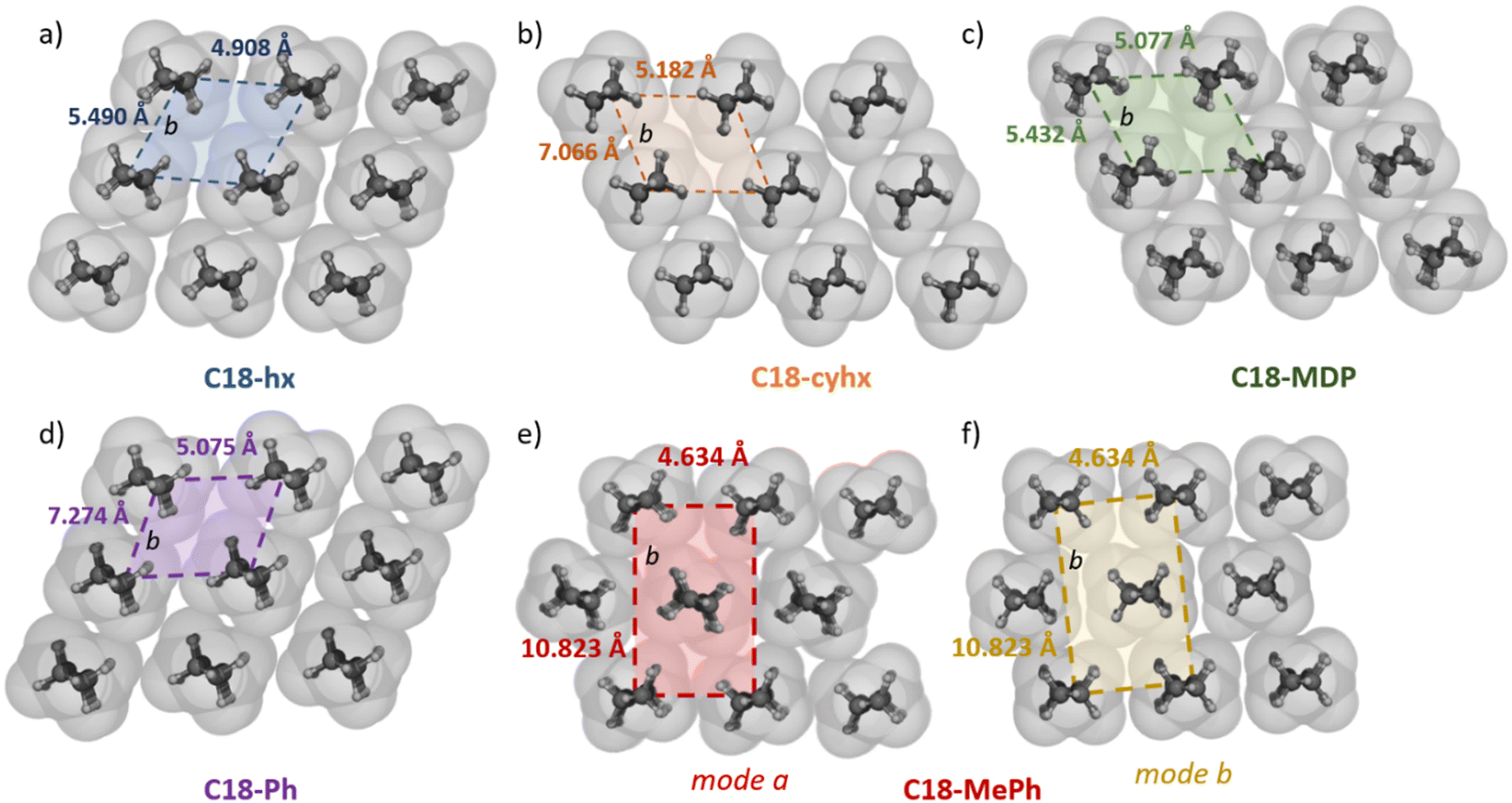 | ||
| Fig. 5 Packing of the hydrocarbon chains in C18-hx (a), C18-cyhx (b), C18-MDP (c), C18-Ph (d), and C18-MePh packing mode a and b (e) and (f), with van der Waals spheres of the atoms shown in grey. The b axis of the methylene subcells is highlighted to show the axis representing interactions between hydrocarbon chains from neighbouring H-bonded sheets, which is elongated by inter-sheet offsetting. The corresponding a axis represents interactions between hydrocarbon chains within the same H-bonded sheet. | ||
Interactions between hydrocarbon chains in C18-MePh are more complex. There are two modes of hydrocarbon chain packing in this structure as a result of the two half molecules that make up the asymmetric unit. In both cases, hydrocarbons pack with orthorhombic symmetry, with the same subcell dimensions (Fig. 5e). The key difference between these packing modes is in the orientation of the hydrocarbon chains relative to those from neighbouring sheets – planes between hydrocarbon chains are at an angle of 28° in mode a, and near-linear in the second packing arrangement, denoted mode b in Fig. 5. Interactions are therefore tilted edge-to-face in mode a, and edge-on and face-on in packing mode b. The volume of the methylene subcells for C18-MePh is 12.7 Å3, which is the largest subcell volume of the five compounds. Correlating these packing arrangements with molecular structures suggests that core groups that can have strong interactions in two dimensions – i.e. with neighbouring core groups within H-bonded sheets and from adjacent sheets – encourages the alignment of sheets such that hydrocarbon interactions are optimised. This results in closer chain packing, as reflected in the lower volume of the methylene subcells, which correlates with increasing ΔHf.
3. Powder X-ray diffraction and polymorphism
While single-crystal XRD is unmatched in its ability to depict solid-state structures and visualise corresponding molecular interactions, powder X-ray diffraction (PXRD) should also be used to ensure single-crystal structures are representative of the structure of bulk samples; this is especially necessary for the analysis of materials that are prone to polymorphism.This technique is also commonly used to elucidate molecular packing in materials that are difficult to crystallise. For example, in waxes and related aliphatic compounds – for which single crystals are typically challenging to obtain – PXRD is often used to probe the packing arrangements of aliphatic chains. These compounds are known to exhibit three basic symmetries of the methylene subcells, which results in characteristic diffraction peaks in the wide-angle (WAXD) region. These polymorphic symmetries are commonly denoted as α, where chain symmetry is hexagonal, β′, where chain symmetry is orthorhombic (as seen in the crystal structure of C18-MePh), and β, where chain symmetry is triclinic (as seen in the crystal structures of C18-hx, C18-MDP, C18-cyhx and C18-Ph). The varying free energies of these different crystalline arrangements can lead to vastly different melting properties (e.g. Tm and ΔHf). It has been reported that stability increases in the order α < β′ < β, and that melting points and enthalpies of fusion typically follow in the same order.28
The experimental PXRD patterns of the five dicarbamates studied here are shown in Fig. 6, alongside the patterns calculated from the single-crystal structures. The similarity between the calculated and experimental patterns of C18-MDP and C18-hx indicate that the crystal structures are representative of the bulk samples for these compounds. This is also likely the case for C18-Ph, as the single broad peak observed at ∼24° can presumably be attributed to the two peaks observed in the calculated pattern overlapping due to increased thermal motion of the hydrocarbon chains at room temperature (relative to the −173 °C used for single-crystal data collection). Nevertheless, this may indicate weaker interactions between chains and contribute to the lower ΔHf of C18-Ph relative to C18-hx and C18-MDP. These PXRD patterns therefore indicate that C18-hx, C18-MDP and C18-Ph pack with β-polymorphic symmetry in the bulk samples, as indicated by the single-crystal structures.
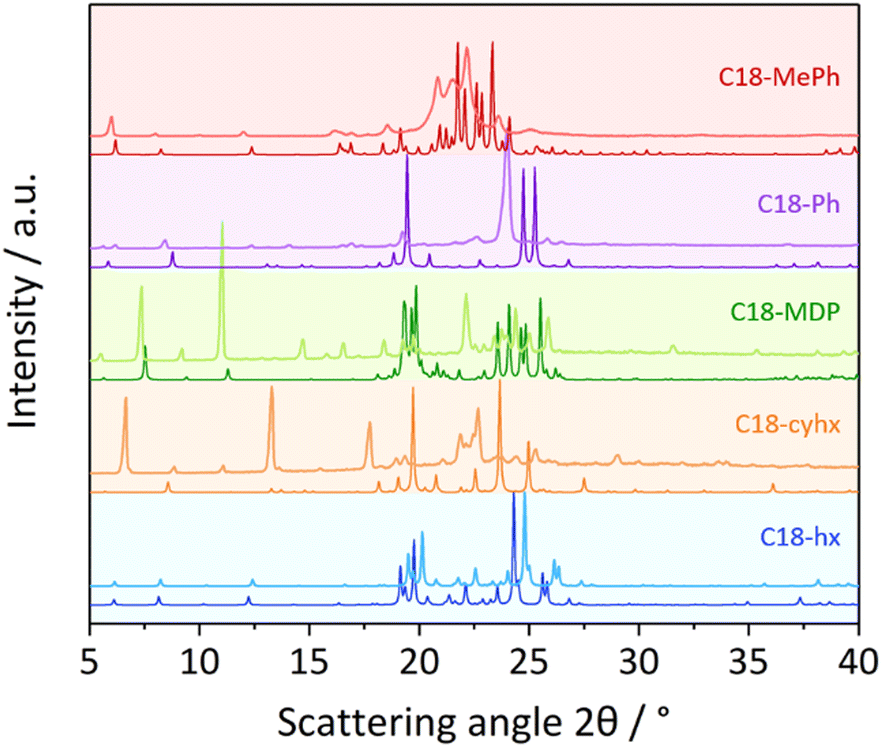 | ||
| Fig. 6 Experimental (lighter coloured lines) and calculated (darker lines) PXRD patterns of C18-hx, C18-cyhx, C18-MDP, C18-Ph and C18-MePh. Experimental patterns were collected at room temperature, while calculated patterns are predicted from crystal structures collected at −173 °C; some shifting of peaks to lower angles in the experimental patterns, particularly peaks in the lower angle region, is therefore expected. | ||
The breadth of the peaks in the experimental PXRD pattern of C18-MePh (Fig. 6, light red line) make it challenging to compare this pattern with the pattern calculated from the crystal structure (dark red line). The consistency of some peak positions, particularly in the lower angle region, indicate that there is some consistency between the crystal structure and the packing in the bulk sample. The breadth of the peaks nevertheless indicates some disorder in the sample – possibly a mixture of polymorphs, or dynamic disorder as a result of increased thermal motion at room temperature.
Similarly, there are clear differences between the calculated and experimental PXRD patterns of C18-cyhx, in this case in the peak positions. This indicates that the crystal packing in the bulk sample at room temperature is substantially different to that observed in the single-crystal structure.
Notably, C18-MePh and C18-cyhx are the two compounds that display solid-state transitions prior to melting in DSC experiments (Table 1, Fig. 2b and e). Understanding the nature of these transitions is useful to understand their impact on ΔHf and to determine whether these materials can still be useful for intermediate temperature solid–liquid PCM applications. Furthermore, materials exhibiting solid–solid transitions can often find use as energy storage materials in other applications, for example as solid-state electrolytes.29,30 Elucidating the relationship between molecular structure and solid-state transition behaviour is helpful in many aspects of materials design. Therefore, in the following section, the nature of these transitions is probed with in situ PXRD and variable temperature static solid-state 1H NMR experiments.
4. Probing the nature of the solid–solid transitions in C18-cyhx and C18-MePh
To provide an explanation of the different nature of the solid–solid transitions in C18-cyhx and C18-MePh, the variable temperature PXRD and static solid-state 1H NMR data, which are shown in Fig. 7, are discussed individually for the compounds in the following sections (with reference to the DSC traces of the compounds, Fig. 2b and e).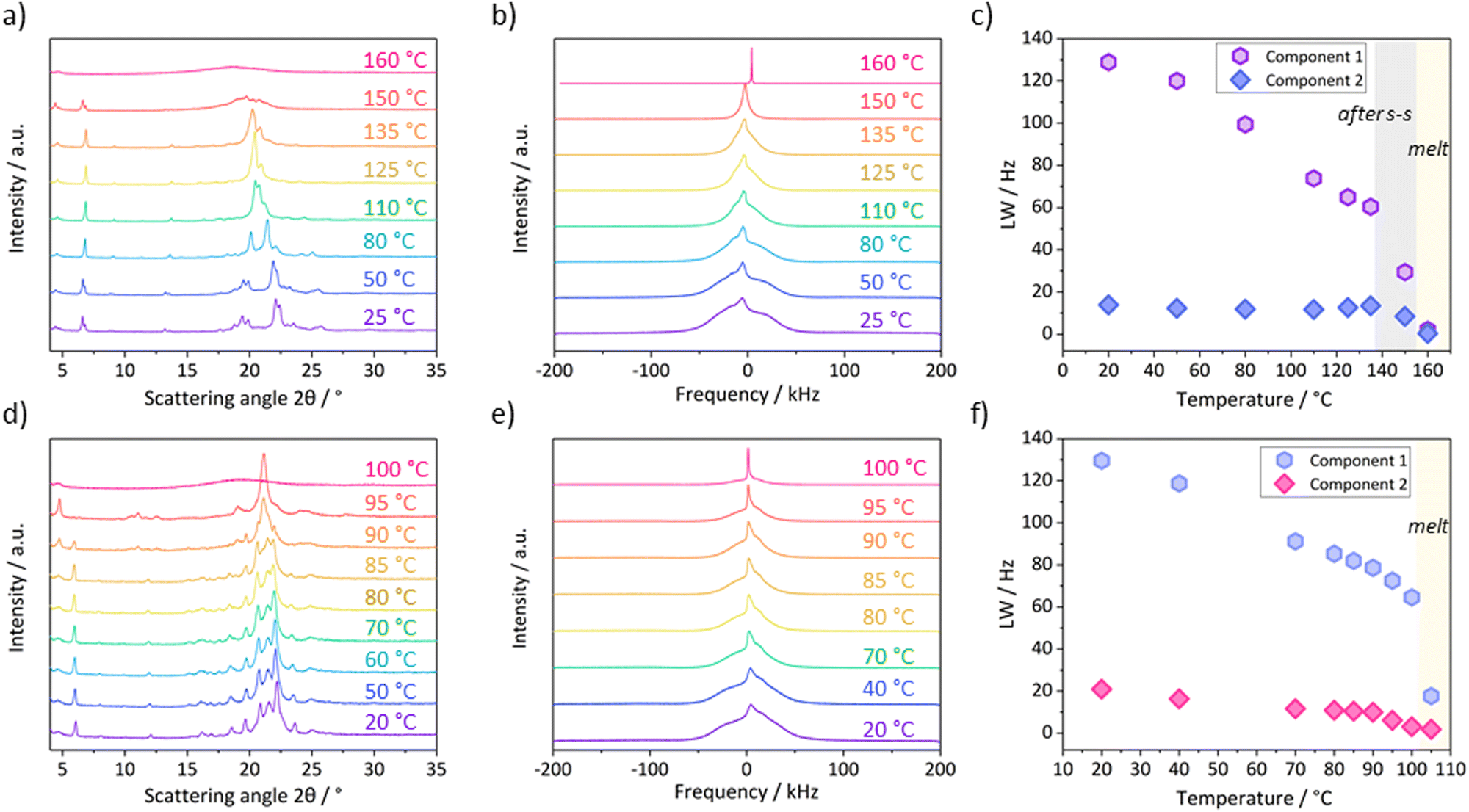 | ||
| Fig. 7 Variable temperature PXRD and static solid-state 1H NMR spectra of C18-cyhx (a) and (b) and C18-MePh (d) and (e). (c) and (f) show the line widths (LW) of the two component fits of the NMR spectra at various temperatures for C18-cyhx and C18-MePh respectively, where the broad component is Component 1 for both fits. | ||
4.1. C18-cyhx
The solid–solid transition (Ts–s = 136°) in C18-cyhx is reversible, indicating a transition between two enantiotropic polymorphs, and not the transition from a higher to lower energy packing arrangement such as α → β′ (as these polymorphs are known to be monotropically related and such a transition should therefore be irreversible). Enantiotropic polymorphism occurs when different polymorphs have the lowest free energy over defined temperature ranges (at constant pressure). Hence, transitions between enantiotropic polymorphs are thermally reversible.Enantiotropic polymorphism is observed for example to occur in cyclohexane, which is known to exist in at least two solid state forms (although additional metastable solid phases have also been identified31). Phase II of cyclohexane is stable below −87 °C, while Phase I is stable between −87 °C and Tm at 6 °C.32–34 Notably, Phase I of cyclohexane is reported to have plastic properties. This plastic phase has been termed a “rotator phase”, where molecules have rotational disorder about the lattice points of the face-centred cubic unit cell.35 Similar polymorphism and plasticity is also common for cyclohexane derivatives.36
It is therefore likely that the solid–solid transition in C18-cyhx represents the onset of a rotator phase, which is broadly defined as a crystalline phase with long range positional order in all three dimensions that lacks rotational order about an axis. Notably, rotator phases are also common in alkanes and their derivatives,37 in which case the rotational disorder is about the long axis of the molecules.38 In most cases, this rotational disorder is thought to arise from waggling motions or vibrations of limited amplitude, rather than free rotation.39
In accordance with what would be expected from the onset of rotational freedom during the transition to a more dynamic phase, the line-width of the broad component of the 1H NMR signal for C18-cyhx decreases substantially following the solid–solid transition (Fig. 7c). This decrease in line-width is attributed to the partial averaging of 1H–1H dipolar interactions due to increased dynamics in the sample.40 This decrease most likely arises through vibrations of the hydrocarbon chains and/or cyclohexane groups. The PXRD patterns collected at temperatures below and above this transition also indicate a clear change in structure, with new broader peaks of reduced intensity appearing in the WAXD region (Fig. 7a, red and pink lines), indicating a decrease in crystallinity across the transition.
Interestingly, the PXRD patterns of C18-cyhx also show changes to peak positions with increasing temperature below Ts–s, where no endothermic transitions are observed on the DSC trace. In the WAXD region, the PXRD patterns show a shifting of peaks towards a dominant broad peak at ∼21° (in the highest temperature pattern collected before Ts–s). This indicates a loss of symmetry in the structure with increasing temperature, consistent with the effects of increased thermal motion resulting in hexagonal (i.e. α-polymorphic) symmetry of the hydrocarbon chains. The gradual decrease of line-widths in the NMR spectra over this temperature range supports the hypothesis that there is increased molecular motion over this temperature range.
These results suggest that there is a significant degree of entropy in C18-cyhx prior to the onset of its melting transition, which explains this compound having the lowest entropy of fusion among the series. Weaker interactions between molecules in this phase likely also account for the low enthalpy of fusion of C18-cyhx (48 kJ mol−1). Notably, the nature of the solid–solid transition in C18-cyhx limits its applicability for solid–liquid PCM applications as a significant portion of the overall ΔH occurs well below Tm.
4.2. C18-MePh
In contrast to the solid–solid transition of C18-cyhx, the transitions prior to melting for C18-MePh are irreversible on cooling, suggesting a monotropic nature (i.e. the transition from lower to higher stability polymorphs, e.g. β′ → β). Monotropic polymorphism occurs when each polymorph has its own Tm, and is often exhibited by lipid crystals and their analogues.41 In the context of PCMs, this type of polymorphism may not be problematic if crystallisation can be controlled to ensure the polymorph with the desired thermal properties forms preferentially in the conditions of the application.Transitions from higher to lower energy monotropic polymorphs may occur through one of two pathways: solid-state transformation or melt-mediated transformation. Solid-state transformation is indicated by an exotherm on the heating cycle with no endotherm prior (as observed in the sample of C18-MePh heated at 1 °C min−1). Melt-mediated transformation occurs when a higher energy/metastable polymorph melts and a lower energy polymorph is subsequently crystallised (e.g. α → melt → β′) – this is indicated by an endotherm on the heating cycle that is followed immediately by an exotherm42 (as observed on the DSC traces of C18-MePh when faster ramp rates of 10 °C min−1 and 20 °C min−1 are used, Fig. 2e).
The rate of solid-state transformation is governed by the magnitude of the free energy barrier of the solid–solid transition (ΔG±s–s), while the rate of melt-mediated transformation is governed by the free energy barriers of melting (ΔG±m) and crystallisation (ΔG±c) (although ΔG±m is expected to be much smaller than ΔG±c and ΔG±c is therefore rate determining for this pathway).28 As such, slower heating rates favour solid-state transformation while melt-mediated transformation is favoured at faster heating rates. This explains the various endo- and exotherms on heating observed by DSC when ramp rates of 10 °C min−1 and 20 °C min−1 are used, compared to the single exo- and endotherm at the slower rate of 1 °C min−1.
As is also common for monotropic polymorphs, the higher energy (lower Tm) polymorphs of C18-MePh crystallise preferentially at faster cooling rates; an exotherm attributed to crystallisation is observed on the cooling cycle at 60 °C with 20 °C min−1 cooling, 70 °C with 10 °C min−1 cooling, and 80 °C at 1 °C min−1 cooling (Fig. 2e). Notably, the PXRD pattern of a sample rapidly cooled from the melt shows only one peak in the WAXD region, which supports the hypothesis that a high energy polymorph with hexagonal (i.e. α-polymorphic) symmetry crystallises preferentially at faster heating rates. Further probing the possibility of isolating different polymorphs through thermal treatment of the material, a sample was tempered at 84 °C for 15 minutes. The PXRD pattern of the sample after this process shows less peaks in the WAXD region when compared to the pattern of the sample crystallised from solution (Fig. S21, ESI†). These results indicate that the phase purity of this material is highly dependent on its thermal treatment, which is consistent with the DSC results discussed above. Given the slow rates of cooling that are expected in application as a PCM, the thermal behaviour of C18-MePh in application is likely to best resemble the 1 °C min−1 DSC curve.
The variable temperature PXRD patterns (Fig. 7d) support the thermal properties described above, with no significant differences in peak positions observed when the temperature is increased at 1 °C min−1 until 90 °C. At this temperature, the WAXD lines merge to form a dominant broad peak at 21°, and a new peak appears in the SAXD region at 5°. By 95 °C, the original SAXD peak at 6° is gone completely, indicating complete transformation into the highest Tm polymorph.
The variable temperature 1H NMR spectra also indicate that the solid–solid transitions in C18-MePh represent transitions between static polymorphs (and not the abrupt introduction of dynamics, i.e. a rotator phase), as there is no significant drop in the line-width of either of the fitted components over the temperature range prior to Tm (Fig. 7f).
For the design of new PCMs, it is valuable to understand the relationship between these solid-state transitions and the various structural features of the molecules. Notably, both the toluene and cyclohexane core groups have limited conformational flexibility, which may hinder the formation of low energy crystal lattices (with correspondingly high ΔHf melting transitions). Nevertheless, the polymorphic behaviour of C18-MePh may not hinder its practical applicability in solid–liquid PCM applications as the highest stability polymorph displays a high ΔHf and forms at the slower ramp rates that would be expected in application.
5. Melting thermodynamics: relationships between ΔSf, ΔHf and Tm
In linear aliphatic molecules, it is well-documented that increasing chain length results in a marked increase in ΔSf.26 This is predominately a result of the exponential increase in thermally accessible conformers as the number of torsionally rotatable (i.e. sp3 C–C) bonds in the molecule increases.43 As rotation is restricted in the solid state, the dynamic freedom introduced to these bonds upon melting thus leads to a vast increase in accessible molecular conformations on melting, which is reflected in higher ΔSf. This increase in ΔSf works to lower Tm, due to the relationship between the two properties, Tm = ΔHf/ΔS, indicated by the first law of thermodynamics.Similarly, the conformational flexibility of the core groups used here is expected to affect ΔSf and Tm. A good example is seen by comparing the Tm of C18-hx and C18-cyhx. Despite the dense packing and strong hydrogen bonds in its solid-state form, C18-hx has a melting temperature > 30 °C lower than C18-cyhx (Tm = 119 °C and 156 °C respectively). This is likely a result of the substantially higher change in entropy when going from the solid to liquid state for C18-hx compared to C18-cyhx (372 J mol−1 K−1 and 164 J mol−1 K−1 respectively), which thermo-dynamically favours a lower Tm.
This difference in ΔSf is attributable to two factors; the enthalpy term, i.e. the strength of the interactions in the solid-state that limit vibrational/rotational freedom (and therefore reduces the entropy of the solid state),44 and the ability of the core group to be multi-conformational, which yields a large number of possible conformations in the liquid state (i.e. increased liquid entropy).
This relationship between core group flexibility and Tm is observed across the series, with the compounds with less flexible core groups demonstrating higher values of Tm (with the exception of C18-MePh, which has the lowest Tm; in this case, the lower symmetry of the molecule likely contributes to lowering Tm12,32).
When considering the effect of the conformational flexibility of the core groups on the entropy of the melting transitions, it is useful to calculate the difference in the degree of disorder between the solid and liquid states from the Boltzmann equation ΔS = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(N). Here, R is the ideal gas constant, and the value of N describes the ratio of the number of possible arrangements/conformations per molecule in the liquid state over the number of possible arrangements/conformations per molecule in the solid-state. The values of N for the melting transitions of each of the compounds are shown in Table 1.
ln(N). Here, R is the ideal gas constant, and the value of N describes the ratio of the number of possible arrangements/conformations per molecule in the liquid state over the number of possible arrangements/conformations per molecule in the solid-state. The values of N for the melting transitions of each of the compounds are shown in Table 1.
For C18-hx, N = 8 × 1018, while for C18-cyhx, N = 7 × 105. This value of N for C18-cyhx, which is the lowest among the series, provides a reasonable explanation for this compound having the highest melting point, and is unsurprising given the high solid-state entropy of C18-cyhx indicated by the PXRD and 1H NMR experiments described in Section 4. Additionally, while there are more rigid core groups in the series, e.g. phenylene, the C–N bond between the urethane and cyclohexane groups cannot freely rotate, which would limit the conformational freedom of the cyclohexane ring and reduce the number of accessible conformations in the liquid state.
This observation of C18-hx and C18-cyhx is consistent with observations of the melting transitions of hexane and cyclohexane. The ΔHf of hexane is much higher than that of cyclohexane (13.1 kJ mol−1 compared to 2.6 kJ mol−1), which has been attributed to the larger surface area of hexane for intermolecular interactions26 and is surely related to the plasticity of cyclohexane prior to melting (see Section 4.1). Nevertheless, hexane melts at a much lower temperature than cyclohexane (Tm = −96 °C and Tm = 6 °C respectively), which is a result of the significantly higher ΔSf of hexane (ΔSf = 74 J mol−1 K−1 compared to 9.7 J mol−1 K−1 for cyclohexane). This is a result of both the high entropy of the solid state of cyclohexane, and the high entropy of the liquid state of hexane (due to its higher number of torsionally rotatable bonds).
Similar to the relationship between conformation flexibility and Tm, the series examined here shows a correlation between conformational flexibility and ΔHf. Omitting C18-MePh from the comparison due to the unusual thermal behaviour of this compound and its unique packing arrangement, the ΔHf of the compounds studied here increase as the conformational flexibility of the core groups increases. This relationship is consistent with the entropy-enthalpy compensation phenomenon, whereby a large increase in molecular motion (ΔSf) leads to large increases in intermolecular distances and therefore a large decrease in intermolecular interactions (ΔHf).26 Presumably, this is also related to the increased surface area of the more conformationally flexible core groups – which results in more intermolecular interactions between molecules – and, relatedly, the ability of the more flexible core groups to adopt solid state arrangements that optimise all core group, hydrogen bonding and van der Waals interactions.
The above discussion indicates the importance of conformational freedom in achieving high ΔHf for analogous materials, which may also serve as a feature that can be used to tailor Tm in novel phase change materials.
Conclusions
An enhanced understanding of the structure–property relationships that govern ΔHf will assist in the design of new phase change materials that meet the diverse property requirements of thermal energy storage applications. Here we have probed the molecular origins of the thermal properties of five aliphatic dicarbamate PCMs which demonstrate excellent practical properties for the storage of renewable thermal energy.Single-crystal and powder X-ray diffraction analyses suggest that low energy packing arrangements are achievable in materials that have sufficient conformational flexibility to optimise multiple types of interactions between molecules – in this case, conformation flexibility of core groups linking aliphatic chains results in favourable hydrogen bonding interactions as well as favourable van der Waals interactions between hydrocarbon chains and core groups. This is aided by the incorporation of functionalities that can adopt various molecular conformations without compromising the free energy of the solid-state structure. When this is not the case, various polymorphs may exist within a bulk sample, or solid-state transitions into disordered high temperature phases can occur, which reduces the strength of interactions in the solid state. This lessens the disruption to intermolecular interactions that occurs upon melting, resulting in low ΔHf materials.
In situ PXRD and static solid-state NMR experiments were used to probe the nature of the solid-state transitions in two of the materials studied here. These results suggest that the solid-state transitions in C18-MePh are between monotropic polymorphs, while the transition in C18-cyhx is between two enantiotropic polymorphs, where the high temperature (Phase I) polymorph has significant dynamic freedom. Correlating these properties to the structures of the core groups, this suggests that the toluene core group – while able to pack efficiently in its highest stability form – is more likely to crystallise into higher energy polymorphs at rapid cooling rates. As well as the rigidity of this core group, this may also be attributed to its asymmetry, indicating symmetry may be a feature that can be used to tailor Tm in the design of new PCMs. The enantiotropic transition in C18-cyhx, which resembles the thermal behaviour of pure cyclohexane, suggests that strong interactions between core groups is crucial for limiting motional freedom in the solid state prior to the melting transition.
Broadly, we provide here the basis for approaches to materials design that will assist the development of novel high ΔHf materials for the storage of renewable thermal energy. In particular, the structure–property relationships established here have the potential to rapidly expand the library of intermediate temperature PCMs when combined with machine learning approaches.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding from the Australian Research Council through its Linkage Project scheme (LP190100522) and collaborating organisation Energy Storage Pty Ltd. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.Notes and references
- J. Robinson, Lectures on the elements of chemistry, delivered in the University of Edinburgh, Philadelphia, 1806.
- V. Bianco, M. De Rosa and K. Vafai, Phase-Change Materials for Thermal Management of Electronic Devices, Appl. Therm. Eng., 2022, 214, 118839 CrossRef.
- Y. Zhao, X. Zhang and X. Xu, Application and Research Progress of Cold Storage Technology in Cold Chain Transportation and Distribution, J. Therm. Anal. Calorim., 2020, 139, 1419–1434 CrossRef CAS.
- M. Song, F. Niu, N. Mao, Y. Hu and S. Deng, Review on Building Energy Performance Improvement Using Phase Change Materials, Energy Build., 2018, 158, 776–793 CrossRef.
- E. Douvi, C. Pagkalos, G. Dogkas, M. K. Koukou, V. N. Stathopoulos, Y. Caouris and M. G. Vrachopoulos, Phase Change Materials in Solar Domestic Hot Water Systems: A Review, Int. J. Thermofluids, 2021, 10, 100075 CrossRef.
- International Renewable Energy Agency. Innovation Outlook: Thermal Energy Storage; Abu Dhabi, 2020.
- K. Matuszek, M. Kar, J. M. Pringle and D. R. Macfarlane, Phase Change Materials for Renewable Energy Storage at Intermediate Temperatures, Chem. Rev., 2023, 123, 491–514 CrossRef CAS PubMed.
- D. Steger, C. Regensburger, B. Eppinger, S. Will, J. Karl and E. Schlücker, Design Aspects of a Reversible Heat Pump-Organic Rankine Cycle Pilot Plant for Energy Storage, Energy, 2020, 208, 118216 CrossRef CAS.
- O. Dumont, G. F. Frate, A. Pillai, S. Lecompte and V. Lemort, Carnot Battery Technology: A State-of-the-Art Review, J. Energy Storage, 2020, 32, 101756 CrossRef.
- K. Yuan, J. Shi, W. Aftab, M. Qin, A. Usman, F. Zhou, Y. Lv, S. Gao and R. Zou, Engineering the Thermal Conductivity of Functional Phase-Change Materials for Heat Energy Conversion, Storage, and Utilization, Adv. Funct. Mater., 2020, 30, 1904228 CrossRef CAS.
- D. Rihani and L. Doraiswamy, Estimation of Heat Capacity of Organic Compounds from Group Contributions, Ind. Eng. Chem. Fundam., 1965, 4, 17–21 CrossRef CAS.
- S. H. Yalkowsky, Rule and the Prediction of Melting Point, J. Pharm. Sci., 2014, 103, 2629–2634 CrossRef CAS PubMed.
- S. H. Yalkowsky and D. Alantary, Estimation of Melting Points of Organics, J. Pharm. Sci., 2018, 107, 1211–1227 CrossRef CAS PubMed.
- R. W. Saalfrank, E. Uller, B. Demleitner and I. Bernt, in Molecular Self-Assembly, Molecular Self-Assembly Organic Versus Inorganic Approaches. Structure and Bonding, ed. M. Fuiita, Springer, Berlin, Heidelberg, 1st edn, 2000, pp. 149–175 Search PubMed.
- S. N. Gunasekara, J. Stalin, M. Marçal, R. Delubac, A. Karabanova, J. N. W. Chiu and V. Martin, Erythritol, Glycerol, Their Blends, and Olive Oil, as Sustainable Phase Change Materials, Energy Procedia, 2017, 135, 249–262 CrossRef CAS.
- S. Höhlein, A. König-Haagen and D. Brüggemann, Thermophysical Characterization of MgCl2· 6H2O, Xylitol and Erythritol as Phase Change Materials (PCM) for Latent Heat Thermal Energy Storage (LHTES), Materials, 2017, 10, 444 CrossRef PubMed.
- K. Matuszek, R. Vijayaraghavan, M. Kar, S. Mahadevan and D. R. MacFarlane, Pyrazolium Phase-Change Materials for Solar-Thermal Energy Storage, ChemSusChem, 2021, 14, 2757–2762 CrossRef CAS PubMed.
- T. Inagaki and T. Ishida, Computational Design of Non-Natural Sugar Alcohols to Increase Thermal Storage Density: Beyond Existing Organic Phase Change Materials, J. Am. Chem. Soc., 2016, 138, 11810–11819 CrossRef CAS PubMed.
- R. Pilar, L. Svoboda, P. Honcova and L. Oravova, Study of Magnesium Chloride Hexahydrate as Heat Storage Material, Thermochim. Acta, 2012, 546, 81–86 CrossRef CAS.
- R. D. McGillicuddy, S. Thapa, M. B. Wenny, M. I. Gonzalez and J. A. Mason, Metal–Organic Phase-Change Materials for Thermal Energy Storage, J. Am. Chem. Soc., 2020, 142, 19170–19180 CrossRef CAS PubMed.
- P. Dixit, V. J. Reddy, S. Parvate, A. Balwani, J. Singh, T. K. Maiti, A. Dasari and S. Chattopadhyay, Salt Hydrate Phase Change Materials: Current State of Art and the Road Ahead, J. Energy Storage, 2022, 51, 104360 CrossRef.
- M. M. Kenisarin, Thermophysical Properties of Some Organic Phase Change Materials for Latent Heat Storage. A Review, Sol. Energy, 2014, 107, 553–575 CrossRef CAS.
- Z. Zhang, Y. Yuan, N. Zhang and X. Cao, Thermophysical Properties of Some Fatty Acids/Surfactants as Phase Change Slurries for Thermal Energy Storage, J. Chem. Eng. Data, 2015, 60, 2495–2501 CrossRef CAS.
- Y. Han, Y. Shokef, A. M. Alsayed, P. Yunker, T. C. Lubensky and A. G. Yodh, Geometric Frustration in Buckled Colloidal Monolayers, Nature, 2008, 456, 898–903 CrossRef CAS PubMed.
- T. Inagaki and T. Ishida, Computational Analysis of Sugar Alcohols as Phase-Change Material: Insight into the Molecular Mechanism of Thermal Energy Storage, J. Phys. Chem. C, 2016, 120, 7903–7915 CrossRef CAS.
- D. H. Williams, D. P. O’Brien and B. Bardsley, Enthalpy/Entropy Compensation as a Competition between Dynamics and Bonding: The Relevance to Melting of Crystals and Biological Aggregates, J. Am. Chem. Soc., 2001, 123, 737–738 CrossRef CAS PubMed.
- S. L. Piper, C. M. Forsyth, M. Kar, C. Gassner, R. Vijayaraghavan, S. Mahadevan, K. Matuszek, J. M. Pringle and D. R. MacFarlane, Sustainable Materials for Renewable Energy Storage in the Thermal Battery, RSC Sustainable, 2023, 1, 470–480 RSC.
- C. Himawan, V. Starov and A. Stapley, Thermodynamic and Kinetic Aspects of Fat Crystallization, Adv. Colloid Interface Sci., 2006, 122, 3–33 CrossRef CAS PubMed.
- V. Armel, M. Forsyth, D. R. MacFarlane and J. M. Pringle, Organic Ionic Plastic Crystal Electrolytes; a New Class of Electrolyte for High Efficiency Solid State Dye-Sensitized Solar Cells, Energy Environ. Sci., 2011, 4, 2234–2239 RSC.
- H. Zhu, D. R. MacFarlane, J. M. Pringle and M. Forsyth, Organic Ionic Plastic Crystals as Solid-State Electrolytes, Trends Chem., 2019, 1, 126–140 CrossRef CAS.
- J. Dumas and N. Chanh, New Aspects of the Polymorphism of Cyclohexane Dispersed within an Emulsifying Medium, J. Therm. Anal., 1988, 34, 1083–1092 CrossRef CAS.
- J. Wei, Molecular Symmetry, Rotational Entropy, and Elevated Melting Points, Ind. Eng. Chem. Res., 1999, 38, 5019–5027 CrossRef CAS.
- R. J. Obremski, C. W. Brown and E. R. Lippincott, Vibrational Spectra of Single Crystals. Polymorphic Solids of Cyclohexane, J. Chem. Phys., 1968, 49, 185–191 CrossRef CAS.
- Y. Sataty and A. Ron, Far-Infrared Spectrum and Phase Transitions of Solid Cyclohexane, Chem. Phys. Lett., 1974, 25, 384–386 CrossRef CAS.
- N. P. Funnell, M. T. Dove, A. L. Goodwin, S. Parsons and M. G. Tucker, Local Structure Correlations in Plastic Cyclohexane—a Reverse Monte Carlo Study, J. Phys.: Condens. Matter, 2013, 25, 454204 CrossRef PubMed.
- G. Kabo, A. Kozyro, M. Frenkel and A. Blokhin, Solid Phase Transitions of the Cyclohexane Derivatives and the Model of Energy States of Molecules in Plastic Crystals. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect, Mol. Cryst. Liq. Cryst., 1999, 326, 333–355 CrossRef CAS.
- P. K. Mukherjee, Phase Transitions among the Rotator Phases of the Normal Alkanes: A Review, Phys. Rep., 2015, 588, 1–54 CrossRef CAS.
- E. B. Sirota, Supercooling, Nucleation, Rotator Phases, and Surface Crystallization of n-Alkane Melts, Langmuir, 1998, 14, 3133–3136 CrossRef CAS.
- D. Cholakova and N. Denkov, Rotator Phases in Alkane Systems: InBulk, Surface Layers and Micro/Nano-Confinements, Avd. Colloid Interface Sci., 2019, 269, 7–42 CrossRef CAS PubMed.
- H. Zhu and L. A. O’Dell, Nuclear Magnetic Resonance Characterisation of Ionic Liquids and Organic Ionic Plastic Crystals: Common Approaches and Recent Advances, Chem. Commun., 2021, 57, 5609–5625 RSC.
- K. Sato, L. Bayés-García, T. Calvet, M. À. Cuevas-Diarte and S. Ueno, External Factors Affecting Polymorphic Crystallization of Lipids, Eur. J. Lipid Sci. Technol., 2013, 115, 1224–1238 CrossRef CAS.
- N. Garti and K. Sato, Crystallization and Polymorphism of Fats and Fatty Acids, Marcel Dekker, New York, 1st edn, 2000, p. 1988 Search PubMed.
- L. Chan, G. M. Morris and G. R. Hutchison, Understanding Conformational Entropy in Small Molecules, J. Chem. Theory Comput., 2021, 17, 2099–2106 CrossRef CAS PubMed.
- M. S. Searle and D. H. Williams, The Cost of Conformational Order: Entropy Changes in Molecular Associations, J. Am. Chem. Soc., 1992, 114, 10690–10697 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2259815–2259817. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ma00434a |
| This journal is © The Royal Society of Chemistry 2023 |