DOI:
10.1039/D3MA00462G
(Paper)
Mater. Adv., 2023,
4, 4943-4953
Enhanced catalytic activity of ZnO–CuO–Co3O4 composites achieved using a mechanochemical method for effective Fenton-like dye removal: the generation and catalytic mechanism of various superficial active sites†
Received
25th July 2023
, Accepted 13th September 2023
First published on 15th September 2023
Abstract
Mechanochemical methods are effective in creating active sites of solid catalysts, and have great potential to improve the activity of heterogenous catalysts in advanced oxidative processes of organic wastewater. Herein, ZnO–Co3O4–CuO composites were treated using three kinds of grinding methods to adjustably expose the superficial oxygen vacancies and Cu/Co–O dangling bonds according to the grinding strength. In the Fenton-like treatment of organic dye wastewater, the catalytic activity of the composites was accordingly improved, especially in the deep mineralization of wastewater. After the intense trituration of the composites, the formed oxygen vacancies worked as effective sites for the chemisorption of H2O2 molecules, while the adjacent Co/Cu sites were simultaneously activated to facilitate the redox cycle of the Fenton-like reactions. In this way, the interfacial electron transfer was dramatically accelerated to transform H2O2 molecules into hydroxyl radicals and singlet oxygen which were confirmed as the contributing oxidation species in the degradation of organic dyes. The aforementioned results clarified that the mechanochemical treatment of solid catalysts has a dual function of both the generation of oxygen vacancies and activation of Co/Cu redox sites, which could provide a simple and universal strategy for the catalytic enhancement of Fenton-like catalysts.
1. Introduction
Fenton-like techniques are recognized as feasible and effective advanced oxidation processes (AOPs) for organic wastewater treatment, which involves the catalytic generation of oxidative species from H2O2 and decomposition of hydrosoluble organics.1 Recently, researchers paid more attention to the development and application of heterogenous Fenton-like systems because of their significant advantages, including the convenient recycling of solid catalysts, a widely applicable pH range and controllable ion leaching over the traditional Fenton reaction catalysed by Fe2+/Fe3+ ions.2,3 The efficiency of a heterogenous Fenton-like system is significantly influenced by the surface microstructure and active sites of a solid catalyst that facilitate the chemisorption and dissociation of H2O2 molecules. Therefore, fine-tuning of the surface chemistry of solid catalysts is emerging into an effective strategy used for enhancing their catalytic activity in Fenton-like reactions.
Previous research revealed that oxygen vacancies (OVs) on the surface of a catalyst play a vital role in heterogeneous Fenton-like reactions.4–11 The surface OVs have the chemical characteristics of Lewis active sites, which can facilitate the bond cleavage of H2O2 molecules through intense chemisorption.12–14 In addition, one or two dissociative electrons are adsorbed near the OVs to achieve the charge balance of the catalyst framework. The localized electrons not only contribute to the conductivity and band structure modulation of the catalyst but also participate in the redox of catalytic Fenton-like reactions directly.15,16 In the heterogenous Fenton-like treatment of organic wastewater over FeCeOx, OVs could accelerate the Fe(III)/Fe(II) redox through efficient electron migration so as to continuously decompose H2O2 molecules into hydroxyl radicals (˙OH).17 Significantly, the OVs with localized electrons on the surface of bismuth oxychloride (BiOCl) were demonstrated to be new types of “Fenton-catalytic centers” to decompose H2O2 into ˙OH in spite of the poor catalysis of BiOCl.12
Various methods, including heteroatom doping,15,18–21 heterojunction formation,22–24 element non-stoichiometry and morphological control,25–28 have been explored to construct OVs on a catalyst surface to ameliorate their catalytic activity in AOPs. As a typical mechanochemical method, ball milling has been widely applied in material engineering because of its low-cost operation and feasible up-scaling application.29,30 This method was applied to expose the superficial defects of molybdenite materials so as to enhance their catalytic activity in the homogeneous Fenton treatment of organic dyes.31 In previous research, the enhanced catalytic activity was mainly attributed to the formation of OVs through the ball milling, whereas the contribution of other types of exposed superficial sites received less attention. Actually, the OVs and transition metal sites (M–O dangling bonds) are simultaneously generated when the original chemical bonds of transition metal oxides are forcibly broken in the mechanochemical treatment. In comparison with the OVs, the formed redox sites (transition metal sites) probably play a nonnegligible even dominant role in the activation of H2O2 molecules. Therefore, it is significant to clarify the interfacial catalytic mechanism of the different active sites generated by the mechanochemical treatment in the Fenton-like reactions.
In this work, three kinds of grinding methods were applied to reduce the particle size of ZnO–Co3O4–CuO composites, thus to expose the superficial OVs and activate Co/Cu redox sites. In the Fenton-like treatment of organic dye wastewater, the catalytic activity of the composites was gradually improved along with the mechanochemical treatment strength. The interfacial reaction mechanism was further investigated to reveal the crucial role of the exposed Co/Cu sites in addition to the OVs. This work aims to provide a simple and effective method to enhance the heterogeneous catalysis of transition metal oxides in the Fenton-like wastewater treatment through in-depth analysis of the catalytic activity of the exposed active sites.
2. Experimental
2.1 Preparation of ZnO–Co3O4–CuO catalysts
The chemicals used in the experiments are presented in the ESI† Section S1.1. In a typical synthesis, Co(Ac)2·4H2O (0.50 g), Zn(Ac)2·2H2O (0.44 g) and Cu(Ac)2·H2O (0.19 g) were uniformly mixed using a manual mortar to obtain a molar ratio of n(Co)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(Zn):n(Cu) = 1.0:1.0:0.5. The mixed reactants were placed in a porcelain crucible and calcined in a muffle furnace at 800 °C for 2 h with a heating rate of 2 °C min−1 to obtain a ZnO–Co3O4–CuO composite. Three kinds of grinding technologies were employed to smash the sintered composite. The composite was manually ground using a glass mortar for 30 min to obtain the sample I. It was ground using a planetary miller (XGB04, BYT corporation, China) for 180 min and using a high-speed vibrating ball miller (MSK-SFM-3, KJ corporation, China) for 10 min to obtain the samples II and III, respectively. Twenty-four agate balls with a diameter of 3 mm were used to improve the comminution effect in each ball milling. The contrast samples including ZnO, Co3O4 and CuO were synthesized following the same procedure as the sample III.
:n(Zn):n(Cu) = 1.0:1.0:0.5. The mixed reactants were placed in a porcelain crucible and calcined in a muffle furnace at 800 °C for 2 h with a heating rate of 2 °C min−1 to obtain a ZnO–Co3O4–CuO composite. Three kinds of grinding technologies were employed to smash the sintered composite. The composite was manually ground using a glass mortar for 30 min to obtain the sample I. It was ground using a planetary miller (XGB04, BYT corporation, China) for 180 min and using a high-speed vibrating ball miller (MSK-SFM-3, KJ corporation, China) for 10 min to obtain the samples II and III, respectively. Twenty-four agate balls with a diameter of 3 mm were used to improve the comminution effect in each ball milling. The contrast samples including ZnO, Co3O4 and CuO were synthesized following the same procedure as the sample III.
2.2 Characterization
An X-ray diffraction spectrometer (XRD, D8Advance, Bruker), a transmission electron microscope (TEM, 100SX, Japan Electron) and a scanning electron microscope (SEM, SU8010, Hitachi) were employed to determine the crystal phase, composition and microstructure. An X-ray photoelectron spectrometer (XPS, Axis Ultra, Kratos), a Raman spectrometer (inVia, Renishaw) and an electron spin resonance (ESR, A300, Bruker) spectrometer were used to analyse their chemical states. CO2-temperature programmed desorption (NH3-TPD), O2-temperature programmed oxidation (O2-TPO), H2-temperature programmed reduction (H2-TPR) and NH3-temperature programmed desorption (NH3-TPD) detections were carried out on a chemisorption analyser (AutoChem II, Micromeritics). The specific surface areas of the samples I to III were detected on a physisorption apparatus (ASAP 2460, Micromeritics) at 77 K. A Fourier transform infrared spectrometer (FT-IR, Tensor27, Bruker) was used to obtain the IR spectra of the samples.
2.3 Catalyst performance evaluation
In a typical procedure, 25 mg of catalyst was added to 50 mL of methyl orange (MO) aqueous solution (20 mg L−1) under vigorous stirring to get a homogenous sol. After stirring for 30 min, 150 μL of H2O2 was added to the sol to trigger the Fenton-like reactions. At an interval of 15 min, 1 mL of aliquots was taken out and separated using a filter membrane. The residual dye in the filtrate was immediately analysed using a UV-vis spectrometer (U-3900, Hitachi) at a maximum absorption wavelength (464 nm). Each experiment was repeated three times. The experimental details and measurement of ˙OH, singlet oxygen (1O2) relative concentration, electrochemical data, ESR spectrum, total organic carbon (TOC) content, GC-MS data, curve fitting of the XPS data and artemia salina acute toxicity tests are presented in the ESI† (Sections S1.2–S1.5).
3. Results and discussion
3.1 Fabrication of ZnO–Co3O4–CuO catalysts using the mechanochemical method
The XRD patterns and detailed data of the samples are presented in Fig. S1 and Table S1 (ESI†), respectively. For the samples I–III, apparent diffraction peaks located at 2θ = 31.9, 34.5, 36.4, 47.6, 56.7, 62.9 and 68.1° were indexed exactly to the (100), (002), (101), (102), (110), (103) and (112) lattice planes of the hexagonal ZnO crystals (PDF no. 36-1451), and the peaks at 2θ = 31.3, 37.0, 44.9, 55.7, 59.5 and 65.4° were indexed to the (220), (311), (400), (422), (511) and (440) planes of cubic Co3O4 (PDF no. 43-1003). In addition, the residual diffraction peaks at 2θ = 35.6, 38.8 and 48.8° could be assigned to the (002), (111) and (−202) planes of the monoclinic CuO crystals (PDF no. 45-0937). Compared with those of the pristine metal oxidates (Table S1, ESI†), the characteristic diffraction peaks of the composites shifted by less than 0.3°, indicating that the composites were composed of ZnO, Co3O4 and CuO crystals. In Fig. S1A (ESI†), the XRD peaks of the samples II and III broadened gradually in comparison with that of the sample I, thereby suggesting the decrease in the particle size. The Scherrer formula was used to calculate the average size of the obtained catalysts. In Table 1, the average size of the grains treated by manual grinding (sample I) was calculated to be 1164 nm, but it decreased to 875 nm and 374 nm after two kinds of ball milling. Accordingly, the special surface areas of the samples were enhanced from 2.6 to 8.2 m2 g−1 along with the improvement of grinding strength. As shown in the insets of Fig. S1 (ESI†), the pristine product stuck together into a tablet because of a high-temperature sintering effect, whereas it was crushed into powder after the mechanical treatment.
Table 1 Partial properties of the catalysts treated using various mechanochemical methods
| Sam. |
Composition |
d/nm |
S
BET/m2 g−1 |
|
d: average size of the catalyst, SBET: special surface area. |
| I |
ZnO–Co3O4–CuO |
1164 |
2.6 |
| II |
ZnO–Co3O4–CuO |
875 |
6.7 |
| III |
ZnO–Co3O4–CuO |
374 |
8.2 |
The FT-IR spectra of the composites (Fig. S2A, ESI†) displayed three distinct bands centred at 678, 600 and 480 cm−1 that are attributed to the stretching vibrations of M–O bonds in comparison with that of the pristine (Fig. S2B, ESI†). The bands at 678 and 600 cm−1 could be assigned to the stretching vibrations of Co(II)–O bonds in the tetrahedral structure and Co(III)–O in the octahedral coordination, respectively. The broad band at around 480 cm−1 was ascribed to Zn–O stretching vibrations. By increasing the grinding strength, the shoulder bands at 620 and 583 cm−1 disappeared, and the Co–O stretching bands with a higher wavenumber became sharper. The changes in the IR spectra hint the generation of surface OVs in the samples II and III according to previous literature.28
SEM, TEM and energy dispersive X-ray spectroscopy (EDS) mapping were used to analyse the microstructures and element distribution of the catalysts. In Fig. 1(A–C), irregular particles with a size beyond 500 nm were observed in the sample I just dispersed by manual grinding, but those were broken into nano-particles after ball milling. A localized TEM image (Fig. 1D) of the sample III demonstrated that some of the agglomerates consisted of nano-particles with a size below 50 nm. The EDS mapping results revealed that Co, Zn, Cu and O elements were distributed uniformly in the agglomerates produced by high-speed ball milling.
 |
| | Fig. 1 SEM (A)–(C), TEM (D) images and EDS maps (E)–(I) of the samples I–III: (A)–(C) SEM images of the samples I–III, respectively, (D) TEM image of the sample III, and (E)–(I) EDS map of the sample III. | |
Based on the above analysis, the mechanochemical treatment plays an observable role in enlarging the surface area of the composites through the grain size reduction. In principle, grinding the materials provides mechanical force that induces the potential energy accumulation through atomic migration and lattice distortions. When this energy is released via chemical bond breakage and heating, the particle size is reduced, accompanied by other relevant effects: amorphization, generation of defects and even chemical reactions.32
3.2 Formation of superficial active sites using mechanochemical methods
Various active sites formed by the mechanochemical treatment were systematically investigated. Firstly, we focused on the changes in the exposed crystal planes after trituration. The primary XRD peak intensity ratios of various lattice planes of ZnO, CuO and Co3O4 were calculated and listed in Table S2 (ESI†). Although the characteristic diffraction peak intensity ratios of ZnO crystals varied obviously from I110:I002:I101:I102:I110:I103:I112 = 0.54:0.43:1:0.24:0.36:0.33:0.28 to 0.60:0.46:1:0.24:0.33:0.29:0.24 (sample I), then to 0.73:0.66:1:0.23:0:0.26:0 (sample III), after the trituration, it had little effect on their catalysis due to the inherent catalytic inertia of ZnO in the activation of H2O2. Furthermore, the intensity ratios of Co3O4 in the treated composites were close to those of the pristine material, indicating insignificant changes in the exposed Co3O4 lattice planes. Compared with the pristine CuO, some of the diffraction peaks with a high Miller index, including the (−110), (020), (202) and (−113) planes, were further weakened after the trituration. This means the mechanochemical treatment for the composites is not conducive to exposing highly active planes of the CuO crystals because the high Miller index planes always have high surface energy and preferable catalytic activity. In short, the contribution of exposing various lattice planes toward the potential catalysis could be ignored, such that we paid more attention to the microscopic active sites produced by the mechanochemical treatment.
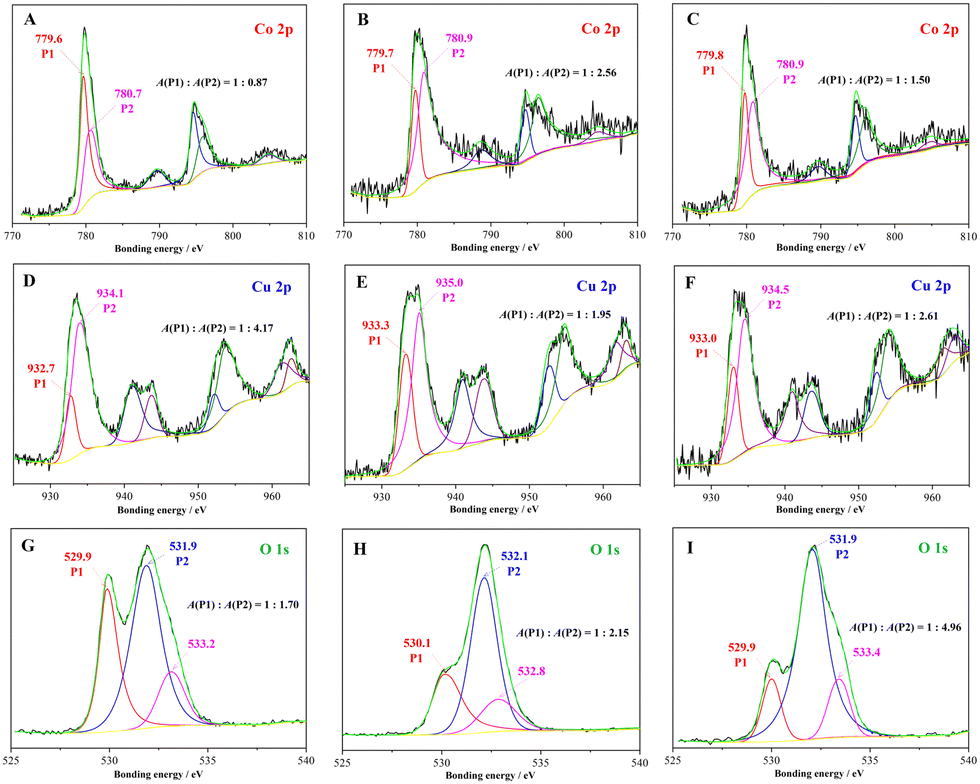
XPS was used to distinguish the chemical state variation of the catalysts after the mechanochemical treatment. As shown in Fig. S2 (ESI†), the three typical samples had similar Zn 2p XPS spectra with two symmetric peaks located at 1021.5 and 1044.6 eV which were ascribed to Zn(II) 2p3/2 and Zn(II) 2p1/2. In Fig. 2(A–C), two asymmetric peaks corresponding to spin–orbit splitting of Co 2p3/2 and Co 2p1/2 appeared in the Co 2p XPS spectra of the samples I–III with bonding energy near 780 and 795 eV, respectively. After multi-curve fitting, the asymmetric peak of Co 2p3/2 was separated into two peaks with a bonding energy of 779.7 ± 0.1 and 780.8 ± 0.1 eV, which were assigned to Co(III) 2p3/2 (P1) and Co(II) 2p3/2 (P2), respectively. The fitting peak area ratio (P1:P2) was used to quantitatively analyse the Co(III):Co(II) molar ratio.33 The molar ratio was calculated to be 1:0.87, but it decreased obviously after the mechanochemical treatment. The results hint that more Co(II) sites instead of Co(III) were exposed on the surface of the composites after the intense trituration. In the Cu 2p XPS spectra (Fig. 2D–F) of the three samples, four peaks could be clearly distinguished as Cu 2p3/2, Cu satellite peak (I), Cu 2p1/2 and Cu satellite peak (II) with the increasing bonding energy.34,35 The Cu 2p3/2 peak was separated and fitted as two kinds of Cu valence states, namely Cu(I) 2p3/2 (933.0 ± 0.3 eV) and Cu(II) 2p3/2 (934.5 ± 0.5 eV), and the fitting peak area ratios of Cu(I):Cu(II) were improved to 0.51 (sample II) and 0.38 (sample III) after the mechanical trituration in contrast to 0.24 (sample I) after the manual trituration. Notably, the above XPS results suggest the increasing mechanochemical treatment has prominent effects on the exposure of low valence metal sites including Co(II) and Cu(I) in the trituration process. In Fig. 2(G–I), the O 1s XPS spectra of the samples show three associated peaks localized near 530.0 ± 0.1 (P1), 532.0 ± 0.1 (P2) and 533.1 ± 0.3 eV that were assigned to the lattice oxygen, the oxygen of surface hydroxyl and adsorbed oxygen species, respectively. The XPS peak area ratio of P1/P2 has always been used to quantify the OV concentration of the catalysts.16,36 The ratios of P1/P2 declined gradually from 1:1.70 (sample I) to 1:2.51 (sample II), then to 1:4.56 (sample III) with the decrease in the particle size, indicating a corresponding increase of OVs formed on the surface of the catalysts through the mechanochemical treatment.
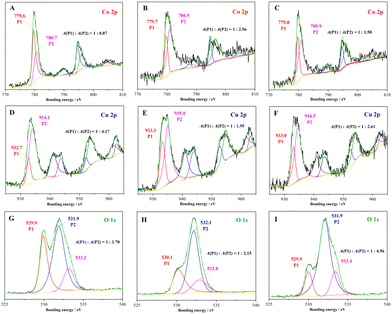 |
| | Fig. 2 XPS graphs of the samples: (A)–(C) Co 2p XPS of the samples I–III, (D)–(F) Cu 2p XPS of the samples I–III, and (G)–(I) O 1s XPS of the samples I–III, respectively. | |
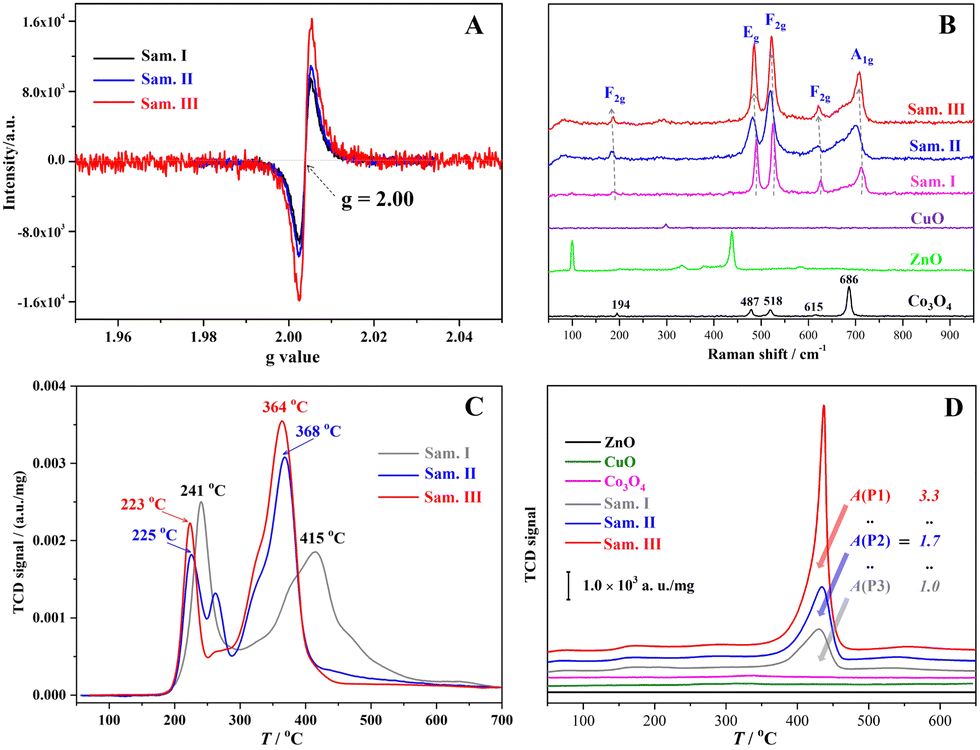
A series of structure characterization tests were carried out to distinguish the active sites formed on the catalysts. In Fig. 3(A), the ESR signals of unpaired electrons appeared at g = 2.00, which directly confirmed the formation of the OVs in the three samples. The intensity of the ESR signals increased gradually along with vigorous grinding, thereby confirming the enhanced OV concentration from the samples I to III. The ESR results with regard to the enhanced OV concentration are consistent with that confirmed by the O 1s XPS data (Fig. 2G–I). In Fig. 3(B), the Raman spectra of the samples I to III presented five distinguishable signals located near 194, 487, 518, 615 and 686 cm−1 that corresponded to F2g(1), Eg, F2g(2), F2g(3) and Ag symmetry in Co3O4, respectively.37 The Raman-activated characters similar to that of the pristine Co3O4 suggested that more Co3O4 crystals exposed on the surface of the composites might facilitate laser scattering in comparison with ZnO and CuO. Significantly, the Raman spectra of the samples I to III depicted observable red-shifted and broader features, which are closely related to the crystal stress and particle size. Usually, the red-shift and peak broadening phenomenon can be ascribed to an optical phonon size effect that is caused by the particle size-dependent effect and formation of crystal stress in the grinding processes.38,39
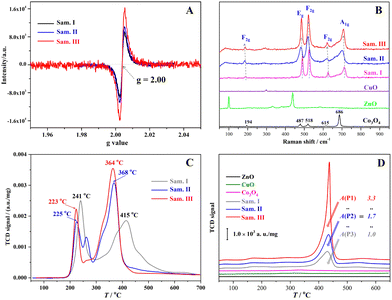 |
| | Fig. 3 Active site characterization results of the samples: (A) ESR spectra of the samples I–III, (B) Raman spectra of the samples, (C) H2-TPR curves of the samples, and (D) CO2-TPD curves of the samples. | |
A chemisorption analyser was used to determine the active sites of the samples treated using ball milling. H2-TPR curves (Fig. 3C) of the composites presented two redox peaks in the temperature ranges of 200–300 and 300–500 °C that corresponded to the reduction of CuO and Co3O4 in comparison with the H2-TPR chart (Fig. S3, ESI†) of the pure transition metal oxides. After the mechanochemical treatment, CuO and Co3O4 in the composites were greatly activated because their reduction temperatures declined more than 16 and 47 °C. In Fig. 3(D), the CO2-TPD curves of the three composites had evident desorption peaks in the temperature range from 380 to 470 °C in comparison with the insignificant signal of independent or individual transition metal oxidates, indicating the formation of basic sites through ternary material combination. Moreover, the alkali densities of the composites after the intense trituration enhanced 1.7 and 3.3 times in comparison with that of the sample I. Given that the mechanochemical treatment for the composites breaks the original M–O–M bonds in essence, that is, the generation of OVs and M–O dangling bonds occurs simultaneously. The increased alkali densities could be attributed to the formation of M–O dangling bonds in the mechanochemical treatment because the surface M–O dangling bond is a kind of proton acceptor. In Fig. S4 (ESI†), no acidic site of the composites was distinguished in the NH3-TPD, and O2-TPO curves demonstrates that the mechanochemical treatment had a preferable effect upon the exposure of the oxidative sites (high valence cations) rather than the reductive sites. According to the above results, the mechanochemical treatment for the composites not only increases their surface OV concentration but creates more redox active sites including Cu–O and Co–O dangling bonds, which would be beneficial to the heterogeneous Fenton-like reactions through more effective chemisorption and interfacial electron transfer.
3.3 Catalytic Fenton-like treatment of organic dye wastewater
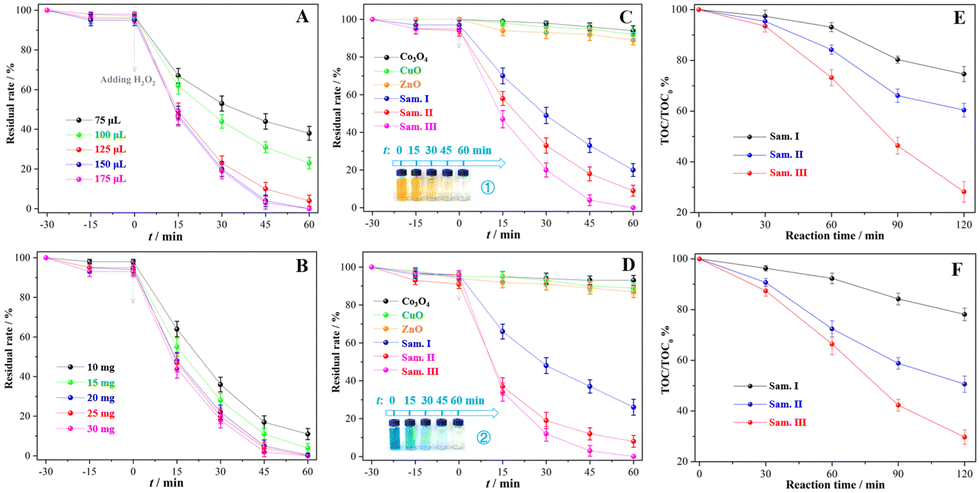
The Fenton-like treatment of MO aqueous solution catalysed by the sample III was utilized to optimize subsequent reaction conditions. In Fig. 4(A) and (B), the usage of H2O2 and catalyst was optimized as 150 μL and 25 mg to achieve the balance between the cost and efficiency of wastewater treatment, and MO solution was completely discolored after 60 min under the corresponding conditions. The oxidative discoloration rates of the MO solution were 80.4%, 91.2% and 100.0% (Fig. 4C) catalysed by the samples I, II and III, respectively, which were much higher than the oxidative discoloration rates of those catalysed by pure transition metal oxides. In the Fenton-like treatment of methylene blue (MB) solution, 74.7%, 92.6% and 100.0% MB molecules were discolored oxidatively after 60 min reactions catalysed by the samples I, II and III (Fig. 4D), respectively, whereas the discoloration rates were less than 10% in the presence of ZnO, Co3O4 or CuO. Insets 1 and 2 depict the rapid discoloration of MO and MB solution in the Fenton-like systems with the sample III. The TOC degradation rate was utilized to estimate the mineralization degree of the dye solution. After 120 min of the catalytic reaction, the TOC degradation rate of the MO solution was only 25.4% catalysed by the sample I, but it increased dramatically to 39.6% and 71.8%, in the presence of the samples II and III (Fig. 4E), respectively. The mineralization of MB solution had a tendency similar to that of MO (Fig. 4F). Compared with other systems in the presence of Cu/Co-containing catalysts (Table S3, ESI†), the current Fenton-like system with the composites treated using the mechanochemical methods showed an observable effect in improving the deep mineralization of organic dye solution in addition to the ordinary discoloration.
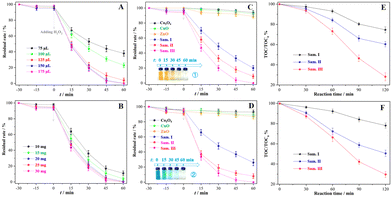 |
| | Fig. 4 Organic dye degradation in various catalytic systems: (A) Fenton-like treatment of MO catalysed by the sample III with various amounts of H2O2, (B) Fenton-like treatment of MO catalysed by the sample III with various catalysts, (C) Fenton-like treatment of MO catalysed by various catalysts, (D) Fenton-like treatment of MB catalysed by various catalysts, (E) TOC removal efficiency during the Fenton-like treatment of MO catalysed by the sample III, (E) TOC removal efficiency during the Fenton-like treatment of MB catalysed by the sample III. Insets: The discoloration process of MO or MB during the Fenton-like reactions. Optimized reaction conditions: [MO or MB] = 20.0 mg L−1, [H2O2] = 0.9 g L−1, [catalyst] = 5.0 g L−1, and room temperature. | |
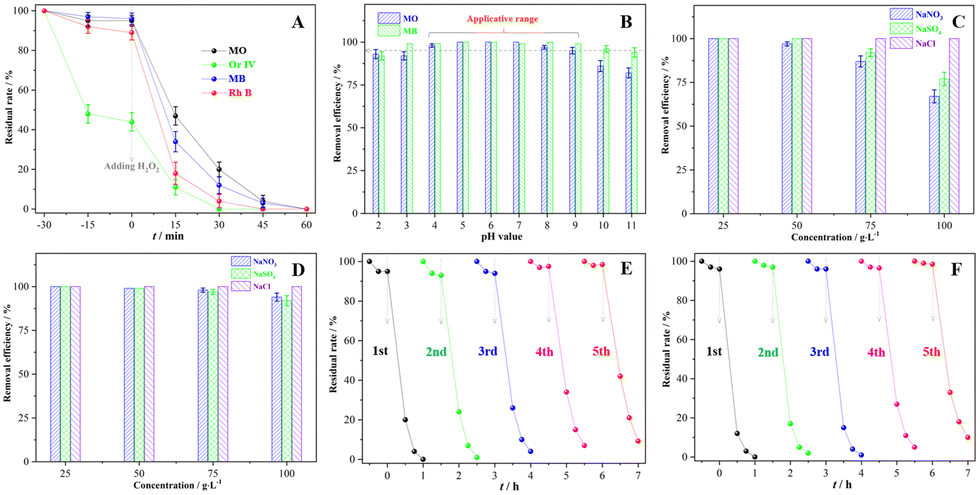
Fig. 5 presents availability and tolerance of the Fenton-like system catalysed by the sample III in the dye wastewater treatment under complicated conditions. In Fig. 5(A), four types of organic dye solutions were completely discolored after 60 min of reaction regardless of whether there was an adsorption or not. Adjusting the pH value range from 4 to 9, the discoloration rates of MO and MB solution were both higher than 95.0% (Fig. 5B), confirming that the corresponding system had a wide pH application scope. More than 90.0% MO or MB were effectively decomposed in 60 min so long as the inorganic salt concentration was below 50.0 g L−1 (Fig. 5C and D), indicating that the system has great potential for the treatment of hypersaline organic wastewater. In the Fenton-like treatment of MO or MB solution, the catalytic activity of the sample III declined less than 10.0% after 5 cycles, hence reflecting its desirable stability. The XRD pattern (Fig. S5, ESI†) of the recycled catalyst retained the same crystallographic features as those of the pristine one in spite of the declined crystallinity. Moreover, the Fenton-like system catalysed by the sample III was applied to treat other kinds of organic wastewater. After 90 min treatment, more than 80% tetracycline and humic acid were degraded (Fig. S6A, ESI†), and the TOC removal rates of those were higher than 32% (Fig. S6B, ESI†). The conspicuous properties of the sample III verified that the Fenton-like system can be expanded to much wider applications.
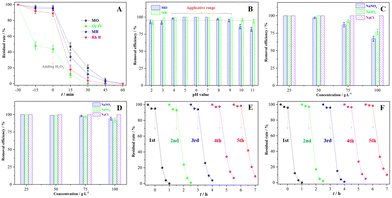 |
| | Fig. 5 Availability and tolerance of the Fenton-like system catalysed by the sample III: (A) the degradation of various organic dyes, (B) the degradation of MO or MB with various pH values, (C) the degradation of MO with various inorganic salts, (D) the degradation of MB with various inorganic salts, (E) reusability of the sample III in the degradation of MO solution, and (F) reusability of the sample III in the degradation of MB solution. | |
3.4 Catalytic mechanism of the ZnO–Co3O4–CuO composite in the Fenton-like treatment of organic dye wastewater
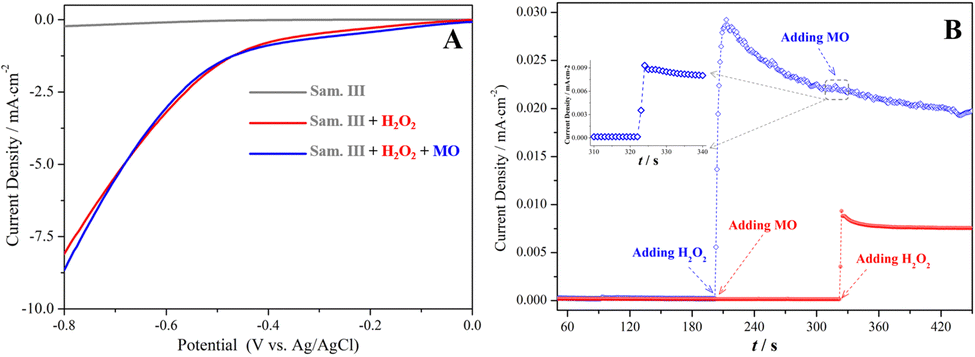
Electrochemical tests, including linear sweep voltammetry (LSV) and electric current–time (i–t) curves, were carried out to verify the electron transfer between the reactants and the sample III. In Fig. 6(A), the current density in the LSV curves was dramatically enhanced only if H2O2 existed in the system. In addition, the electron transfer kinetics was further investigated using the chronoamperometry test. As shown in Fig. 6(B), the current density did not change at all after MO was initially added into the system, suggesting that MO molecules and the catalyst only have a slight physisorption not related to the electron transfer. However, the current output was substantially elevated once H2O2 was added. The results suggested the chemisorption and activation of H2O2 molecules on the surface of the catalyst essentially involved the electron transfer. In the presence of H2O2, another slight elevation of the current density occurred after adding MO, suggesting that MO molecules could participate in the interfacial electron transfer in the Fenton-like reactions.
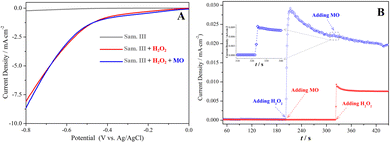 |
| | Fig. 6 (A) LSV curves of the ZnO–Co3O4–CuO (sample III) electrode detected in 0.5 mol L−1 Na2SO4 solution, (B) i–t curves of the ZnO–Co3O4–CuO (sample III) electrode detected in 0.5 mol L−1 Na2SO4 solution. | |
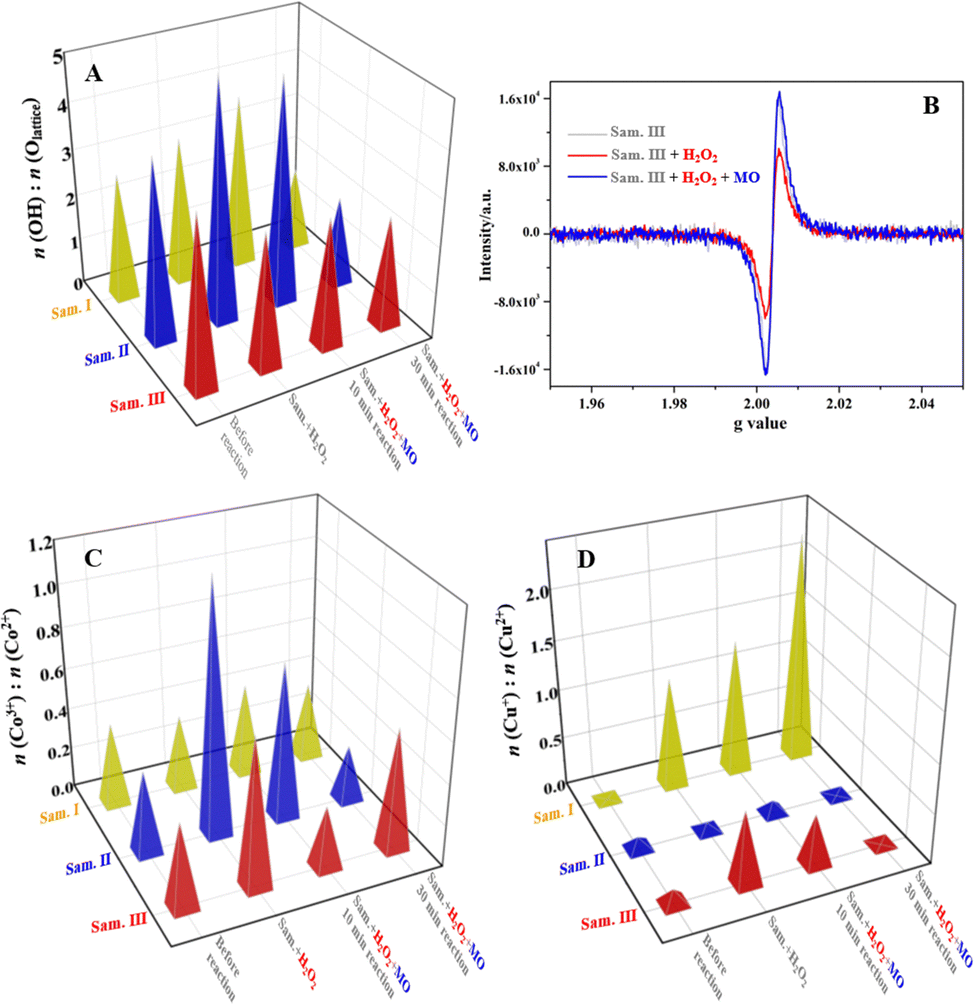
Curve fitting of the Co 2p, Cu 2p and O 1s XPS spectra was carried out using professional software (ESI,† Section S1.4). As shown in Table S4 (ESI†), the bonding energies of the fitting peaks could be assigned to the corresponding chemical states of Co, Cu and O elements in spite of ±0.5 eV deviation. The O 1s XPS spectra of the three samples were fitted into Olattice (P1) and –OH (P2) peaks in addition to Oadsorption, and the peak area ratios of P1/P2 were calculated to distinguish the superficial O chemical state evolution in the Fenton-like reactions. In Fig. 7(A), the ratios (Olattice/–OH) of all samples varied significantly in the Fenton-like reaction process, which indicated the active O atoms or OVs participated in the competitive chemisorption of H2O2 molecules and the organics. ESR was further used to confirm the participation of the OVs in the Fenton-like reactions. Taking the sample III as an example, the ESR signal (Fig. 7B) of the sample III recycled from the system in the presence of H2O2 decreased in comparison with that of the pristine one. Given that the EPR technique detects the unpaired electron near the OVs in essence, it confirms that the unpaired electron transferred from the OVs to the adsorbed H2O2 molecules, which can facilitate the reduction of H2O2 molecules to produce more ˙OH radicals. Relevant work reported that the OVs on the catalyst surface always worked as a type of “O trap” for the chemisorption of H2O2 molecules.35 The catalysis of the unpaired electrons adjacent to the OVs was confirmed by H2O2 activation on the inert BiOCl.12 Most noteworthily, the OV concentration of the catalyst recycled from the Fenton-like system recovered as before, indicating that the reductive organic species formed in the degradation could work as electron donors for the regeneration of the OVs.
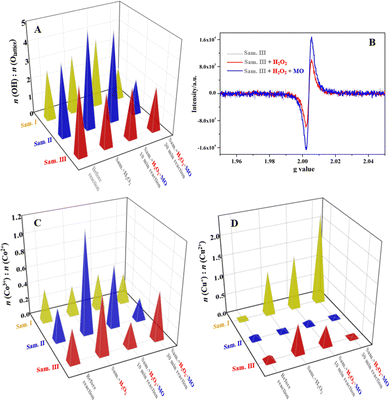 |
| | Fig. 7 XPS and ESR charts of the samples recycled from various systems: (A) relative value analysis of fitting O 1s XPS peaks, (B) ESR graphs of the sample III recycled from various systems, (C) relative value analysis of fitting Co 2p XPS peaks, and (D) relative value analysis of fitting Cu 2p XPS peaks. | |
The Co 2p3/2 XPS peak was separated as two fitting peaks that corresponded to Co(III, P1, 779.7 ± 0.2 eV) and Co(II, P2, 780.8 ± 0.2 eV), and the peak area ratio (Fig. 7C) of P1/P2 was calculated to investigate the Co valence state variation of the samples in the Fenton-like reactions. For the sample I, the Co(III)/Co(II) ratio of the sample I was 1.15, while it varied in a small range of 0.72–0.93 for the recycled composite after H2O2 chemisorption or actual Fenton-like reactions. The inconspicuous valence state variation implies the superficial Co sites on the sample I participated deficiently in the interfacial electron transfer of the redox, illustrating the ineffective activation of Co sites through the manual grinding. Differently, the Co(III)/Co(II) ratios of the samples II and III changed significantly in the range of 0.39–1.16 and 0.67–1.79, respectively, which indicates the Co sites of those two take an active part in the superficial electron transfer. Taking the sample III as an example, the Co(III)/Co(II) ratio of the recycled one after H2O2 chemisorption increased from 0.67 to 1.79, which suggests electrons transferred from the surface Co(II) sites to H2O2 molecules, inducing the generation of ˙OH radicals. When MO molecules participated in the superficial Fenton-like reactions, the ratio declined to 0.55 again. It indicates that the electron-rich organic species formed in the degradation process could facilitate the reduction of Co(III), which has been verified by the electrochemical tests indirectly (Fig. 6).
Similarly, the Cu 2p3/2 peak of the recycled samples was fitted as Cu(I, P1, 933.0 ± 0.4 eV) and Cu(II, P2, 934.5 ± 0.5 eV) peaks to reveal the Cu valence state variation in the Fenton-like reactions. In Fig. 7(D), the Cu(I)/Cu(II) ratio of the recycled sample I increased more than 0.4 compared with that of the pristine one, which implies Cu sites could be the primary catalytic center of the sample I. Analogously, the Cu(I)/Cu(II) ratios of the samples II and III experienced a large valence state fluctuation in the range of 0.37–1.16 and 0.33–1.29, respectively, indicating the efficient electron transfer between the Cu redox sites and the reactants (H2O2 and organics) over the Fenton-like reaction process. On the basis of the above results, Cu–O/Co–O dangling bonds in addition to the OVs are verified as the redox active sites for the catalysis of the Fenton-like reactions through the efficient interfacial electron transfer, and the mechanochemical treatment of the ternary composites plays a critical role in exposing the superficial active sites, especially Co sites, through breaking the original chemical bonds.
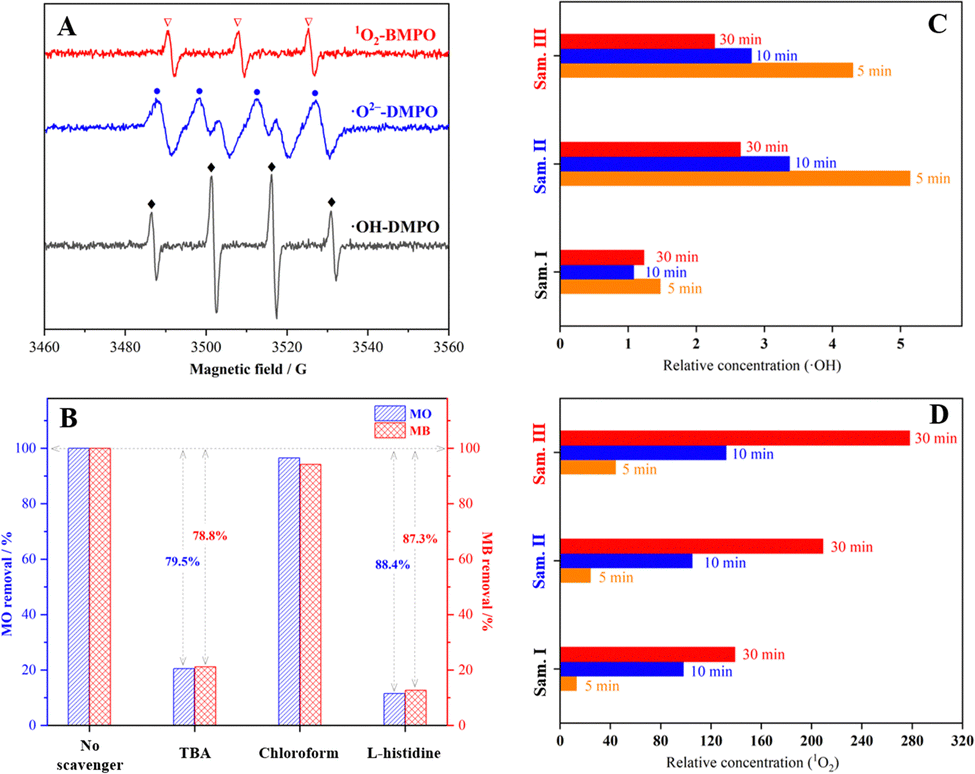
ESR spin-trap technique was employed to distinguish the oxidative species formed in the decomposition of H2O2 catalysed by the sample III. Fig. 8(A) depicts quartet peaks with a 1:2:2:1 intensity ratio which were assigned to the ESR signals of ˙OH/DMPO adducts, indicating the generation of ˙OH radicals in the system with the sample III. Deionized water was substituted by methanol for the ESR detection of ˙O2− radicals in view of the extreme instability of ˙O2− radicals in aqueous solution.40,41 Four peaks with equal intensity were distinguished as the characteristic ESR signals of ˙O2−/DMPO adducts, suggesting the formation of ˙O2− radicals. In addition, triple signals belonging to 1O2/BMPO adducts with equal intensity were detected to confirm the existence of 1O2. The results of quenching experiment are presented in Fig. 8(B). When tertiary butanol (TBA) was added to the system to selectively quench ˙OH radicals, only 20.5–21.2% MO or MB was degraded after 60 min. However, the inhibiting effect for dye degradation could be ignored in the presence of an ˙O2− scavenger (chloroform). After 1O2 molecules were quenched by L-histidine, the degradation efficiency of MO and MB decreased by 88.4% and 87.3%, respectively. The above results suggest that ˙OH radicals and 1O2 dominated the oxidative decomposition of organic dyes, which could be classified as a mixed radical/non-radical degradation pathway.2,3,42
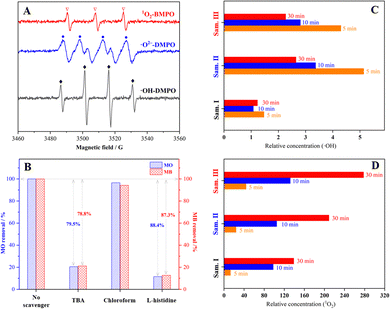 |
| | Fig. 8 Free radical studies in the Fenton-like reactions: (A) ESR graphs for oxidative species generated in the Fenton-like system with the sample III, (B) quenching experiment in the Fenton-like degradation of MO/MB, (C) ˙OH relative concentration in various systems determined by fluorescence spectrometry of TA oxidation, and (D) 1O2 relative concentration in various systems determined by UV-vis spectrometry of ABDA degradation. | |
Considering that ˙OH radicals and 1O2 are the primary contributing species, fluorescence spectrometry of terephthalic acid (TA) oxidation (ESI,† Section S1.2) and UV-vis spectrometry of 9,10-anthracenediyl-bis(methylene) dimalonic acid (ABDA) degradation (ESI,† Section S1.2) were applied to the semiquantitative analysis of ˙OH and 1O2 relative concentration, respectively. As shown in Fig. 8(C and D), the accumulated concentration of ˙OH and 1O2 increased with the progress of the catalytic reaction. The relative concentration of ˙OH generated in the system catalysed by the samples II and III was 1.8–2.1 times higher than that of the system catalysed by the sample I. After 30 min, the relative concentration of 1O2 in the system with the sample II was 1.5 times higher than that with the sample I, and it was further doubled with the sample III. The above demonstrates Cu/Co–O dangling bonds and the OVs formed in the mechanochemical treatment facilitate the decomposition of H2O2 into ˙OH and 1O2 that facilitate the oxidative decomposition of organic dyes substantively.
A series of contrast experiments were conducted to distinguish the catalysis of the OVs and Cu/Co–O dangling bonds in H2O2 activation. In Fig. S7(A) (ESI†), the generated ˙OH concentration of H2O2 solution catalysed by the ground ZnO crystals for 5 min was 1.5 times higher than that catalysed by the pristine material. Considering that ZnO is catalytically inert for the H2O2 activation, the formed OVs in the trituration play a primary role in the catalytic transformation of H2O2 into ˙OH. Similar catalysis of the OVs was also verified on the inert BiOCl previously.12 Unfortunately, the mechanochemical treatment for the pristine Co3O4 and CuO made no contribution to the catalysis promotion in the transformation of H2O2 into ˙OH. However, the generated ˙OH concentrations of the systems with ZnO–CuO or ZnO–Co3O4 binary composites (Fig. S8, ESI†) regardless of being ground were all enhanced in comparison to those with individual metal oxide, which indicates Cu/Co–O dangling bonds on the surface of CuO or Co3O4 were activated through combining with ZnO. In this case, it is understandable that the ZnO–Co3O4–CuO composite after the intense trituration (sample III) had the best catalytic activity in the transformation of H2O2 into ˙OH. In Fig. S7(B) (ESI†), pure ZnO or Co3O4, even being ground, was ineffective in the catalytic generation of 1O2, which suggests that the simple OVs or Co–O dangling bonds cannot facilitate the transformation of H2O2 into 1O2. Differently, the 1O2 concentrations in the corresponding systems catalysed by the binary or ternary composites were enhanced more than 40 times in comparison with that catalysed by the individual ZnO or Co3O4, and they increased even more when the composites were intensely ground. This means that the exposed Cu/Co–O dangling bonds on the surface of the ZnO–Co3O4–CuO composite were responsible for the catalytic transformation of H2O2 into 1O2, even though the OVs barely worked in the transformation.
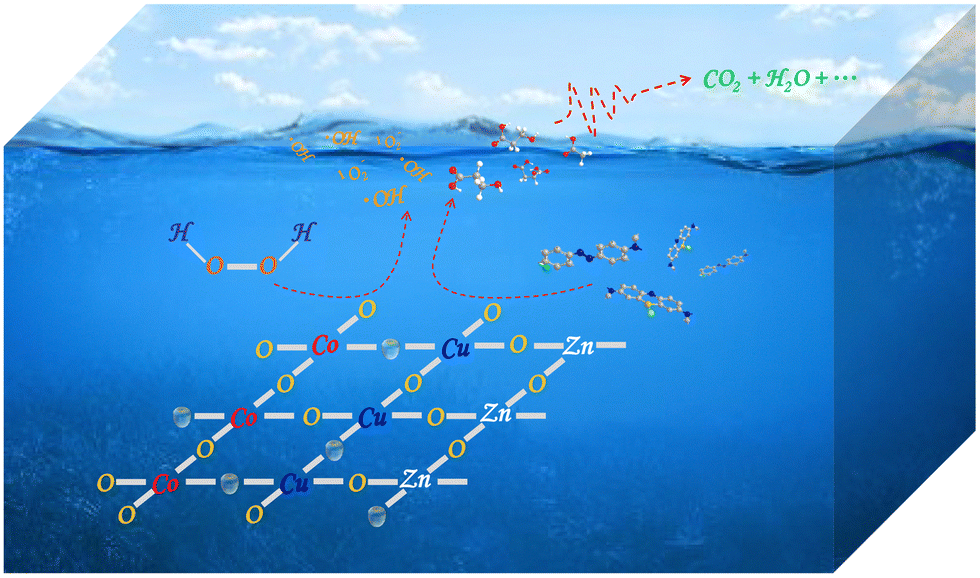
On the basis of the above discussion, a probable mechanism was protocoled and summarized in Fig. 9 and eqn (1)–(7). Above all, the pristine ZnO–Co3O4–CuO composite could be smashed into pieces in a violent ball milling process where the active sites including the OVs and superficial Cu/Co–O dangling bonds were produced through breaking of the original chemical bonds. Therefore, the superficial active sites of the composites were enhanced along with more intense treatment (Table 1 and Fig. 3). Since transition metal elements have alterable chemical valences in nature, the exposed Co/Cu sites directly participated in the interfacial redox to transform the adsorbed H2O2 molecules into ˙OH and ˙O2− radicals catalytically. In this way, an effortless Fenton-like reaction cycle could be established with the assistance of the superficial Co/Cu sites as shown in the eqn (1)–(4). On the other hand, the OVs on the surface of the composites also worked as new types of catalytic sites for the activation of H2O2. As shown in eqn (5), the unpaired electrons adjacent to the OVs could transfer to the adsorbed H2O2 molecules, leading to the reductive decomposition of H2O2 into ˙OH radicals. The previous research demonstrated that the OVs of the solid catalysts can effectively adsorb H2O2 molecules, thus facilitating the homolytic cleavage of H2O2 into free radicals.12,17,21,26,36,43–46 Moreover, the exposed active sites of the composites after the mechanochemical treatment might have stronger chemical interaction with the electron-rich organic species formed in the oxidative degradation than with H2O2 molecules. In eqn (6)–(9), the electron transfer from organic species to the active sites could establish a new and effortless reaction pathway for the reduction of Co(III)/Cu(II) ions to accelerate the rate-limiting step and facilitate the recovery of OVs. Although the catalytic decomposition of H2O2 molecules cannot produce 1O2 directly, 1O2 could be generated in the subsequent self-decay between ˙O2− and ˙OOH radicals (eqn (10)).
| | ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Co(II)–O + H2O2 → Co(III)–O + ˙OH + OH− Co(II)–O + H2O2 → Co(III)–O + ˙OH + OH− | (1) |
| | | Co(III)–O + H2O2 → Co(II)–O + ˙O2− + 2H+ | (2) |
| | | Cu(I)–O + H2O2 → Cu(II)–O + ˙OH + OH− | (3) |
| | | Cu(II)–O + H2O2 → Cu(I)–O + ˙O2− + 2H+ | (4) |
| | | VO-e + H2O2 → VO + ˙OH + OH− | (5) |
| | | ˙OH + MO/MB → ˙R/˙OR + … | (6) |
| | | Co(III)–O + ˙R/˙OR → Co(II)–O…R/ORads | (7) |
| | | Cu(II)–O + ˙R/˙OR → Cu(I)–O…R/ORads | (8) |
| | | VO + ˙R/˙OR → VO-e…R/ORads | (9) |
| | | ˙O2− + ˙OOH → 1O2 + HOO− | (10) |
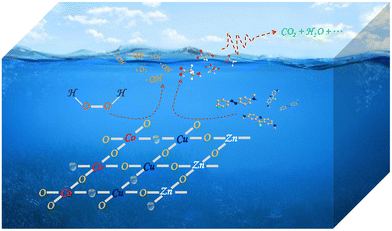 |
| | Fig. 9 Catalytic mechanism of the ZnO–Co3O4–CuO composite treated by high-speed ball milling in the Fenton-like treatment of organic wastewater. | |
3.5 Fenton-like degradation pathway and biological toxicity of MO and MB wastewater
The intermediates formed in the Fenton-like degradation of MO and MB were analysed by GC-MS. Fig. S9 (ESI†) depicts the possible degradation pathways of MO and MB. Generally, the degradation processes could be mainly divided into three steps: (1) dealkylation and hydroxylation of MO or MB molecules attacked by the oxidative active species (˙OH radicals and 1O2), (2) ring-opening reactions of benzene rings to form organic acids, and (3) deep mineralization of the micromolecular organic products to CO2 and H2O. Significantly, GC-MS data indicated that the degradation of the dyes produced a number of organic acids that could be adsorbed on the basic sites of the composites formed through the mechanochemical treatment.
Table S5 (ESI†) presents the acute toxicity results of MO and MB solutions for artemia salina. The average mortality of artemia salina in the original MO (20 mg L−1) and MB (20 mg L−1) was 59.0% and 48.7%, respectively, but it decreased by more than 10.0% in the corresponding effluents treated for 4 hrs. Additionally, the median effective concentrations (EC50) in the original MO and MB solution were 7.11 and 11.7 mg L−1, respectively, and they were drastically enhanced to 21.2 and 150.3 mg L−1 in the treated solutions. The above results confirm that the biological toxicity of organic dye solutions could be significantly reduced after the Fenton-like treatment.
4. Conclusions
In this work, three kinds of grinding methods were applied to reduce the particle size and expose the redox active sites of ZnO–Co3O4–CuO composites. In the ball milling process, the OVs and superficial Cu/Co–O dangling bonds were simultaneously formed through breaking the original chemical bonds, and the exposed active site concentration was increased with the grinding strength accordingly. In the Fenton-like system with the composite treated using high-speed ball milling, the mineralization degree of organic dye wastewater was improved by more than 45%, compared to that with the composite treated using manual grinding. The corresponding system had a wide pH application range from 4 to 9, outstanding tolerance for the organic salt and acceptable stability. After the mechanochemical treatment, the exposed OVs and Co/Cu sites of the composite were confirmed as the primary active sites which could facilitate the decomposition of H2O2 molecules mainly into aggressive free radicals to mineralize the organics through the effective chemisorption and interfacial electron transfer. In this way, an effortless Fenton-like cycle was established through the efficient electron transfer between the active sites and the reactants. The current work provides a simple strategy for the catalytic enhancement of Fenton-like catalysts by systematically investigating the interfacial catalytic mechanism of the ZnO–Co3O4–CuO composite.
Author contributions
Xueping Li, Kangkang Miao and Sifan Guo: conceptualization, methodology, data curation, and formal analysis; Nan Wang, Qian Zhuang and Huaming Qian: data curation and formal analysis; Xiaolin Luo: conceptualization, methodology, project administration, writing – original draft, writing – review and editing, and validation; and Guodong Feng: conceptualization and funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors appreciate the financial support from the National Natural Science Foundation of China (no. 22271229), the Natural Science Foundation of Shaanxi Province (no. 2022JM-074), Shaanxi Province Technology Innovation Guidance Special Project (no. 2023YFBT-29-02), Key Research and Development Program of Shaanxi Province (no. 2021GY-159) and Key Laboratory Project of Education Department of Shaanxi Province (no. 20JS006). The authors would like to thank Dr Ping Wen, Le Wang and Lin Ma for their XPS, GC-MS and acute toxicity experimental assistance.
References
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Evaluation of advanced oxidation processes for water and wastewater treatment: A critical review, Water Res., 2018, 139, 118–131 CrossRef CAS PubMed.
- R. Anjali and S. Shanthakumar, Insights on the current status of occurrence and removal of antibiotics in wastewater by advanced oxidation processes, J. Environ. Manage., 2019, 246, 51–62 CrossRef CAS PubMed.
- D. Zhou, L. Chen, J. Li and F. Wu, Transition metal catalyzed sulfite auto-oxidation systems for oxidative decontamination in waters: A state-of-the-art minireview, Chem. Eng. J., 2018, 346, 726–738 CrossRef CAS.
- A. D. Bokare and W. Choi, Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- P. Gao, X. J. Chen, M. J. Hao, F. Xiao and S. X. Yang, Oxygen vacancy enhancing the Fe2O3-CeO2 catalysts in Fenton-like reaction for the sulfamerazine degradation under O2 atmosphere, Chemosphere, 2019, 228, 521–527 CrossRef CAS PubMed.
- J. H. Huang, J. T. Chen, T. Yao, J. F. He, S. Jiang, Z. H. Sun, Q. H. Liu, W. R. Cheng, F. C. Hu, Y. Jiang, Z. Y. Pan and S. Q. Wei, CoOOH nanosheets with high mass activity for water oxidation, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- H. Jin, X. K. Tian, Y. L. Nie, Z. X. Zhou, C. Yang, Y. Li and L. Q. Lu, Oxygen vacancy promoted heterogeneous Fenton-like degradation of ofloxacin at pH 3.2–9.0 by Cu substituted magnetic Fe3O4@FeOOH nanocomposite, Environ. Sci. Technol., 2017, 51, 12699–12706 CrossRef CAS PubMed.
- L. Lyu, L. Zhang, G. He, H. He and C. Hu, Selective H2O2 conversion to hydroxyl radicals in the electron-rich area of hydroxylated C-g-C3N4/CuCo–Al2O3, J. Mater. Chem. A, 2017, 5, 7153–7164 RSC.
- L. Lyu, G. Yu, L. Zhang, C. Hu and Y. Sun, 4-phenoxyphenol-functionalized reduced graphene oxide nanosheets: A metal-free Fenton-like catalyst for pollutant destruction, Environ. Sci. Technol., 2018, 52, 747–756 CrossRef CAS PubMed.
- H. Zhang, C. Li, L. Lyu and C. Hu, Surface oxygen vacancy inducing peroxymonosulfate activation through electron donation of pollutants over cobalt-zinc ferrite for water purification, Appl. Catal. B, 2020, 270, 118874–118885 CrossRef CAS.
- Y. Zhu, R. Zhu, Y. Xi, J. Zhu, G. Zhu and H. He, Strategies for enhancing the heterogeneous Fenton catalytic reactivity: A review, Appl. Catal. B, 2019, 255, 117739–117755 CrossRef CAS.
- H. Li, J. Shang, Z. P. Yang, W. J. Shen, Z. H. Ai and L. Z. Zhang, Oxygen vacancy associated surface Fenton chemistry: Surface structure dependent hydroxyl radicals generation and substrate dependent reactivity, Environ. Sci. Technol., 2017, 51, 5685–5694 CrossRef CAS PubMed.
- J. Paier, C. Penschke and J. Sauer, Oxygen defects and surface chemistry of ceria: quantum chemical studies compared to experiment, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- Y. Wang, P. Zhang, T. Li, L. Lyu, Y. Gao and C. Hu, Enhanced Fenton-like efficiency by the synergistic effect of oxygen vacancies and organics adsorption on FexOy-d-g-C3N4 with Fe–N complexation, J. Hazard. Mater., 2021, 408, 124818–124828 CrossRef CAS PubMed.
- L. Fu, D. Wu, M. Wen, Y. Zhu, Q. Wu, T. Zhou and Y. Fu, 2D hetero-nanostructured reduced-CuNiFe-oxides with self-produced H2O2 Fenton-like photo-catalysis for tetracycline degradation, Inorg. Chem. Front., 2023, 10, 567–578 RSC.
- M. Wang, C. Liu, H. Shi, T. Long, C. Zhang and B. Liu, Facile synthesis of chitosan-derived maillard reaction productions coated CuFeO2 with abundant oxygen vacancies for higher Fenton-like catalytic performance, Chemosphere, 2021, 283, 131191–131200 CrossRef CAS PubMed.
- N. Zhang, E. P. Tsang, J. Chen, Z. Fang and D. Zhao, Critical role of oxygen vacancies in heterogeneous Fenton oxidation over ceria-based catalysts, J. Colloid Interf. Sci., 2020, 558, 163–172 CrossRef PubMed.
- W. Xie, F. Zhou, X. Bi, D. Chen, Z. Huang, Y. Li, S. Sun and J. Liu, Decomposition of nickel(II)−ethylenediaminetetraacetic acid by Fenton−like reaction over oxygen vacancies-based Cu−doped Fe3O4@γ−Al2O3 catalyst: A synergy of oxidation and adsorption, Chemosphere, 2019, 221, 563–572 CrossRef CAS PubMed.
- X. Yang, C. Lai, L. Li, M. Cheng, S. Liu, H. Yi, M. Zhang, Y. Fu, F. Xu, H. Yan, X. Liu and B. Li, Oxygen vacancy assisted Mn-CuO Fenton-like oxidation of ciprofloxacin: Performance, effects of pH and mechanism, Sep. Purif. Technol., 2022, 287, 120517–120526 CrossRef CAS.
- Y. Yu, H. Chen, L. Yan and C. Jing, Oxygen vacancy modulated interface chemistry: identifying iron(IV) in heterogeneous Fenton reaction, Environ. Sci.: Nano, 2021, 8, 978–985 RSC.
- N. Zhang, C. Xue, K. Wang and Z. Fang, Efficient oxidative degradation of fluconazole by a heterogeneous Fenton process with Cu-V bimetallic catalysts, Chem. Eng. J., 2020, 380, 122516–122524 CrossRef CAS.
- C. Song, Q. Zhan, F. Liu, C. Wang, H. Li, X. Wang, X. Guo, Y. Cheng, W. Sun, L. Wang, J. Qian and B. Pan, Overturned loading of inert CeO2 to Active Co3O4 for unusually improved catalytic activity in Fenton-like reactions, Angew. Chem., Int. Ed., 2022, 134, 406–414 Search PubMed.
- Q. Xia, Z. Yao, D. Zhang, D. Li, Z. Zhang and Z. Jiang, Rational synthesis of micronano dendritic ZVI@Fe3O4 modified with carbon quantum dots and oxygen vacancies for accelerating Fenton-like oxidation, Sci. Total Environ, 2019, 671, 1056–1065 CrossRef CAS.
- Y. Zhang, R. Xiao, S. Wang, H. Zhu, H. Song, G. Chen, H. Lin, J. Zhang and J. Xiong, Oxygen vacancy enhancing Fenton-like catalytic oxidation of norfloxacin over prussian blue modified CeO2: Performance and mechanism, J. Hazard. Mater., 2020, 398, 122863–122871 CrossRef CAS PubMed.
- N. Zhang, Y. Yi, J. Lian and Z. Fang, Effects of Ce doping on the Fenton-like reactivity of Cu-based catalyst to the fluconazole, Chem. Eng. J., 2020, 395, 124897–124905 CrossRef CAS.
- M. A. Younis, S. Lyu, C. Lei, B. Yang, Z. Li, Q. He, J. Lu, L. Lei and Y. Hou, Efficient mineralization of sulfanilamide over oxygen vacancy-rich NiFe-LDH nanosheets array during electro-Fenton process, Chemosphere, 2021, 268, 129272–129280 CrossRef CAS PubMed.
- T. J. Fisher, Y. Zhou, T. S. Wu, M. Wang, Y. L. Soo and C. L. Cheung, Structure–activity relationship of nanostructured ceria for the catalytic generation of hydroxyl radicals, Nanoscale, 2019, 11, 4552–4561 RSC.
- W. Xu, W. Xue, H. Huang, J. Wang, C. Zhong and D. Mei, Morphology controlled synthesis of α-Fe2O3-x with benzimidazole-modified Fe-MOFs for enhanced photo-Fenton-like catalysis, Appl. Catal. B, 2021, 291, 120129–120142 CrossRef CAS.
- M. Wu, H. Li, S. Ma, S. Chen and W. Xiang, Boosting the surface oxygen activity for high performance Iron-based perovskite oxide, Sci. Total Environ, 2021, 795, 148904–148915 CrossRef CAS PubMed.
- H. Zou, J. Zhao, F. He, Z. Zhong, J. Huang, Y. Zheng, Y. Zhang, Y. Yang, F. Yu, M. A. Bashir and B. Gao, Ball milling biochar iron oxide composites for the removal of chromium (Cr(VI)) from water: Performance and mechanisms, J. Hazard. Mater., 2021, 413, 125252–125261 CrossRef CAS PubMed.
- F. Jiang, L. Zhang, T. Yue, H. Tang, L. Wang, W. Sun, C. Zhang and J. Chen, Defect-boosted molybdenite-based co-catalytic Fenton reaction, Inorg. Chem. Front., 2021, 8, 3440–3449 RSC.
- Z. Yin, Q. Zhang, S. Li, G. Cagnetta, J. Huang, S. Deng and G. Yu, Mechanochemical synthesis of catalysts and reagents for water decontamination: Recent advances and perspective, Sci. Total Environ., 2022, 825, 153992–154008 CrossRef CAS PubMed.
- G. H. Major, N. Farley, P. M. A. Sherwood, M. R. Linford, J. Terry, V. Fernandez and K. Artyushkova, Practical guide for curve fitting in x-ray photoelectron spectroscopy, J. Vac. Sci. Technol., 2020, 38, 061203–061223 CrossRef CAS.
- H. Hu, K. Miao, X. Luo, S. Guo, X. Yuan, F. Pei, H. Qian and G. Feng, Efficient Fenton-like treatment of high-concentration chlorophenol wastewater catalysed by Cu-Doped SBA-15 mesoporous silica, J. Clean. Prod., 2021, 318, 128632–128641 CrossRef CAS.
- K. Miao, X. Luo, W. Wang, J. Guo, S. Guo, F. Cao, Y. Hu, P. Chang and G. Feng, One-step synthesis of Cu–SBA-15 under neutral condition and its oxidation catalytic performance, Microporous Mesoporous Mater., 2019, 289, 109640–109647 CrossRef CAS.
- S. Zhan, H. Zhang, X. Mi, Y. Zhao, C. Hu and L. Lyu, Efficient Fenton-like process for pollutant removal in electron-rich/poor reaction sites induced by surface oxygen vacancy over cobalt−zinc oxides, Environ. Sci. Technol., 2020, 54, 8333–8343 CrossRef CAS PubMed.
- S. Zhao, X. Jin, P. Wu, Y. Zhao, G. Chen, Y. Li, A. Li, D. Ye and Y. Qiu, Cu2+-decorated porous Co3O4 nanosheets for photothermocatalytic oxidation of toluene, ACS Appl. Nano Mater., 2020, 3, 10454–10461 CrossRef CAS.
- X. X. Guo, T. T. Hu, B. Meng, Y. Sun and Y. F. Han, Catalytic degradation of anthraquinones-containing H2O2 production effluent over layered Co-Cu hydroxides: Defects facilitating hydroxyl radicals generation, Appl. Catal. B, 2020, 260, 118157–118166 CrossRef CAS.
- I. Lorite, J. J. Romero and J. F. Fernández, Effects of the agglomeration state on the Raman properties of Co3O4 nanoparticles, J. Raman Spectrosc., 2012, 43, 1443–1448 CrossRef CAS.
- X. Luo, H. Hu, Z. Pan, F. Pei, H. Qian, K. Miao, S. Guo, W. Wang and G. Feng, Efficient and stable catalysis of hollow Cu9S5 nanospheres in the Fenton-like degradation of organic dyes, J. Hazard. Mater., 2020, 396, 122735–122747 CrossRef CAS PubMed.
- L. Lyu, L. Zhang, Q. Wang, Y. Nie and C. Hu, Enhanced Fenton catalytic efficiency of γ-Cu−Al2O3 by σ-Cu2+−ligand complexes from aromatic pollutant degradation, Environ. Sci. Technol., 2015, 49, 8639–8647 CrossRef PubMed.
- M. Kohantorabi, G. Moussavi and S. Giannakis, A review of the innovations in metal- and carbon-based catalysts explored for heterogeneous peroxymonosulfate (PMS) activation, with focus on radical vs. non-radical degradation pathways of organic contaminants, Chem. Eng. J., 2021, 411, 127957–127982 CrossRef CAS.
- X. Luo, X. Yuan, F. Pei, H. Hu, H. Qian, K. Miao, S. Guo, W. Wang and G. Feng, Fabrication of ZnO/Au@Cu2O heterojunction towards deeply oxidative photodegradation of organic dyes, Sep. Purif. Technol., 2021, 262, 118301–118309 CrossRef.
- X. Li, Y. Guo, L. Yan, T. Yan, W. Song, R. Feng and Y. Zhao, Enhanced activation of peroxymonosulfate by ball-milled MoS2 for degradation of tetracycline: Boosting molybdenum activity by sulfur vacancies, Chem. Eng. J., 2022, 429, 132234–132244 CrossRef CAS.
- X. Y. Jiang, E. Kwon, J. C. Wen, J. Bedia, B. X. Thanh, S. Ghotekar, J. Lee, Y. C. Tsai, A. Ebrahimi and K. Y. A. Lin, Direct growth of nano-worm-like Cu2S on copper mesh as a hierarchical 3D catalyst for Fenton-like degradation of an imidazolium room-temperature ionic liquid in water, J. Colloid Interf. Sci., 2020, 638, 39–53 CrossRef PubMed.
- L. Lyu, K. Deng, J. Liang, C. Lu, T. Gao, W. Cao and C. Hu, The interaction of surface electron distribution-polarized Fe/polyimide hybrid nanosheets with organic pollutants driving a sustainable Fenton-like process, Mater. Adv., 2020, 1, 1083–1091 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Guodong
Feng
*b
*a and
Guodong
Feng
*b