
Fragment screening libraries for the identification of protein hot spots and their minimal binding pharmacophores†
Rebecca L.
Whitehouse‡
 a,
Wesam S.
Alwan§
a,
Olga V.
Ilyichova
abc,
Ashley J.
Taylor¶
a,
Indu R.
Chandrashekaran
acd,
Biswaranjan
Mohanty||
a,
Bradley C.
Doak
*acd and
Martin J.
Scanlon
*acd
a,
Wesam S.
Alwan§
a,
Olga V.
Ilyichova
abc,
Ashley J.
Taylor¶
a,
Indu R.
Chandrashekaran
acd,
Biswaranjan
Mohanty||
a,
Bradley C.
Doak
*acd and
Martin J.
Scanlon
*acd
aMedicinal Chemistry, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia. E-mail: martin.scanlon@monash.edu
bAustralian Synchrotron, ANSTO, Clayton, VIC 3168, Australia
cARC Training Centre for Fragment Based Design, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia
dMonash Fragment Platform, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia
First published on 29th November 2022
Abstract
Fragment-based drug design relies heavily on structural information for the elaboration and optimisation of hits. The ability to identify neighbouring binding hot spots, energetically favourable interactions and conserved binding motifs in protein structures through X-ray crystallography can inform the evolution of fragments into lead-like compounds through structure-based design. The composition of fragment libraries can be designed and curated to fit this purpose and herein, we describe and compare screening libraries containing compounds comprising between 2 and 18 heavy atoms. We evaluate the properties of the compounds in these libraries and assess their ability to probe protein surfaces for binding hot spots.
Introduction
Fragment-based drug design (FBDD) has emerged as a popular method of identifying hit compounds as starting points for medicinal chemistry campaigns. Due to their small size, fragments sample chemical space more effectively than larger compounds,1,2 which means that relatively small libraries of compounds can be screened to find hits.A simple model developed by Hann and co-workers3 describes the relationship between the complexity of a molecule and its ability to make complementary interactions with a protein surface, suggesting that simple compounds are more likely to obtain a singular, unique and therefore “useful” binding event. This represents a central tenet of fragment screening, in that fragments make protein interactions of high quality,4 albeit often with an intrinsically low affinity. Consequentially, highly sensitive techniques are required for screening to identify binding and synthetic strategies are essential for optimising fragments into lead-like compounds. X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy are powerful techniques for fragment screening and binding characterisation due to their ability to detect ligands which bind with equilibrium dissociation constants (KD) in the range of millimolar or above. In addition to their sensitivity, they can provide site-specific binding information which is essential to support structure-based drug design (SBDD).
Alanine scanning, where single residues within a protein binding surface are systematically replaced by alanine, has previously shown that there are regions on proteins which disproportionately contribute to binding energy.5 Characterising these regions, referred to as “hot spots”, can identify which protein interactions are the most energetically favourable for binding. Protein hot spots can be characterised through alanine scanning and site mutagenesis,5 computational modelling,6–8 or screening small probes by NMR9 and X-ray crystallography.10–13
Screening high concentrations of low molecular weight probes has been utilised to combine FBDD and protein hot spot characterisation. Organic solvents and libraries of compounds that are even smaller than those typically found in fragment libraries, such as the MiniFrag11 and FragLite12 libraries, have been employed to identify protein binding hot spots and to assess the overall druggability of protein targets. Analysis of data obtained using these approaches allows the identification of neighbouring or overlapping binding pockets, which can be used to drive the design and synthesis of higher affinity compounds.8,12 One of the challenges with screening very small compounds is that they usually bind with low affinity, which means that high concentrations of ligand are required to achieve sufficient occupancy to observe a binding event. In addition, the Hann complexity model suggests that there is an “optimal” size and complexity to provide useful binding information. In the current work we have screened three libraries containing compounds of different size and complexity against a bacterial enzyme involved in oxidative protein folding – DsbA.
Escherichia coli DsbA (EcDsbA) is a bacterial oxidoreductase responsible for the introduction of disulfide bonds into many bacterial virulence factors.14 Inhibition of this pathway presents a novel mechanism for the treatment of drug resistant microbial infections,15,16 however, it requires disruption of a protein–protein interaction. EcDsbA is thought to have over 300 substrates which bind within a shallow and hydrophobic groove.17,18 Although small molecules have been developed to inhibit EcDsbA via this hydrophobic groove (Fig. 1A), they have thus far failed to obtain high affinities and potency.19–22
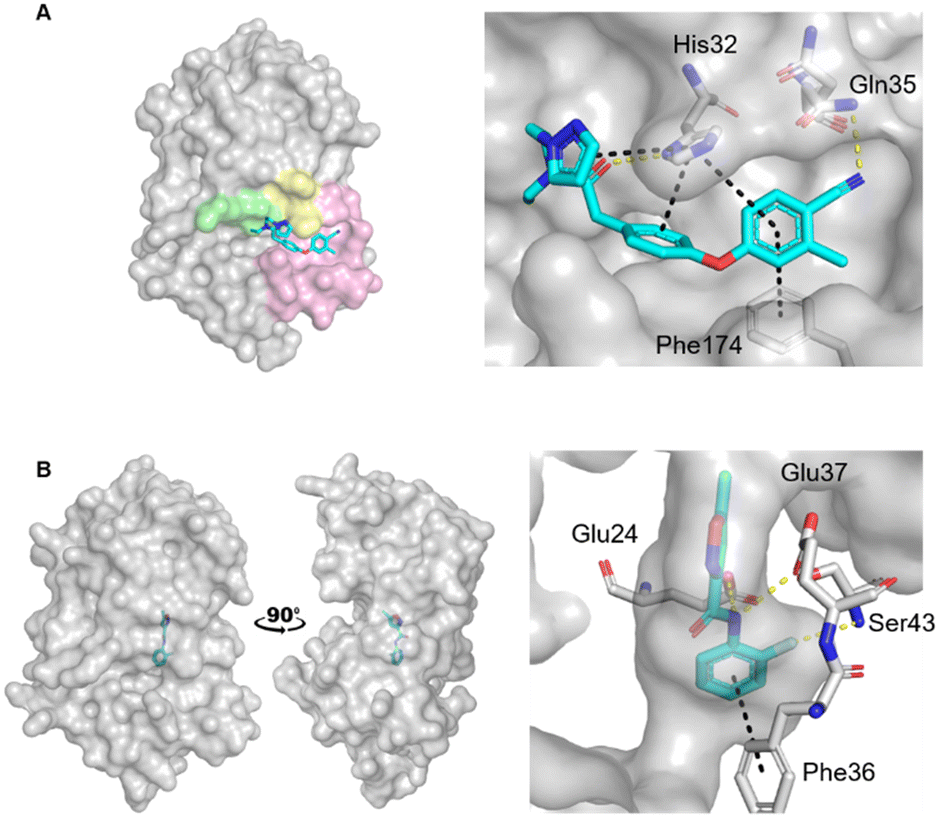 | ||
| Fig. 1 Crystal structures of fragments bound to previously identified EcDsbA hot spots. Fragment hits bound to A) the hydrophobic groove binding site (PDB ID: 6PDH)20 with zoom of interactions of a diphenyl ether fragment 1 and B) the internal cryptic pocket (PDB ID: 8DN0)23 with zoom of interactions of isoxazole fragment 2. EcDsbA is shown as a grey surface, fragments as cyan sticks, hydrogen bonds as yellow dashes and π-stacking interactions as black dashes. Known features of EcDsbA annotated in A) with the catalytic 30CPHC33 motif shown in yellow, the hydrophobic groove in pink and the substrate peptide binding cis-Pro loop in green. | ||
Recently we screened an in-house library of 1148 fragments by 1D 1H saturation transfer difference (STD) NMR to identify compounds that bind to oxidised EcDsbA and these fragments were validated by 15N–1H heteronuclear single quantum coherence (HSQC) NMR.23 In this way, 26 fragments were identified as validated hits. During this screen, two distinct chemical shift perturbation (CSP) profiles were observed by HSQC. The first was consistent with ligands binding to a hydrophobic groove that is adjacent to the active site of EcDsbA, whereas the second profile was different, suggesting a distinct and previously unreported binding site. A crystal structure of oxidised EcDsbA in complex with isoxazole 2 (PDB ID: 8DN0, Fig. 1B) revealed that the fragment was bound within an internal “cryptic” pocket.23 This cryptic pocket is entirely enclosed within the protein structure, and it is located behind the helix containing the active site cysteine residues of EcDsbA. The pocket is only created upon ligand binding, and this suggested that access to the internal cavity required the protein to undergo a conformational change.
The fragment screen identified that EcDsbA has two distinct fragment binding sites and each presents significant challenges for further elaboration. The two sites have different properties and requirements for binding where one is flat, broad and hydrophobic and the other is narrow and polar. Furthermore, fragments that bind at the hydrophobic groove are more easily identified whereas very few hits were found in the internal cryptic pocket. Consequently, we used oxidised EcDsbA as a model to test the ability of different compound libraries to identify binding hot spots. We conducted screens against oxidised EcDsbA using a computational approach, as well as X-ray crystallography using both an organic solvent library and a “MicroFrag” library with compound properties lying between fragments and solvents. We sought to use these screens to explore whether the libraries could identify protein binding hot spots, recapitulate known interactions with EcDsbA and provide useful information for implementation in SBDD.
Results and discussion
Computational and crystallographic solvent screening
An FTMap analysis6,7 was conducted against the structure of oxidised EcDsbA (PDB ID: 1FVK24) with structural waters removed. This identified 12 probe clusters on the protein surface, characterised across seven potential binding hot spots (Fig. 2A and S1†).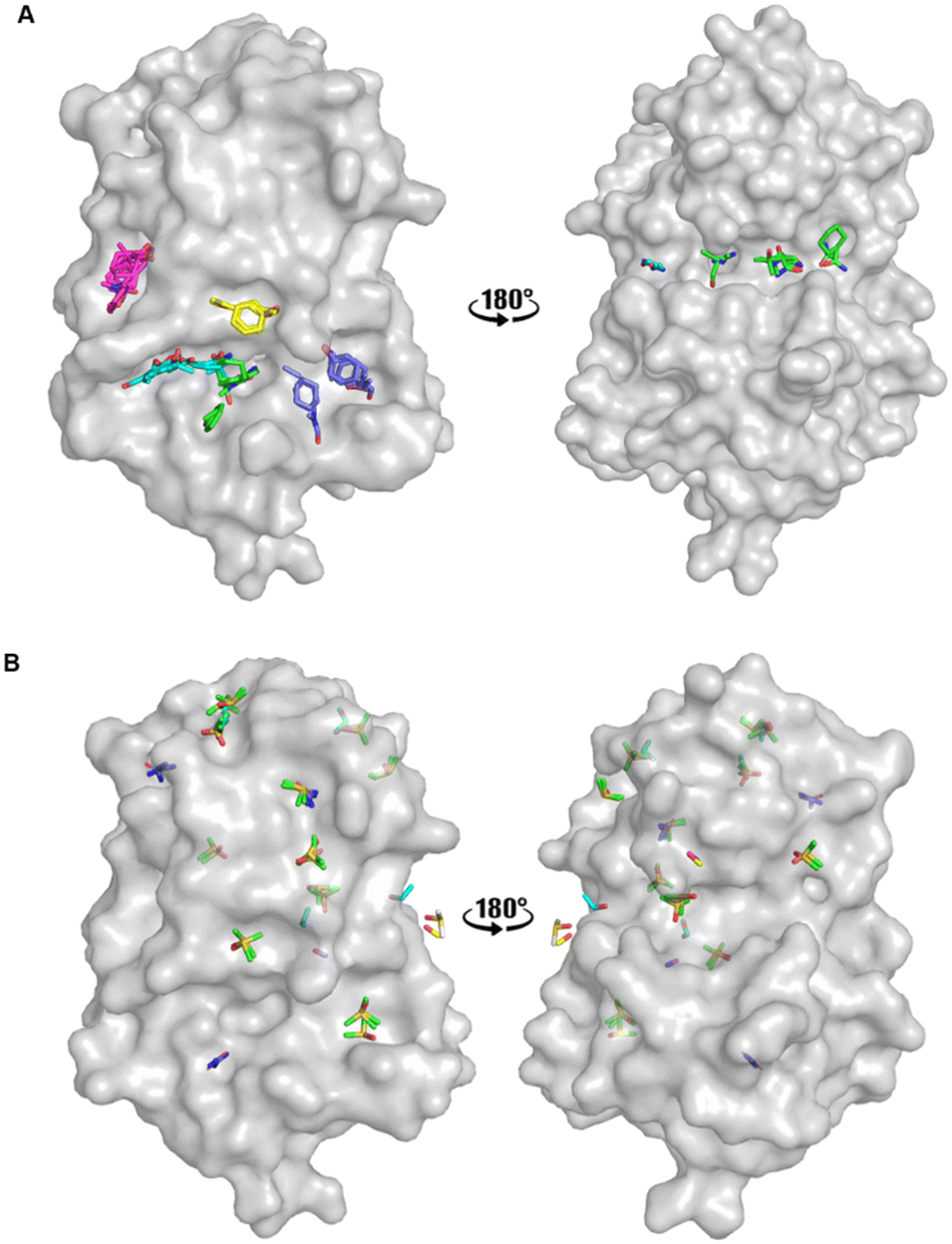 | ||
| Fig. 2 Organic solvent screens against EcDsbA. A) Clusters identified by FTMap.6,7EcDsbA is shown as a grey surface (PDB ID: 1FVK24) with each binding hot spot shown as a different colour. B) Clusters identified by a crystallographic organic solvent screen. All poses found at crystal contact interfaces, and those which did not make interactions with the protein or did not show neighbouring or overlapping solvents have been removed for clarity. EcDsbA is shown as a grey surface, solvents are shown as coloured sticks where DMSO is shown in green, urea in purple, ethanol in blue, acetonitrile in pink and methanol in yellow. | ||
FTMap predicted that the probes were able to bind within the hydrophobic groove, adjacent to the active site and along the cis-Pro loop where peptides interact with EcDsbA.25–27 One cluster was shown to occupy the area where a number of structural waters had been removed from the structure. The structural waters form more hydrogen bonds than the probes at this position, casting doubt on their ability to displace these water molecules.
Fragments that have been reported to bind within the hydrophobic groove all form at least one π-stacking interaction with His32 of the active site.19–22 Notably, this interaction was not formed by any of the FTMap probes. Furthermore, some of the clusters within the hydrophobic groove were amongst the lowest ranked by FTMap (Fig. S1†). The remaining hot spots were found on the opposing face of the protein, well away from the main fragment binding site on EcDsbA.
A high-concentration solvent screen was conducted by X-ray crystallography using common organic solvents (Fig. 2B). Pre-grown crystals of oxidised EcDsbA were dehydrated and soaked in 50 and 80% (v/v) of a single organic solvent for 0.5–5 minutes. Data were collected on the MX2 beamline at the Australian Synchrotron and processed with the automated data-processing pipeline implemented at the beamline.28–30 Briefly, each data set was indexed, integrated, and scaled with xdsme and Aimless.31–33 The output statistical description of the data was manually inspected, and data sets with poor statistics were rejected at this stage (see Experimental section). In all, nine high-resolution crystal structures of six solvents were obtained.
Binding poses which made interactions with more than one protomer in the asymmetric unit or which made no interactions with the protein were removed, and the remaining solvent poses were used in the hot spot analysis. The remaining crystal structures were aligned. Hot spots were considered as binding pockets where two or more solvent probes had overlapping poses or formed conserved interactions with the protein. Using these criteria, we identified seven protein binding hot spots occupied by more than one solvent (Fig. 2B). Another five clusters were found to have multiple copies of the same solvent when multiple crystal structures or the two protomers within the asymmetric unit were aligned. These additional five clusters were considered as weak hot spots.
The two known fragment binding sites were identified; however, the hydrophobic groove was only identified as a weak protein hot spot. DMSO was the only ligand to be found in this pocket. No aromatic solvents yielded crystal structures with density for the ligands and so the π-stacking interactions observed for fragments with His32, Phe36 and Phe174 were not identified. Furthermore, previously characterised polar interactions with His32, Pro163, Gln164 and Thr168 were not identified in this screen.
No strong hot spots were found adjacent to either fragment binding site, however, two weak binding pockets were detected in proximity to the hydrophobic groove. DMSO bound adjacent to the active site and acted as a hydrogen bond acceptor for an interaction with the backbone amide of Val150, and this interaction has previously been identified in EcDsbA-fragment structures. Urea bound in a more polar region adjacent to the hydrophobic groove where it participated in hydrogen bonds with Thr10, Gln160 and Leu161. The pseudo-symmetry of these molecules created difficulty in unambiguously assigning the ligand orientation and thereby decreasing the confidence of their role in the hydrogen bonding interaction.
MicroFrag library design
The MicroFrag library was designed to be diverse using the criteria described below, and to be suitable for use in high concentration X-ray crystallography assays. Furthermore, it was intended to identify all potential polar and non-polar protein–ligand interactions within a binding hot spot. For these reasons, a MicroFrag had to meet the following criteria: five to eight heavy atoms, one 5- or 6-membered ring, a minimum of one heteroatom, and a Clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P between −2 and 2. The library was also biased to contain motifs which were more commonly found in the PDB database34 as well as substructures of compounds within the DrugBank database.35
P between −2 and 2. The library was also biased to contain motifs which were more commonly found in the PDB database34 as well as substructures of compounds within the DrugBank database.35
Tanimoto coefficients are frequently used to compare and describe the similarity of small molecules. These similarity values can differ greatly dependent on the molecular fingerprint used, especially for compounds with a low heavy atom count (HAC). Therefore, the library used a combination of circular and extended fingerprints and Molecular ACCess System (MACCS) keys to calculate Tanimoto coefficients.36,37 Diversity in the MicroFrag library was also measured by calculating the coverage of 2D 2-point pharmacophore fingerprints (Fig. 3). For this determination, a pharmacophore was described as either a positive or negative charge, hydrogen bond donor or acceptor, 5- or 6-membered aromatic ring and their 2D connectivity was considered as the distance in bonds between the pharmacophore pair.
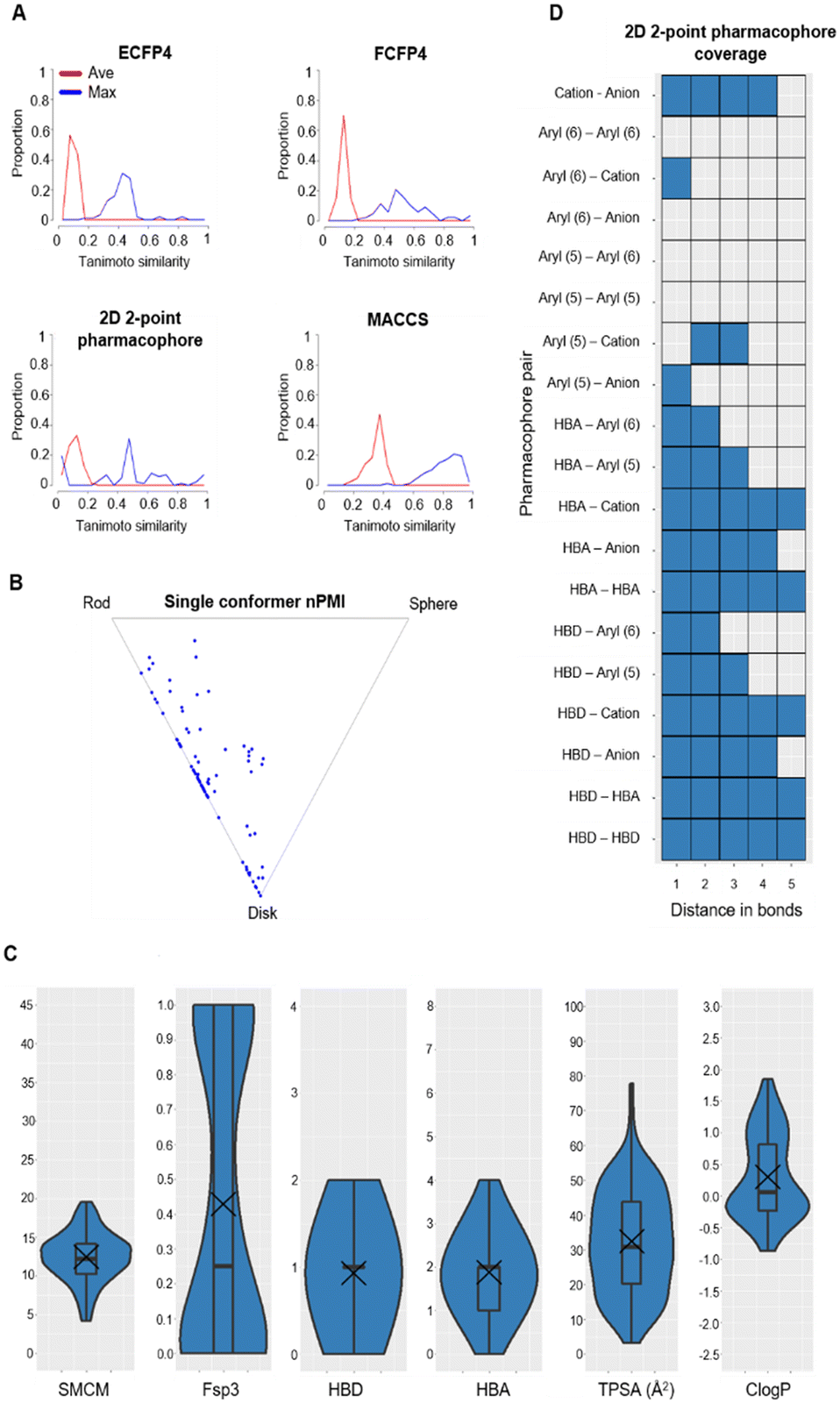 | ||
| Fig. 3 Diversity and property analysis of the MicroFrag library. A) Tanimoto similarities of the library based on the calculated fingerprint profiles. B) Principal moments of inertia as a 2D triangular plot with each dot representing a single compound of the library. C) Physicochemical properties of the MicroFrag library visualised as violin plots. Abbreviations used: number of principal moments of inertia (nPMI),38 synthetic and molecular complexity model (SMCM),39 fraction of sp3 hybridised carbons (Fsp3), hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), topological polar surface area (TPSA) and calculated octanol–water partition coefficient (ClogP). D) Coverage of 2D 2-point pharmacophores by all compounds in the library as a function of the number of bonds separating the pharmacophore pairs. Pharmacophore pairs are described using the following abbreviations: 6-membered aromatic ring (aryl (6)), 5-membered aromatic ring (aryl (5)), hydrogen bond acceptor (HBA), hydrogen bond donor (HBD). | ||
With these considerations an iterative selection protocol which compared physicochemical properties, Tanimoto coefficients and pharmacophore coverage was used to select 91 compounds for the MicroFrag library.
MicroFrag screening
The MicroFrag library was screened against oxidised EcDsbA at a ligand concentration of 1 M by X-ray crystallography. The datasets were collected, filtered, and analysed as described in the organic solvent screen. Where the datasets did not meet the required collection statistics the MicroFrag was soaked at half concentration and collected again. This resulted in crystal structures with electron density for 47 MicroFrags with resolutions between 1.47–1.88 Å. These compounds provided 180 binding poses and identified 12 hot spots occupied by ≥2 different MicroFrags (Fig. 4A).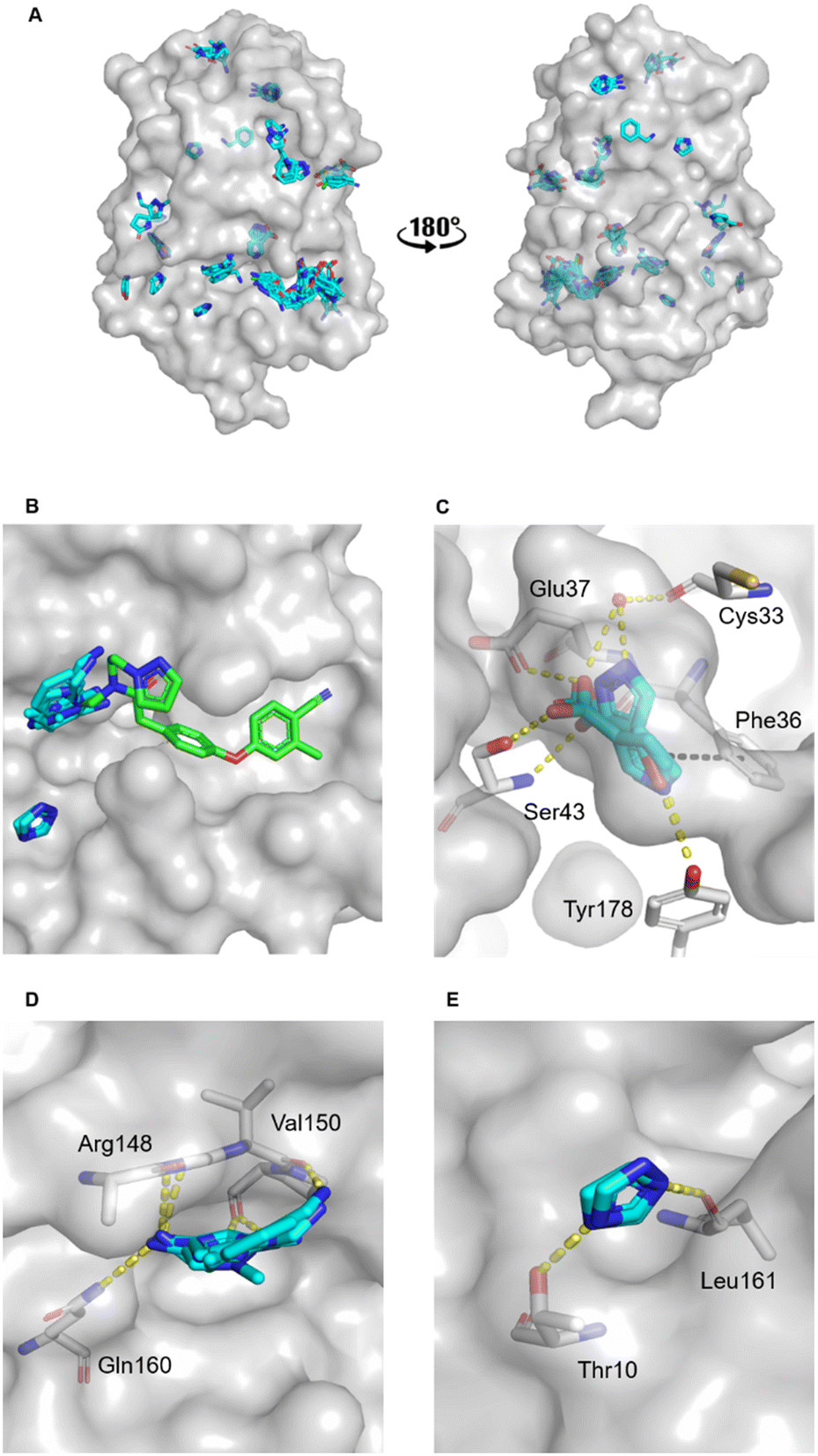 | ||
| Fig. 4 MicroFrag screen against oxidised EcDsbA. A) Clusters identified by a crystallographic MicroFrag screen. All poses found at crystal contact interfaces, as well as those which did not make interactions with the protein or did not show neighbouring or overlapping MicroFrags have been removed for clarity. EcDsbA is shown as a grey surface, MicroFrags are shown as cyan sticks. B) Overlay of a known fragment binder (PDB ID: 6PDH20) and neighbouring MicroFrag clusters. C) Interactions made by MicroFrags within the internal cryptic pocket D and E) interactions made by MicroFrag clusters adjacent to the known hydrophobic groove hot spot. EcDsbA is shown as a grey surface and residues as grey sticks, MicroFrags are shown as cyan sticks, water as red spheres, conserved hydrogen bond interactions are shown as yellow dashes, and π-stacking interactions as black dashes. The disulfide bond between Cys30 and Cys33 is not shown. | ||
The hot spots included the hydrophobic groove, the internal cryptic pocket, and the known peptide binding sites that have previously been reported for structures of EcDsbA in complex with known substrates. In this dataset, the hydrophobic groove had the largest cluster of MicroFrag poses. All interactions in this groove that have previously been identified in fragment co-structures were also observed for at least two MicroFrags. The most common interactions observed were between the MicroFrag ligands and His32, Gln35, Phe36, Gln164, Thr168 and Phe174, which is consistent with what has previously been observed in the reported series of fragments hits.
Furthermore, two MicroFrags were found to occupy the internal cryptic pocket. These structures showed the MicroFrag binding poses overlapped with the binding mode of isoxazole fragment 2, identified in the primary fragment screen. As in the original crystal structure, the MicroFrags identified hydrogen bonds with Glu37 and Ser43 and an edge-face π-stacking interaction with Phe36 (Fig. 4C). However, the hydrogen bond formed by isoxazole 2 to Glu24 was not identified in the MicroFrag screen. The isoxazole ring of the initial fragment hit extends higher into the cryptic pocket than the MicroFrags and a water molecule is present in the corresponding location in the MicroFrag structures. The MicroFrags identified a conserved water mediated hydrogen bond to the backbone of the catalytic Cys33 as well as the potential for a direct interaction with the sidechain of Tyr178 at the bottom of the pocket.
Two clusters of MicroFrags were found adjacent to the hydrophobic groove. In all, seven hydrogen bonds were conserved at these sites across the MicroFrag structures (Fig. 4D and E). These clusters highlighted a preference for 5-membered aromatic rings with two or more aromatic nitrogens. The most favourable pharmacophore pairs allowed for the MicroFrag to participate in multiple hydrogen bonds at once, often with a primary amine substituent which participated in two interactions itself.
Clusters such as these – at previously unidentified pockets adjacent to well characterised ligand binding sites suggest strategies for fragment elaboration. These motifs could potentially be incorporated into future compound designs. The clusters identified in Fig. 4D and E appear to be the most promising in this regard due to the position of these hot spots and the identification of conserved interactions, pharmacophores, and scaffolds.
Comparison of screening libraries for hot spot identification
The Hann model provides a rationale for the observation that libraries of smaller and simpler compounds can give higher hit rates. This is exemplified by the current data, where a fragment library which contains 1148 compounds identified 26 validated hits, whilst the 91-member MicroFrag library identified 47 (Fig. 5). To further explore relationships between hit rates and compound properties we analysed the features of the ligands within the libraries.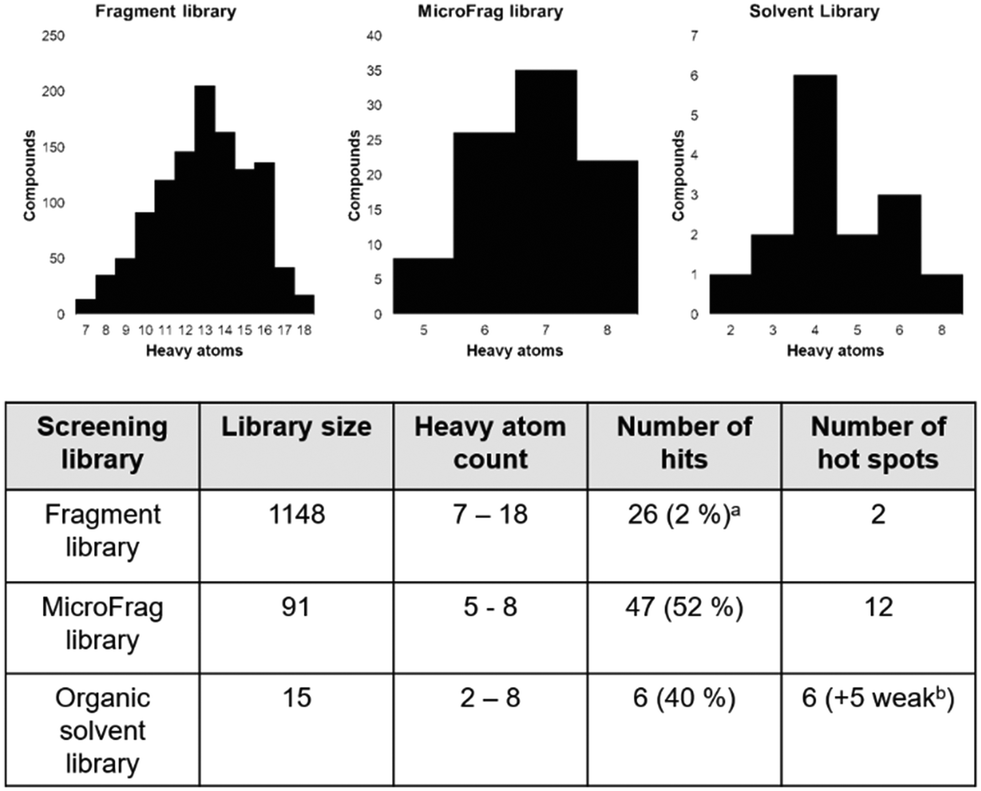 | ||
| Fig. 5 Screening library sizes (upper panel), their relative hit rates and the number of hot spots identified (lower panel). aHits identified by 1D 1H STD NMR and validated by 15N–1H HSQC NMR bhot spots identified by multiple copies of one solvent are considered weak hot spots. | ||
Concatenation of the profiles of the screening libraries and their hits highlighted the preferred range for many of the compound properties (Fig. 6). Ligands which were between five and eight heavy atoms, had synthetic and molecular complexities (SMCM) between 10 and 15, ≤3 2D 2-point pharmacophores, ≤1 hydrogen bond acceptor, topological polar surface areas (TPSA) between 20 and 40 Å2 and ClogP values between −1 and 0.5 were significantly enriched among the hit compounds. This comparison indicates the profile of the MicroFrag library is best suited for the identification of protein–ligand hot spots, as hinted at in other screens of low molecular probes.11,12
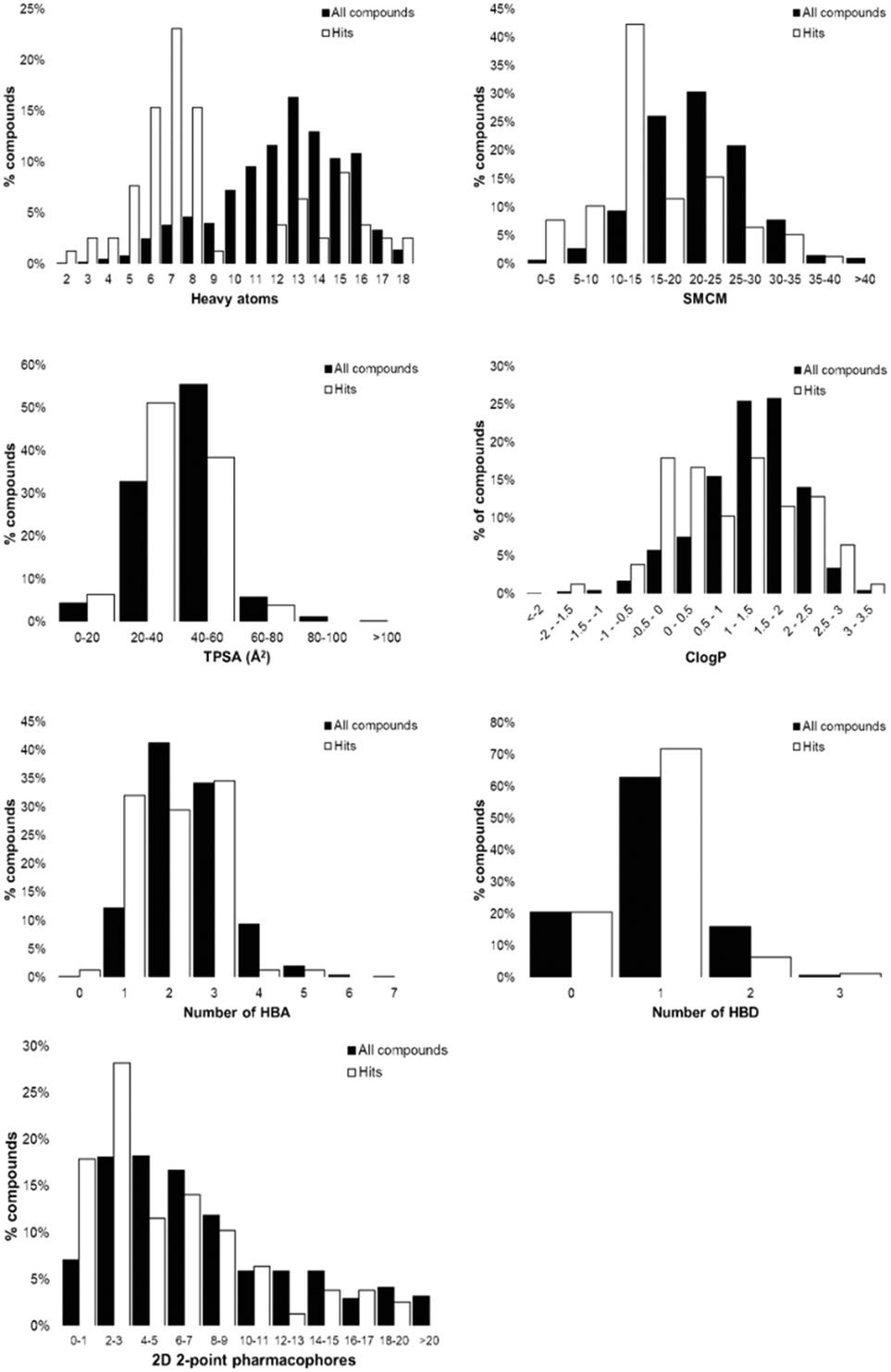 | ||
| Fig. 6 Comparison of library and hit compound property profiles. Bar graphs are calculated as a percentage of all library members across all three screening libraries (black) and as the percentage of all hit compounds across all three screening libraries (white). Profile abbreviations used; SMCM – synthetic and molecular complexity model, TPSA – popological polar surface area, ClogP – calculated logP, HBA – hydrogen bond acceptors, HBD – hydrogen bond donors. | ||
While the preference for MicroFrag compounds could be in part skewed by the higher screening concentrations, this did not hold true for the organic solvents. Increasing ligand concentration is used to increase ligand occupancy, and higher occupancy can result in clearer electron density for ligands in crystal structures. It would, therefore, be reasonable to assume that the concentrations used in the organic solvent screen would result in the best structures for a given ligand. Benzyl alcohol was tested under the conditions for both the organic solvent screen and the MicroFrag screen. We were able to obtain a crystal structure with five poses in the MicroFrag screen, however the solvent screen failed to yield a structure with good diffraction and ligand density. This is likely due to the tolerance of EcDsbA to the extreme conditions of the solvent screen, highlighting that the lower concentrations used in the MicroFrag screen offer some advantages over the organic solvent screen.
15N–1H HSQC dose response titrations were collected for some members of each screening library against oxidised EcDsbA (Fig. S2†). We were able to obtain titration data for solvents which indicated they bound weakly to hot spots, even in cases where the solvent did not give electron density in the crystal structure. The binding affinity observed for the ligands differed between hot spots; therefore we attempted to compare the range of KD values observed for ligands of each library across multiple hot spots. Affinities were estimated based on CSP observed in the 15N–1H HSQC titrations (Fig. S3†). Due to uncertainty in the mode of binding of the solvents and MicroFrags, the affinities were loosely clustered based on the saturating concentration observed in the titration as >100 mM, 10–100 mM and <10 mM for solvents, MicroFrags and fragments respectively. Together, the NMR and crystallography datasets suggest that organic solvents do not bind with a high enough affinity to achieve saturation, which makes it more challenging to characterise protein binding hot spots.
Access of isoxazole 2 to the cryptic pocket requires substantial conformational changes adjacent to the internal binding site. Although MicroFrags were smaller than the originally identified isoxazole fragments, they were still able to access the pocket by inducing the conformation change that allows ligand entry. The MicroFrag library recapitulated previous fragment binding data and showed a clear bias for binding within the hydrophobic groove. Moreover, the specific protein–fragment interactions observed in co-structures with EcDsbA were also found in the MicroFrag data. Although the organic solvents occupied the hydrophobic groove of EcDsbA, and adjacent sites, no common interactions with the protein were conserved between different solvents, and interactions known to mediate fragment binding were not observed. Furthermore, the solvent screen identified hot spots on the opposite face of the protein to the catalytic cysteines, which were not observed as validated ligand binding sites in any other screen. In co-structures of EcDsbA bound to fragments, these sites are occupied by water molecules or metal ions, and there is currently no data to suggest that they would be able to bind to larger ligands. Together, these observations suggest that the MicroFrag screen was more successful in identifying and characterising protein binding hot spots for use in drug development.
The organic solvent screen was able to identify new protein hot spots and interactions, however, the size of the probes and the ambiguous binding poses they provide make it unclear how these data could be incorporated into fragment elaboration strategies with confidence. Conversely, it was reassuring that the MicroFrag screen identified the known binding pockets and interactions with EcDsbA. Since MicroFrags bind very weakly to the protein, it is unlikely that they would provide useful starting points for medicinal chemistry. However, it is more evident how these minimal pharmacophores could be used to inform structure-based drug design. Together, these screening data represent an experimental reflection of the Hann complexity model which suggest that smaller, simpler compounds are more likely to bind to a protein target. However, there is a point at which compounds can become too small and simple for useful experimental data to be acquired.
Conclusions
Protein binding hot spots can be identified by screening an array of fragment or fragment-like compounds by X-ray crystallography. Furthermore, by designing libraries which differ in terms of compound complexity, physicochemical properties and molecular size, different kinds of information can be obtained. In the current study, a traditional fragment library was able identify the major protein binding hot spots. The organic solvent screen was able to identify hot spots that were not observed in the fragment screen, however due to their simplicity, size and affinity did not provide information with a clear application to ligand development. Conversely, we have shown that the properties of the MicroFrag screening library make it uniquely placed to identify secondary binding sites, their preferred interactions and minimal pharmacophores. This library identified the highest number of hits and hot spots alike with these data suggesting that the MicroFrag library may represent the lower end of ligands which can provide useful binding information. As in Hann's complexity model, these data reaffirmed that, to a certain point, screening small, low complexity fragments results in higher protein complementarity and highlighted the ability of these ligands to obtain meaningful binding events for structure-based design.Experimental procedures
Protein production and crystallisation
Unlabelled and uniformly 15N-labelled EcDsbA was expressed by autoinduction and purified as described previously.20,21 Previously published crystallisation conditions were used to obtain apo-EcDsbA crystals. Briefly, 1 μL of 30 mg mL−1EcDsbA was mixed with an equal volume of crystallisation buffer (11–13% PEG8000, 5–7.5% glycerol, 1 mM CuCl2, 100 mM sodium cacodylate pH 6.1) and equilibrated against 500 μL of reservoir buffer at 20 °C using hanging drop vapour diffusion.Organic solvent soaking experiments
Pre-grown EcDsbA crystals were dehydrated and transferred into 2 μL drops of 50 or 80% organic solvent diluted in 13% PEG8000, 6.5% glycerol, 1 mM CuCl2, 100 mM sodium cacodylate pH 6.1 for 0.5–5 min. Crystals were mounted on cryo loops and flash-cooled in liquid nitrogen. Datasets were collected at the MX2 beamline at the Australian Synchrotron.29 For all datasets, the resolution cut-off used was based on the following criteria in the highest resolution range: CC1/2 was at least 0.6, 〈I/σ(I)〉 was greater than 1.0, and completeness was greater than 90%.40 In cases where data collection statistics did not fulfil these quality requirements or crystals diffracted to lower than 2.5 Å resolution, the structure was not refined.MicroFrag soaking experiments
Pre-grown EcDsbA crystals were transferred into 2 μL drops of 13% PEG8000, 6.5% glycerol, 1 mM CuCl2, 100 mM sodium cacodylate pH 6.1, 9% MeOH and the MicroFrag of interest at a concentration of 1 M and incubated for 0.5–5 minutes. Crystals were mounted on cryo loops and flash-cooled in liquid nitrogen. If the addition of compound caused cracking or dissolution of the crystal, the final MicroFrag concentration was adjusted to 0.5 M, adjusted to the pH range 4–9 and the soaking experiment was repeated. Datasets were collected at the MX1 and MX2 beamlines at the Australian Synchrotron.29,30 For all datasets, the resolution cut-off used was based on the following criteria in the highest resolution range: CC1/2 was at least 0.6, 〈I/σ(I)〉 was greater than 1.0, and completeness was greater than 90%.40 In cases where data collection statistics did not fulfil these quality requirements or crystals diffracted to lower than 2.5 Å resolution, the crystallographic experiment was repeated at 0.5 M and adjusted to the pH range 4–9.Refinement
Crystal structures were refined as described previously.20 Briefly, the data was processed using the automated data processing pipeline implemented at the MX1 and MX2 beamlines at the Australian Synchrotron,29,30 where data were indexed, integrated, and scaled with xdsme and AIMLESS.31–33 Phasing was performed by MR with Phaser41 using chain A of EcDsbA (PDB: 1FVK24) as a search model, and ligand restraints were generated in phenix.eLBOW42 and using the Grade web server.43 Iterative cycles of manual model building and refinement were performed using Coot44 and phenix.refine.45NMR titrations
A reference 15N–1H HSQC spectrum of uniformly labelled 15N oxidised EcDsbA (100 μM protein, 10% D2O) was acquired for each screening condition. Titrations were collected as 5-point, 2-fold serial dilutions. Fragment hit titrations were conducted using 50 mM HEPES, 50 mM NaCl, 2 mM EDTA, pH 6.8, 10% D2O, 2% d6-DMSO, MicroFrag titrations using 100 mM phosphate, 50 mM NaCl, pH 6.8, 10% D2O, and organic solvent titrations using 50 mM phosphate, 25 mM NaCl, pH 6.8, 10% D2O. Weighted chemical shift perturbations (CSP) and equilibrium dissociation constants (KD) were determined as previously reported.21Library design
All analysis and design were conducted using KNIME Analytic Platform version 4.0.2 (ref. 46) with nodes developed by KNIME Analytic Platform, RDKit, CDK toolkits, R, Vernalis and Indigo (EPAM Systems).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would to acknowledge Gaurav Sharma and Martin L. Williams for production of the protein used in these studies. This research was undertaken in part using the facilities of the Monash Fragment Platform. X-ray crystallography was undertaken on the MX1 and MX2 beamline at the Australian Synchrotron, part of ANSTO. This research was under-taken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. We acknowledge the CSIRO Collaborative Crystallisation Centre (https://www.csiro.au/C3). We thank the Australian Research Council (ARC grant ID IC180100021 and DP200100796) for funding.References
- L. Ruddigkeit, R. van Deursen, L. C. Blum and J.-L. Reymond, J. Chem. Inf. Model., 2012, 52, 2864–2875 CrossRef CAS PubMed.
- R. J. Hall, P. N. Mortenson and C. W. Murray, Prog. Biophys. Mol. Biol., 2014, 116, 82–91 CrossRef CAS.
- M. M. Hann, A. R. Leach and G. Harper, J. Chem. Inf. Comput. Sci., 2001, 41, 856–864 CrossRef CAS.
- I. D. Kuntz, K. Chen, K. A. Sharp and P. A. Kollman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9997–10002 CrossRef CAS.
- T. Clackson and J. A. Wells, Science, 1995, 267, 383–386 CrossRef CAS.
- R. Brenke, D. Kozakov, G. Y. Chuang, D. Beglov, D. Hall, M. R. Landon, C. Mattos and S. Vajda, Bioinformatics, 2009, 25, 621–627 CrossRef CAS PubMed.
- D. Kozakov, L. E. Grove, D. R. Hall, T. Bohnuud, S. E. Mottarella, L. Luo, B. Xia, D. Beglov and S. Vajda, Nat. Protoc., 2015, 10, 733–755 CrossRef CAS.
- A. Miranker and M. Karplus, Proteins: Struct., Funct., Bioinf., 1991, 11, 29–34 CrossRef CAS.
- E. Liepinsh and G. Otting, Nat. Biotechnol., 1997, 15, 264–268 CrossRef CAS.
- K. N. Allen, C. R. Bellamacina, X. Ding, C. J. Jeffery, C. Mattos, G. A. Petsko and D. Ringe, J. Phys. Chem., 1996, 100, 2605–2611 CrossRef CAS.
- M. O'Reilly, A. Cleasby, T. G. Davies, R. J. Hall, R. F. Ludlow, C. W. Murray, D. Tisi and H. Jhoti, Drug Discovery Today, 2019, 24, 1081–1086 CrossRef.
- D. Wood, J. D. Lopez-Fernandez, L. Knight, I. Al-Khawaldeh, C. Gai, S. Lin, M. P. Martin, D. Miller, C. Cano, J. Endicott, I. Hardcastle, M. Noble and M. J. Waring, J. Med. Chem., 2019, 62, 3741–3752 CrossRef CAS PubMed.
- A. C. English, S. H. Done, L. S. D. Caves, C. R. Groom and R. E. Hubbard, Proteins: Struct., Funct., Bioinf., 1999, 37, 628–640 CrossRef CAS.
- J. C. Bardwell, K. McGovern and J. Beckwith, Cell, 1991, 67, 581–589 CrossRef CAS.
- B. Heras, M. J. Scanlon and J. L. Martin, Br. J. Clin. Pharmacol., 2015, 79, 208–215 CrossRef CAS.
- B. Heras, S. R. Shouldice, M. Totsika, M. J. Scanlon, M. A. Schembri and J. L. Martin, Nat. Rev. Microbiol., 2009, 7, 215–225 CrossRef CAS.
- R. J. Dutton, D. Boyd, M. Berkmen and J. Beckwith, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11933–11938 CrossRef CAS.
- A. Hiniker and J. C. A. Bardwell, J. Biol. Chem., 2004, 279, 12967–12973 CrossRef CAS PubMed.
- B. Doak, R. Whitehouse, K. Rimmer, M. Williams, B. Heras, S. Caria, O. Ilyichova, M. Vazirani, B. Mohanty and J. Harper, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-262lh.
- M. R. Bentley, O. V. Ilyichova, G. Wang, M. L. Williams, G. Sharma, W. S. Alwan, R. L. Whitehouse, B. Mohanty, P. J. Scammells, B. Heras, J. L. Martin, M. Totsika, B. Capuano, B. C. Doak and M. J. Scanlon, J. Med. Chem., 2020, 63, 6863–6875 CrossRef CAS.
- L. A. Adams, P. Sharma, B. Mohanty, O. V. Ilyichova, M. D. Mulcair, M. L. Williams, E. C. Gleeson, M. Totsika, B. C. Doak, S. Caria, K. Rimmer, J. Horne, S. R. Shouldice, M. Vazirani, S. J. Headey, B. R. Plumb, J. L. Martin, B. Heras, J. S. Simpson and M. J. Scanlon, Angew. Chem., Int. Ed., 2015, 54, 2179–2184 CrossRef CAS PubMed.
- L. F. Duncan, G. Wang, O. V. Ilyichova, M. J. Scanlon, B. Heras and B. M. Abbott, Molecules, 2019, 24, 3756 CrossRef PubMed.
- W. S. Alwan, PhD Thesis, Monash University, 2020, DOI:10.26180/5e4b83c8e6e6a.
- L. W. Guddat, J. C. Bardwell, R. Glockshuber, M. Huber-Wunderlich, T. Zander and J. L. Martin, Protein Sci., 1997, 6, 1893–1900 CrossRef CAS.
- J. J. Paxman, N. A. Borg, J. Horne, P. E. Thompson, Y. Chin, P. Sharma, J. S. Simpson, J. Wielens, S. Piek, C. M. Kahler, H. Sakellaris, M. Pearce, S. P. Bottomley, J. Rossjohn and M. J. Scanlon, J. Biol. Chem., 2009, 284, 17835–17845 CrossRef CAS.
- K. Inaba, S. Murakami, M. Suzuki, A. Nakagawa, E. Yamashita, K. Okada and K. Ito, Cell, 2006, 127, 789–801 CrossRef CAS.
- W. Duprez, L. Premkumar, M. A. Halili, F. Lindahl, R. C. Reid, D. P. Fairlie and J. L. Martin, J. Med. Chem., 2015, 58, 577–587 CrossRef CAS.
- T. M. McPhillips, S. E. McPhillips, H. J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. Kuhn, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204–1214 CrossRef CAS.
- P. Legrand, GitHub repository, 2017, DOI:10.5281/zenodo.837885.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS PubMed.
- P. W. Rose, A. Prlić, A. Altunkaya, C. Bi, A. R. Bradley, C. H. Christie, L. D. Costanzo, J. M. Duarte, S. Dutta, Z. Feng, R. K. Green, D. S. Goodsell, B. Hudson, T. Kalro, R. Lowe, E. Peisach, C. Randle, A. S. Rose, C. Shao, Y.-P. Tao, Y. Valasatava, M. Voigt, J. D. Westbrook, J. Woo, H. Yang, J. Y. Young, C. Zardecki, H. M. Berman and S. K. Burley, Nucleic Acids Res., 2017, 45, D271–D281 CrossRef CAS PubMed.
- D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, N. Assempour, I. Iynkkaran, Y. Liu, A. Maciejewski, N. Gale, A. Wilson, L. Chin, R. Cummings, D. Le, A. Pon, C. Knox and M. Wilson, Nucleic Acids Res., 2018, 46, D1074–D1082 CrossRef CAS PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef CAS PubMed.
- W. H. B. Sauer and M. K. Schwarz, J. Chem. Inf. Comput. Sci., 2003, 43, 987–1003 CrossRef CAS.
- T. K. Allu and T. I. Oprea, J. Chem. Inf. Model., 2005, 45, 1237–1243 CrossRef CAS.
- P. A. Karplus and K. Diederichs, Science, 2012, 336, 1030–1033 CrossRef CAS.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS.
- N. W. Moriarty, R. W. Grosse-Kunstleve and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 1074–1080 CrossRef CAS PubMed.
- O. S. Smart, T. O. Womack, A. Sharff, C. Flensburg, P. Keller, W. Paciorek, C. Vonrhein and G. Bricogne, Grade, version 1.2.20, https://www.globalphasing.com Search PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- D. Liebschner, P. V. Afonine, M. L. Baker, G. Bunkóczi, V. B. Chen, T. I. Croll, B. Hintze, L. W. Hung, S. Jain, A. J. McCoy, N. W. Moriarty, R. D. Oeffner, B. K. Poon, M. G. Prisant, R. J. Read, J. S. Richardson, D. C. Richardson, M. D. Sammito, O. V. Sobolev, D. H. Stockwell, T. C. Terwilliger, A. G. Urzhumtsev, L. L. Videau, C. J. Williams and P. D. Adams, Acta Crystallogr., Sect. D: Struct. Biol., 2019, 75, 861–877 CrossRef CAS.
- M. R. Berthold, N. Cebron, F. Dill, T. R. Gabriel, T. Kotter and T. Meinl, et al., KNIME: The Konstanz Information Miner, in Data Analysis Machine Learning and Applications (Studies in Classification Data Analysis and Knowledge Organization), Springer, Berlin Heidelberg, 2008, pp. 319–326 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Organic solvent and MicroFrag library SMILES, library design details, affinity characterisation, and crystallography statistics. See DOI: https://doi.org/10.1039/d2md00253a |
| ‡ Current address: Cell Biology, Blavatnik Institute, Harvard Medical School, Boston, MA 02115, United States of America. |
| § Current address: Agilent Technologies, Mulgrave, VIC 3170, Australia. |
| ¶ Current address: Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, TN 37232 USA. |
| || Current address: Sydney Analytical Core Research Facility, The University of Sydney, Sydney, NSW 2006, Australia. |
| This journal is © The Royal Society of Chemistry 2023 |