
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePost-lithiation: a way to control the ionic conductivity of solid-state thin film electrolyte†
Jixi
Chen
 *,
Alessandro
Palliotto
,
Shinhee
Yun
,
Dennis Valbjørn
Christensen
,
Vincenzo
Esposito
and
Nini
Pryds
*
*,
Alessandro
Palliotto
,
Shinhee
Yun
,
Dennis Valbjørn
Christensen
,
Vincenzo
Esposito
and
Nini
Pryds
*
Department of Energy Conversion and Storage, Technical University of Denmark, Fysikvej, Building 310, 2800 Kgs, Lyngby, Denmark. E-mail: jixich@dtu.dk; nipr@dtu.dk
First published on 18th November 2023
Abstract
Ionic conductivity is pivotal for solid-state battery performance. While the garnet oxide electrolyte Li7La3Zr2O12 (LLZO) boasts high ionic conductivity due to its distinct crystal structure and lithium-ion mobility, lithium loss during fabrication hampers its potential. In this study, we introduce a method that merges synthesis optimization with a post-lithiation process, enhancing LLZO's ionic conductivity. This approach compensates lithium loss with a gas-phase diffusion process, which stabilizes the cubic LLZO phase and amplifies its ionic conductivity by more than three orders of magnitude compared to electrolytes without post-lithiation. Through our comprehensive experimental procedure, we have conclusively determined that the film deposited at 700 °C and subsequently annealed at 700 °C with LiOH exhibits the highest conductivity, with a notable value of 1.11 × 10−2 S cm−1 at 200 °C. This is a significant boost compared to the as-deposited film (3.54 × 10−6 S cm−1 at 200 °C). Our findings present an additional approach to boosting lithium ion diffusion. The approach employed in this work has the potential to be applicable to films produced through other deposition methods, as it addresses the prevalent issue of lithium loss, a significant barrier to the utilization of lithium-rich thin films.
1. Introduction
The expansion of the internet of things (IoT) is followed by increasing demands for the miniaturisation of energy storage and harvesting devices.1 Many IoT edge devices are battery-powered, particularly sensors which are placed in decentralised locations. Solid-state batteries (SSBs) are an emerging option for powering the IoT, offering safe alternatives to conventional lithium-ion batteries due to the absence of flammable liquid electrolytes, in addition to low cost, scalability and high performance with both high-energy (Wh kg−1) and high-power (W kg−1) densities.2,3 The use of thin-film technology in SSBs offers various advantages, such as: (i) being well suited for the design of small devices; (ii) reducing the thickness of the solid electrolyte film allowing to increase in the ionic conductivity of the electrolyte materials; (iii) reducing the charge-transfer resistance of the electrolyte–electrode interface; (iv) manufacturing micro-batteries using the same techniques that are currently used in the microelectronics industry.4 Typical SSB batteries are formed by planar layers consisting of cathode, electrolyte, anode and current collectors. Recently, SSBs have been reported to be composed of thin film materials such as (1) Cathodes: Li2CoO3 (LCO),5 Li(NixMnyCoz)O2 (NMC),6 LiNi0.5Mn1.5O4 (LNMO),7 and LiFePO4 (LFP);8 (2) electrolytes: Li3−xPO4−yN2 (LiPON),9 LixLa2/3+yTiO3−d (LLTO),10 Li1.3Al0.3Ti1.7(PO4)3 (LATP),11 and Li7La3Zr2O12 (LLZO);12,13 (3) anodes: Li4Ti5O12 (LTO),14 silicon,15 and lithium metal.16,17 The challenges are to find a way to meet the demanding power budgets while achieving the desired battery size, lifetime, charging time and cost. However, it is hard to construct SSBs that simultaneously meet all performance targets required for commercialization.Lithium garnet Li7La3Zr2O12 is one of the most promising solid-state electrolyte (SSE) materials because of its high ionic conductivity (1 mS cm−1 at ambient temperature),18 low electronic conductivity and wide electrochemical stability range.19,20 It has also displayed the potential of preventing Li-dendrite growth owing to its high Young's modulus.21 Thin-film processing of lithium garnet electrolytes allows the: (1) formation of desired material phases that would otherwise be thermodynamically unfavourable and (2) a high degree of thickness and surface control, yielding high-density films, which is beneficial for device integration.22
To date, thin film ionic conductivities in literature lag behind bulk values, notably for films made via pulsed laser deposition (PLD).22,23 For instance, LLZO's Li conductivity in its stable tetragonal phase (t-LLZO, space group I41/acd) at room temperature (RT) is 10−6 S cm−1. To reach conductivities around 10−3 S cm−1, the cubic (c-LLZO, Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d) or acentric cubic phase (I
d) or acentric cubic phase (I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d) of LLZO must be stabilized through doping or other methods.24–28 Reducing LLZO film thickness or particle size can also stabilize the cubic phase due to size-dependent phase transformation.29 Another challenge is lowering the production temperature while maintaining high conductivity, making it more compatible with cathode materials like NMC and LFP, typically processed at 600–800 °C.8,30
3d) of LLZO must be stabilized through doping or other methods.24–28 Reducing LLZO film thickness or particle size can also stabilize the cubic phase due to size-dependent phase transformation.29 Another challenge is lowering the production temperature while maintaining high conductivity, making it more compatible with cathode materials like NMC and LFP, typically processed at 600–800 °C.8,30
One of the main drawbacks of thin-film architectures is the high amount of lithium losses that occur during film fabrication and the subsequent annealing. This has a negative impact on the formation of the phases, density, and crystallinity of the LLZO films.22,31 Therefore, extensive studies have been carried out to compensate for the loss of lithium by adding extra Li during or after the deposition processes.23,29
To offset the Li depletion during deposition, the common approaches involve using two targets for sputtered films or/and using over-lithiated ceramic targets for PLD films.22 However, stabilizing crystalline SSE and electrode materials in highly lithiated phases is challenging for PLD films using only over-lithiated ceramic pellets. In a recent work by Pfenninger et al, the authors suggest a method to produce cubic, thin Li-garnet SSB electrolytes at minimal processing temperatures by creating internal lithiation reservoirs to preserve the essential phase. This approach significantly lowers the processing temperatures for cubic Li-garnets, yielding thin ceramics with high Li+ conduction.23 Rawlence et al have employed a post-annealing technique annealing LLZO films with different thicknesses inside of a sealed crucible containing Li2O powder.29
These findings underscore that enhancing the conductivity of LLZO thin films hinges on stabilizing the cubic phase through precise control of film stoichiometry and microstructure. Without compensating for Li loss, the film's ionic conductivity is significantly compromised.
In this study, we meticulously examine how deposition temperature and post-annealing conditions influence the structural and electrochemical attributes of LLZO thin films, crafted using pulsed laser deposition on a single-crystalline MgO (100) substrate. Our research aim was to contribute to the understanding of the intrinsic properties of the undoped LLZO: (1) Explore the intrinsic ionic conductivity of pure LLZO at room temperature, without the influence of dopants, offering insights into its fundamental properties and potential pathways for enhancing conductivity without additives. (2) Assess the performance of pure LLZO in its pristine state, serving as a benchmark to discern any deviations introduced by doping. (3) Investigate whether dopant-free alternatives for SSE are viable, as doping, while often enhancing conductivity, can introduce stability issues and manufacturing complexities. Our findings suggest that utilizing as-deposited amorphous LLZO or polycrystalline La2Zr2O7 (LZO), followed by post-lithiation annealing, emerges as a potent strategy to produce high-conductivity sub-micron c-LLZO thin films.
2. Results and discussion
2.1. Deposition of LLZO by PLD under different conditions
Lithium garnet thin films were fabricated on MgO (100) substrates by PLD from a stoichiometric Li7La3Zr2O12 (LLZO) target, as schematically shown in Fig. 1a (the detail of the experimental procedure is given in the Experimental part). We first deposited the films under various deposition conditions with different laser energy fluences (0.5, 1, 2, and 3 J cm−2), oxygen pressures (10−4, 10−3, 10−2, and 10−1 mbar), and substrate temperatures (Td = 50, 300, 500, 600 and 700 °C). In all the experiments reported in this paper, the thickness was kept constant at approximately 100 nm to avoid a broader range of parameter variations. Future research should also be focused on systematically varying the film thickness. The resulting phases of the films were then investigated using grazing-incidence X-ray diffraction (GIXRD) with detailed information given in the Supplementary Information (Fig. S1–S4, ESI†) and a summary provided in Fig. 1b. The overall observation indicates that: (1) Films deposited at 700 °C show a lithium deficient La2Zr2O7 (LZO) pyrochlore phase regardless of the fluences and background pressure. (2) Films deposited under pO2 of 10−2 mbar and fluence of 1 J cm−2, and deposition temperature of 600 °C show the formation of the LZO phase as well as a residual amorphous phase. (3) At a temperature below 600 °C, the XRD patterns show only the amorphous phase.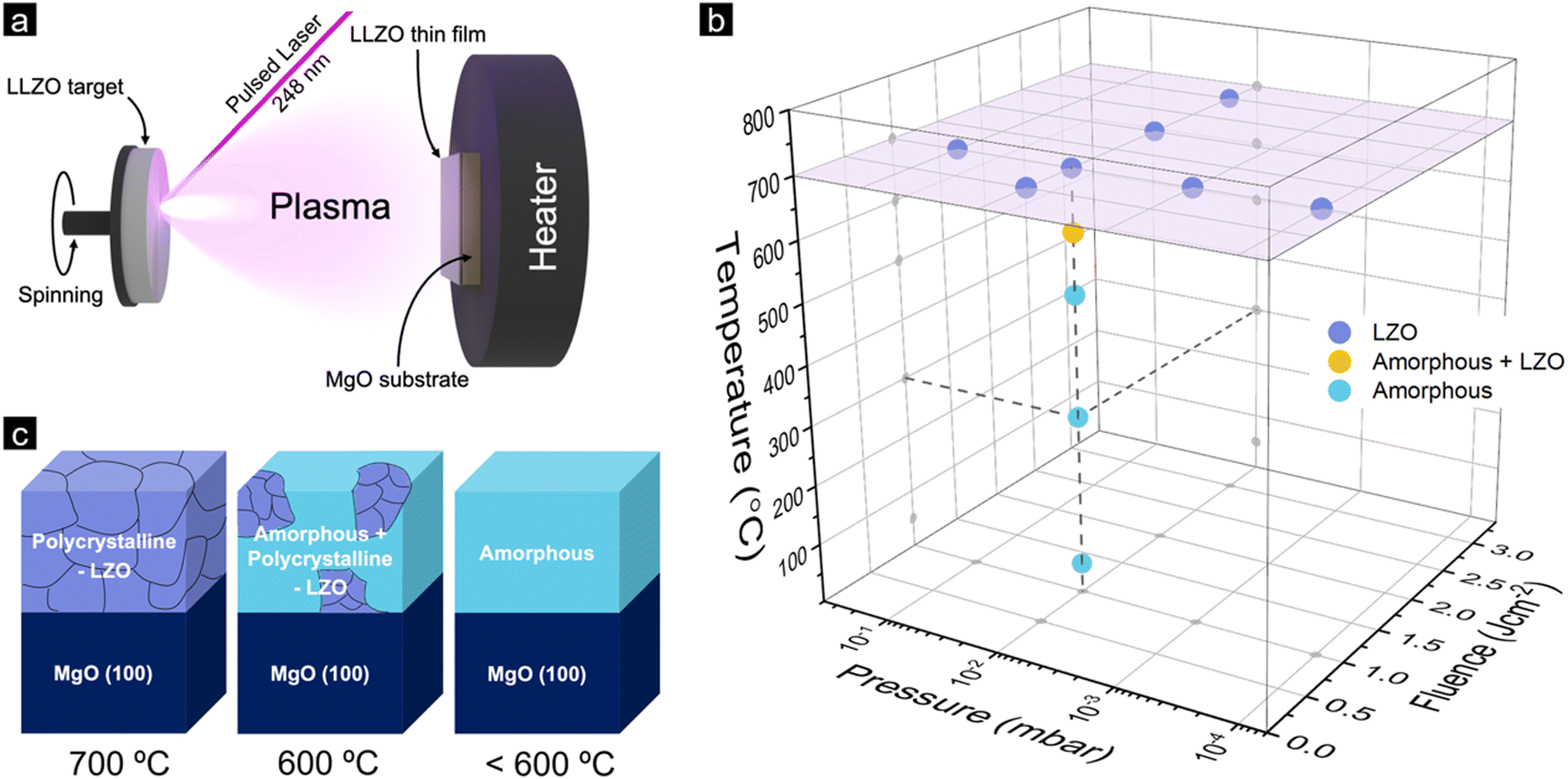 | ||
| Fig. 1 (a) Schematic illustration of pulsed laser deposition (PLD) of LLZO. (b) Phase diagram of Li-garnet films deposited at different conditions by PLD. (c) Schematic view of the composition/structure of the films constructed using the grazing-incidence X-ray diffraction data. | ||
At a deposition temperature of 50 °C, we further found indications of the LLZO phase that can be detected with a very small incident angle (∼0.3° with a penetration depth of about 10 nm) using GIXRD (Fig. S4, ESI†). This indicates that a very thin layer of LLZO films may form on top of the films deposited by PLD, which was also reported by Park et al.13 Overall, it is worth noting that during our investigation of the deposition process parameters, e.g., including the laser energy, we observed that regardless of the changes in the deposition parameters the film did not exhibit the cubic phase of LLZO. A schematic illustration of the phases formed during the as-deposited films at different deposition temperatures is shown in Fig. 1c.
2.2. Post-annealing of the as-deposited Li-garnet thin films
After thorough characterisation of the as-deposited films, we decided to shift our focus to the second part of this study, which involves a post-annealing process to ensure that the light element lithium was incorporated into the film, compensating for the deficiencies of these elements during the deposition.32 Li-garnets particularly severely suffer from these composition deficiencies due to a large mass ratio of Li vs. La.33 Even for the fabrication of bulk LLZO materials, Li evaporates during the annealing processes due to its high volatility and therefore needs to be compensated.34 This is more pronounced for vacuum thin-film processes because of their large surface-to-volume ratio and lower background gas pressure associated with the growth process.35 At an elevated deposition temperature, the lithium landing on the substrate is re-sputtered into the chamber due to the increased vapour pressure of Li, resulting in a non-stoichiometric element transfer in the forming film during deposition. Therefore, films deposited at 700 °C display a lithium-deficient LZO phase (Fig. 1b and c).In order to compensate for the loss of Li during the deposition at high temperatures (700 °C), we employ a post-annealing process with the following steps (Fig. 2a): (1) deposition of LLZO at RT or 700 °C, (2) Post annealing with an excess of Li2O or LiOH powder placed next to the sample in an oxygen atmosphere at 1 bar for 24 hours (see the Experimental section for more detailed) and (3) cooling to RT. After the annealing, the films were studied for their phase purity using GIXRD. The weight ratio of LLZO vs. LZO of the film deposited at different temperatures (Td = 50 and 700 °C) in the presence of either Li2O or LiOH powders are shown in Fig. 2b–e (for more details, see Fig. S5–S8, ESI†). Notably, Xiang et al. have addressed lithium loss and proposed a compensatory method.40 However, key distinctions exist between our work and theirs: (1) We used LLZO, not LLZTO as they did. (2) Our doping techniques differ significantly. Xiang et al. coated LLZTO films with a LiOH solution, while we employed a gas-phase diffusion process for Li insertion. (3) Our study focused on 100 nm thick films deposited using PLD, whereas their research used films from tape casting with a thickness of 40 μm.
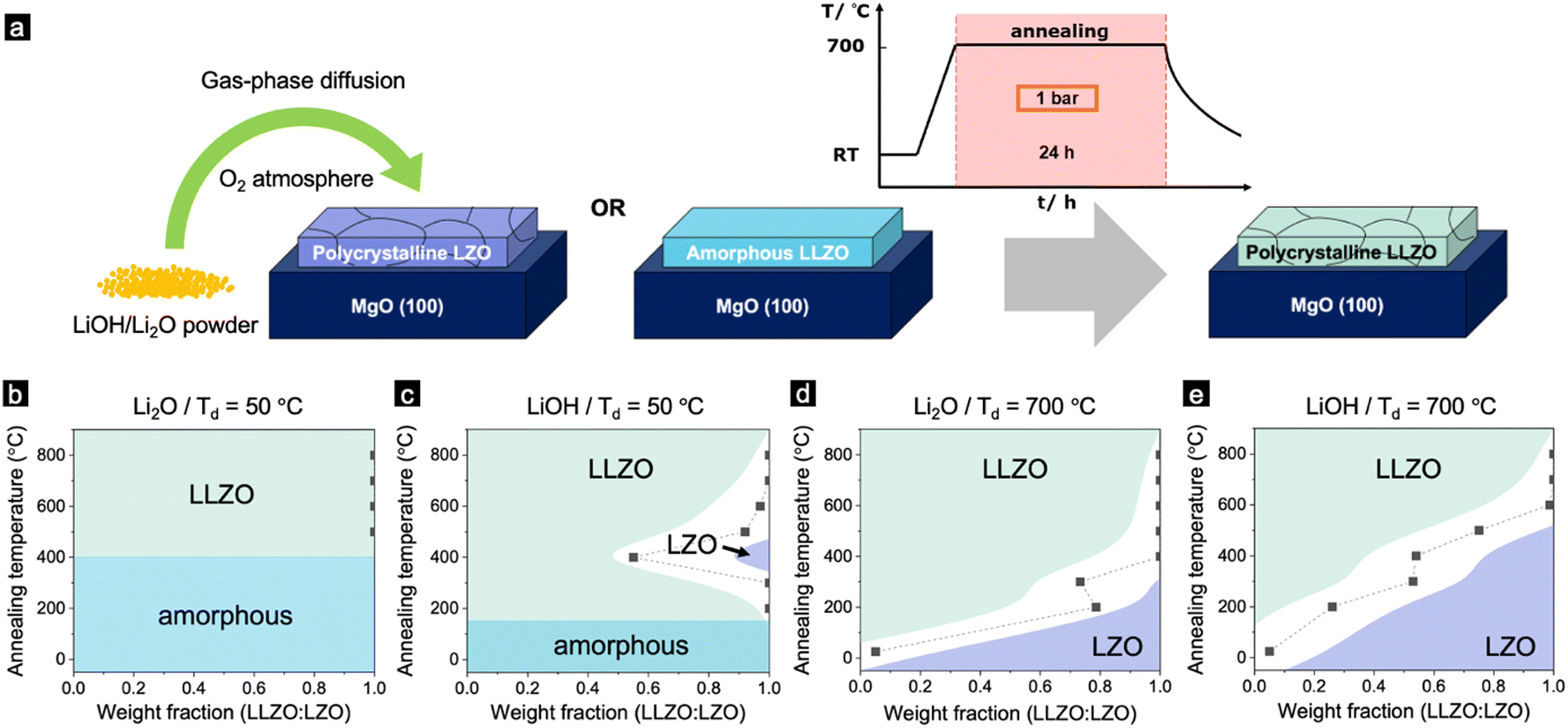 | ||
| Fig. 2 (a) Schematic illustration of the annealing process for electrolyte thin films. (b)–(e) Representations of the annealed Li-garnet films displaying the phases and weight ratio of LLZO to LZO prepared under different deposition and annealing temperatures, as constructed using the grazing-incidence X-ray diffraction data. Here, samples in (b) are deposited at 50 °C and annealed with Li2O, (c) deposited at 50 °C then annealed with LiOH, (d) deposited at 700 °C then annealed with Li2O, and (e) deposited at 700 °C then annealed with LiOH. | ||
Fig. 2b and c display films deposited at 50 °C and subsequently post-annealed using either LiOH or Li2O. As the annealing temperature rises, the films transition directly from an amorphous to a polycrystalline LLZO phase. The LZO phase appears only when LiOH powder is added during post-annealing (see Fig. 2c). Conversely, for films deposited at 700 °C and then annealed, a combination of LLZO and LZO is evident for both annealing powders (Fig. 2d and e). As the annealing temperature increases, the weight ratio of LLZO to LZO also rises (refer to Fig. 2d and e). Notably, samples annealed with Li2O have a higher LLZO content than those annealed with LiOH (Fig. 2d and e).
The difference in the evolution of the phases between the films annealed with Li2O and LiOH can be attributed to their different melting point. The melting point of LiOH is 462 °C, which is significantly lower than Li2O (1,438 °C).36 This explains why the film started to crystallize at a lower temperature for the film deposited at 50 °C (Fig. 2c). However, it is surprising that the LZO films annealed with Li2O fully converted to an LLZO dominant phase at a lower temperature than the films annealed with LiOH. A similar phase transformation from LZO pyrochlore to LLZO garnet was also observed in bulk materials, via a molten salt synthesis (MSS) in a highly basic flux.37
Fig. 3a shows the GIXRD patterns of the films deposited at 50 °C and 700 °C without post-annealing, as well as films deposited at 700 °C after post-annealing with LiOH or Li2O for 24 hours under an oxygen atmosphere. As we mentioned before, the films deposited at 50 °C and 700 °C without annealing show mainly the amorphous phase and LZO polycrystalline phase, respectively. The films deposited at 700 °C and annealed with LiOH or Li2O at 700 °C did not show any LZO phase. The main phase of both films was c-LLZO. Furthermore, there are several Li2CO3 peaks for both samples. This is likely due to some Li2O powder settling on the films and subsequently reacting with H2O and CO2 in the atmosphere (the exposure of the samples to air was minimised for the impedance test but was impossible during the XRD measurement).
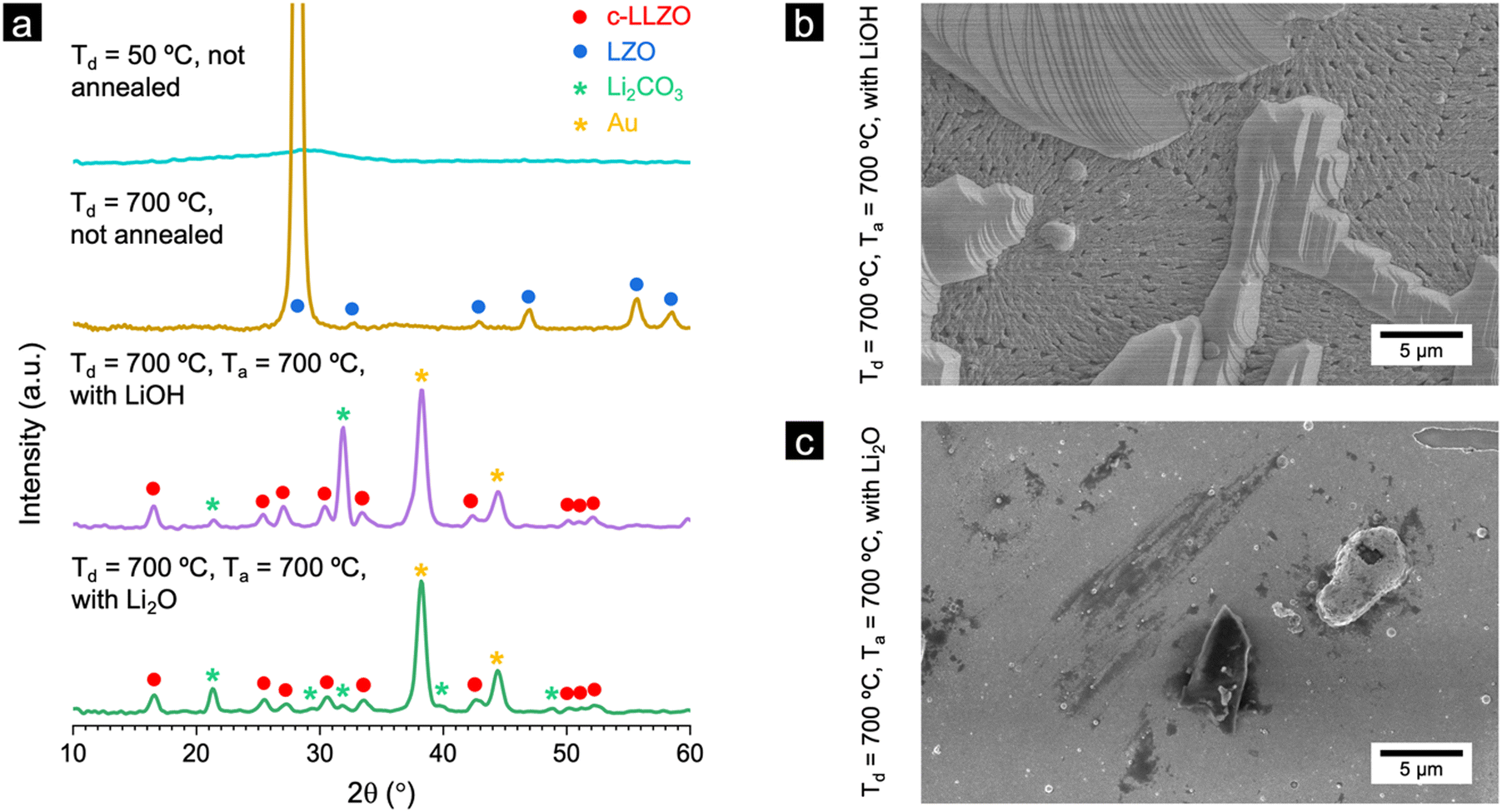 | ||
| Fig. 3 (a) Grazing-incidence X-ray diffraction patterns of as-deposited (at 50 °C and 700 °C) LLZO films, and Li-garnet films deposited at 700 °C and annealed at 700 °C in presence of Li2O or LiOH. Top-view SEM images of LLZO films deposited at 700 °C and annealed at 700 °C with (b) LiOH and (c) Li2O. | ||
The surface morphology of the film annealed with LiOH (Td = 700 °C and Ta = 700 °C) is found to contain two layers. The bottom layer covers the surface of the MgO and has a porous structure. The top layer is comprised of lamellar-like structures (Fig. 3b), which are rich in carbon and oxygen as identified by energy dispersive x-ray spectroscopy (EDX, Fig. S9, ESI†). In contrast, when annealing with Li2O (Td = 700 °C and Ta = 700 °C), the film is dense and crack-free, with fewer topograhic features on top of the surface (Fig. 3c). This difference in surface morphology could result from the differences in the diffusion mechanism of LiOH and Li2O owing to the vastly different melting point and vapour pressure of these materials. The difference in surface morphology could be an indicator of the effectiveness of lithium in diffusing from an external source.
2.3. Ionic conductivity of lithium garnet thin films
The electrochemical impedance spectroscopy (EIS) measurements of the films deposited at 700 °C and annealed with Li2O and LiOH at 700 °C are illustrated in Fig. 4 using the electrode configuration displayed in the inset. The impedance spectra were fitted with the equivalent circuit to calculate the resistance and capacitance that control the ionic dynamics in the thin film (inset of Fig. 4 with a measurement temperature of 150 °C). The model includes the contact resistance (Rcontact), This equivalent circuit is used to model ionic conductors,38 and it includes the contact resistance (Rcontact), the geometric capacitance (Cgeom), the bulk Resistance (Rbulk), and the electrode–electrolyte interfacial polarization (CPEint). For this measurement, a capacitor (Cgeom) in parallel with a resistor (Rbulk) represents the total ionic conductivity of the LLZO films, which is shown in the enlarged schematic in Fig. 4. The total ionic conductivity accounts for both the bulk and grain boundary contributions, which cannot be separated because the two processes overlap with each other. The calculated ionic conductivities (σ) and activation energies (Ea) are shown in Table 1 for a measurement temperature of 200 °C for all the samples deposited and annealed at different conditions. Here, the activation energies were calculated from an Arrhenius plot as described next with the σT relation. We can see substantial increases in the ionic conductivities from the as-deposited films to the films deposited at 700 °C (crystalline LZO) and post-annealed with either LiOH or Li2O at 700 °C.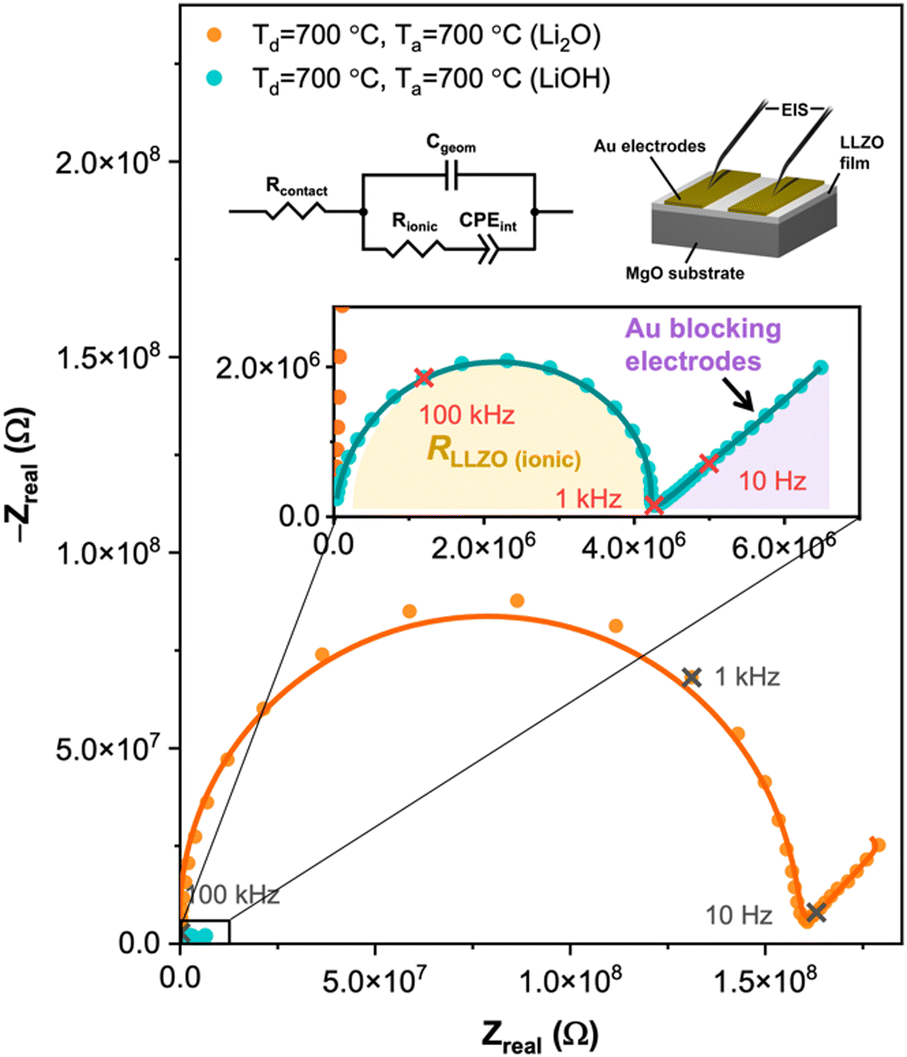 | ||
| Fig. 4 Schematic of the equivalent circuit used to fit the EIS data and extract the ionic conductivities; and the measurement configuration; and Nyquist plots (measured at 150 °C) of the films deposited at 700 °C and annealed with Li2O and LiOH at 700 °C. | ||
| Deposition condition | Annealing condition | Target material | Crystalline phase | Conductivity [S cm−1] | Activation energy [eV] |
|---|---|---|---|---|---|
| — | — | LLZO target | Crystalline Li7La3Zr2O12 | 5.12 × 10−3 | 0.63(4) |
| 50 °C | — | Li7La3Zr2O12 | Amorphous | 3.54 × 10−6 | 0.884(2) |
| 50 °C | 700 °C | Li7La3Zr2O12 | Amorphous | 2.71 × 10−6 | 0.782(3) |
| 50 °C | 700 °C with LiOH | Li7La3Zr2O12 | Crystalline Li7La3Zr2O12 | 9.61 × 10−6 | 0.77(5) |
| 50 °C | 700 °C with Li2O | Li7La3Zr2O12 | Crystalline Li7La3Zr2O12 | 1.64 × 10−3 | 0.81(3) |
| 700 °C | 700 °C with LiOH | Li7La3Zr2O12 | Crystalline Li7La3Zr2O12 | 1.11 × 10−2 | 0.712(4) |
| 700 °C | 700 °C with Li2O | Li7La3Zr2O12 | Crystalline Li7La3Zr2O12 | 6.04 × 10−4 | 0.85(4) |
There is a significant difference in the conductivity of films annealed with LiOH and films annealed with Li2O. Our findings demonstrate that films deposited at room temperature exhibit greater conductivity when subjected to annealing with Li2O, whereas films deposited at 700 °C achieve superior conductivity when annealed with LiOH. This result might be due to the different diffusion mechanisms of Li2O and LiOH. The low melting point of LiOH aligns effectively with the phase transition temperature of crystalline LZO. In contrast, when dealing with amorphous LLZO, Li2O remains stable throughout the 24-hour annealing period, making it a more enduring source of lithium. However, more work should be done to reveal the detailed mechanism of the gas-phase lithium diffusion.
Fig. 5 shows the Arrhenius plots of the LLZO thin films deposited at 50 °C without post-annealing, and thin films deposited at 700 °C and post-annealed with LiOH and Li2O at 700 °C. For comparison, we have included data from the literature for the undoped LLZO thin films deposited by PLD as well as data from the bulk LLZO target used for fabricating the film. Interestingly, the film deposited at 700 °C and post-annealed with LiOH at 700 °C shows an ionic conductivity comparable to the bulk target material, which is about 9 × 10−3 S cm−1 at 100 °C. This value is more than 3 orders of magnitudes higher than the as-deposited thin film. The ionic conductivity of the current results outperformed the values reported in the literature for undoped thin film LLZO deposited by PLD.
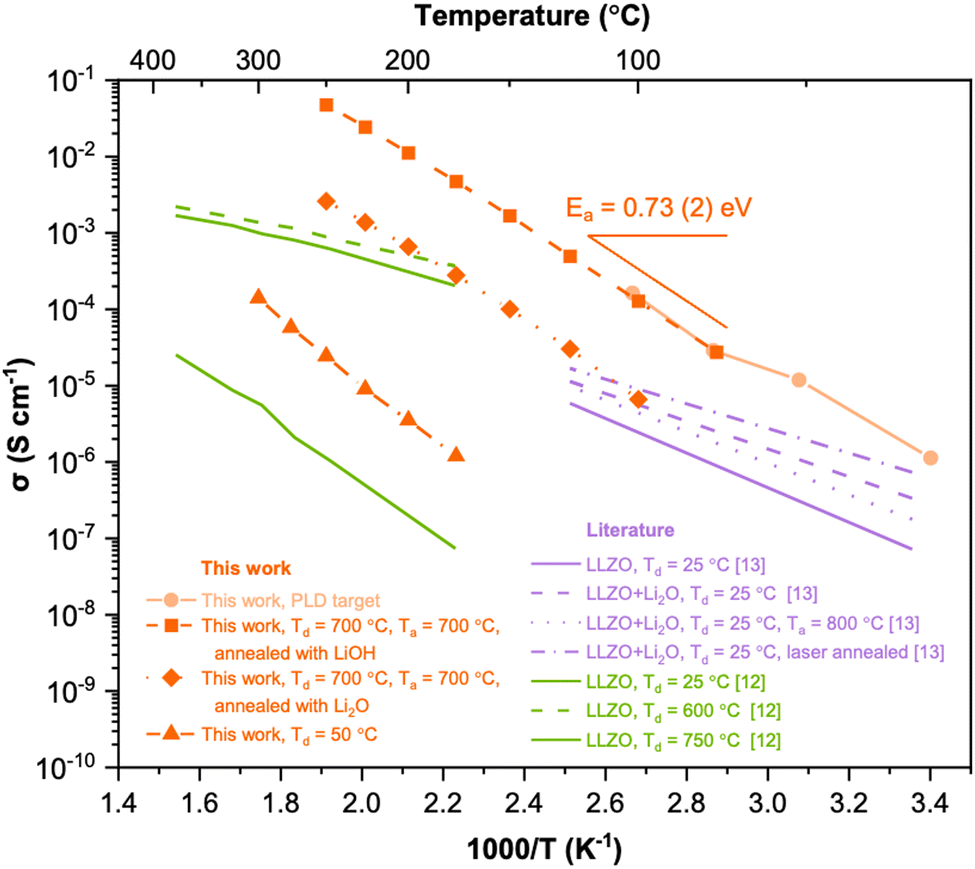 | ||
| Fig. 5 Arrhenius representation of Li+ conductivity of LLZO thin films fabricated in this work compared with literature (undoped LLZO films deposited by PLD).12,13 | ||
In summary, we have effectively offset the lithium loss from thin film deposition using a post-lithiation technique, where lithium is transferred from a neighbouring powder to the film. This method significantly boosts Li+ conductivity. The highest conductivity was achieved in an LLZO film deposited at 700 °C and post-annealed at the same temperature with LiOH. As a result, the conductivity of the sub-micron Li-garnet oxide surpassed previously reported values for PLD-grown undoped LLZO (at 100 °C) by over an order of magnitude and was three to four orders of magnitude higher than without post-lithiation. This underscores the efficacy of the post-lithiation annealing approach in producing LLZO thin films with outstanding ionic conductivity.
3. Experimental
3.1. Thin film process by PLD and post-lithiation of LLZO electrolyte
The Li7La3Zr2O12 target was provided by Toshima Manufacturing Co. (Japan). The phase purity of the target LLZO was confirmed by XRD (Fig. S10, ESI†). Thin films were deposited by PLD (TSST) equipped with a KrF excimer laser (248 nm) at a pulse frequency of 5 Hz. The target-to-substrate distance was kept constant at 40 mm. The deposition was performed at a fixed temperature (700 °C) with background O2 pressure ranging from 10−4 mbar to 10−1 mbar and laser energy densities of 0.5, 1.0, 2.0, and 3.0 J cm−2, or deposited at different substrate temperatures, Td = 50, 300, 500, 600 and 700 °C, with a fixed background O2 pressure of 0.01 mbar and laser energy density of 1 J cm−2. The films had a constant thickness of 100 nm throughout the work and were deposited on 5 × 5 mm2 (100) oriented single-crystal MgO substrates (Crystal GmbH, Germany).For the post-annealing process, the films were annealed using a ceramic plate covered with an alumina crucible with the presence of ca. 50 mg of LiOH (Alfa Aesar, 98%) or Li2O powder (Alfa Aesar, 99.5%) placed next to the sample, to create a lithium-rich atmosphere to maintain/introduce lithium content. The samples were annealed up to 800 °C for 24 h under a continuous flow of oxygen, with a ramping rate of 200 °C h−1 (Fig. 2a). A default annealing temperature of 700 °C was used if not stated otherwise. Pictures of how the powder is dispersed in the alumina crucible can be found in the supplementary information (Fig. S11 and S12, ESI†).
3.2. Structural characterization of the target and thin films
Grazing-incidence X-ray diffractometry (GI-XRD) was carried out to investigate the crystallographic phase of the films using a Rigaku Smartlab X-ray diffractometer with Cu Kα radiation; the incident angle Ω = 1°, measuring from 10–60° 2θ angle. Phase identification and weight fraction calculation were conducted using Rigaku Smartlab Studio II.The surface morphology of the thin films was examined using a scanning electron microscope (Zeiss Merlin) at a voltage of 5 kV with an in-lens secondary electron detector. Films were coated with 10 nm carbon to mitigate charging from the electron beam. The films used for cross-sectional imaging were cleaved using a cleaving plier and then coated with 10 nm carbon with a carbon coater.
3.3. Electrochemical characterization of the target and thin films
To study the ionic transport processes of the films, we employed an in-plane 2-probe geometry with an Au/LLZO/Au cell configuration for the electrochemical impedance spectroscopy. 50 nm thick parallel gold electrodes were deposited onto the films by sputtering (Leica EM ACE600) through custom-made shadow masks with an electrode separation of 200 μm (Fig. 4). The exposure of the films to the atmosphere was kept minimum (films were stored in a vacuum box right after deposition; a continuous flow of Ar gas was fed into the chamber during the impedance measurement). Impedance was measured from 1 Hz to 1 MHz with an amplitude of 50 mV using a Solartron 1255B frequency analyzer coupling with a Solartron 1296 dielectric interface. The samples were measured on a Linkam HFS-600E heating stage at different temperatures, with Au-coated tungsten microprobes. The measurements were carried out under a constant flow of Ar gas. The impedance data were fitted and analysed using ZView 4 software.4. Conclusions
In our study, we successfully fabricated Li-garnet thin films with notable ionic conductivity using a post-lithiation method with LiOH and Li2O, creating a Li-rich environment. Through post-annealing, we established a lithium vapour pressure around the films, enhancing Li+ mobility and boosting lithium diffusion. We have also demonstrated that the decomposition of LLZO to LZO can be reversed using the post-lithiation technique.39 This approach effectively addresses the lithium loss issue common in LLZO films made via physical vapour deposition (PVD). This novel avenue for producing ultra-thin LLZO films has also the potential to lower the ionic conductivity and offer economic benefits by reducing the quantity of materials required. It also paves the way for crafting other lithium oxide films or volatile ions, potentially revolutionizing the production of stoichiometric micro batteries/electronics.Author contributions
J. C., V. E. and N. P. designed the experimental concept, J. C. and S. Y. prepared the films, J. C. and A. P. conducted the transport measurements. J. C. performed the structural characterization. J. C. and A. P. performed data analysis. J. C. and N. P. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. P., D. V. C., V. E. and J. C. acknowledge the Danish Council for Independent Research Technology and Production Sciences for the DFF- Research Project 3 (grant No 00069B). V. E. acknowledges the DFF-Research Project 2 (grant No 1032-00261A). N. P. also acknowledge the support from the ERC Advanced “NEXUS” Grant 101054572. D. V. C. also acknowledges the support of the Novo Nordisk Foundation NERD Programme: New Exploratory Research and Discovery, Superior Grant NNF21OC0068015.References
- A. Chatterjee, C. N. Lobato, H. Zhang, A. Bergne, V. Esposito, S. Yun, A. R. Insinga, D. V. Christensen, C. Imbaquingo, R. Bjørk, H. Ahmed, M. Ahmad, C. Y. Ho, M. Madsen, J. Chen, P. Norby, F. M. Chiabrera, F. Gunkel, Z. Ouyang and N. Pryds, J. Phys. Energy, 2023, 5, 022001 CrossRef.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- S. Randau, D. A. Weber, O. Kötz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffée, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Nat. Energy, 2020, 5, 259–270 CrossRef CAS.
- C. M. Julien and A. Mauger, Coatings, 2019, 9, 386 CrossRef CAS.
- M. Antaya, J. R. Dahn, J. S. Preston, E. Rossen and J. N. Reimers, J. Electrochem. Soc., 1993, 140, 575–578 CrossRef CAS.
- C. Jacob, T. Lynch, A. Chen, J. Jian and H. Wang, J. Power Sources, 2013, 241, 410–414 CrossRef CAS.
- H. Konishi, K. Suzuki, S. Taminato, K. Kim, S. Kim, J. Lim, M. Hirayama and R. Kanno, J. Power Sources, 2014, 246, 365–370 CrossRef CAS.
- Z. G. Lu, H. Cheng, M. F. Lo and C. Y. Chung, Adv. Funct. Mater., 2007, 17, 3885–3896 CrossRef CAS.
- J. B. Bates, G. R. Gruzalski and C. F. Luck, Proc. - IEEE Micro Electro Mech. Syst., 1993, 82–86 Search PubMed.
- M. Morcrette, A. Gutiérrez-Llorente, A. Laurent, J. Perrière, P. Barboux, J. P. Boilot, O. Raymond and T. Brousse, Appl. Phys. A: Mater. Sci. Process., 1998, 67, 425–428 CrossRef CAS.
- K. Hoshina, K. Yoshima, M. Kotobuki and K. Kanamura, Solid State Ionics, 2012, 209, 30–35 CrossRef.
- J. Tan and A. Tiwari, ECS Solid State Lett., 2012, 1, Q57–Q60 CrossRef CAS.
- J. S. Park, L. Cheng, V. Zorba, A. Mehta, J. Cabana, G. Chen, M. M. Doeff, T. J. Richardson, J. H. Park, J.-W. Son and W.-S. Hong, Thin Solid Films, 2015, 576, 55–60 CrossRef CAS.
- M. Hirayama, K. Kim, T. Toujigamori, W. Cho and R. Kanno, Dalton Trans., 2011, 40, 2882–2887 RSC.
- J. Graetz, C. C. Ahn, R. Yazami and B. Fultz, Electrochem. Solid-State Lett., 2003, 6, A194 CrossRef CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- B. J. Neudecker, N. J. Dudney and J. B. Bates, J. Electrochem. Soc., 2000, 147, 517 CrossRef CAS.
- L. Buannic, B. Orayech, J.-M. L. D. Amo, J. Carrasco, N. A. Katcho, F. Aguesse, W. Manalastas, W. Zhang, J. Kilner and A. Llordés, Chem Mater, 2017, 29, 1769–1778 CrossRef CAS.
- T. Thompson, S. Yu, L. Williams, R. D. Schmidt, R. Garcia-Mendez, J. Wolfenstine, J. L. Allen, E. Kioupakis, D. J. Siegel and J. Sakamoto, ACS Energy Lett., 2017, 2, 462–468 CrossRef CAS.
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- S. Yu, R. D. Schmidt, R. Garcia-Mendez, E. Herbert, N. J. Dudney, J. B. Wolfenstine, J. Sakamoto and D. J. Siegel, Chem. Mater., 2016, 28, 197–206 CrossRef CAS.
- M. Balaish, J. C. Gonzalez-Rosillo, K. J. Kim, Y. Zhu, Z. D. Hood and J. L. M. Rupp, Nat. Energy, 2021, 6, 227–239 CrossRef CAS.
- R. Pfenninger, M. Struzik, I. Garbayo, E. Stilp and J. L. M. Rupp, Nat. Energy, 2019, 4, 475–483 CrossRef CAS.
- J. Awaka, A. Takashima, K. Kataoka, N. Kijima, Y. Idemoto and J. Akimoto, Chem. Lett., 2011, 40, 60–62 CrossRef CAS.
- R. Wagner, G. J. Redhammer, D. Rettenwander, A. Senyshyn, W. Schmidt, M. Wilkening and G. Amthauer, Chem. Mater., 2016, 28, 1861–1871 CrossRef CAS PubMed.
- D. Rettenwander, G. Redhammer, F. Preishuber-Pflügl, L. Cheng, L. Miara, R. Wagner, A. Welzl, E. Suard, M. M. Doeff, M. Wilkening, J. Fleig and G. Amthauer, Chem. Mater., 2016, 28, 2384–2392 CrossRef CAS PubMed.
- W. G. Zeier, S. Zhou, B. Lopez-Bermudez, K. Page and B. C. Melot, ACS Appl. Mater. Interfaces, 2014, 6, 10900–10907 CrossRef CAS PubMed.
- Y. Li, J.-T. Han, C.-A. Wang, H. Xie and J. B. Goodenough, J. Mater. Chem., 2012, 22, 15357–15361 RSC.
- M. Rawlence, I. Garbayo, S. Buecheler and J. L. M. Rupp, Nanoscale, 2016, 8, 14746–14753 RSC.
- S. Lobe, A. Bauer, S. Uhlenbruck and D. Fattakhova-Rohlfing, Adv. Sci., 2021, 8, 2002044 CrossRef CAS PubMed.
- J. Sastre, T.-Y. Lin, A. N. Filippin, A. Priebe, E. Avancini, J. Michler, A. N. Tiwari, Y. E. Romanyuk and S. Buecheler, ACS Appl. Energy Mater., 2019, 2, 8511–8524 CrossRef CAS.
- J. Schou, Appl. Surf. Sci., 2009, 255, 5191–5198 CrossRef CAS.
- A. Ojeda-G-P, C. W. Schneider, M. Döbeli, T. Lippert and A. Wokaun, Appl. Surf. Sci., 2016, 389, 126–134 CrossRef CAS.
- M. Rosen, R. Ye, M. Mann, S. Lobe, M. Finsterbusch, O. Guillon and D. Fattakhova-Rohlfing, J. Mater. Chem. A, 2021, 9, 4831–4840 RSC.
- G. Bimashofer, S. Smetaczek, E. Gilardi, C. W. Schneider, A. Limbeck, T. Lippert and J. Stahn, Appl. Phys. Mater. Sci. Process., 2021, 127, 473 CrossRef CAS PubMed.
- M. A. Urusova and V. M. Valyashko, Russ. J. Phys. Chem. B, 2015, 9, 1148–1152 CrossRef CAS.
- J. M. Weller and C. K. Chan, J. Mater. Chem. A, 2020, 8, 17405–17410 RSC.
- R. A. Huggins, Ionics, 2002, 8, 300–313 CrossRef CAS.
- S. Vema, F. N. Sayed, S. Nagendran, B. Karagoz, C. Sternemann, M. Paulus, G. Held and C. P. Grey, ACS Energy Lett., 2023, 8, 3476–3484 CrossRef CAS PubMed.
- W. Xiang, R. Ma, X. Liu, X. Kong, S. Shen, L. Wang, Z. Jin, Z. Zhan, C. Chen and C. Wang, Nano Energy, 2023, 116, 108816 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00894k |
| This journal is © The Royal Society of Chemistry 2023 |