
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSquare-wave pulse electrodeposition of gold nanoparticles for ethanol electrooxidation
Setia
Budi
 *a,
Annisa
Auliya
ab,
Suci
Winarsih
c,
Mohammad Hamzah
Fauzi
c and
Yusmaniar
a
*a,
Annisa
Auliya
ab,
Suci
Winarsih
c,
Mohammad Hamzah
Fauzi
c and
Yusmaniar
a
aDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Negeri Jakarta, Jl Rawamangun Muka, Jakarta Timur 13220, Indonesia. E-mail: setiabudi@unj.ac.id
bThe Centre for Science Innovation, Jakarta Timur 13120, Indonesia
cResearch Center for Quantum Physics, National Research, and Innovation Agency (BRIN), South Tangerang 15314, Indonesia
First published on 14th October 2023
Abstract
Gold nanoparticles (AuNPs) were synthesized as an electrocatalyst using a square-wave pulse deposition technique. By adjusting the applied upper potentials, we were able to control the size and distribution of the Au NPs. Analysis using the X-ray photoelectron spectroscopy technique confirmed the formation of metallic-phase Au particles on the fluorine-doped tin oxide (FTO) coated glass. Morphological studies show that at an upper potential of 0.00 V, fine particles were uniformly dispersed on the FTO substrate. With increasing potential, the particles underwent growth and coalescence, forming short chain-like structures. At potentials exceeding 0.25 V, the particle number decreased, and larger spherical particles were obtained, particularly at 1.00 V. Electrochemical impedance spectroscopy measurements exhibited low charge transfer resistance for the Au NPs synthesized at 0.25 V, producing high catalytic activity for the ethanol electrooxidation. The Au NPs electrocatalyst electrodeposited at low potential also shows a high poisoning resistance as indicated by the low jb/jf ratio value that resulted in catalytic stability as indicated by a slow current decay from the chronoamperometry measurements. The results contribute to the advancement of efficient and stable Au NPs electrocatalysts, particularly in ethanol electrooxidation.
Introduction
The utilization of metal catalysts in ethanol electrooxidation has garnered significant attention due to its potential application in direct ethanol fuel cells (DEFCs) for electricity generation. DEFCs are considered a promising energy source for portable electronic devices and public transportation systems.1 Several non-precious metals, including Sn, Bi, and Ni, have been investigated for their performance in ethanol electrooxidation. However, to address their limited catalytic properties and stability, these metals need to be combined with other metals, such as PtSn,2 PtSnRh,3 PtBi,4 RhBi,5 PdPtNi,6 CoNiB,7 CoNi,8 to improve their low catalytic properties and stability. Previous studies identified Platinum9–11 and Palladium12 as the efficient electrocatalysts for low-temperature DEFC applications. Nevertheless, the high cost, limited availability, and susceptibility to poisoning by intermediate species like carbon monoxide (CO) have impeded the widespread commercialization of these precious metal catalysts.13–15 In light of these challenges, gold (Au) has emerged as a viable alternative non-Pt catalyst, exhibiting favorable catalytic activity for ethanol oxidation.16 Moreover, Au catalysts demonstrated remarkable resistance to poisoning caused by adsorbed carbon monoxide (COads).17The field of catalyst development places significant emphasis on morphological control, given the pivotal role played by factors such as particle size, shape,18 facet energy,19 and atomic coordination20 in influencing catalyst performance. Consequently, various techniques have been devised to manipulate the morphology of nanoparticle-based catalysts, including chemical vapor deposition,21 ultrasound,22 hydrothermal,23 sol–gel,24 and electrodeposition.25 Among these methods, electrodeposition offers several advantages for synthesizing metal nanoparticles, including its cost-effectiveness, rapidity,26 and the ability to adjust various parameters to tailor deposit characteristics.
Gold nanoparticle synthesis can be accomplished through potentiostatic deposition, a technique that maintains a constant electrode potential, with the inclusion of various additives including hydrophilic polymer,27 phosphate buffer solution,28 tetramethylammonium tetrafluoroborate,29 dan L-cysteine.30 In this approach, particle size is carefully regulated by adjusting the applied potential and deposition time.31,32 Alternatively, granular gold particles can be fabricated in the presence of additives by sweeping the electrode potential using cyclic voltammetry technique.33 Conversely, the galvanostatic technique, which operates at a fixed current, has been employed to fabricate porous gold-nanocomposites utilizing sacrificial anodes.34,35 In comparison to the aforementioned electrodeposition techniques, the square-wave pulse technique offers a unique approach by controlling two different potentials that are continuously switched during the deposition process. This technique has been successfully utilized to modify the structure of Pt nanoparticles, transforming them from spherical to cube-like36 or dendritic shapes.37 Furthermore, it has been demonstrated that the square-wave pulse deposition technique enable precise control over the size and distribution of Pt nanocrystals, leading to enhanced catalytic activity for methanol electrooxidation.38 In the present study, we employed the square-wave pulse deposition technique to synthesize gold nanoparticles (Au NPs). Our approach involved the use of an additive-free electrolyte, enabling precise control over the size and distribution of the Au NPs by adjusting the upper deposition potential. This parameter adjustment had a profound impact on the catalytic activity of the ethanol electrooxidation.
Materials and methods
Materials
The materials used in this study were tetrachloroauric(III) acid (HAuCl4·3H2O), sulfuric acid (H2SO4), ethanol (C2H5OH, 96%), sodium hydroxide (NaOH), and potassium chloride (KCl) procured from Merck. The substrate used to deposit AuNPs was NSG Pilkington fluorine-doped tin oxide (FTO) coated glass with a sheet resistance of 7 Ω/sq.Methods
The AuNPs were electrodeposited onto the FTO using the square-wave pulse technique. An electrolyte solution containing 0.5 mM HAuCl4·3H2O and 0.5 M H2SO4 was used for the sample deposition. The electrodeposition process was controlled using an ER466 potentiostat connected to a three-electrode system. The FTO-coated glass substrate served as a working electrode, the Pt plate acted as the counter electrode, and Ag/AgCl (KCl 3 M) acted as a reference electrode. The upper potentials (Eu) were varied from 0.00 V and 1.00 V, while the lower potential was maintained at −0.50V. A pulse potential frequency of 2 Hz and pulse potential duration of 250 ms were employed as illustrated in the schematic diagram of the pulse potential waveform (Fig. 1). Prior to the electrodeposition process, the FTO substrate was thoroughly rinsed with ethanol and distilled water. The electrodeposition process was conducted at room temperature with a duration of 25 minutes. The as-prepared samples were rinsed with distilled water and dried. | ||
| Fig. 1 Schematic waveform of the pulse potential controlled during Au NPs deposition. | ||
The morphology of Au NPs deposits was examined by a field emission scanning electron microscope (FESEM, Thermo Scientific Quattro S). Elemental analysis of the samples was conducted using an energy-dispersive X-ray spectroscope (EDX, EDAX Apollo X). Structural study of the electrodeposited Au NPs was performed by X-ray diffractometer (XRD, PANalytical AERIS). The chemical state of the samples was determined by X-ray photoelectron spectroscope (XPS, PHI Quantera II). Impedance characteristic was analyzed by the electrochemical impedance spectroscopy technique using an electrochemical workstation (Corrtest CS310). The EIS measurements were carried out in a solution containing 0.5 M KCl with a frequency of 50 kHz to 0.1 Hz. The reference and counter electrode used for the EIS test were Ag/AgCl (KCl 3 M) and Pt wire, respectively. Then, the catalytic activity of the Au NPs in ethanol electrooxidation was determined using cyclic voltammetry (CV) and chronoamperometry (CA) techniques. The measurements were performed in a solution containing 1.0 M ethanol with 0.1 M NaOH as a supporting electrolyte. The CV dan CA measurements were controlled by an ER466 potentiostat connected to the Ag/AgCl electrode (3 M KCl) as a reference and Pt plate 8 × 7 mm as a counter electrode. During the CV measurements, the potential range used was −0.75 to 0.75 V, and the scan rate was set at 25 mV s−1. The CA measurements were carried out at 0.25 V for a duration of 9000 seconds.
Results and discussion
Micrographs of AuNPs, along with their particle size distribution from the samples synthesized with different upper potentials (Eu), are presented in Fig. 2. When the upper potential is set at 0 V, the synthesized Au NPs exhibit a distribution of very fine particles on the substrate surface (Fig. 2a). Upon increasing the potential to 0.25 V, the particles undergo growth and coalescence, forming short-chain-like structures with a spherical base shape (Fig. 2c). At 0.50 V, although the particles’ shape remains relatively consistent, their size increases, while the number of particles decreases (Fig. 2e). To further validate this analysis, FESEM examinations were also performed by tilting the sample at 50°. The obtained micrographs confirm the occurrence of particle coalescence with the large number of Au particles at 0.25 V (Fig. 3a), and an increase in particle size with a higher upper potential (Fig. 3b). Furthermore, at 1.00 V, the particle number decreases further, and larger nanosphere are observed (Fig. 2g). These results suggest that at higher upper potentials, nucleation decreases, resulting in an increased reaction rate due to the more positive potential, which promotes particle growth. Conversely, a relatively low potential leads to the formation of more nuclei, producing a larger number of fine particles, as depicted in Table 1.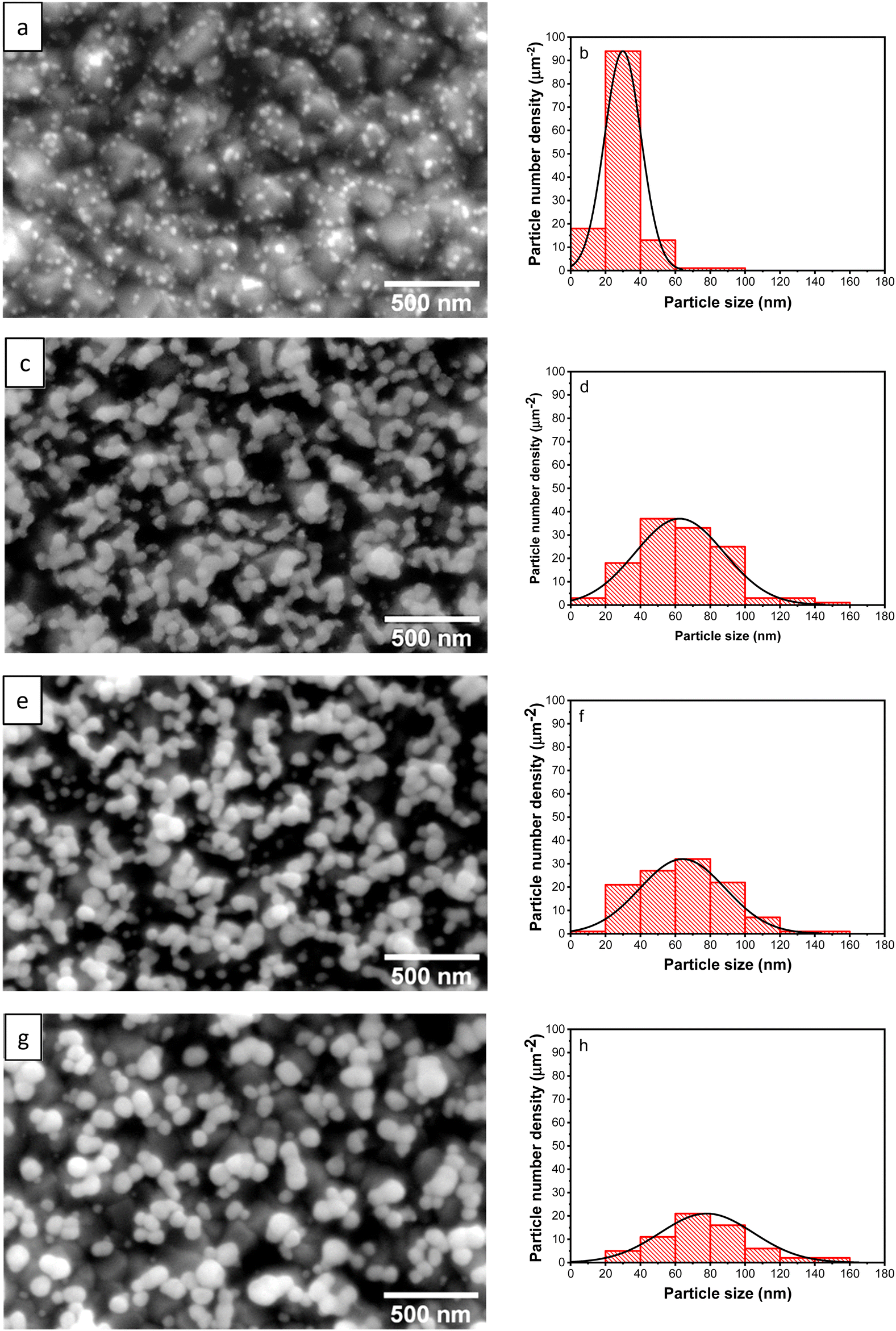 | ||
| Fig. 2 FESEM micrographs and particles size distribution of the Au NPs synthesized with different upper potentials 0.00 V (a) and (b), 0.25 V (c) and (d), 0.50 V (e) and (f), and 1.00 V (g) and (h). | ||
 | ||
| Fig. 3 Tilted FESEM micrographs of the Au NPs synthesized at upper potential 0.25 V (a) and 0.5 V (b); point analysis EDX spectrum (c) and Au mapping of the sample at 0.50 V (d). | ||
| E u (V) | Particles number (mm−2) | Average particle size (nm) | Crystallite size (nm) |
|---|---|---|---|
| 0.00 V | 1.013 × 1010 | 29.9 | 21.1 |
| 0.25 V | 1.018 × 1010 | 63.2 | 21.3 |
| 0.50 V | 9.102 × 109 | 64.7 | 22.6 |
| 1.00 V | 5.192 × 109 | 76.5 | 34.2 |
Fig. 3c represents the EDX spectrum of the deposited nanoparticles deposit, displaying prominent peaks corresponding to the presence of Au elements. Simultaneously, observable peaks corresponding to C, Sn, O, Al, and Si elements can be attributed to their existence in the FTO-glass substrate. This spectrum confirms the successful growth of the particles distributed over the substrate, as illustrated by the elemental mapping of the Au element shown in Fig. 3d.
To investigate the chemical state of deposited Au, the sample was further examined under an XPS. Fig. 4 presents the XPS core level spectrum, revealing peaks at binding energies of 83.7 and 87.4 eV, corresponding to the spin–orbit of 4f7/2 and 4f5/2 of Au0, respectively. This spectrum indicates that the particles consist of the metallic phase of Au.
 | ||
| Fig. 4 XPS spectrum of AuNPs synthesized at 0.50 V. | ||
Fig. 5 shows XRD patterns of the Au NPs deposits synthesized at different upper potentials. The observed peaks correspond to the face-centered cubic (FCC) structure of Au are identified at 2θ 38.15°, 44.35°, 64.63°, and 77.52°, corresponding to the (111), (200), (220), and (311) crystallographic planes, respectively (ICDD Au-00-004-0784). Other peaks in the pattern originate from the FTO-coated glass substrate. The crystallite size of the Au NPs was determined using the Scherrer eqn (1) based on the peak at 2θ 38.15° (111).
 | (1) |
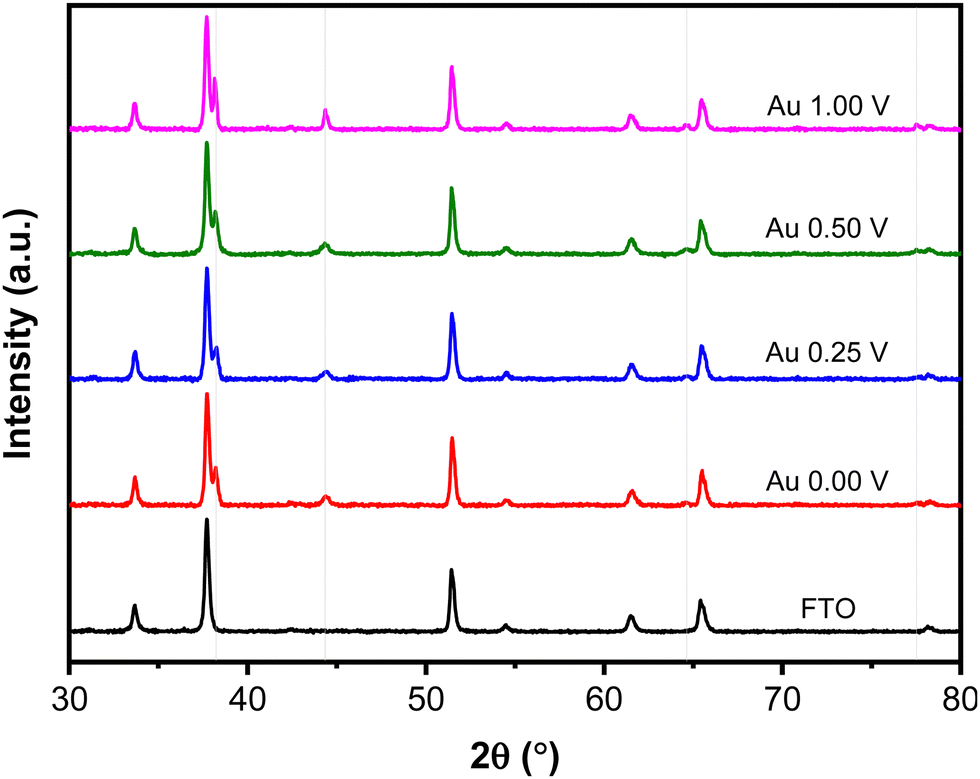 | ||
| Fig. 5 XRD patterns of Au NPs synthesized with different upper potentials. The grey lines indicate Au NPs peaks. | ||
The impedance characteristic of the Au NPs was investigated using the electrochemical impedance spectroscopy (EIS) technique. The Nyquist plots shown in Fig. 6 exhibit different arc radii, indicating the difference in charge transfer resistance (Rct) among the sample prepared with different upper potentials. Notably, the sample prepared at 0.25 V shows the lowest Rct value, while the highest Rct is observed from the sample prepared at 1.00 V. The presence of large numbers that coalesced and formed a short chain-like structure at 0.25 V is believed to enhance electron transfer kinetics within the system. Conversely, the sparse distribution of particles at 1.00 V hampered the electron transfer, resulting in an increase in charge transfer resistance on the surface. This hindered electron transfer can be attributed to the reduced number of active sites available for catalyzing the reaction, leading to less efficient charge transfer in the system.
 | ||
| Fig. 6 Nyquist plots of Au NPs synthesized with different upper potentials. | ||
The electrocatalytic activity of the electrodeposited Au NPs was evaluated in an alkaline electrolyte. The measured potentials versus the Ag/AgCl reference electrode were converted to the reversible hydrogen electrode (RHE) using eqn (2).
| ERHE = EAg/AgCl + 0.0591 × pH + EoAg/AgCl | (2) |
Fig. 7a shows the cyclic voltammograms recorded using Au NPs and FTO in an electrolyte containing ethanol. During the forward scan, a peak at 0.25 V, attributed to the oxidation of ethanol, was observed when employing the Au NPs electrode. In contrast to the bare FTO-glass substrate showing a flat curve, the presented voltammograms demonstrate the electrocatalytic activity toward ethanol oxidation within the system. To confirm that the observed peak is indeed attributed to the ethanol oxidation reaction, a CV measurement was conducted in an electrolyte without ethanol. As shown in Fig. 7b, the voltammogram exhibits a nearly flat response, indicating the absence of an oxidation peak. This unequivocally validates that the anodic peak observed in the presence of ethanol is intricately linked to the ethanol oxidation process facilitated by the Au NPs. During the backward scan, another peak appears at 0.07 V, indicating the oxidation of toxic intermediate species that are generated in the forward scan. These oxidized species could potentially include COads or other adsorbed species, which are known to form during the electrooxidation of ethanol.17
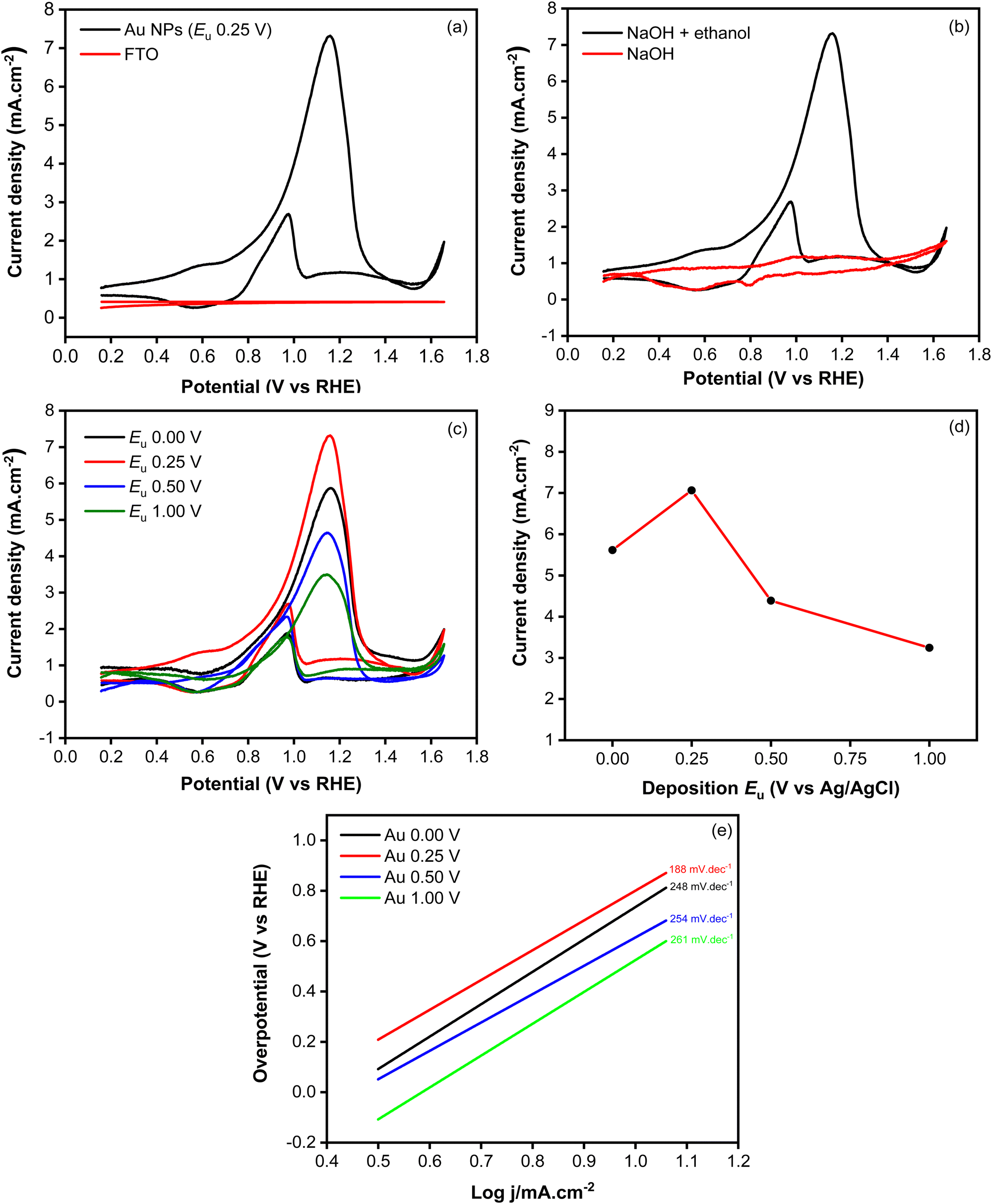 | ||
| Fig. 7 Cyclic voltammograms measured in the electrolyte containing 0.1 M NaOH and 1 M ethanol using Au NPs synthesized at 0.25 V and FTO (a); cyclic voltammograms measured in the absence and presence of ethanol using Au NPs synthesized at 0.25 V (b); and cyclic voltammograms (c), anodic current density peaks (d), and Tafel plots (e) measured in the electrolyte containing 0.1 M NaOH and 1 M ethanol using of Au NPs synthesized at different upper potentials. | ||
To investigate the influence of morphological changes on the catalytic activity of Au NPs for ethanol electrooxidation, cyclic voltammetry measurements were carried out using samples synthesized at different upper potentials (Fig. 7c). As shown in Fig. 7d, the highest oxidation peak, characterized by an anodic current of 7.06 mA cm−2, was observed at the upper potential of 0.25 V. This was followed by the samples synthesized at 0.00 V, 0.50 V and 1.00 V, which exhibited anodic currents of 5.62, 4.39 and 3.24 mA cm−2, respectively. The kinetic behavior of the catalytic reaction involving the Au NPs electrocatalysts was investigated through Tafel plots, as depicted in Fig. 7e. The Tafel slope of the Au NPs electrodeposited at 0.25 V was calculated to be 188 mV dec−1, which is notably lower comparison to those prepared at 0 V (248 mV dec−1), 0.50 V (254 mV dec−1), and 1.00 V (261 mV dec−1). The lowest Tafel slope observed at 0.25 V signifies the most favorable kinetic properties, which can be attributed to faster charge-transfer kinetics. The superior catalytic activity of Au NPs electrodeposited at 0.25 V can be attributed to the presence of a large number of particles, providing more active sites which are responsible for catalyzing the reaction. Additionally, the coalescence of particles resulted in the formation of a film that reduced charge transfer resistance, facilitating enhanced electron transfer that could promote reaction in the interface of electrode and electrolyte. During the backward scan, a cathodic current at 0.07 V, indicating the electrochemical oxidation of COads which is known to be absorbed and poisoned the catalyst.39,40 This has been a challenge in ethanol electrooxidation because it worsens catalyst performance with low poisoning resistance. To evaluate the poisoning resistance of the Au NPs catalyst, jb/jf ratio was determined. It is known that a smaller peak ratio of the backward current density scan (jb) to forward scan (jf) signifies superior poisoning resistance of the catalyst towards intermediate species of COads. Based on the data summarized in Table 2, lower jb/jf ratios were obtained at relatively lower applied potentials, indicating a good poisoning resistance improved at potential which can produce higher stability of the electrodeposited Au NPs.
| E u (V) | j b/jf | E onset (V vs. RHE) | Tafel slope (mV dec−1) |
|---|---|---|---|
| 0.00 V | 0.289 | 0.703 | 248 |
| 0.25 V | 0.303 | 0.621 | 188 |
| 0.50 V | 0.470 | 0.725 | 254 |
| 1.00 V | 0.471 | 0.773 | 261 |
To further investigate these findings, chronoamperometry measurements were conducted over a duration of 9000 seconds in an electrolyte solution containing NaOH (0.10 M) and ethanol (1.00 M). The recorded chronoamperometry curves exhibit an initial rapid decay in current at the early stage of measurements (the inset of Fig. 8), which can be attributed to the adsorption of CO on the active site, resulting in a decrease in the catalytic activity of the electrocatalyst.41 However, the curve obtained using Au NPs deposited at an upper potential of 0 V shows the lowest current attenuation, indicating a more stable catalytic behavior for the ethanol oxidation reaction. In accordance with jb/jf ratio, the remaining curves demonstrate a rapid drop in current, with higher potential resulting in faster current decay. After approximately 2500 s, the current reached a steady state and remained constant suggesting that the diffusion-controlled reaction predominated during the ethanol electrooxidation.39
 | ||
| Fig. 8 Chronoamperometry curves of AuNPs synthesized with different upper potentials. Inset shows current decay at the early stage of measurements. | ||
Conclusions
Au NPs deposits with varying particle sizes and distribution were successfully synthesized using the square-wave pulse deposition technique by adjusting the applied upper potentials. At low potentials, a large number of fine Au NPs were obtained, while at potential 0.25 V, the particles underwent coalescence and formed short chain-like structures. Increasing the potential produced larger particle sizes. Notably, the highest number of Au NPs was observed at 0.25 V. These morphological changes indicate that the nucleation decreased, and the reaction led to the particle growth that produced sparse particles over the FTO substrate. The applied upper potential was found to have a crucial role in determining the poisoning resistance and catalytic stability of the Au NPs. Specifically, lower potential exhibited higher resistance to poisoning and greater electrocatalyst stability. The Au NPs electrodeposited at a potential of 0.25 V demonstrate high catalytic activity which can be associated with the abundance of Au NPs providing numerous active sites for catalyzing the reaction within the system. Additionally, the coalescence of particles, leading to the formation of short chain-like structures, played a crucial role in reducing the charge transfer resistance, thereby facilitating an increased current transfer that promoted reaction on the catalyst surface.Author contributions
Setia Budi: conceptualization, formal analysis, funding acquisition, methodology, supervision, validation, visualization, writing – original draft, writing – review & editing. Annisa Auliya: writing – original draft, investigation, project administration. Suci Winarsih: resources, funding acquisition. Mohammad Hamzah Fauzi: resources, funding acquisition. Yusmaniar: supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Research and Innovation Agency (BRIN) and Lembaga Pengelola Dana Pendidikan (LPDP) Ministry of Finance, Republic of Indonesia for supporting this research under Riset dan Inovasi Untuk Indonesia Maju (RIIM) scheme batch 2 with contract number 1/RIIM/II/LPPM/IV/2023 (110/IV/KS/11/2022).References
- S. P. S. Badwal, S. Giddey, A. Kulkarni, J. Goel and S. Basu, Appl. Energy, 2015, 145, 80–103 CrossRef CAS.
- D. S. B. L. Oliveira, F. Colmati, E. R. Gonzalez and R. de Sousa Jr, Int. J. Hydrogen Energy, 2023, 48, 24481–24491 CrossRef CAS.
- F. E. Teran, D. M. Santos, J. Ribeiro and K. B. Kokoh, Thin Solid Films, 2012, 520, 5846–5850 CrossRef CAS.
- X. Di Tang, Y. Yang Wang, C. Y. Wang, L. Yuan and Y. Yi, J. Alloys Compd., 2023, 938, 168398 CrossRef.
- B. Q. Miao, B. Sun, T. J. Wang, F. Shi, P. Chen, P. J. Jin, D. S. Li, F. M. Li and Y. Chen, Appl. Catal., B, 2023, 337, 122967 CrossRef CAS.
- D. Wang, Y. Zhang, K. Zhang, X. Wang, C. Wang, Z. Li, F. Gao and Y. Du, J. Colloid Interface Sci., 2023, 650, 350–357 CrossRef CAS PubMed.
- M. Braun, M. Chatwani, P. Kumar, Y. Hao, I. Sanjuán, A. A. Apostoleri, A. C. Brix, D. M. Morales, U. Hagemann, M. Heidelmann, J. Masa, W. Schuhmann and C. Andronescu, J. Phys. Energy, 2023, 5, 1–16 Search PubMed.
- H. Syafei and D. G. Kurniawan, Chem. Mater., 2023, 2, 14–18 CrossRef.
- J. Flórez-Montaño, G. García, O. Guillén-Villafuerte, J. L. Rodríguez, G. A. Planes and E. Pastor, Electrochim. Acta, 2016, 209, 121–131 CrossRef.
- A. Sabella, R. Syifa and N. A. Dwiyana, Chem. Mater., 2022, 1, 88–92 CrossRef.
- A. Sabella and A. Auliya, Chem. Mater., 2023, 2, 46–50 CrossRef.
- M. Wei, L. Chen, X. Wang, A. Zhu, Q. Zhang and Q. Liu, J. Colloid Interface Sci., 2023, 633, 932–947 CrossRef CAS PubMed.
- M. T. Darby, E. C. H. Sykes, A. Michaelides and M. Stamatakis, Top. Catal., 2018, 61, 428–438 CrossRef CAS PubMed.
- I. M. Al-Akraa, B. A. Al-Qodami and A. M. Mohammad, Int. J. Electrochem. Sci., 2020, 15, 4005–4014 CrossRef CAS.
- E. Torralba, N. Blanchard, C. Cachet-Vivier, D. Muller-Bouvet, J. González and S. Bastide, Electrochim. Acta, 2020, 354, 136739 CrossRef CAS.
- V. A. Online, P. Rodriguez and M. T. M. Koper, Phys. Chem. Chem. Phys., 2014, 16, 13583–13594 RSC.
- A. Zhang, Y. Chen, Z. Yang, S. Ma, Y. Huang, G. Richter, P. Schützendübe, C. Zhong and Z. Wang, ACS Appl. Energy Mater., 2019, 3, 336–343 CrossRef.
- S. Hebié, K. B. Kokoh, K. Servat and T. W. Napporn, Gold Bull., 2013, 46, 311–318 CrossRef.
- C. Chiu, P. Chung, K. Lao, C. Liao and M. H. Huang, J. Phys. Chem., 2012, 116, 23757–23763 CAS.
- A. I. Svalova and P. V. Stishenko, Procedia Eng., 2016, 152, 67–72 CrossRef CAS.
- S. Manna, W. Kim, Y. Takahashi and O. G. Shpyrko, J. Appl. Phys., 2016, 174301 CrossRef.
- J. Park, M. Atobe and T. Fuchigami, Ultrason. Sonochem., 2006, 13, 237–241 CrossRef CAS.
- Y. Liu, J. Zhou, X. Yuan, T. Jiang and L. Petti, RSC Adv., 2015, 5, 68668–68675 RSC.
- A. E. Rue and M. M. Collinson, Int. J. Electrochem., 2012, 2012, 1–9 CrossRef.
- F. Yang, K. Cheng, K. Ye, X. Xiao, F. Guo, J. Yin, G. Wang and D. Cao, Electrochim. Acta, 2013, 114, 478–483 CrossRef CAS.
- M. Mundotiya and W. Ullah, Novel Metal Electrodeposition and the Recent Application, IntechOpen, 2018, pp. 1–17 Search PubMed.
- J. M. Lázaro-Martínez, A. J. Byrne, E. Rodríguez-Castellón, J. M. Manrique, L. R. Jones and V. Campo Dall’ Orto, Electrochim. Acta, 2019, 301, 126–135 CrossRef.
- H. Chen, T. Yang, F. Liu and W. Li, Sens. Actuators, B, 2019, 286, 401–407 CrossRef CAS.
- M. M. Rahman, X. B. Li, N. S. Lopa and J. J. Lee, Bull. Korean Chem. Soc., 2014, 35, 2072–2076 CrossRef CAS.
- N. Sakai, Y. Fujiwara, M. Arai, K. Yu and T. Tatsuma, J. Electroanal. Chem., 2009, 628, 7–15 CrossRef CAS.
- X. Zhan, S. Hu, J. Wang, H. Chen, X. Chen, J. Yang, H. Yang and Z. Su, Sens. Actuators, B, 2021, 346, 130499 CrossRef CAS.
- S. P. Babu and P. Elumalai, Mater. Res. Express, 2017, 4, 1–10 Search PubMed.
- G. Zhao and G. Liu, Nanomaterials, 2019, 41(9), 1–13 Search PubMed.
- S. P. Nayak, L. K. Ventrapragada, S. S. Ramamurthy, J. K. Kiran Kumar and A. M. Rao, Nano Energy, 2022, 94, 1–13 CrossRef.
- S. Prasad Nayak, L. K. Ventrapragada, A. M. Rao and J. K. Kiran Kumar, Mater. Lett., 2023, 330, 8–11 CrossRef.
- W. Fu, B. Liu, J. Liu, X. Han, Y. Deng, C. Zhong and W. Hu, Int. J. Electrochem. Sci., 2021, 16, 1–16 Search PubMed.
- J. Liu, X. Wang, Z. Lin, Y. Cao, Z. Zheng, Z. Zeng and Z. Hu, Electrochim. Acta, 2014, 136, 66–74 CrossRef CAS.
- L. Zheng, J. Sun, L. Xiong, R. Jin, J. Li, X. Li, D. Zheng, Q. Liu, L. Niu, S. Yang and J. Xia, Fuel Cells, 2010, 10, 384–389 CrossRef CAS.
- S. Babu and P. Elumalai, Mater. Res. Express, 2016, 4, 0–10 CAS.
- M. Hasan, S. B. Newcomb, J. F. Rohan and K. M. Razeeb, J. Power Sources, 2012, 218, 148–156 CrossRef CAS.
- H. Rostami, A. A. Rostami and A. Omrani, Int. J. Hydrogen Energy, 2015, 40, 10596–10604 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |