DOI:
10.1039/D3MA00432E
(Paper)
Mater. Adv., 2023,
4, 6673-6681
rGO nickel matrix composites with high ozone degradation efficiency under high humidity†
Received
18th July 2023
, Accepted 31st October 2023
First published on 13th November 2023
Abstract
Low stability or deactivation of materials under high humidity limits the development of ozone removal via catalytic degradation. To address this challenge, a novel material comprising Ni2(CO3)(OH)2 (NiCH) that encapsulates a reduced graphene oxide (rGO) microsphere (NiCH–rGO) with excellent ozone removal was studied herein. The postprocessing catalyst exhibits a stable ozone degradation efficiency of 99% and excellent stability over 12 h under a relative humidity of 60% at a space velocity of 900 L h−1. The mechanism for promoting ozone removal can be summarized as follows: On the one hand, rGO promotes the degradation of ozone via a bond formed between rGO and NiCH, which increases the internal charge transfer of NiCH–rGO. On the other hand, the introduction of rGO decreases the average valency of Ni from 2.42 to 2.37, which enhances the adsorption behavior of ozone. Further, density functional theory and quartz crystal microbalance demonstrated that an appropriate rGO concentration reduced the adsorption energy for ozone and water adsorption capacity. Therefore, the method for improving the properties of catalysts has provided a new approach to designing high-efficiency catalysts.
Introduction
Ozone is a primary air pollutant,1 which mainly exists in the stratosphere. Although it can absorb 220–300 nm ultraviolet rays to protect organisms from harmful ultraviolet radiation, inhalation of high concentrations of ozone damages the respiratory system and further impairs lung function due to its strong oxidation capacity.2–4 Therefore, the Chinese government promulgated “indoor air quality standards” (GB/T 18883-2002), which stipulate that indoor ozone average concentration should not exceed 0.15 mg m−3. At present, ozone removal methods mainly include thermal decomposition, adsorption, and catalytic decomposition.5–7 Room-temperature ozone catalytic decomposition is an environmentally friendly and convenient ozone removal method. Among catalytic materials, manganese-based materials are widely used because of their high ozone removal rate, but their activity is inhibited by water coexisting with ozone.8 However, in a practical application environment, ozone often coexists with water. Therefore, it is necessary to develop materials that can maintain stable and efficient catalytic properties in high-humidity environments.
In traditional catalysts, environmental humidity can inhibit ozone catalytic degradation because water molecules are easy to occupy the active center to prevent ozone from adsorbing on the catalyst surface. By increasing the number of oxygen vacancies on the surface of the material through usual methods such as morphology control,8 doping,9–12 and surface modification,13–15 a material with high catalytic efficiency can be obtained. Another way to enhance the efficiency of catalysts is to build a carrier, such as reduced graphene oxide (rGO), for the formation of fine grains that grow on rGO.16,17 Gong et al. found that Cu2O/rGO presents high catalytic activity, attributing to a synergistic effect between the rGO and ultrafine Cu2O solid phases, which facilitates necessary electron density transfer in the catalytic process.16,17 Furthermore, they found that the electron transfer from NiO to Ni in the ozone decomposition via Ni/NiO facilitates electron transfer from negatively charged active oxygen to the surface and benefits their release.17 All these greatly promote the progress of the reaction and increase the reaction rate.
In the ozone decomposition, metal atoms are the active sites. The valence of these atoms changes during the reaction, becoming oxidized as they adsorb ozone. Further, the desorption of oxygen reduces the active site, forming an oxidation/reduction cycle during the reaction.17–19 Widely used metal oxides, such as MnOx, have been found to undergo irreversible oxidization. Mn3+ is oxidized to Mn4+ by ozone, causing deactivation owing to structural or valence change.4
Ni2(CO3)(OH)2 (NiCH) has flexible oxidation states and is generally used in electrochemical energy storage materials. Compared with other metal oxides, the Ni2+/Ni3+ pairs in NiCH show high reversibility during the reaction improving the stability and performance in long-term ozone decomposition tests.20–22 Therefore, NiCH is advantageous for ozone catalytic decomposition.
Based on previous studies, building a catalyst with rGO is an effective approach that is less impacted by humidity. On this basis, we synthesized NiCH–rGO catalyst via the hydrothermal method, which was then investigated as an ozone catalyst for the first time. NiCH showed high performance in ozone decomposition with the flexible valence of Ni; the addition of rGO enhanced surface ozone adsorption, enhanced electron transfer of ozone to the Ni atom, and decreased the impact of humidity on active site ensuring stable catalytic activity for ozone decomposition. Moreover, when the NiCH–rGO catalyst was loaded on the melamine foam for reaction measurement, the composite material presented steady activity during the 12-h-long test, which indicated that the obtained catalyst had preferable potential for ozone removal under real conditions.
Experimental
Synthesis
All chemical reagents and solvents were purchased from Kelong Chemical Reagent Co., Ltd (Chengdu, China) and used as received without further purification. Deionized water was obtained from a Milli-Q water purification system. Aqueous graphene oxide (GO) with a concentration of 8.0 mg mL−1 was prepared using the modified Hummers' method.23,24 The NiCH–rGO was prepared using the hydrothermal method. Nickel(II) chloride hexahydrate (NiCl2·6H2O, 0.015 mol) and urea (CH4N2O, 0.06 mol) were dissolved in 120 mL of deionized water, then a GO solution with a certain mass ratio was added and stirred well. Note: Depending on the amount of graphene added, the resulting catalyst is Ni2(CO3)(OH)2-rGO x wt%, where x is the mass ratio of GO to NiCl2·6H2O, (x = 1, 2, 5, 10, 25). The mixture was sealed and heated to 120 °C for 16 h in a Teflon-lined stainless-steel autoclave with a capacity of 150 mL and cooled to room temperature. The precipitate was collected after filtration, washed with distilled water and ethanol, and dried in an oven at 60 °C. Pure rGO and NiCH were made using the same solution, which did not contain NiCl2·6H2O or GO solution. The preparation process is shown in Fig. 1.
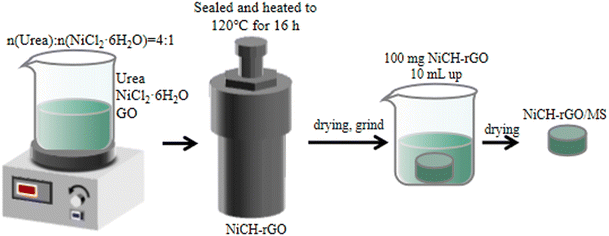 |
| | Fig. 1 Schematic diagram for the synthesis of NiCH–rGO. | |
Characterization
X-Ray diffraction (XRD) was used to analyze the patterns of the samples by an X Pert PRO MPD with 40 kV/40 mA Cu kα radiation. Scanning electron microscopy (SEM) was performed on a ZEISS EV0 MA15 with a voltage of 20 kV, and transmission electron microscopy (TEM) was conducted on a FEI Tecnai G2 F20. Selected area electron diffraction (SAED) patterns were used to determine the composition of the samples. A microscopic confocal Raman spectrometer (Thermo Fischer Dxr, 532 nm laser) was employed to probe the structure of samples. X-ray photoelectron spectroscopy (XPS) was obtained using Thermo Fisher Scientific ESCALAB 250Xi, with all binding energies referenced to the C 1s level at 284.8 eV. Nitrogen adsorption–desorption isotherms were recorded on a specific surface and porosity analyzer (ASAP 2460) at −195.85 °C. The electrochemical measurements were performed with a CHI 660E electrochemistry workstation using an Ag/AgCl electrode as the reference electrode in a three-electrode system, with an electrolyte solution of 0.1 M (nBu4N)PF6/MeCN. A quartz crystal microbalance (QCM, SRS 200, Deshang Co., Ltd, China) circular wafer was used for water adsorption and desorption analysis. The samples were dispersed in ethanol with a concentration of 0.01 g mL−1. A 20 μL suspension was coated on the chip, which had a 500 kHz base frequency and dried at 60 °C for 24 h. For dry or high relative humidity (RH) values, air (80%) was used in this experiment with a gas flow of 5 mL min−1.
Ozone removal activity measurement
The reaction conditions were set at 25 °C, and the total gas flow was kept at 1.5 L min−1. Ozone generated from a low-pressure mercury lamp was kept at a concentration of 40 ± 2 ppm. RH was controlled by bubbling air into deionized water. The ozone removal rate of catalysts was evaluated using a self-made device. Furthermore, 100 mg of catalyst was dispersed in 10 mL of deionized water and stirred until a suspension was formed. This was then dispersed on a melamine sponge with a size of ∅4 × 4 cm and dried at 60 °C for 24 h. The sponge was placed into the pipe ∅3 for test. The inlet and outlet concentrations, Cin and Cout, respectively, were measured using an ozone detector (model 106L, 2B Technologies). Finally, the ozone removal rate was calculated using eqn (1):| | | Ozone removal rate = 100% × (Cin − Cout)/Cin | (1) |
where Cin (ppm) and Cout (ppm) are the initial and final ozone concentrations, respectively.
Theoretical calculation method
Density functional theory (DFT) calculations were undertaken using the Cambridge Sequential Total Energy Package program25 using a planewave pseudopotential approach with a cutoff energy of 380 eV. The NiCH cell with k-points of 2 × 2 × 1 was employed for the pristine sample and a vacuum slab of 15 Å. All calculation models were optimized with force and energy thresholds of 0.05 eV Å−1 and 1 × 10−5 eV, respectively. The main aim of this calculation was to provide an understanding of the adsorption abilities of NiCH and NiCH–rGO and determine the most possible adsorption site and active site for ozone decomposition. The adsorption energy was evaluated using eqn (2):| | | Ead = E(composite+adsorbate) − E(composite) − E(adsorbate) | (2) |
E(composite+adsorbate) represents the total energy of each sample and adsorbate molecules (O3, O2, or H2O).
Results and discussion
Catalytic performance
The structure of NiCH before and after the addition of GO was analyzed using XRD (Fig. 2a). The peaks at 17.5°, 49.6°, and 60.5° could be assigned to the (1 2 0), (2 6 0), and (5 2 1) facets, respectively. In addition, there is an obvious wide peak at approximately 35°, which may have resulted due to low crystallinity of the samples. Moreover, for pure rGO, the wide peak near 22° is due to doping of nitrogen in rGO.26 No diffraction peaks relevant to rGO could be observed, which probably resulted from the low content or being completely encased in NiCH.26–29 All obtained samples have same diffraction peaks, corresponding to NiCH (Nullaginite, PDF#35-0501, Fig. 2a), which means that the crystalline phase was maintained during the treatment. To confirm the influence of processing on the catalytic activity of the samples, we used a self-made device to detect their activity. Then, the catalytic performance of the prepared samples was evaluated via the degradation of air pollutant ozone at 40 ppm. We found that when RH was <10%, all samples could remove ozone efficiently within 30 min (Fig. 2b). With the rGO/NiCH mass ratio range 0–25%, catalytic activity first dropped rapidly and then became steady approximately at 80% RH. After the addition of 10 wt% rGO, catalytic performance significantly improved in a high-humidity environment, and the ozone removal rate increased from 70% to 99% compared to pure NiCH (Fig. 2c), indicating that the catalytic activity was enhanced. Pure rGO does not exhibit catalytic activity (Fig. 2d), which demonstrates that rGO may be a supporting material or that a synergistic effect between rGO and NiCH yields improved ozone degradation efficiency of NiCH. Moreover, the structure of the material before and after the reaction did not change noticeably, illustrating that the structure of the material is stable (Fig. S1, ESI†).
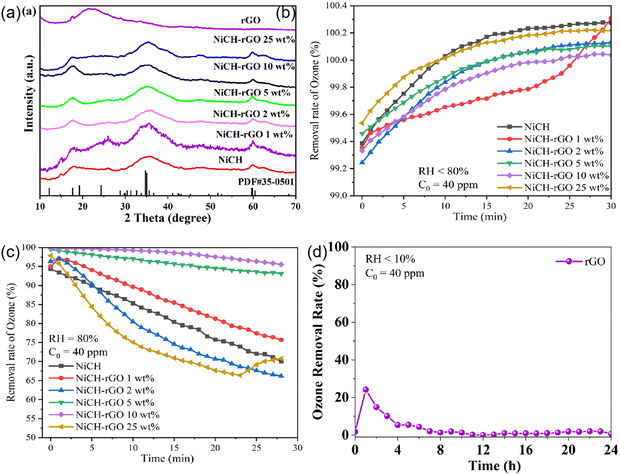 |
| | Fig. 2 (a) XRD patterns of NiCH before and after the addition of rGO and rGO; (b) ozone removal rate of the sample when RH is less than 10%; (c) ozone removal rate of the sample when RH is 80%; (d) test for rGO. | |
Through a series of screenings, the best samples were obtained. To study its application value, NiCH–rGO 10 wt% and the widely used α-MnO2 were detected at 60% RH. The performance of α-MnO2 dropped to 20%, whereas NiCH–rGO 10 wt% remained at 99% for 12 h under 60% RH (Fig. 3a).8 To explore the stability of NiCH–rGO 10 wt%, we performed a catalytic recycling test under 80% RH. NiCH and NiCH–rGO 10 wt% were tested for 30 min at a RH of less than 10%, and the RH was adjusted to 80% for another 30 min. Following this order, the samples were tested five times (Fig. 3b). The ozone removal ratio of NiCH–rGO 10 wt% did not drop after five cycles. With the same quality of the catalyst and reaction conditions, the removal ratio of NiCH was much lower than that of NiCH–rGO 10 wt% at 80% RH, which demonstrated that rGO may be more than a supporting material in the composite and that there is indeed a synergistic effect between rGO and NiCH. This may be attributed to the doping of rGO increasing the active sites on the catalyst surface; thus, accelerating the degradation of ozone. To further explore the catalytic decomposition mechanism of ozone, the physical structure and chemical structure of the material were analyzed.
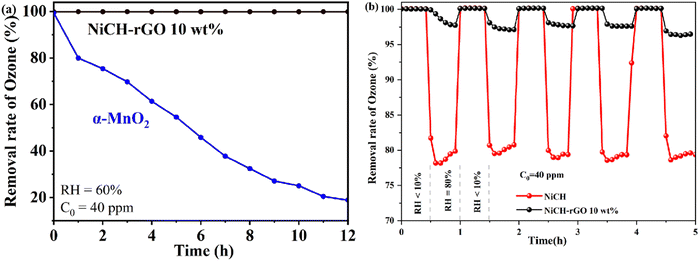 |
| | Fig. 3 (a) The ozone removal rate of the sample compared with α-MnO2 when RH is 60%; (b) a five-cycles test for NiCH and NiCH–rGO 10 wt%. | |
Physical structure of NiCH–rGO
To confirm the effect of rGO on the morphology of the samples, the morphologies of NiCH and NiCH–rGO 10 wt% were observed employing SEM and TEM. NiCH has an urchin-like agglomeration in the microsphere with a diameter of 1–2 μm (Fig. 4a), which resembles pristine NiCH as reported before, and nanosheets were found at the edge of NiCH (Fig. 4b and c).27 As shown in Fig. S2, ESI,† the rGO sheet has specific folds on the surface, and some of the layers are connected to each other to form a three-dimensional network framework. The NiCH–rGO 10 wt% composite prepared in Fig. 4e was self-assembled from rGO micrometer tablets decorated with NiCH particles. It was clearly seen that the NiCH microsphere is densely cluttered on the surface of the folded graphene sheets. The morphology of NiCH–rGO 10 wt% illustrated the morphological changes of the sample due to rGO introduction. The particle diameter was significantly reduced to approximately 250 nm with the addition of rGO (Fig. 4e); the images of NiCH–rGO 10 wt% (Fig. 4e and f) presented a flower-like morphology with accumulated nanosheets, and rGO was covered by NiCH. This was also proved via TEM images of NiCH–rGO 10 wt% (Fig. 4f and g), which depicted larger size and thinner structure compared to pure phase alkaline nickel carbonate; further amplification of the TEM image of the material can observe the same sheet at approximately 5–10 nm. The SAED of two catalysts (Fig. 4d–h) showed two diffraction rings corresponding to (610) and the (201) and (−201) lattice plane of NiCH, which confirmed that the crystal structure of NiCH and NiCH–rGO 10 wt% is nullaginite. According to the XRD results, the surface of NiCH–rGO 10 wt% was mostly covered with NiCH, which increased the specific surface area, thus affecting ozone adsorption and active site exposure. To study the reasons for improvement of the catalytic performance of materials, we then researched the specific surface area of materials.
 |
| | Fig. 4 SEM and TEM images and SAED patterns of pure NiCH (a)–(d) and NiCH–rGO 10 wt% (e)–(h). | |
For the catalytic reaction, one of the key factors affecting the efficiency of the reaction is the number of active sites. Therefore, the change in specific surface area of NiCH–rGO 10 wt% was studied. Fig. 5 shows the N2 adsorption–desorption isotherms and the corresponding pore size distribution curves of NiCH and NiCH–rGO 10 wt%. All samples exhibit the representative isotherms of type IV and hysteresis loop of type H3 at high relative pressure (P/P0). The pore distribution of NiCH ranged broadly from 2.2 to 50 nm, and the NiCH–rGO 10 wt% ranged from 1.7 to 50 nm. Moreover, Brunner−Emmet−Teller (BET) tests show that the surface area of NiCH and NiCH–rGO 10 wt% is 207.6 and 267.6 m2 g−1, respectively. The increased surface area caused by the morphology change consistent with SEM and TEM. The increase in specific surface area provides more active sites, which is an important reason for the improvement of catalytic performance. However, the larger surface area can also result in water molecules being adsorbed, so the specific surface area may not be the determinate factor in increased catalytic performance.
 |
| | Fig. 5 N2 adsorption–desorption isotherms of (a) NiCH and (b) NiCH–rGO 10 wt%. Insets are the corresponding pore size distribution curves of the corresponding sample. | |
Chemical structure of NiCH–rGO
Based on the physical structure analysis, the introduction of rGO can generate a larger surface area for ozone decomposition. Hence, we analyzed the chemical structure of the material. The existence of rGO in the system was further confirmed by Raman spectroscopy (Fig. 6a) and thermogravimetric analysis (Fig. 6b and c). As shown in Fig. 6a, the peak at 1100 cm−1 was assigned to the CO32− symmetric stretching vibration.30 For pure rGO, the D-band located at 1351 cm−1 was attributed to disorder-induced scattering resulting from imperfections, and the G-band located at 1580 cm−1 arose from the emission of zone-center optical phonons.31 As shown in Fig. S3, ESI,† in the NiCH–rGO10 wt% material, there were two peaks with low intensity at 1350 cm−1 and 1600 cm−1, corresponding to D and G peaks26 in the Raman spectrum of graphene, respectively, which indicated that GO is reduced to reduce GO during the synthesis. The analysis of the peak intensity showed that ID/IG =1.07 and graphene material was doped with N.32 This may be related to the precipitant urea added during the material preparation. After the catalytic material underwent an ozone-catalyzed reaction, D and G peaks were significantly enhanced; the G peak located at 2700 cm−1 was broadened, corresponding to rGO on the surface of the more exposed material. The thermal analysis was performed using a DSC823 TGA/SDTA85/e system (METTLER TOLEDO) at a heating rate of 10 °C min−1 from 40 °C to 600 °C under 50 mL min−1 flowing nitrogen. The TGA curves of NiCH–rGO showed a new weight loss peak near 520 °C compared to NiCH. According to the results, the composite of NiCH and rGO was successfully synthesized.
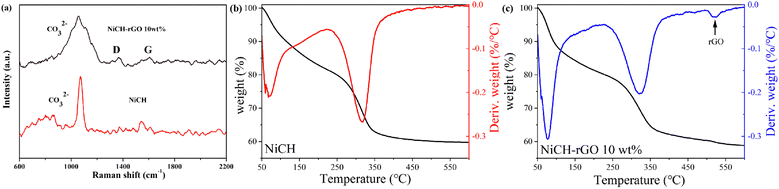 |
| | Fig. 6 (a) Raman spectra of NiCH and NiCH–rGO 10 wt%; TGA curves of (b) NiCH and (c) NiCH–rGO 10 wt%. | |
The valence transition of metals is an important factor determining the ability of ozone removal. To explore the chemical state of the elements in NiCH–rGO 10 wt% and the influence of introducing rGO on NiCH–rGO 10 wt%, XPS of NiCH–rGO 10 wt% and NiCH was measured. As shown in Fig. 7a, after calibration of the binding energy using the C 1s peak at 284.8 eV, the C 1s peaks at 284.8, 285.9, 288.9, and 289.8 eV are attributed to C–C, C–OH, C–OOH, and carbonate groups in NiCH and NiCH–rGO 10 wt%,33 whereas the N 1s peaks at 399.9 eV correspond to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds,34 respectively; nitrogen is found in the XPS data of NiCH and NiCH–rGO 10 wt% (Fig. S4, ESI†), indicating that samples contain nitrogen.35 For NiCH, the two peaks with binding energies at 532.7 and 531.6 eV and separated by 1.1 eV are characteristic of O 1 s with lattice oxygen at 531.6eV and surface oxygen at 532.7eV (Fig. 7b). Compared to NiCH, the peaks of NiCH–rGO 10 wt% at 532.7 eV and 531.6 eV shift to 532.3 and 531.3 eV, respectively (Fig. 7b). Further calculation of the peak area and binding energy of the two samples found that the addition of rGO reduced the content of surface oxygen from 85% to 68%. The decrease in surface oxygen and the binding energy change of lattice oxygen indicate that the addition of rGO changed the bonding in NiCH. This is confirmed by the decrease in binding energy that is observed in Ni 2p3/2 spectra of these two samples in Fig. 7c. Ni 2p3/2 spectra could be split into three peaks at 855 eV, 857 eV, and 861.5 eV, ascribed to Ni2+, Ni3+, and the satellite peak, respectively.30,36 We found that the addition of rGO reduced the content of Ni2+ from 58.4% to 63.3% after calculating the peak area and binding energy of the two samples. The binding energy of Ni2+ and Ni3+ decreases by 0.3 eV and 0.4 eV in NiCH–rGO, respectively, and the Ni2+: Ni3+ ratio causes the average valence of Ni to change from 2.42 to 2.37. The enhancement of electron flow was proved by an electrochemical impedance test (Fig. S5, ESI†). This suggests that NiCH compounds with rGO may form Ni–O binding between the Ni atom and the oxygen group on rGO. The binding reduces the valence of Ni atoms, and at the same time, according to the characteristics of graphene, it may enhance the electron transport between NiCH and rGO.16
N bonds,34 respectively; nitrogen is found in the XPS data of NiCH and NiCH–rGO 10 wt% (Fig. S4, ESI†), indicating that samples contain nitrogen.35 For NiCH, the two peaks with binding energies at 532.7 and 531.6 eV and separated by 1.1 eV are characteristic of O 1 s with lattice oxygen at 531.6eV and surface oxygen at 532.7eV (Fig. 7b). Compared to NiCH, the peaks of NiCH–rGO 10 wt% at 532.7 eV and 531.6 eV shift to 532.3 and 531.3 eV, respectively (Fig. 7b). Further calculation of the peak area and binding energy of the two samples found that the addition of rGO reduced the content of surface oxygen from 85% to 68%. The decrease in surface oxygen and the binding energy change of lattice oxygen indicate that the addition of rGO changed the bonding in NiCH. This is confirmed by the decrease in binding energy that is observed in Ni 2p3/2 spectra of these two samples in Fig. 7c. Ni 2p3/2 spectra could be split into three peaks at 855 eV, 857 eV, and 861.5 eV, ascribed to Ni2+, Ni3+, and the satellite peak, respectively.30,36 We found that the addition of rGO reduced the content of Ni2+ from 58.4% to 63.3% after calculating the peak area and binding energy of the two samples. The binding energy of Ni2+ and Ni3+ decreases by 0.3 eV and 0.4 eV in NiCH–rGO, respectively, and the Ni2+: Ni3+ ratio causes the average valence of Ni to change from 2.42 to 2.37. The enhancement of electron flow was proved by an electrochemical impedance test (Fig. S5, ESI†). This suggests that NiCH compounds with rGO may form Ni–O binding between the Ni atom and the oxygen group on rGO. The binding reduces the valence of Ni atoms, and at the same time, according to the characteristics of graphene, it may enhance the electron transport between NiCH and rGO.16
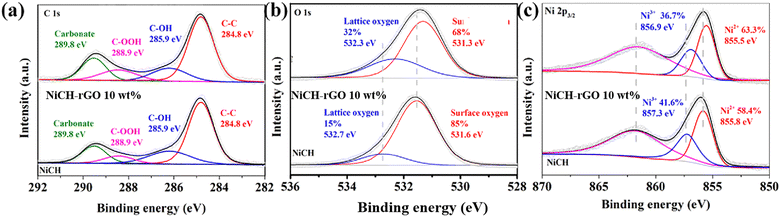 |
| | Fig. 7 (a) C 1s, (b) O1 s, and (c) Ni 2p3/2 XPS spectra of NiCH and NiCH–rGO 10 wt%. | |
Water resistance ability of NiCH–rGO
In addition, water molecules are also one of the factors affecting the catalytic decomposition process. Therefore, we studied the adsorption properties of water molecules on the material surface. The adsorption of water molecules on catalysts was determined by both surface structure and adsorption energy. The weight of adsorbed molecules can translate into a change in frequency, as observed in QCM.37–39 First, dry air was used to clean the surface of the sample for 5 min, then high-RH air was introduced into the chip chamber for 5 min. The weight change caused by water vapor is shown in Fig. 8. NiCH maintains stable quality in a dry environment, and the weight increased by 0.15 g g−1 after being treated in 80% RH air. The surface water adsorption was decreased to 0.05 g g−1 upon the addition of rGO to NiCH; there is only one-third of pure NiCH, and water molecules desorbed from samples after 5 min of dry airflow. The result suggests that the adsorption performance of water on the NiCH surface will be reduced with the introduction of rGO.
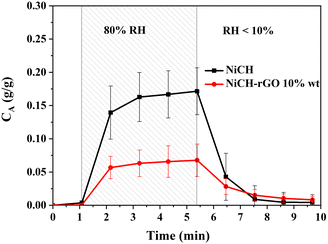 |
| | Fig. 8 Surface adsorption and desorption of water molecules on NiCH and NiCH–rGO 10 wt%. | |
Free radicals of NiCH–rGO
To study the mechanism of the reaction process, the EPR (electronic paramagnetic resonance spectrometer) spin-trap technique was used to analyze the ozone decomposition species generated in suspensions and to further investigate the possible active radicals in the reaction process. In general, superoxide radical (˙O2−) and hydroxyl radical (˙OH) are intermediate products of ozone-catalyzed reactions. Neither ˙O2− nor ˙OH signals were detected in a solution only containing ozone. As illustrated in Fig. 8, NiCH and NiCH–rGO 10 wt% exhibited a typical four-line signal with a relative intensity ratio close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 for the DMPO-˙OH adduct (Fig. 9a), and the signal intensities of the four peaks of the DMPO-˙O2− adduct were almost identical (Fig. 9b). However, NiCH–rGO 10 wt% revealed the higher intensity of the ˙O2-signal. Existing literature indicates that superoxide radicals in aqueous ozone solutions originate from the decomposition of ozone.40 This means that more ozone decomposes, which is conducive to the formation of ˙O2−. Combining the analyses, it can be concluded that ozone reacted with Ni2+ on the surface to form ˙O2− and O–Ni3+.41 At the same time, due to the presence of water molecules, the oxygen on the surface of the Ni atom can react with H2O and generate ˙OH. Water molecules will occupy active sites and react with Ni atoms to produce ˙OH. The more ˙OH NiCH–rGO 10 wt% produced, the more active sites it had. This result was consistent with the ozone decomposition rate.42,43
:2:2:1 for the DMPO-˙OH adduct (Fig. 9a), and the signal intensities of the four peaks of the DMPO-˙O2− adduct were almost identical (Fig. 9b). However, NiCH–rGO 10 wt% revealed the higher intensity of the ˙O2-signal. Existing literature indicates that superoxide radicals in aqueous ozone solutions originate from the decomposition of ozone.40 This means that more ozone decomposes, which is conducive to the formation of ˙O2−. Combining the analyses, it can be concluded that ozone reacted with Ni2+ on the surface to form ˙O2− and O–Ni3+.41 At the same time, due to the presence of water molecules, the oxygen on the surface of the Ni atom can react with H2O and generate ˙OH. Water molecules will occupy active sites and react with Ni atoms to produce ˙OH. The more ˙OH NiCH–rGO 10 wt% produced, the more active sites it had. This result was consistent with the ozone decomposition rate.42,43
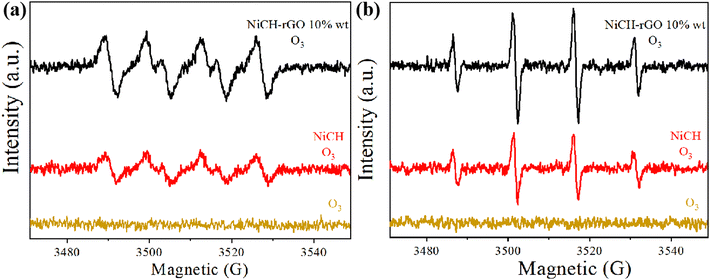 |
| | Fig. 9 DMPO EPR spin-trapping of NiCH and NiCH–rGO for (a) ˙O2− radicals and (b) –OH radicals with 2 mg L−1 ozone concentration. | |
DFT theoretical calculations
According to the effects of the introduction of rGO on the adsorption properties and valence state changes, we verified the conclusion by DFT theoretical calculations. Combining XRD data and TEM images, the ozone adsorption model of monolayer NiCH and NiCH–rGO 10 wt% was established, and the adsorption structures are shown in Fig. S6, ESI.† As shown in Table 1, the adsorption energies of H2O of Ni atoms in NiCH and NiCH–rGO 10 wt% are −0.14 eV and −0.24 eV, respectively, and the adsorption energies of O2 in NiCH and NiCH–rGO 10 wt% are −3.07 eV and −3.21 eV, respectively. Such a small difference between both samples shows that the adsorption energies of H2O and O2 are not the main factor for the enhanced catalytic activity. However, the adsorption energies of O3 of Ni atoms in NiCH and NiCH–rGO 10 wt% are −0.36 eV and −1.10 eV, respectively. The DFT calculation results point out that O3 molecules show a positive tendency to adsorb on NiCH–rGO 10 wt% compared with NiCH. This indicates that the introduction of rGO changed the adsorption site of ozone on the material surface; the adsorption site of ozone in NiCH changed from the O atom to the Ni atom. Combining the Ni 2p data from XPS, it can be determined that the Ni atom with reduced binding energy is more conducive to the adsorption of ozone. Although ozone is more readily adsorbed on the surface of all catalysts than water, the adsorption of ozone on the surface of NiCH–rGO 10 wt% has a greater advantage, which means that the challenge of water molecules occupying the active sites and slowing down the ozone decomposition reaction on the surface has been addressed.
Table 1 Adsorption energy of O3, H2O, and O2 on the Ni atom in NiCH and NiCH–rGO 10 wt%
|
|
O3 (eV) |
H2O (eV) |
O2 (eV) |
| NiCH |
−0.36 |
−0.14 |
− 3.07 |
| NiCH–rGO 10 wt% |
−1.10 |
−0.24 |
−3.21 |
The charge transfer in the reaction process is also a factor affecting the reaction rate of ozone catalytic decomposition. To further study the internal charge transfer in the material adsorption process and the mechanism of the catalytic reaction, the charge transfer between ozone and Ni atoms on the surface of NiCH and NiCH–rGO 10 wt% was analyzed by theoretical calculations. Electrons transfer from the ozone molecule to Ni atoms was Δq = 0.54 e for NiCH, as shown in Fig. S7a, ESI;† this increased to Δq = 0.59 e for NiCH–rGO 10 wt%, as shown in Fig. S7b, ESI,† which confirmed the change in binding energy in XPS data. Higher electrons transfer rate improves the ozone adsorption performance of NiCH, which was also macroscopically reflected in the adsorption behavior tests. As shown in Fig. S7c, ESI,† the electrons transferred from rGO to the Ni atom in NiCH with Δq = 0.44 e for NiCH–rGO. This contributed to the lower average valence of the transition metal atom at the active site, facilitating ozone adsorption, similar to other metal atoms like Mn and Fe.8,13 Increased electron transfer can enhance the oxidation and reduction reactions of Ni atoms, which facilitates the redox reaction during the reaction of ozone on Ni atoms.
Mechanism
According to the characterization analysis and DFT theoretical calculation, ozone is more likely to be adsorbed on Ni atoms on the surface of NiCH–rGO 10 wt%, and the reaction of ozone and Ni atoms increases the valence of Ni atoms (Ni2+ → Ni3+). According to the study on the mechanism of ozone decomposition on NiCH,44 the green divalent nickel material is converted to a black trivalent nickel material after the reaction. We achieved self-reduction of the catalyst by exposing the reacted catalyst to the air, and the corresponding catalyst was converted from black to green (Ni3+ → Ni2+). This is consistent with the UV-vis spectrum data of the catalyst (Fig. S8, ESI†) for different reaction times. In Fig. S8 (ESI†), the red line indicates ESI† as the sample after being regenerated, and its color is nearly the same as that of the fresh sample. Combining the conclusions of the existing literature,45–47 the reaction process of ozone on NiCH is given in eqn (3)–(5):| | | O3 + Ni2+ → O2 + O−Ni3+ | (3) |
| | | O−Ni3+ + O3 → O2 + O2−Ni3+ | (4) |
The Ni atoms on NiCH were active sites. Ozone reacted with Ni2+ and formed Ni–O–O–O, which decomposed into ˙O2− and Ni3+–O; ˙O2− decomposed into O2 and then desorbed from the surface; another O3 attached Ni3+–O and soon became ˙O2− and O2–Ni3+; then ˙O2− continued to decompose into O2. Finally, O2–Ni3+ decomposed into O2 and a reproducible active site.
Conclusions
We synthesized a NiCH–rGO composite via the hydrothermal method, which possesses a low degree of crystallization. The structure and morphology of NiCH–rGO was maintained in the NiCH composite. Based on the characterization and DFT calculation results, addition of rGO first decreased the average valence of the Ni, which increased the ozone adsorption of NiCH and enhanced the electron transfer of ozone to the Ni atom. Moreover, a mechanism based on the cycle of the Ni2+/Ni3+ pair was proposed. All of this caused the ozone removal rate to improve at 60% RH for 12 h. Finally, this study provides a novel approach toward ozone catalytic degradation.
Author contributions
Qian Zhang: conceptualization, writing – original draft, revision Wenyan Xiao, Bangxin Li: experiment, writing – original draft, revision Yu Lin, Linyu Huang, Jifei Liao: analysis with constructive discussions Huiguo Han, Jie Zhu, Yan Fu: data curation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (U1862111), the Sichuan Science and Technology Program (2020ZDZX0042), the Cheung Kong Scholars Program of China, and the State Key Laboratory of the Industrial Vent Gas Reuse Fund Project (SKLIVGR-SWPU-2020-06).
References
- M. Lippmann, J. Air Waste Manage. Assoc., 2012, 39, 672–695 Search PubMed
 .
.
- T. Batakliev Todor, V. Georgiev, M. Anachkov, S. Rakovsky and G. Zaikov, NBI-Technol., 2014, 6, 36–46, DOI:10.15688/jvolsu10.2014.6.4 .
- D. Farmanzadeh and N. A. Askari, Appl. Surf. Sci., 2018, 444, 642–649 CrossRef CAS .
-
K. Ikehata and Y. Li, Advanced oxidation processes for waste water treatment, Academic Press, 2018, pp. 115–134 DOI:10.1016/B978-0-12-810499-6.00005-X .
- W. D. Ellis and P. V. Tometz, Atmos. Environ., 1967, 1972(6), 707–714 Search PubMed .
- J. V. Michael, J. Chem. Phys., 2003, 54, 4455–4459 CrossRef .
- Y. Takeuchi and T. Itoh, Sep. Technol., 1993, 3, 168–175 CrossRef CAS .
- J. Jia, P. Zhang and L. Chen, Appl. Catal., B, 2016, 189, 210–218 CrossRef CAS .
- J. Jia, W. Yang, P. Zhang and J. Zhang, Appl. Catal., A, 2017, 546, 79–86 CrossRef CAS .
- Y. Yang, J. Jia, Y. Liu and P. Zhang, Appl. Catal., A, 2018, 562, 132–141 CrossRef CAS .
- Y. Yang, P. Zhang and J. Jia, Appl. Surf. Sci., 2019, 484, 45–53 CrossRef CAS .
- G. Zhu, J. Zhu, W. Li, W. Yao, R. Zong, Y. Zhu and Q. Zhang, Environ. Sci. Technol., 2018, 52, 8684–8692 CrossRef CAS PubMed .
- R. Cao, P. Zhang, Y. Liu and X. Zheng, Appl. Surf. Sci., 2019, 495, 143607 CrossRef CAS .
- Y. Liu, W. Yang, P. Zhang and J. Zhang, Appl. Surf. Sci., 2018, 442, 640–649 CrossRef CAS .
- Y. Liu and P. Zhang, J. Phys. Chem. C, 2017, 121, 23488 CrossRef CAS .
- S. Gong, J. Chen, X. Wu, N. Han and Y. Chen, Catal. Commun., 2018, 106, 25–29 CrossRef CAS .
- S. Gong, A. Wang, Y. Wang, H. Liu, N. Han and Y. Chen, ACS Appl. Nano Mater., 2020, 3, 597–607 CrossRef CAS .
- Z. Lian, J. Ma and H. He, Catal. Commun., 2015, 59, 156 CrossRef CAS .
- C. Wang, J. Ma, F. Liu, H. He and R. Zhang, J. Phys. Chem. C, 2015, 119, 23119–23126 CrossRef CAS .
- S. Dai, Z. Zhang, J. Xu, W. Shen, Q. Zhang, X. Yang, T. Xu, D. Dang, H. Hu, B. Zhao, Y. Wang, C. Qu, J. Fu, X. Li, C. Hu and M. Liu, Nano Energy, 2019, 64, 103919 CrossRef CAS .
- C.-W. Hu, Y. Yamada and K. Yoshimura, Sol. Energy Mater. Sol. Cells, 2018, 177, 120–127 CrossRef CAS .
- J. Katić, M. Metikoš-Huković, R. Peter and M. Petravić, J. Power Sources, 2015, 282, 421–428 CrossRef .
- W. Wan, R. Zhang, W. Li, H. Liu, Y. Lin, L. Li and Y. Zhou, Environ. Sci.: Nano, 2016, 3, 107–113 RSC .
- H. Sun, Z. Xu and C. Gao, Adv. Mater., 2013, 25, 2554–2560 CrossRef CAS PubMed .
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. – Cryst. Mater., 2005, 220, 567–570 CrossRef CAS .
- E. Asedegbega-Nieto, M. Perez-Cadenas, M. V. Morales, B. Bachiller-Baeza, E. Gallegos-Suarez, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Diamond Relat. Mater., 2014, 44, 26–32 CrossRef CAS .
- C. Xi, G. Zhu, Y. Liu, X. Shen, W. Zhu, Z. Ji and L. Kong, Colloids Surf., A, 2018, 538, 748–756 CrossRef CAS .
- Z. Yu, X. Su, D. Wei, S.-J. Yao, H. Ma, H. Zhao, X. Pu, W. Li and J. Wang, Nano, 2019, 14 Search PubMed .
- G. Zhu, C. Xi, M. Shen, C. Bao and J. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 17208–17214 CrossRef CAS PubMed .
- Y.-J. Shih, Y.-H. Huang and C. P. Huang, Electrochim. Acta, 2018, 263, 261–271 CrossRef CAS .
- C.-M. Chen, Q. Zhang, X.-C. Zhao, B. Zhang, Q.-Q. Kong, M.-G. Yang, Q.-H. Yang, M.-Z. Wang, Y.-G. Yang, R. Schlögl and D. S. Su, J. Mater. Chem., 2012, 22 Search PubMed .
- A. B. Dongil, B. Bachiller-Baeza, A. Guerrero-Ruiz, I. Rodríguez-Ramos, A. Martínez-Alonso and J. M. D. Tascón, J. Colloid Interface Sci., 2011, 355, 179–189 CrossRef CAS PubMed .
- Y. Jia, T. Luo, X.-Y. Yu, J.-H. Liu and X.-J. Huang, New J. Chem., 2013, 37, 534–539 RSC .
- O. J. Achadu, I. Uddin and T. Nyokong, J. Photochem. Photobiol., A, 2016, 317, 12–25 CrossRef CAS .
- T. H. Lee and J. W. Rabalais, J. Electron Spectrosc. Relat. Phenom., 1977, 11, 123–127 CrossRef CAS .
- C. Lin, Y. Zhao, H. Zhang, S. Xie, Y.-F. Li, X. Li, Z. Jiang and Z.-P. Liu, Chem. Sci., 2018, 9, 6803–6812 RSC .
- S. Heydari and G. H. Haghayegh, J. Sens. Technol., 2014, 04, 81–100 CrossRef .
- K. Spinella, L. Mosiello, G. Palleschi and F. Vitali, Open J. Appl. Biosens., 2013, 02, 112–119 CrossRef .
- S. K. Vashist and P. Vashist, J. Sens., 2011, 2011, 571405 Search PubMed .
- S. T. Oyama, Catal. Rev., 2000, 42, 279–322 CrossRef CAS .
- J. Lin, A. Kawai and T. Nakajima, Appl. Catal., B, 2002, 39, 157–165 CrossRef CAS .
- J. Bing, C. Hu, Y. Nie, M. Yang and J. Qu, Environ. Sci. Technol., 2015, 49, 1690–1697 CrossRef CAS PubMed .
- L. Yan, J. Bing and H. Wu, Sci. Rep., 2019, 9, 14752 CrossRef PubMed .
- Lin Yu, Xiao Wenyan and Li Bangxin,
et al.
, Mater. Rep., 2023, 37, 22120067 Search PubMed .
- W. Li, G. V. Gibbs and S. T. Oyama, J. Am. Chem. Soc., 1998, 120, 9041–9046 CrossRef CAS .
- W. Li and S. T. Oyama, J. Am. Chem. Soc., 1998, 120, 9047–9052 CrossRef CAS .
- M. Stoyanova, P. Konova, P. Nikolov, A. Naydenov, S. Christoskova and D. Mehandjiev, Chem. Eng. J., 2006, 122, 41–46 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Wenyan
Xiao
ab,
Bangxin
Li
ab,
Yu
Lin
ab,
Lingyu
Huang
ab,
Jifei
Liao
ab,
Huiguo
Han
c,
Jie
Zhu
ab and
Yan
Fu
ab
*a,
Wenyan
Xiao
ab,
Bangxin
Li
ab,
Yu
Lin
ab,
Lingyu
Huang
ab,
Jifei
Liao
ab,
Huiguo
Han
c,
Jie
Zhu
ab and
Yan
Fu
ab








