
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA Zn-doped Fe3O4 nanoparticle@N, S and P doped Elaeagnus angustifolia gum derived carbon hybrid electrocatalyst: synthesis, characterization and electrochemical sensing of acetaminophen†
Xamxikamar
Mamat
,
Zehong
Gao
and
Longyi
Chen
 *
*
Key Laboratory of Chemistry of Plant Resources in Arid Regions, State Key Laboratory Basis of Xinjiang Indigenous Medicinal Plants Resource Utilization, Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi, 830011, China. E-mail: chenly@ms.xjb.ac.cn
First published on 13th September 2023
Abstract
Over use or long term exposure of acetaminophen (APAP) could cause severe damage to human health. Currently APAP related events are still a global burden. Therefore, secure management of APAP is important for avoiding possible APAP induced incidents. Rapid sensing of APAP in human body fluids can help in the better monitoring of health conditions and providing information for necessary medical aid. Electrochemical sensors utilizing functional nanomaterials and nanostructures have been proved to be a facile tool for sensing APAP in human body fluids. Among various functional nanomaterials, transition element doped Fe3O4 nanoparticle decorated carbon materials have found extensive applications in the areas of catalysis, energy, environmental, etc. In order to explore the versatility of Zn doped Fe3O4 nanoparticles incorporated with a biomass carbon electrocatalyst in the electrochemical sensing area, we prepared a N, S and P doped biomass carbon material (NSP-BC) from the biomass of Elaeagnus angustifolia gum through a pyrolysis approach. Then, a facile one-pot polyol solvothermal synthesis method was adopted for deposition of Zn doped Fe3O4 nanoparticles onto the NSP-BC. Among the synthesized electrocatalysts, the electrocatalyst with a Zn/Fe ratio of 0.132 was found to show the highest Ipa response current towards the APAP molecule. The electrocatalysts were systematically characterized by scanning electron microscopy, energy-dispersive X-ray spectroscopy, element mapping, X-ray photoelectron spectroscopy, X-ray powder diffraction, Raman spectroscopy, inductively coupled plasma-optical emission spectroscopy and electrochemical analysis. The electrocatalyst modified glassy carbon electrode sensor achieved a wide linear sensing range of APAP from 2.5 to 500 μM covering the urinary physiological APAP concentration range with a limit of detection of 63.9 nM and a sensitivity of 0.64 μA μM−1 cm−2. The electrocatalyst modified GCE sensor also exhibited satisfactory recovery rates of APAP in urine samples. This facile synthesized Zn-doped Fe3O4 nanoparticle@NSP-BC electrocatalyst has potential application in clinical diagnosis and pharmaceutical analysis.
Introduction
Acetaminophen (N-acetyl-p-aminophenol, APAP, also commonly known as paracetamol) is a pharmaceutical molecule regularly used in antipyretic and analgesic drugs. Nowadays, over 100 products containing APAP are sold over the counter around the world. Overuse or long-term exposure of APAP could lead to harmful and unknown effects on human health.1–3 Among the undesirable impacts, liver damage is one of the notorious adverse injuries of APAP.4–7 The current maximum adult dose of APAP is 4 grams per day as recommended by the US Food & Drug Administration. One harmful effect of APAP is potentially related to pregnancy. A study8 on analysis of urinary APAP concentration and pregnancy probability found that a higher concentration of APAP in male urine would require the couple spending more time on having a baby (or having a child?). Interestingly the concentration of APAP in female urine did not show a correlation with pregnancy probability. Besides the mentioned underlying damage, APAP might have other unclear influences on human health as well. In a study9 of the APAP's impact on the efficacy of immunotherapy in cancer patients, researchers found that APAP is a potential suppressor of antitumor immunity and should be used with caution on patients treated with immune checkpoint blockers.In this regard, related APAP events and investigation results are summarized here to visualize the importance of secure management and monitoring of APAP globally. An investigation report published in 2008 pointed out that APAP overdoses are the number one cause (nearly 50%) of acute liver failure and result in 30% mortality in the US.10 In another investigation report published in 2015, researchers summarized trends in the rates of adverse events attributable to APAP use in the United States. After considering drug sales and other factors, they came to the conclusion that APAP-related adverse events continue to be a public health burden.11 APAP overdose is also a problem in the military health system in the US.12 A recent investigation report in Switzerland surveying the overuse of APAP reveals that 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 790 APAP-related poisoning calls were identified from January 1, 2000, to December 31, 2018.13 In another report, researchers examined trends in emergency department presentation rates for APAP-related poisonings across Canada from April 2011 to February 2019; 13.7% were related to APAP use.14 In a review article, researchers suggested that albeit the fact that APAP is an over-the-counter drug, potential risks should be taken into consideration on using APAP-related medicines.15 Besides rapid sensing of APAP, various techniques are developed for the treatment of APAP-caused accidents.16–18
790 APAP-related poisoning calls were identified from January 1, 2000, to December 31, 2018.13 In another report, researchers examined trends in emergency department presentation rates for APAP-related poisonings across Canada from April 2011 to February 2019; 13.7% were related to APAP use.14 In a review article, researchers suggested that albeit the fact that APAP is an over-the-counter drug, potential risks should be taken into consideration on using APAP-related medicines.15 Besides rapid sensing of APAP, various techniques are developed for the treatment of APAP-caused accidents.16–18
So far, centralized laboratories have satisfactorily measured APAP in various samples by high-performance liquid chromatography, mass spectrometry, etc. However such analysis requires skilled personnel and instruments. On the other hand, the electrochemical sensing method has the advantages of being rapid, in situ, having easy operation, being miniaturized, being integrated with internet, etc. Hence electrochemical sensing technology possesses potential for revolutionizing many aspects in academic research and societal need, and has promising future in clinical diagnosis and pharmaceutical analysis areas.19
Iron oxide nanoparticles are considered as a versatile electrode modifying material that has catalytic activity and has been widely studied in the electrochemical sensing sector due to the multiple electrons and multiple valences of Fe2+ and Fe3+.20 Many other transition elements, like Mg, Ni, Co, Zn etc.,21–26 can be co-doped into each other's metal oxide crystal lattice as they share a similar atom radius and physiochemical properties. The transition element doped Fe3O4 nanoparticles could display many novel special catalytic, optical, electronic and magnetic properties due to the interaction between the metal elements. One of the specialties of transition metal elements is that they usually have multiple d orbital electrons which could interact with each other's d electrons. As a result of the good mixing of the doped transition metal elements, the doped nanoparticle surface would present different metal elements by varied metal ratios and they would show different catalytic functions towards different molecules. All in all, incorporating transition element dopants into Fe3O4 nanoparticles is a good study paradigm for discovering novel nanomaterials for electrochemical catalytic sensing and miscellaneous applications.
Biomass derived carbon materials have extensive potential applications in various areas, like energy, sensing, environment and catalysis.27–30 Firstly, biomass is widely present in nature and easily accessed. For example, tree leaves, grass, fruit peels, chitin, etc. are all vast biomass resources. Secondly, the major chemical composition of biomass is carbon based materials. Plant is majorly composed of lignin, cellulose and hemicellulose. Thirdly, the preparation of biomass carbon can be realized by versatile physiochemical approaches. Most regularly used procedures include hydrothermal and pyrolysis methods. Different composited and structured biomass can form different functional biomass carbon materials, which share commonly a high specific surface area and strong conductivity, and are a good matrix for integration with countless molecules and nanomaterials. Especially, in recent years, biomass carbon materials have been widely used in the electrochemical sensor platform, demonstrating the practicability of biomass carbon as a recyclable and economically electrode material.31,32 Besides biomass carbon, some studies applied the biomass materials of fibrous nanocomposite materials for toxic dye removal and remediation utilising the large specific surface area offered by the nanomaterials and biomass. Cotton cloth was adopted as the base for deposition of chitosan nanomaterials. An improved removal property of the fibrous nanocomposite was achieved for methyl orange.33 Another research utilized the biodegradable natural rice flour and Graham flour for methyl orange adsorption.34
In the nanomaterial/carbon hybrid composite research area, various nanomaterials were integrated with graphene materials for bioapplications.35–37 Other nanocarbon materials like graphene materials require complicated physical and chemical processes to be obtained from graphite. Moreover, graphene materials are majorly prepared from non-recyclable fossil materials. Therefore, the vast and easily processed biomass carbon gained more attention now. Not only because biomass carbon has a sufficient natural source and is recyclable, but also biomass carbon could be prepared facilely with favourable performance in many applications.
Herein, we adopted the pyrolysis approach to prepare the biomass carbon material from Elaeagnus angustifolia gum, which is majorly composed of polysaccharide. The electron-rich elements of N, S and P are co-doped into the biomass carbon matrix in a simple manner to enhance the biomass carbon's conductivity. Then, a facile solvothermal polyol method is used to deposit Zn doped Fe3O4 nanoparticles onto the biomass carbon material to form the electrocatalyst. Various material and electrochemical characterization studies were done to characterize the electrocatalyst and electrocatalyst modified GCE sensor. The electrochemical sensing analysis of APAP was performed and a satisfactory recovery rate of detecting APAP in urine samples was achieved.
Results and discussion
Electrochemical characterization
The chemical formula of APAP and NAPQI is C8H9NO2 and C8H7NO2, respectively. As shown in Fig. 1, the APAP molecule is electrochemically catalysed at the electrocatalyst surface, along with losing 2 hydrogen and 2 electrons, and becomes N-acetyl-4-benzoquinone imine (NAPQI). In this electrochemical reaction, the oxidation peak position is at about 0.4 V and the reduction peak position is at about 0.2 V. In this study, several varied compositional electrocatalysts (Zn-doped Fe3O4 nanoparticle@NSP-BC) were synthesized, namely S1 to S6 with the Zn/Fe ratio varying from 0 to 0.3. The electrocatalyst modified GCE sensor was characterized by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) as shown in Fig. 2(A) and (B). The Ipa response current and Rct resistance values of electrocatalyst modified GCE sensors are, respectively, 13.55 μA, 2159 ohm (GCE), 6.41 μA, 490.1 ohm (S1), 17.62 μA, 309.7 ohm (S2), 19.18 μA, 210.7 ohm (S3), 23.60 μA, 629.1 ohm (S4), 20.55 μA, 1481 ohm (S5), and 21.13 μA, 893.2 ohm (S6). Among the prepared electrocatalysts, electrocatalyst S4 (Zn/Fe molar ratio of 0.13) has the highest Ipa response current among the prepared electrocatalysts (Zn/Fe molar ratio from 0 to 0.3), while electrocatalyst S3 has the lowest Rct resistance. In the composition of the electrocatalyst, the NSP-BC acted as the electron transport enhancing intermediate and conductive matrix for the decoration of Zn-doped Fe3O4 nanoparticles. The Zn dopant ratio could modulate the nanoparticle's physiochemical properties, like crystal lattice, electron conductivity, nanoparticle surface electronic state etc. The Zn element is considered to have special catalytic properties, therefore the distribution and concentration of the Zn element on the Fe3O4 nanoparticle surface would endow doped nanoparticles with different catalytic interactions towards varied molecules.38 Although electrocatalyst S3 has the overall faster electron transport (lowest Rct resistance), electrocatalyst S4 showed a much favourable electrocatalytic sensing of the APAP molecule (highest Ipa response current). Therefore electrocatalyst S4 is selected as the electrocatalyst for the electrochemical sensing of APAP. A study investigated on the carbon nanotubes accelerated electrochemical sensing towards APAP and found that it was the carbonaceous impurities in the CNT that caused the acceleration.39 In the biomass carbon experiments, the enhanced electrochemical catalytic sensing towards APAP probably comes from the Zn-doped molar ratio. Fig. 2(C) shows the electrocatalyst S4 modified GCE's CV scan with varying scan rates (100, 150, 200, 250, 300, 350, 400, 450 and 500 mV s−1). The redox pair [Fe(CN)6]3−/4− shows the redox peaks’ potential positions at about 0.5 V and 0.1 V. Larger scan rate induced stronger Ipa current response.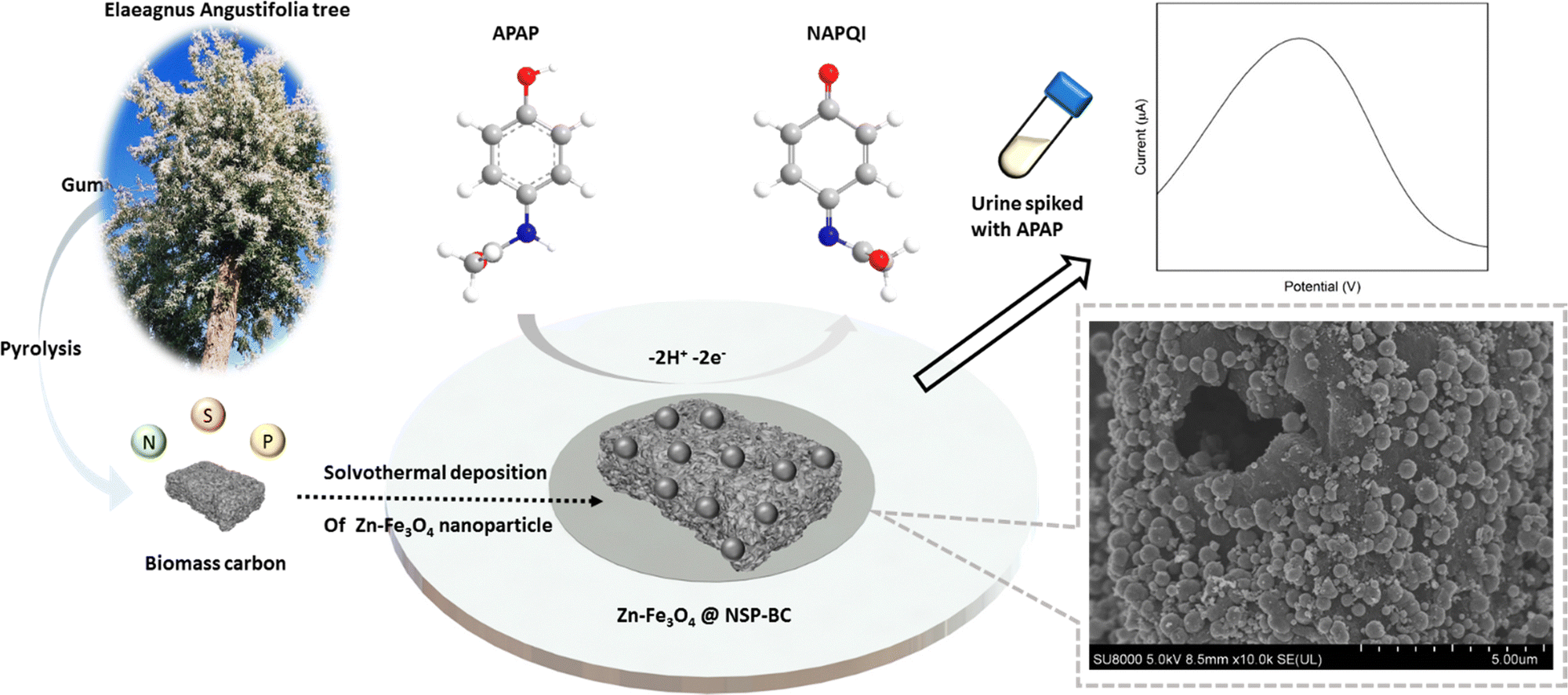 | ||
| Fig. 1 Schematic illustration of the preparation of the Zn doped Fe3O4 nanoparticle@NSP-BC, the electrochemical catalytic reaction of APAP's oxidation into NAPQI on the surface of the electrocatalyst modified GCE and the SEM image of the typical nanoparticle decorated NSP-BC electrocatalyst. | ||
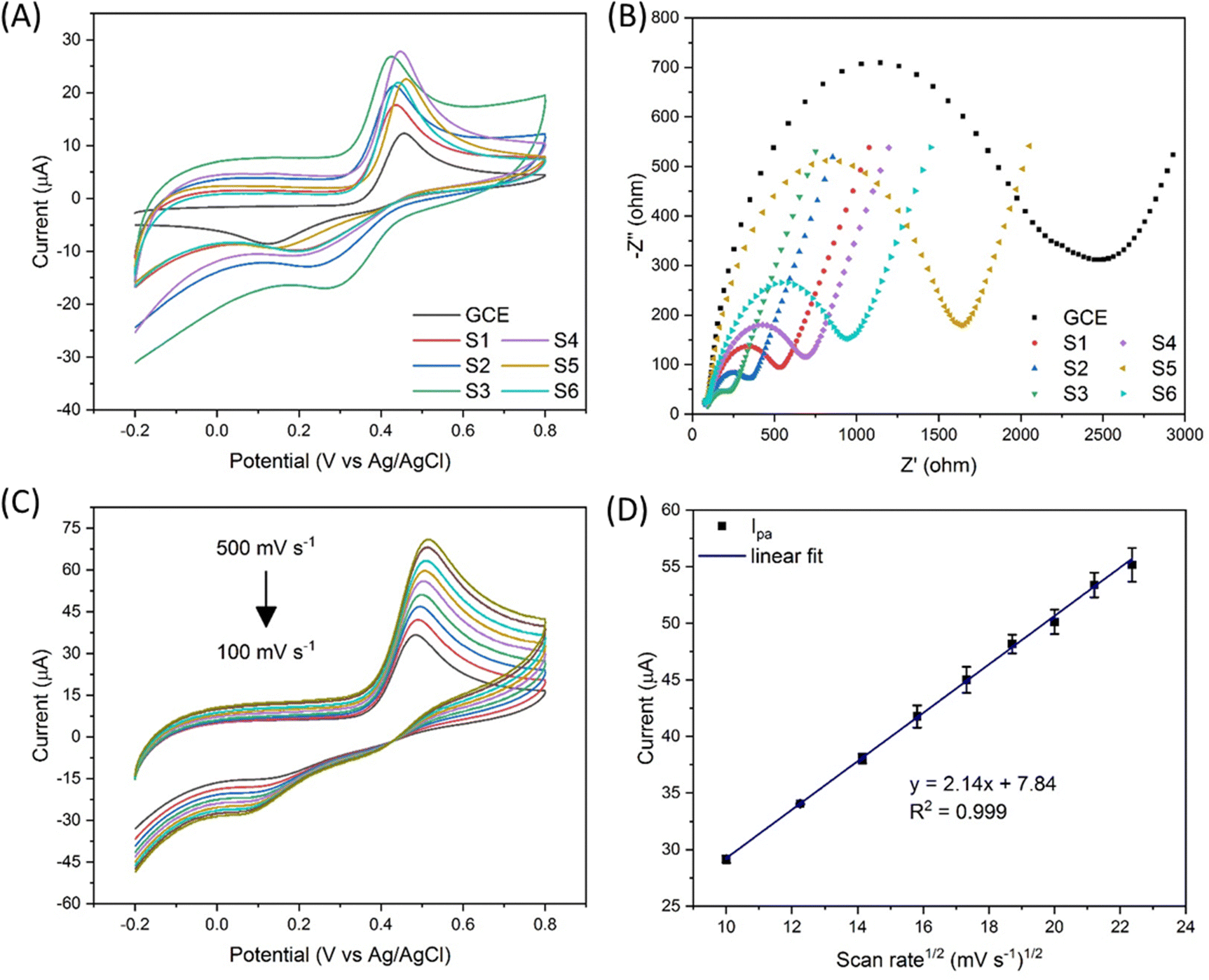 | ||
| Fig. 2 (A) CV spectra of the GCE and electrocatalyst modified GCEs in APAP solution (500 μM in 0.01 M PBS, scan rate 50 mV s−1), (B) EIS spectra of the GCE and electrocatalyst modified GCEs in 5 mM [Fe(CN)6]3−/4−, 0.1 M KCl (frequency range from 100 KHz to 0.1 Hz), (C) electrocatalyst S4 modified GCE's CV spectrum in 5 mM [Fe(CN)6]3−/4−, 0.1 M KCl with changing scan rate, and (D) linear fit of the Ipa current versus the square root of the scan rate in (C). | ||
In Fig. 2(D), a linear fit relationship between the Ipa current and the square root of the scan rate in Fig. 2(C) was obtained with R2 = 0.999, which indicated that the redox reaction of [Fe(CN)6]3−/4− is a diffusion-controlled process catalysed by the Zn-doped Fe3O4 nanoparticle@NSP-BC electrocatalyst. The Randles–Sevcik equation (eqn (1)) was used to estimate the electrochemical active surface area of the electrocatalyst S4 modified GCE.40
| Ip = 2.69 × 105A·D1/2·n3/2·v1/2C | (1) |
I p: peak current (A);
A: the electrochemical active surface area (cm2);
D: the diffusion coefficient of the molecule in the bulk solution (6.67 × 10−6 cm2 s−1 for potassium ferricyanide);
n: the number of electrons that participated in the reaction;
v: the scan rate (V s−1);
C: the concentration of the bulk solution (M).
The electrochemically active surface area of the electrocatalyst S4 modified GCE sensor was calculated to be 0.0225 cm2. For the bare GCE used in the experiments, the glassy carbon electrode's electrochemically active surface area is calculated to be 0.0174 cm2, and the electrocatalyst S4 modified GCE has a 29.3% increase in active surface area compared to the bare GCE. And all the electrocatalyst modified GCE has a higher electrochemical sensing signal than the bare GCE. All the Zn alloyed electrocatalyst has a higher electrochemical signal than the pure Fe3O4 nanoparticle@NSP-BC electrocatalyst. Especially, the electrocatalyst S4 modified GCE has a 74.2% increase of Ipa compared to the bare GCE's electrochemical sensing signal, which indicated the strong electrochemical catalytic performance of Zn doped Fe3O4 nanoparticle@NSP-BC towards the APAP molecule. As discussed in references,41–43 the addition of nanomaterials or an embedded ligand would lead to a decrease of matrix's specific surface area, but the catalytic or absorptive functions of the added material would offer desirable applications towards sensing or adsorption. And in this study, the electrocatalytic properties of the electrocatalyst comes from the Zn doped Fe3O4 nanoparticle, whose electrocatalytic performance can be modulated by controlling the Zn dopant molar ratio or by using other transitional elements or by using more element co-dopants together.
Material characterization
As shown in Fig. 3(A), the Raman spectroscopy measurement was carried out to study the molecular structure of the NSP-BC and the nanoparticle@NSP-BC electrocatalyst. The NSP-BC spectrum shows the typical D band and G band of most carbon materials at around 1337 cm−1 and 1591 cm−1. The electrocatalyst shows weaker D band and G band signals, confirming the presence of the NSP-BC material in the electrocatalyst. And the D/G ratio in the NSP-BC spectrum is calculated to be 1.56, confirming the heteroatom structures (doping of N, S and P) of the biomass carbon. In the nanoparticle@NSP-BC electrocatalyst, the D/G ratio for S1 to S6 is calculated to be 1.48, 1.54, 1.50, 1.55, 1.60 and 1.59, respectively, which indicates that the solvothermal polyol synthesis method of decorating nanoparticle did not show obvious influence on the NSP-BC heteroatom structures.44,45 The peaks of the nanoparticle@NSP-BC electrocatalyst between 250 and 750 cm−1 (light pink square) are considered to come from the Fe3O4 nanoparticle and the Zn doped Fe3O4 nanoparticle in the hybrid nanoparticle@biomass carbon nanocomposite.46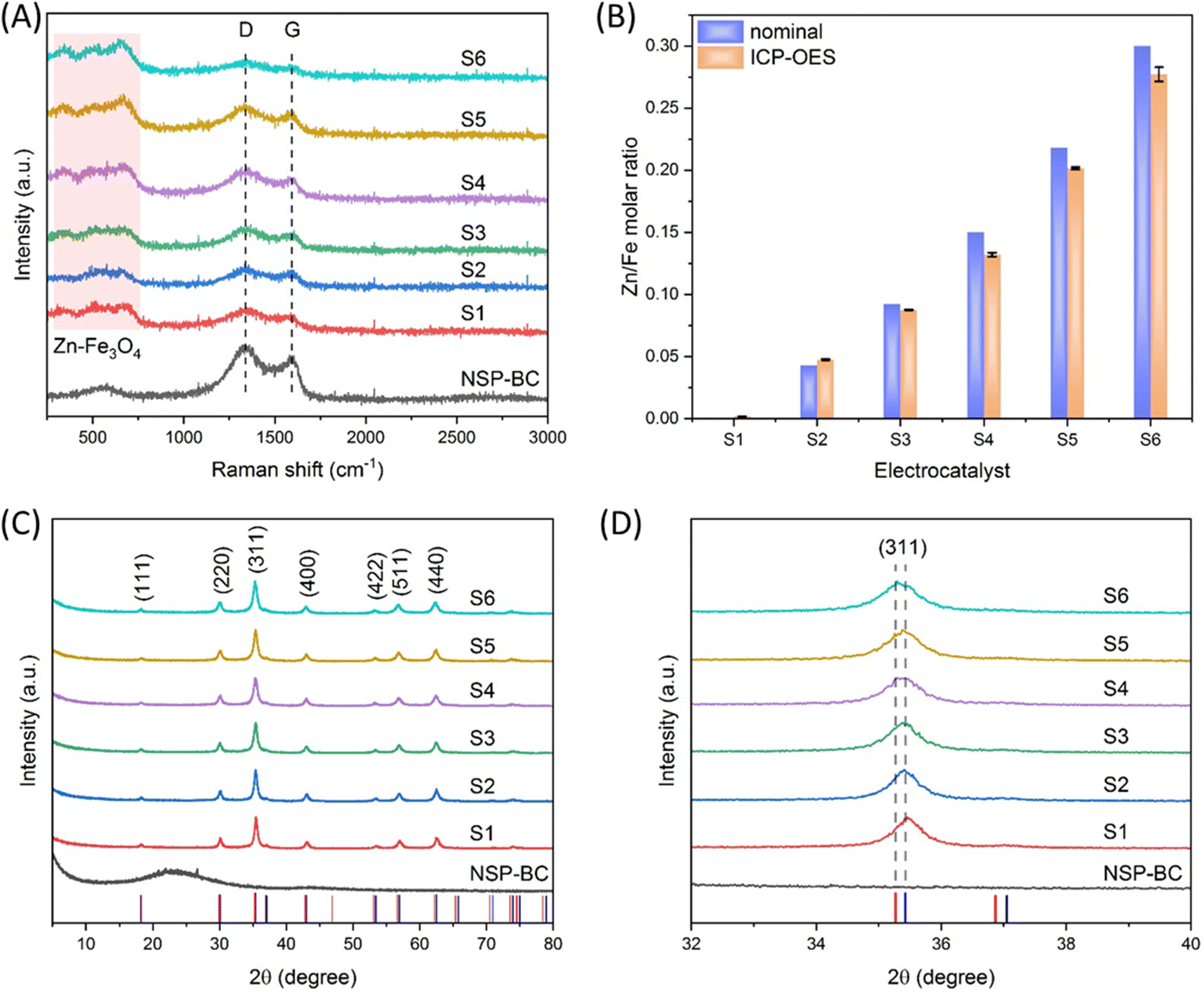 | ||
| Fig. 3 (A) Raman spectra of the NSP-BC and nanoparticle@NSP-BC electrocatalyst; the laser wavelength is 632.81 nm, (B) comparison of the Zn/Fe molar ratio of nominal value and ICP-OES results, (C) XRD spectra of the NSP-BC and nanoparticle@NSP-BC electrocatalyst, Cu-Kα radiation, λ = 1.5406 Å, and (D) zoom in view of the (311) peak in (C), the red vertical droplines are from ZnFe2O4 (PDF#22-1012) and the blue vertical droplines are from Fe2+Fe23+O4, synthetic (PDF#19-0629). | ||
In order to quantify the actual Zn/Fe molar ratio in the electrocatalyst, the ICP-OES measurement for electrocatalysts S1 to S6 were performed. As shown in Fig. 3(B), the experimental nominal Zn/Fe ratio and the ICP-OES results were drawn in adjacency in the graph for comparison. The nominal value of Zn/Fe molar ratios for electrocatalysts S1 to S6 are, respectively, 0, 0.043, 0.092, 0.15, 0.218 and 0.3. The ICP-OES results of the Zn/Fe molar ratios for electrocatalysts S1 to S6 are, respectively, 0.000018, 0.0475, 0.0875, 0.132, 0.201 and 0.277. The nominal and actual Zn/Fe molar ratios are very close with small deviation. One reason may be that some Zn ions are adsorbed into the NSP-BC during the synthesis and thus resulted in the lowering of the Zn/Fe molar ratio.
Fig. 3(C) shows the XRD spectra of the NSP-BC and nanoparticle@NSP-BC electrocatalyst samples. The red vertical droplines and the blue vertical droplines are from the standard XRD card of ZnFe2O4 (PDF#22-1012) and Fe2+Fe23+O4, synthetic (PDF#19-0629). The NSP-BC's XRD spectrum shows a similar curve to other biomass carbon's XRD spectrum.44,45 The XRD spectra of the Zn doped Fe3O4 nanoparticle@NSP-BC electrocatalysts all displayed similar peaks, which are in accordance with the standard XRD cards and reference.47 The characteristic diffraction peaks of (111), (220), (311), (400), (422), (511) and (440) are marked in the graph. As the Zn2+ ion has a radius of 74 pm, the Fe2+ ion has a radius of 61 pm, and the Fe3+ ion has a radius of 49 pm. The doped Zn2+ ion replaces the Fe2+ ion, thus causing the enlargement of the crystal lattice parameters. The lattice parameters of the nanoparticle@NSP-BC electrocatalysts S1 to S6 are calculated to be 8.3987, 8.3990, 8.4029, 8.4133, 8.4173 and 8.4329 Å respectively. As the Zn alloying ratio increases, the lattice parameter increases accordingly. The calculated lattice parameter values are rational in comparison with references (near 8.3 to 8.4 Å range) and in accordance with references with little changes.38,48 The presence of NSP-BC in the reaction solution may induce some difference compared to the pure nanoparticle synthesis. The calculation is also dependent on the XRD patterns and peak simulation results.
As a result of the larger radius of the Zn2+ ion, the doped Fe3O4 nanoparticle displays a slight peak shift compared with a pure Fe3O4 nanoparticle. Fig. 3(D) shows the zoom in view of the (311) peak in Fig. 3(C), as the Zn alloying ratio increases, and the (311) peak slowly shifts from the pure Fe3O4 nanoparticle peak (2θ = 35.422) to near the ZnFe2O4 (PDF#22-1012) peak (2θ = 35.264). The peak shifting trend confirms the successful alloying of the Zn element into the crystal structure of the Fe3O4 nanoparticle.
As shown in Fig. 4, SEM, EDX and element mapping were used to characterize the (A) NSP-BC, (B) electrocatalyst S1 and (C) electrocatalyst S4. In the SEM images, NSP-BC shows porous structures, which were made by the pore-forming chemical of MnSO4 during the biomass carbon preparation process. Electrocatalyst S1 (pure Fe3O4 nanoparticle@NSP-BC) and electrocatalyst S4 (Zn–Fe3O4 nanoparticle@NSP-BC) shows that the nanoparticles are well attached with the biomass carbon material. As in the solvothermal process, the biomass carbon works as a nanoparticle deposition and adhesion substrate and the nanoparticles grow on the biomass carbon's surface. Therefore, these well hybrid nanostructures are formed. The element mapping images show the successful doping of N, S and P into the biomass carbon. The element mapping of Fe and Zn shows the presence of Fe and Zn elements. Fig. S1(A) (ESI†) shows the EDX spectra of the tested samples, and the results are in accordance with the element mapping results. The biomass carbon shows the elements C, O, N, S and P. Electrocatalyst S1 (pure Fe3O4 nanoparticle@NSP-BC) shows the presence of C, O, N, S, P and Fe. And electrocatalyst S4 (Zn–Fe3O4 nanoparticle@NSP-BC) shows the presence of C, O, N, S, P, Fe and Zn. The EDX spectrum also provides a Zn/Fe atomic ratio of 0.133 for electrocatalyst S4, which matches very well with the ICP-OES result of 0.132 from Fig. 3(B). The nanoparticle's size distribution is analysed as shown in Fig. S1(B) (ESI†). The nanoparticles are majorly distributed from the 400 to 600 nm diameter range. Although the pure Fe3O4 nanoparticle in electrocatalyst S1 seems to take more proportion in the 500 nm range, the Zn–Fe3O4 nanoparticle in electrocatalyst S4 seems a bit smaller than the pure Fe3O4 nanoparticle in S1. This may come from the sampling nanoparticles by using software rather than using dynamic light scattering analysis, which is commonly used for colloid's diameter analysis. However in this case, the nanoparticle@biomass carbon hybrid nanostructure is not fit for the DLS analysis.
 | ||
| Fig. 4 (A) SEM and element mapping images of NSP-BC, (B) electrocatalyst S1, and (C) electrocatalyst S4; scale bars are all 1 μm. | ||
In order to analyse the atom valence state on the surface of the NSP-BC, electrocatalyst S1 and electrocatalyst S4 samples, the XPS characterization was performed. Fig. 5 shows the XPS spectra of NSP-BC, electrocatalyst S1 and electrocatalyst S4. The survey spectra in Fig. 5(A) show the presence of C, O, N, S and P elements’ electrons in all tested samples, confirming the successful hybridization of Fe3O4 and Zn–Fe3O4 nanoparticles with biomass carbon without affecting the biomass carbon's surface properties. Fig. 5(B) shows the C 1s scan of the tested samples, several fitted curves are drawn, and the peaks correspond to different carbon functional groups of C–C, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. The peak at 284.58 eV in NSP-BC shifts to 284.38 eV in Fe3O4 nanoparticle@NSP-BC and 283.68 eV in Zn–Fe3O4 nanoparticle@NSP-BC. Electrocatalyst S1 and electrocatalyst S4 show new peaks at 288.18 eV and 287.58 eV compared with NSP-BC, which is not obvious in NSP-BC C 1s scan. This peak might come from the chemical bonds formed between the biomass carbon material and nanoparticle.
O. The peak at 284.58 eV in NSP-BC shifts to 284.38 eV in Fe3O4 nanoparticle@NSP-BC and 283.68 eV in Zn–Fe3O4 nanoparticle@NSP-BC. Electrocatalyst S1 and electrocatalyst S4 show new peaks at 288.18 eV and 287.58 eV compared with NSP-BC, which is not obvious in NSP-BC C 1s scan. This peak might come from the chemical bonds formed between the biomass carbon material and nanoparticle.
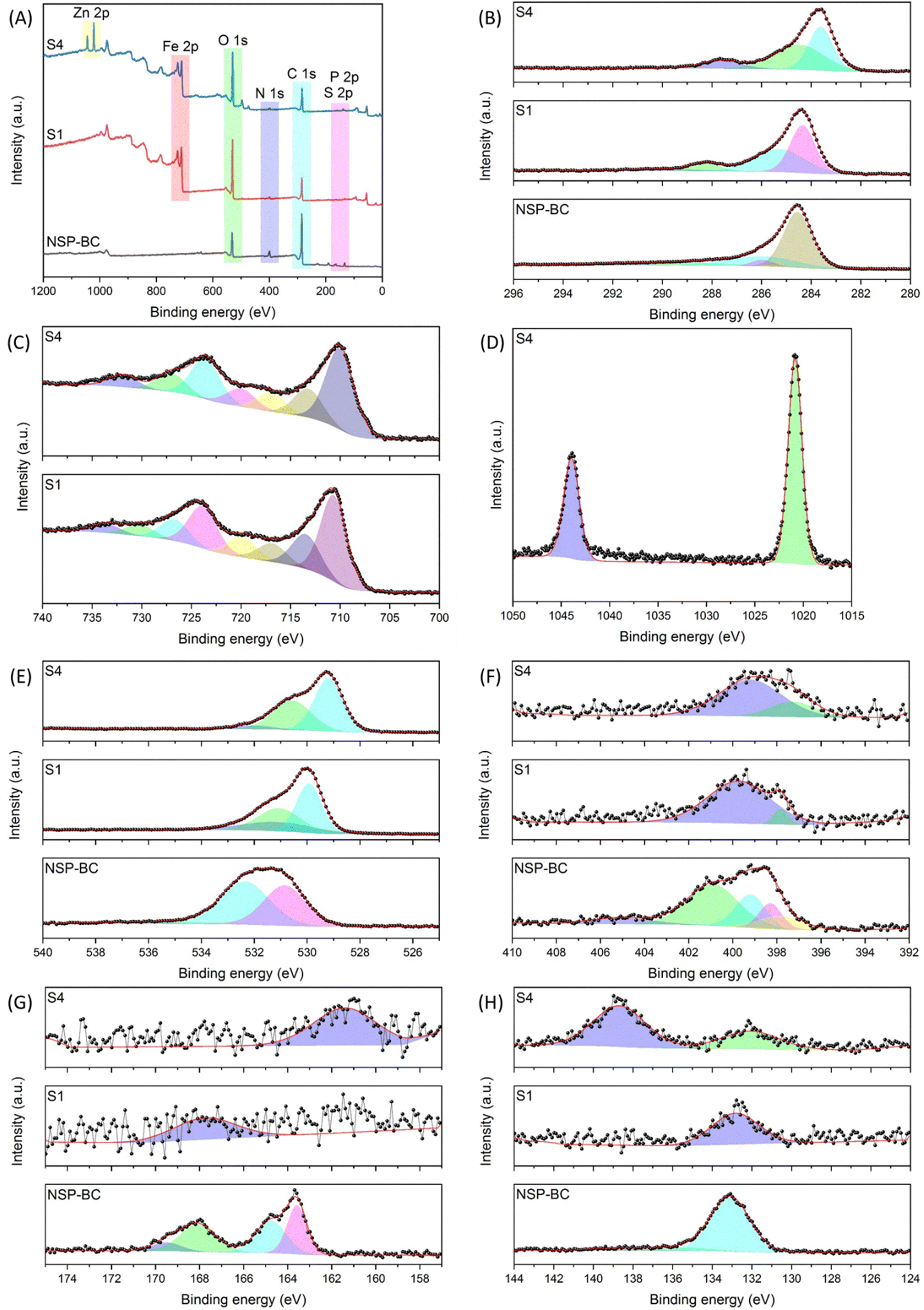 | ||
| Fig. 5 XPS spectra of NSP-BC, electrocatalyst S1 and electrocatalyst S4 samples, (A) survey scan, (B) C 1s scan, (C) Fe 2p scan, (D) Zn 2p scan, (E) O 1s scan, (F) N 1s scan, (G) S 2p scan, and (H) P 2p scan. | ||
Fig. 5(C) shows the Fe 2p scan for electrocatalyst S1 and electrocatalyst S4, and the Fe 2p3/2 and Fe 2p1/2 electrons are designated into the several fitted curves. After the Zn alloying, the peaks in electrocatalyst S1 (724.28 eV and 710.88 eV) shift to the lower binding energy in electrocatalyst S4 (723.68 eV and 710.18 eV). This shift is assumed to have come from the Zn alloying into the crystal lattice.
Fig. 5(D) shows the Zn 2p scan from electrocatalyst S4, and two peaks at 1043.98 eV and 1020.88 eV (ascribed to the binding energy of Zn2+ 2p3/2) are present.47Fig. 5(E) displays the O 1s scan, and the peak at 531.58 eV in NSP-BC also shifts to 529.98 eV in electrocatalyst S1 and 529.28 eV in electrocatalyst S4. Because the metal oxide nanoparticle has plentiful oxygen element, the O 1s scan signal remains very strong in all the measured samples.
Fig. 5(F) displays the N 1s, S 2p and P 2p scan, and the signals of the NSP-BC are much strong and clear, while the signals of electrocatalyst S1 and electrocatalyst S4 are much weaker. This may be due to the covering of metal oxide nanoparticles on the surface of the biomass carbon. Another reason may be that the S and P elements distributed on the NSP-BC surface may form chemical bonds with nanoparticle's metal elements of Fe and Zn, thus bringing down the signal intensity. The solvothermal polyol process may also induce a peak shift, peak emergence and weakening. The N 1s scan is related to pyridinic-N, pyrrolic-N, graphitic-N and oxidized pyridinic-N. Fig. 5(G) shows the S 2p scan with peaks related to S 2p3/2, S 2p1/2 and sulfone like bonds. Fig. 5(H) shows the P 2p scan showing the P–C and P–O bonds. The N 1s, S 2p and P 2p scans are all in accordance with reference, confirming the successful preparation of the designated materials.49
Electrochemical analysis of APAP
The electrocatalyst S4 modified GCE sensor was selected to measure the DPV Ipa signal dependence of APAP concentration. As shown in Fig. 6(A), the measured APAP concentrations are 0, 2.5, 5, 10, 20, 40, 60, 80, 100, 200, 300, 400 and 500 μM respectively. This linear sensing range is selected based on the fact that the urinary APAP concentration is in this range. A study8 by Smarr found that male urinary APAP concentration ranges with the highest quartile (>73.5 ng mL−1, 486 μM) and the lowest quartile (<5.44 ng mL−1, 36 μM). In Smarr's study, the drug intake and diet of the human subjects were not monitored. Therefore, the human subjects may or may not be taking medicines containing APAP. The urinary APAP concentration measured is considered as a representative value. The average male urinary APAP concentration is roughly calculated as 200 μM compared with the calculation result in eqn (2).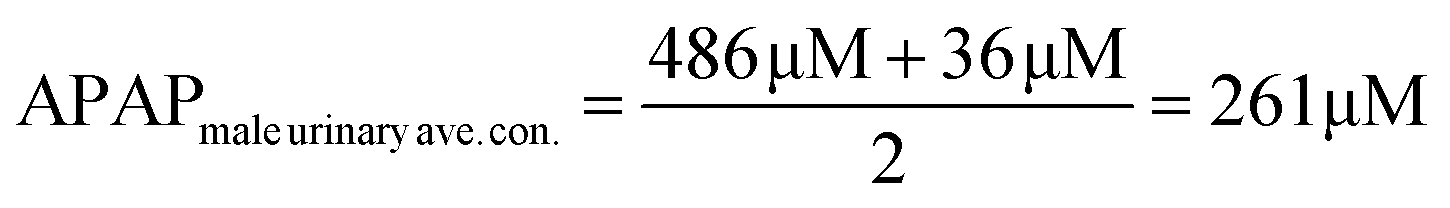 | (2) |
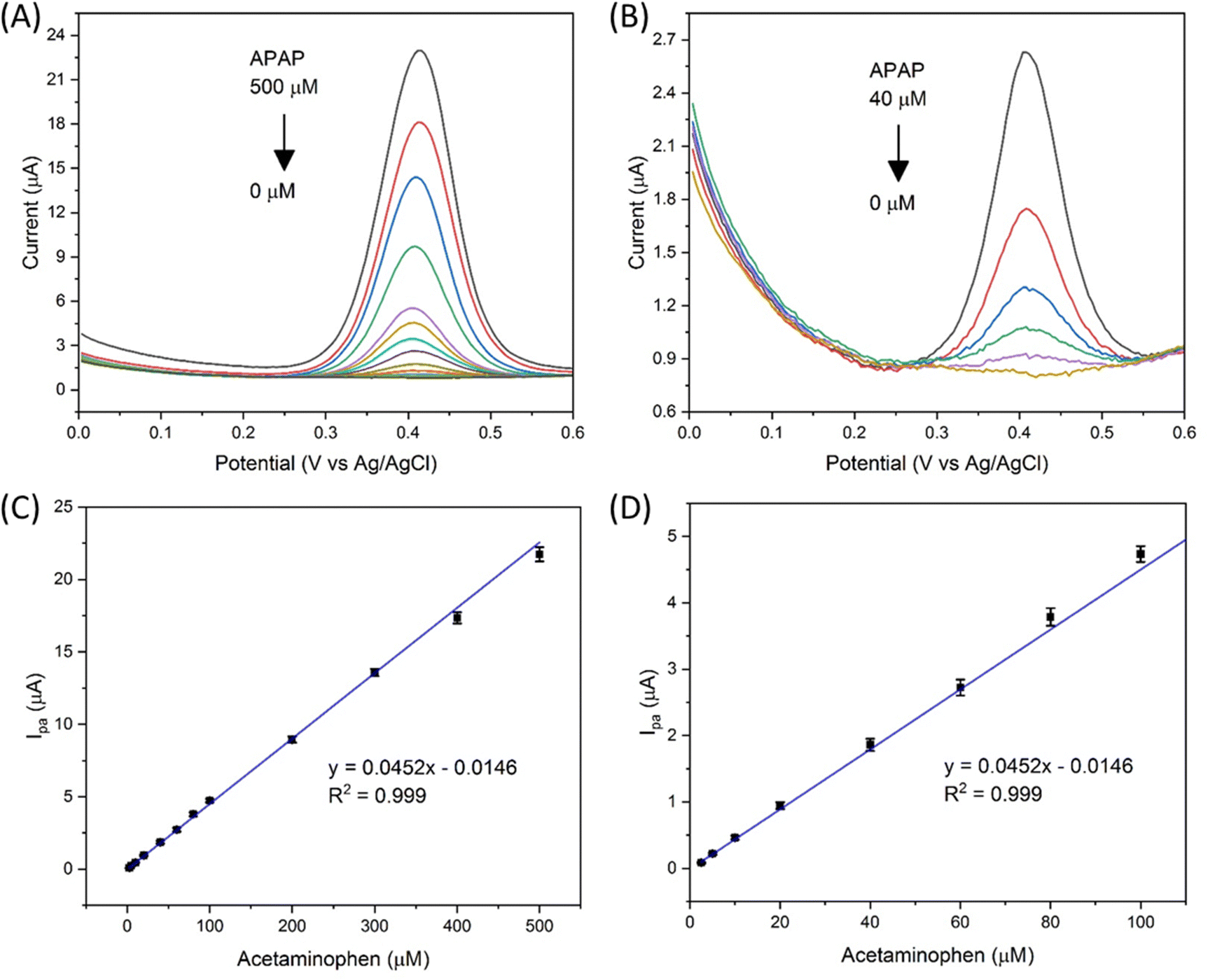 | ||
| Fig. 6 (A) DPV response of the electrocatalyst S4 modified GCE towards APAP (0 to 500 μM in 0.01 M PBS), (B) zoom in view of the DPV response of APAP (0 to 40 μM) in (A), (C) the Ipa dependence of APAP's concentration, and (D) zoom in view of (C) in the range from 2.5 to 100 μM. | ||
A linear fit of the DPV Ipa dependence on APAP concentration was obtained as shown in Fig. 6(C) with the linear range from 2.5 to 500 μM, a limit of detection of 63.9 nmol and a sensitivity of 0.64 μA μM−1 cm−2. The linear fitting equation is listed as eqn (3).
| y = 0.0452x − 0.0146, R2 = 0.999 | (3) |
Fig. 6(D) shows the zoom in view of the APAP concentration from 2.5 to 100 μM. Therefore, the designed Zn doped Fe3O4 nanoparticle@NSP-BC electrocatalyst modified GCE sensor has a wide linear range towards APAP from 2.5 to 500 μM.
The selectivity of the electrocatalyst S4 modified GCE sensor was performed using the interferences of uric acid, ascorbic acid, urea and glucose (UA, AA, urea and GLU). As shown in Fig. 7(A), the APAP and all interferences are prepared at 200 μM concentration in 0.01 M PBS. The mixture is prepared by mixing all the interferences with each interference of 200 μM. Uric acid has a nearby oxidation peak at about 0.337 V, while APAP has an oxidation peak at about 0.410 V. The other interference does not show obvious current response in the 0 to 0.8 V range. The AA DPV curve has a slight uplift in this range but not prominent enough to show obvious influence. The DPV scan curve of the mixture is obviously affected by the UA in Fig. 7(A), while the DPV signal of the mixture (excluding UA) shows a change barely compared with the APAP signal, with a slight peak shift from 0.409 V to 0.428 V and basically unchanged Ipa response in Fig. 7(B). The merged peak in the mixture curve in Fig. 7(A) is difficult to be divided into separate peaks. As summarized and calculated and shown in Fig. 7(C), the mixture (excluding UA) signal shows p value > 0.05 compared with the APAP signal, the mixture signal shows p value < 0.05 compared with the APAP signal, and the mixture signal shows p value < 0.05 compared with the mixture (excluding UA) signal. Under this tested condition, the UA can interfere heavily with the electrocatalyst modified GCE sensor for APAP electrochemical sensing.
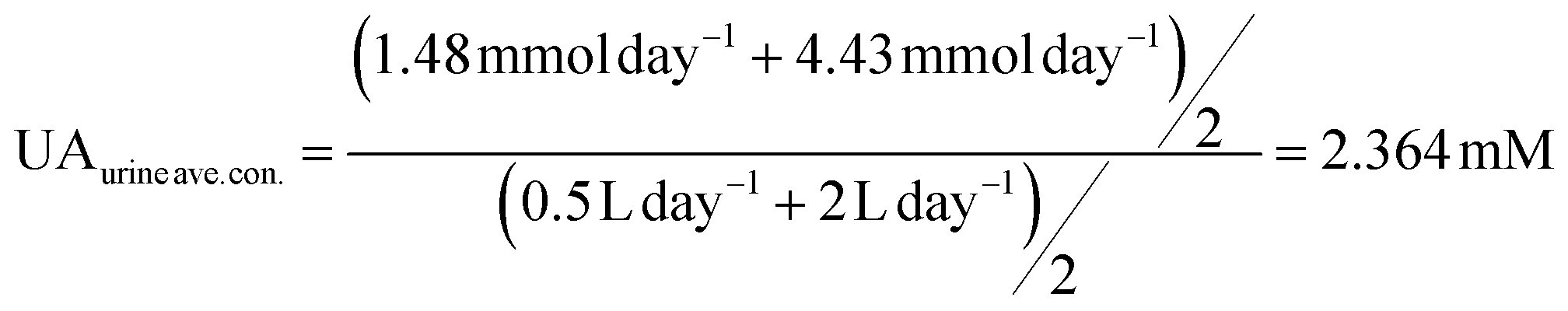 | (4) |
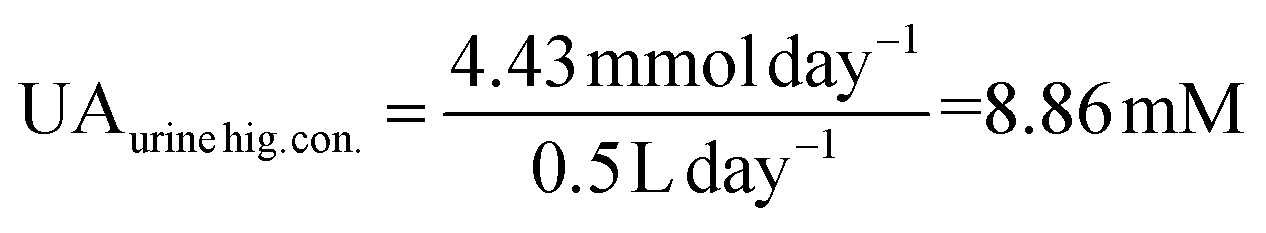 | (5) |
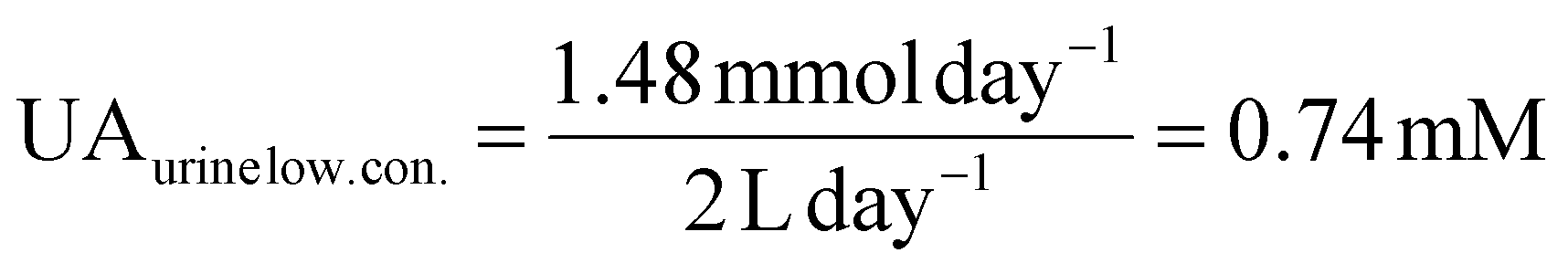 | (6) |
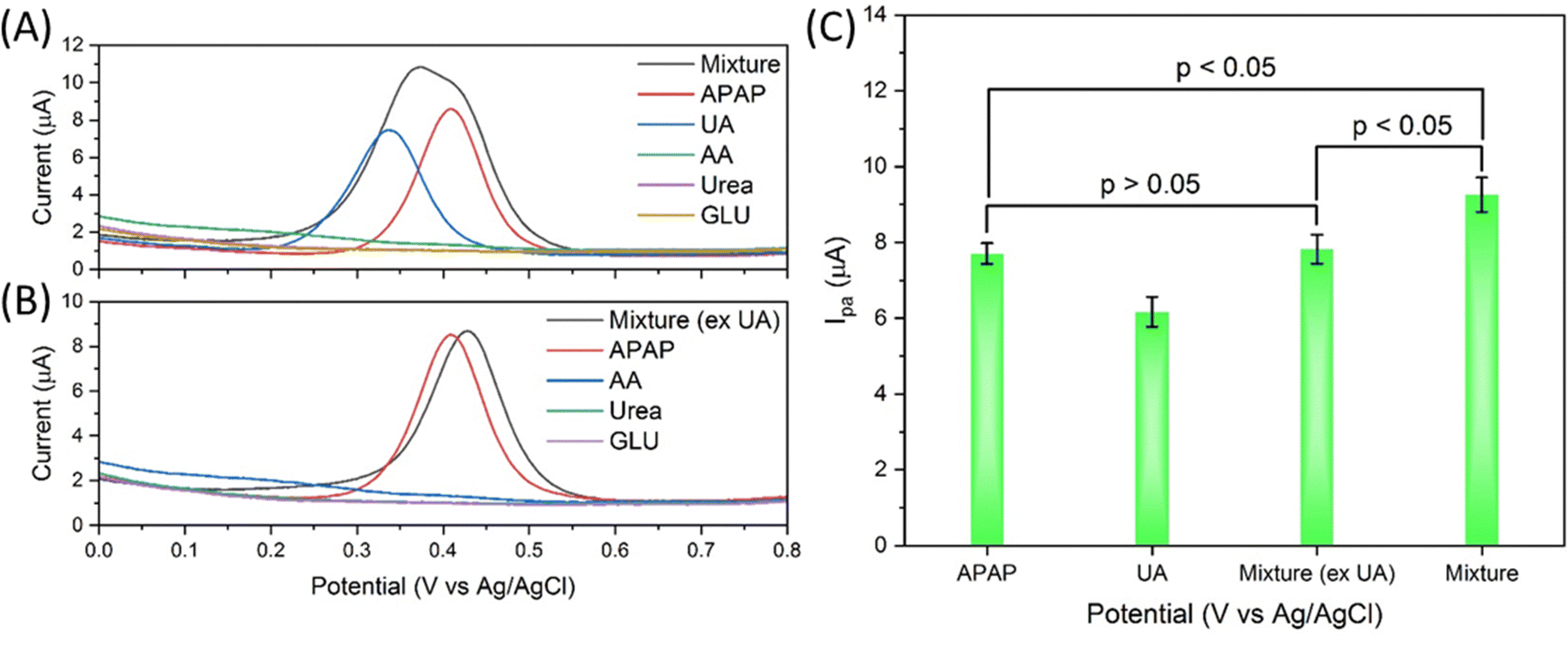 | ||
| Fig. 7 (A) and (B) show selectivity results of the electrocatalyst S4 modified GCE against the interferences of uric acid, ascorbic acid, urea and glucose (UA, AA, urea and GLU), all chemical solution concentrations are 200 μM in 0.01 M PBS, and (B) summary graph of Ipa of the DPV signal from (A) and (B). | ||
UA is excreted in human urine and its concentration depends upon diet heavily. For rough calculation, about 1.48 to 4.43 mmol UA is excreted in urine per 24 hours, and about 0.5 to 2 L urine is produced per 24 hours for healthy people. The possible average, highest and lowest urinary UA concentrations in the healthy range is calculated to be 2.364, 8.86 and 0.74 mM as shown in eqn (4), (5) and (6).
The fluctuation in urinary UA concentration is large and depends on many factors, e.g. weather, sweat amount, drinking amount, diet, exercise, physical condition etc. Therefore, 2 mM UA concentration is selected as the average urinary UA concentration in healthy people's urine sample. Further experiments on the UA and APAP mixture measurement were carried out on the basis of 2 mM UA and 200 μM APAP. In detail, the undiluted urine sample is spiked with 200 μM APAP, and the 10 times diluted urine sample in 0.01 M PBS is spiked with 20 μM APAP. As shown in Fig. 8(A) and (C), the urine and diluted urine samples show the oxidation peaks at about 0.435 V and 0.435 V with Ipa of 8.72 μA and 0.614 μA. The dilution of urine did not change the oxidation potential position. And it is possible that this peak comes from the naturally excreted uric acid in urine. The spiked APAP of 200 μM and 20 μM show the oxidation potential to be about 0.413 V and 0.448 V with Ipa of 9.46 μA and 1.09 μA. The interference peaks in urine sample share almost the same peak position and Ipa with the spiked APAP.
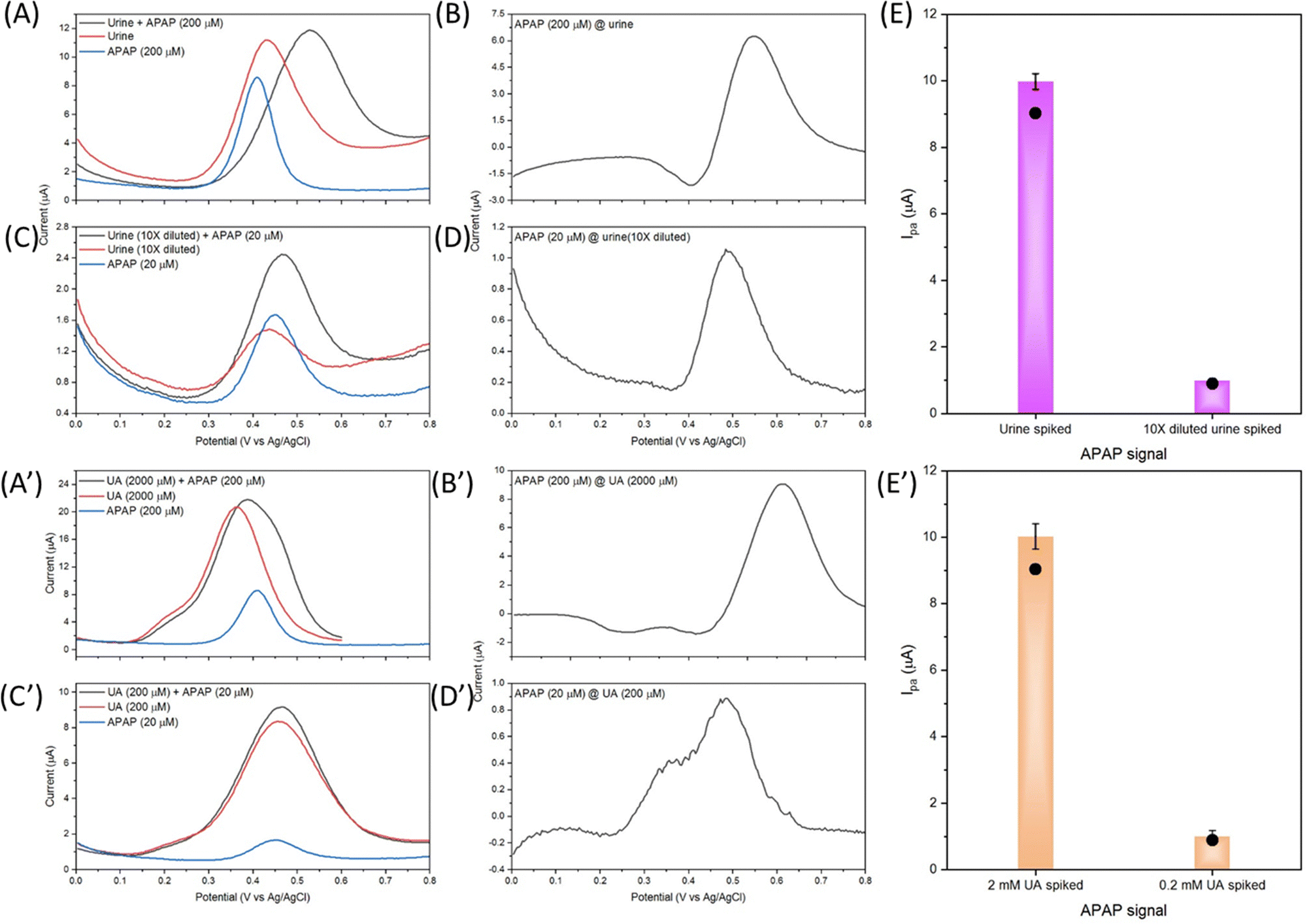 | ||
| Fig. 8 DPV response curves with urine samples spiked with APAP and UA samples spiked with APAP by the electrocatalyst S4 modified GCE, (A) urine, (C) 10 times diluted urine, (B), (D) and (E) processed APAP signal from (A) and (C), (A′) urine comparative UA sample, (C′) 10 times diluted urine comparative UA sample, (B′), (D′) and (E’) APAP signal from (A′) and (C′). The black dots in (E) and (E′) are the corresponding APAP current signals calculated by the standard curve eqn (3). | ||
As discussed in Fig. 7(A), the UA and APAP current response merged into an inseparable peak in the mixture's DPV curve. The same result is observed in Fig. 8(A) and (C), and the urine and diluted urine samples spiked with APAP display peaks with shifted potential and enhanced Ipa of 0.529 V, 9.10 μA and 0.466 V, 1.61 μA. Fig. 8(B) and (D) are obtained through the subtraction of the urine sample signal by the spiked urine sample in CHI-760E software (CH Instruments, Inc.). The Ipa value obtained from the subtracted curves is correspondingly the Ipa value of spiked APAP concentration calculated from eqn (3) as shown in Fig. 8(E). But the peak position shifts to 0.550 V for the urine sample, and 0.488 V for the diluted urine sample.
Fig. 8(A′) to (E′) show graphs from the comparative UA solution spiked with APAP. Similar results are obtained in comparison with Fig. 8(A) to (E). The UA of 2 mM is far more concentrated than the provided urine UA concentration as the UA of 2 mM and 200 μM shows an oxidation peak and Ipa of 0.364 V, 19.43 μA and 0.456 V, 6.60 μA. The spiked APAP signal can be achieved by subtraction of the UA response but the oxidation potential shifts are also present. All in all, Fig. 8 provides an insight into the electrocatalyst modified GCE sensor performance on the urine sample, diluted urine sample and the physiological concentrated UA samples. And the obtained relative good results demonstrate the practical application of the designed Zn doped Fe3O4 nanoparticle@NSP-BC electrocatalyst.
Table 1 summarizes the urine sample test and the comparatively UA sample study of the electrocatalyst S4 modified GCE sensor. Firstly, urine was used directly in the urine sample experiment, and 90% volume of 0.01 M PBS was used in the 10 times diluted urine sample experiment, while in the UA experiments, 0.01 M PBS solution is used. Secondly, in the urine and diluted urine experiments, multiple interferences of biomolecules exist. Thirdly, the UA interference in the urine sample and diluted urine sample shows nearly equal Ipa response, while in the UA sample, the UA shows a much stronger Ipa response than the spiked APAP signal. In principle, the UA experiments should achieve much precise results compared to the urine samples, but due to these discussed factors, the urine samples results (in Table 1, # 1 and # 2) of the urine sample and diluted urine sample measurements are similar to the comparative UA sample results (in Table 1, # 3 and # 4).
| # | Sample | Spiked APAP (μM) | Measured APAP (μM) | Recovery rate (%) |
|---|---|---|---|---|
| 1 | Urine | 200 | 213.86 ± 18.37 | 106.9 |
| 2 | Urine (10× diluted) | 20 | 23.17 ± 1.88 | 115.8 |
| 3 | UA (2 mM) | 200 | 210.96 ± 19.20 | 105.5 |
| 4 | UA (0.2 mM) | 20 | 22.54 ± 3.77 | 112.7 |
The results in Table 1 prove that UA is the major interference in the urinary APAP sensing by the Zn–Fe3O4 nanoparticle@NSP-BC electrocatalyst modified GCE sensor. But in the spiked experiments of urine and UA samples, this electrocatalyst modified GCE sensor is still able to acquire the relatively correct concentration value of the spiked APAP.
Comparison with other work
A survey of related research on nanomaterials’ electrochemical sensing of APAP is listed in Table 2. Their electrocatalyst, detection range and limit of detection are listed for clear comparison with the Zn–Fe3O4 nanoparticle@NSP-BC electrocatalyst in the table. As seen from Table 2, carbon materials are widely used for electrochemical sensing of APAP, like carbon black, biomass carbon (date stone and kelp), carbon nanotubes, graphene, graphene oxide, etc. Multiple kinds of nanomaterials like TiO2, Pd, ZnO/ZnNi2O4, ZnO, Co3O4, and Fe2O3 nanoparticles, metal organic frameworks, etc. are hybridized with carbon based materials. The hybrid electrocatalyst of nanoparticles with carbon materials is widely studied. Many special functionalities come from the anchored nanoparticles and the hybrid nanocomposites. The current study fabricated an electrocatalyst of Zn–Fe3O4 nanoparticle@biomass carbon and achieved a good sensing range covering the physiological urinary APAP concentration range and a good limit of detection for APAP compared to other research studies. One of the obvious advantages of the Zn–Fe3O4 nanoparticle@biomass carbon electrocatalyst is that at the beginning we aimed at measuring the physiological APAP concentration, and this electrocatalyst modified GCE sensor achieved a linear range covering the targeted physiological APAP concentration range with a good recovery rate. Therefore, this electrocatalyst modified GCE sensor could be potentially used to measure the urine sample directly without the need for dilution or other processed steps. Another advantage lies in the materials used for the preparation of the electrocatalyst. Fe and Zn are not expensive materials and their oxides are biocompatible. No expensive or noble metal salts are used. The Elaeagnus angustifolia gum is a natural plant product and its major chemical composition is polysaccharide; after pyrolysis, the biomass carbon is majorly a C element based material, which is also biocompatible, economic and environment friendly. What's more, the electrocatalytic capability of the electrocatalyst could be modulated by switching the dopant element or by using co-doping of more transitional elements. Therefore, this electrocatalyst has above advantages compared to noble metals, toxic elements and unrecyclable material electrocatalysts with complicated preparation procedures.| Electrocatalyst | Sensing range (μM) | Limit of detection (nM) | Ref. |
|---|---|---|---|
| Nitrogen-doped carbon@TiO2 double-shelled hollow spheres | 0.3 to 50 | 0.05 | 50 |
| Strontium vanadate-supported graphitic carbon nitride nanocomposite | 0.019 to 1100 | 26 | 51 |
| Pd nanoparticle@carboxylated graphene oxide | 0.04 to 800 | 12 | 52 |
| ZnO/ZnNi2O4@porous carbon@covalent-organic framework | 0.0485 to 130 | 0.012 | 53 |
| Fe2O3 encapsulated chitosan grafted polyaniline | 5 to 100 | 5700 | 54 |
| Bismuth oxide nanorod | 0.5 to 1250 | 30 | 55 |
| Carbon nanotubes with zinc oxide nanoparticles | 0.000005 to 0.0018 | 0.0000506 | 56 |
| Co3O4-embedded N-doped hollow carbon sphere | 1 to 200 and 1000 to 8000 | 70 and 110 | 57 |
| rGO–PEDOT nanotube composite | 1 to 35 | 400 | 58 |
| Nitrogen-doped carbon-supported bimetal (CuCo-NC) | 0.1 to 400 | 50 | 59 |
| Nafion-coated single-walled carbon nanotube | 1 to 2000 | 800 | 60 |
| Kelp derived activated carbon | 0.01 to 20 | 4 | 61 |
| Bimetallic metal organic framework (Fe and Mg MOF)@multi-layer black phosphorous nanosheets | 0.002 to 30 and 40 to 700 | 0.0007 | 62 |
| Zn–Fe3O4 nanoparticle@NSP-BC | 2.5 to 500 | 63.9 | This work |
Conclusions
We have developed a novel Zn–Fe3O4 nanoparticle@biomass carbon electrocatalyst through the facile solvothermal polyol approach and systematically studied the electrochemical sensing of APAP by the Zn–Fe3O4 nanoparticle@biomass carbon electrocatalyst modified GCE sensor. The Zn dopant ratio could modulate its electrocatalytic performance towards APAP. This Zn–Fe3O4 nanoparticle@NSP-BC electrocatalyst achieved a linear sensing range of 2.5 to 500 μM covering the physiological urinary APAP concentration range, a limit of detection of 63.9 nM and a sensitivity of 0.64 μA μM−1 cm−2. A satisfactory recovery rate was achieved for APAP spiked urine samples. A comparative study of the UA sample spiked with APAP was also performed to prove the useful application of the novel electrocatalyst modified GCE sensor. The influence of the UA interference is also studied and the physiological UA conditions were also researched with good electrochemical sensing results. Therefore, the studied electrocatalyst is ready for real clinical diagnosis. Future study could integrate this electrocatalyst into a paper electrode sensor system for rapid and disposable sensing applications. This study provides a good model for exploring the electrocatalytic performance of transition element doping in nanoparticles and proves the practicability of nanoparticle/biomass carbon electrocatalysts in clinical diagnosis and pharmaceutical analysis application.Experimental section
Chemical and reagents
All chemicals are of analytical grade and used as received. Acetaminophen and phosphate buffered saline tablets are from Aladdin. The PBS solution used throughout the experiments is prepared using the PBS tablet. Sodium hypophosphite, thiocyanuric acid, and uric acid are obtained from Macklin. Manganese sulfate is from Solarbio. Urea is from the Tianjin Tianda Fine Chemical Plant. Potassium chloride is from Tianjin Bodi Chemical Co., Ltd. Ascorbic acid, potassium ferricyanide(III), iron(III) chloride hexahydrate and sodium acetate are from Tianjin Baishi Chemical Co., Ltd. Zinc chloride is from Tianjin Jingjin Chemical Products Sales Co., Ltd. Glucose and potassium hexacyanoferrate(II) trihydrate are from the Tianjin Hedong District Hongyan Reagent Factory. Ethylene glycol is from Tianjin Benchmark Chemical Reagent Co., Ltd. All aqueous solutions were prepared with water (18.2 MΩ cm) from a Millipore system. Dried Elaeagnus angustifolia gum was provided by the Key Laboratory of Chemistry of Plant Resources in Arid Regions, State Key Laboratory Basis of Xinjiang Indigenous Medicinal Plants Resource Utilization. The human urine sample was provided by healthy male volunteer in the lab. The volunteer did not consume any medicine containing APAP for at least a month before providing urine.Synthesis of a Zn doped Fe3O4 NP@NSP-BC electrocatalyst
The NSP-BC was prepared by the following method. Elaeagnus angustifolia gum (0.35 g) and manganese sulfate (0.45 g) were added to 5 mL of deionized water and mixed well and denoted as solution A. Then, urea (0.55 g), sodium hypophosphite (0.45 g), and thiocyanuric acid (0.35 g) were dispersed in another 5 mL of deionized water and denoted as solution B. Solution A and solution B were mixed together and set in an oven at 130 °C for 6 h to obtain a dried yellow powder. Yellow powder was carbonized at 800 °C for 3 h with a heating rate of 5 °C min−1 under N2 flow. The carbonized product was washed with a 15 wt% HCl solution to remove metallic oxide and metal ion residues. The sample was centrifuged, washed several times with deionized water, and dried in a vacuum oven at 60 °C for 24 hours to obtain the NSP-BC.The nanoparticle decorated NSP-BC electrocatalyst was prepared through the solvothermal method. For Zn doped Fe3O4@NSP-BC electrocatalyst synthesis (samples of 1–6, the Zn/Fe ratio of which is 0, 0.043, 0.092, 0.15, 0.22, and 0.30, respectively), NSP-BC (20 mg), zinc chloride (0, 16.4, 32.7, 49.1, 65.4, and 81.8 mg, respectively), iron(III) chloride hexahydrate (810.9, 756.8, 702.8, 648.7, 594.7, and 540.6 mg, respectively) and sodium acetate (1200 mg) were dissolved in ethylene glycol (20 mL), and sonicated and mixed into a homogeneous solution. The mixture was then transferred into an autoclave (50 mL volume) and set in the oven (200 °C). After reaction for 15 hours, the autoclave was taken out and cooled naturally in room environment. The synthesized electrocatalyst was washed with pure ethanol several times and dried in a vacuum oven (60 °C) for 24 hours.
Fabrication of a Zn doped Fe3O4 NP@NSP-BC/GCE sensor
Before modification, the GCE (3 mm in diameter) was polished with 0.5 μm and 0.05 μm alumina slurries to obtain a mirror-like surface. The electrode was then cleaned in ethanol and water by sonication and dried in an ambient environment. Electrocatalyst aqueous solution (2 mg mL−1) was sonicated into a homogeneous solution, and then 5 μL of the solution was dropped onto the GCE surface and dried in an ambient environment.Material and electrochemical characterization
A field-emission scanning electron microscope (Hitachi SU8010) equipped with an energy-dispersive X-ray spectrometer was used to characterize the morphology, elemental composition and elemental mapping of the electrocatalyst. X-ray photoelectron spectroscopy (Thermo Fisher Scientific ESCALAB 250Xi) was used to analyse the electrocatalyst. Raman spectra of the sample were obtained using a spectrometer (LabRAM HR800, Horiba, France). The X-ray powder diffraction measurements were recorded on a diffractometer (Bruker D8 Advance, Germany, Cu-Kα radiation). The elemental analysis was performed by inductively coupled plasma-optical emission spectroscopy (Agilent 5900, US).A CHI-760E electrochemical workstation (Shanghai, Chenhua, China) and a RST 5000C workstation (Zhengzhou Shiruisi Technology Co., Ltd, Zhengzhou, China) were used in the electrochemical experiments. A three-electrode system was used for the CV and DPV scan, with an Ag/AgCl electrode, platinum wire, and GCE. For EIS measurement, a three-electrode system of a Ag/AgCl electrode, platinum plate electrode (10 mm × 10 mm × 0.1 mm), and GCE was used.
Author contributions
Formal analysis, investigation, data curation, writing – original draft, X. M.; methodology, formal analysis, resources, writing – original draft, project administration, Z. G.; conceptualization, writing – review & editing, supervision, funding acquisition, L. C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Key R&D Program of China (No. 2020YFE0205600). This research was funded by the Xinjiang Uygur Autonomous Region's Tianchi Talent – Professor Program awarded to Z. G. This research was funded by the Xinjiang Uygur Autonomous Region's Tianchi Talent – Young Doctor Program awarded to L. C.Notes and references
- J. C. McCrae, E. E. Morrison, I. M. MacIntyre, J. W. Dear and D. J. Webb, Br. J. Clin. Pharmacol., 2018, 84, 2218–2230 CrossRef CAS PubMed.
- S. van den Driesche, J. Macdonald, R. A. Anderson, Z. C. Johnston, T. Chetty, L. B. Smith, C. McKinnell, A. Dean, N. Z. Homer, A. Jorgensen, M. E. Camacho-Moll, R. M. Sharpe and R. T. Mitchell, Sci. Transl. Med., 2015, 7, 288ra280 Search PubMed.
- P. S. C. Leung, K. Lam, M. J. Kurth, R. L. Coppel and M. E. Gershwin, Trends Mol. Med., 2012, 18, 577–582 CrossRef CAS PubMed.
- M. R. McGill and J. A. Hinson, Drug Metab. Rev., 2020, 52, 472–500 CrossRef CAS PubMed.
- R. Ben-Shachar, Y. Chen, S. Luo, C. Hartman, M. Reed and H. F. Nijhout, Theor. Biol. Med. Modell., 2012, 9, 55 CrossRef CAS PubMed.
- E. Kheradpezhouh, L. Ma, A. Morphett, G. J. Barritt and G. Y. Rychkov, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3176–3181 CrossRef CAS PubMed.
- B. Delalat, C. Cozzi, S. Rasi Ghaemi, G. Polito, F. H. Kriel, T. D. Michl, F. J. Harding, C. Priest, G. Barillaro and N. H. Voelcker, Adv. Funct. Mater., 2018, 28, 1801825 CrossRef.
- M. M. Smarr, K. L. Grantz, R. Sundaram, J. M. Maisog, M. Honda, K. Kannan and G. M. Buck Louis, Hum. Reprod., 2016, 31, 2119–2127 CrossRef CAS PubMed.
- A. Bessede, A. Marabelle, J. P. Guégan, F. X. Danlos, S. Cousin, F. Peyraud, N. Chaput, M. Spalato, G. Roubaud, M. Cabart, M. Khettab, A. Chaibi, C. Rey, I. Nafia, F. X. Mahon, J. C. Soria and A. Italiano, Ann. Oncol., 2022, 33, 909–915 CrossRef CAS PubMed.
- W. M. Lee, Hepatol. Res., 2008, 38, S3–S8 CAS.
- J. M. Major, E. H. Zhou, H.-L. Wong, J. P. Trinidad, T. M. Pham, H. Mehta, Y. Ding, J. A. Staffa, S. Iyasu, C. Wang and M. E. Willy, Pharmacoepidemiol. Drug Saf., 2016, 25, 590–598 CrossRef PubMed.
- L. G. Taylor, S. Xie, T. E. Meyer and T. S. Coster, Pharmacoepidemiol. Drug Saf., 2012, 21, 375–383 CrossRef PubMed.
- A. Martinez-De la Torre, S. Weiler, D. S. Bräm, S. S. Allemann, H. Kupferschmidt and A. M. Burden, JAMA Network Open, 2020, 3, e2022897–e2022897 CrossRef PubMed.
- J. Kaur, S. R. McFaull and F. Bang, Health Promot. Chronic Dis. Prev. Can., 2020, 40, 130–133 CrossRef PubMed.
- N. J. Spillers, P. M. Luther, N. C. Talbot, G. H. Ly, E. M. Downs, G. Lavespere, D. Pavlickova, S. Ahmadzadeh, O. Viswanath, G. Varrassi, S. Shekoohi and A. D. Kaye, Cureus, 2023, 15, e41116 Search PubMed.
- Y. Lu, X. Pan, C. Cao, S. Fan, H. Tan, S. Cui, Y. Liu and D. Cui, Adv. Healthcare Mater., 2023, 12, 2300163 CrossRef CAS PubMed.
- Y. He, C. Liu, X. Xia and L. Liu, J. Mater. Chem. B, 2017, 5, 1962–1970 RSC.
- B. Song, C. Zhang, W. Hu, C. Guo, Z. Xia, W. Hu, M. Qin, W. Jiang, J. Lv, D. Xu, S. Zhang and J. Fang, J. Controlled Release, 2021, 331, 350–363 CrossRef CAS PubMed.
- M. Labib, E. H. Sargent and S. O. Kelley, Chem. Rev., 2016, 116, 9001–9090 CrossRef CAS PubMed.
- X. Liu, Y. Yao, Y. Ying and J. Ping, Trends Analyt. Chem., 2019, 115, 187–202 CrossRef CAS.
- J.-t Jang, J. Lee, J. Seon, E. Ju, M. Kim, Y. I. Kim, M. G. Kim, Y. Takemura, A. S. Arbab, K. W. Kang, K. H. Park, S. H. Paek and S. Bae, Adv. Mater., 2018, 30, 1704362 CrossRef PubMed.
- R. Nehru, C.-D. Dong and C.-W. Chen, ACS Appl. Nano Mater., 2021, 4, 6768–6777 CrossRef CAS.
- J. Bai, K. Wang, J. Feng and S. Xiong, ACS Appl. Mater. Interfaces, 2015, 7, 22848–22857 CrossRef CAS PubMed.
- S. M. Babulal, K. Venkatesh, S.-M. Chen, S. K. Ramaraj and C.-C. Yang, J. Alloys Compd., 2021, 876, 160215 CrossRef CAS.
- H. Du, F. Yang, C. Yao, Z. Zhong, P. Jiang, S. G. Stanciu, H. Peng, J. Hu, B. Jiang, Z. Li, W. Lv, F. Zheng, H. A. Stenmark and A. Wu, Small, 2022, 18, 2201669 CrossRef CAS PubMed.
- F. F. H. Aragón, L. Villegas-Lelovsky, J. G. Parizaka, E. G. Zela, R. Bendezu, R. O. Gallegos, D. G. Pacheco-Salazar, S. W. da Silva, R. Cohen, L. C. C. M. Nagamine, J. A. H. Coaquira and P. C. Morais, Mater. Adv., 2023, 4, 1389–1402 RSC.
- Z. Wang, A. T. Smith, W. Wang and L. Sun, Angew. Chem., Int. Ed., 2018, 57, 13722–13734 CrossRef CAS PubMed.
- L. Yuan, M. Gao, H. Xiang, Z. Zhou, D. Yu and R. Yan, ACS Nano, 2023, 17, 6849–6856 CrossRef CAS PubMed.
- Y. Wang, M. Zhang, X. Shen, H. Wang, H. Wang, K. Xia, Z. Yin and Y. Zhang, Small, 2021, 17, 2008079 CrossRef CAS PubMed.
- T. Li, D.-D. Zhi, Z.-H. Guo, J.-Z. Li, Y. Chen and F.-B. Meng, Green Chem., 2022, 24, 647–674 RSC.
- M. Mehmandoust, G. L. Li and N. Erk, Ind. Eng. Chem. Res., 2022, 62, 4628–4635 CrossRef.
- V. S. Bhat and G. Hegde, J. Electrochem. Soc., 2020, 167, 037526 CrossRef CAS.
- A. I. Rehan, A. I. Rasee, M. E. Awual, R. M. Waliullah, M. S. Hossain, K. T. Kubra, M. S. Salman, M. M. Hasan, M. N. Hasan, M. C. Sheikh, H. M. Marwani, M. A. Khaleque, A. Islam and M. R. Awual, Colloids Surf., A, 2023, 673, 131859 CrossRef CAS.
- H. Md. Munjur, M. N. Hasan, M. R. Awual, M. M. Islam, M. A. Shenashen and J. Iqbal, J. Mol. Liq., 2020, 319, 114356 CrossRef CAS.
- P. T. Yin, S. Shah, M. Chhowalla and K.-B. Lee, Chem. Rev., 2015, 115, 2483–2531 CrossRef CAS PubMed.
- S. Zhang, R. Geryak, J. Geldmeier, S. Kim and V. V. Tsukruk, Chem. Rev., 2017, 117, 12942–13038 CrossRef CAS PubMed.
- V. Biju, Chem. Soc. Rev., 2014, 43, 744–764 RSC.
- X. Liu, J. Liu, S. Zhang, Z. Nan and Q. Shi, J. Phys. Chem. C, 2016, 120, 1328–1341 CrossRef CAS.
- L. Wang, A. Ambrosi and M. Pumera, Electrochem. Commun., 2013, 26, 71–73 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, NY, USA, 2nd edn, 2011 Search PubMed.
- M. S. Salman, M. C. Sheikh, M. M. Hasan, M. N. Hasan, K. T. Kubra, A. I. Rehan, M. E. Awual, A. I. Rasee, R. M. Waliullah, M. S. Hossain, M. A. Khaleque, A. K. D. Alsukaibi, H. M. Alshammari and M. R. Awual, Appl. Surf. Sci., 2023, 622, 157008 CrossRef CAS.
- M. R. Awual, M. M. Hasan, G. E. Eldesoky, M. A. Khaleque, M. M. Rahman and M. Naushad, Chem. Eng. J., 2016, 290, 243–251 CrossRef CAS.
- K. T. Kubra, M. M. Hasan, M. N. Hasan, M. S. Salman, M. A. Khaleque, M. C. Sheikh, A. I. Rehan, A. I. Rasee, R. M. Waliullah, M. E. Awual, M. S. Hossain, A. K. D. Alsukaibi, H. M. Alshammari and M. R. Awual, Colloids Surf., A, 2023, 667, 131415 CrossRef CAS.
- P. Veerakumar, V. Veeramani, S.-M. Chen, R. Madhu and S.-B. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 1319–1326 CrossRef CAS PubMed.
- X. Li, H. Li, T. Liu, Y. Hei, M. Hassan, S. Zhang, J. Lin, T. Wang, X. Bo, H.-L. Wang, H. Li and M. Zhou, Sens. Actuators, B, 2018, 255, 3248–3256 CrossRef CAS.
- R. Appiah-Ntiamoah, A. F. Baye and H. Kim, ACS Appl. Nano Mater., 2022, 5, 4754–4766 CrossRef CAS.
- X. S. Nguyen, G. Zhang and X. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 8900–8909 CrossRef CAS PubMed.
- J. Liu, Y. Bin and M. Matsuo, J. Phys. Chem. C, 2012, 116, 134–143 CrossRef CAS.
- N. Yalikun, X. Mamat, Y. Li, X. Hu, P. Wang and G. Hu, J. Electrochem. Soc., 2019, 166, B1131–B1137 CrossRef CAS.
- H. Yang, G. Cao, Y. Huang, Y. Lin, F. Zheng, L. Lin, F. Liu and S. Li, J. Pharm. Anal., 2022, 12, 436–445 CrossRef PubMed.
- N. Nataraj, S.-M. Chen and S. K. Krishnan, Environ. Sci.: Nano, 2022, 9, 3927–3942 RSC.
- S. J. Saleem and M. Guler, Electroanalysis, 2019, 31, 2187–2198 CrossRef CAS.
- Y. Luo, Y. Yang, L. Wang, L. Wang and S. Chen, J. Alloys Compd., 2022, 906, 164369 CrossRef CAS.
- C. S. Kushwaha and S. K. Shukla, ACS Appl. Polym. Mater., 2020, 2, 2252–2259 CrossRef CAS.
- B. G. Mahmoud, M. Khairy, F. A. Rashwan and C. E. Banks, Anal. Chem., 2017, 89, 2170–2178 CrossRef CAS PubMed.
- T. Kokab, A. Shah, M. A. Khan, M. Arshad, J. Nisar, M. N. Ashiq and M. A. Zia, ACS Appl. Nano Mater., 2021, 4, 4699–4712 CrossRef CAS.
- V. Duraisamy, V. Sudha, V. Dharuman and S. M. Senthil Kumar, ACS Biomater. Sci. Eng., 2023, 9, 1682–1693 CrossRef CAS PubMed.
- T.-Y. Huang, C.-W. Kung, H.-Y. Wei, K. M. Boopathi, C.-W. Chu and K.-C. Ho, J. Mater. Chem. A, 2014, 2, 7229–7237 RSC.
- D. Jiang, X. Huang, Q. Zhang, Y. Ge, Y. Ye, C. Xu, Y. Cai, Y. Zhu, M. Yuan, X. Liu, S. Cai, J. Zhu and Y. Wang, Biosens. Bioelectron., 2022, 218, 114773 CrossRef CAS PubMed.
- N. Wester, B. F. Mikladal, I. Varjos, A. Peltonen, E. Kalso, T. Lilius, T. Laurila and J. Koskinen, Anal. Chem., 2020, 92, 13017–13024 CrossRef CAS PubMed.
- D. Kim, J. M. Kim, Y. Jeon, J. Lee, J. Oh, W. Hooch Antink, D. Kim and Y. Piao, Sens. Actuators, B, 2018, 259, 50–58 CrossRef CAS.
- M. Shalauddin, S. Akhter, W. J. Basirun, V. S. Lee, A. R. Marlinda, S. R. Ahmed, A. R. Rajabzadeh and S. Srinivasan, Electrochim. Acta, 2023, 454, 142423 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00480e |
| This journal is © The Royal Society of Chemistry 2023 |