
Expanding the scope of novel 1,2,3-triazole derivatives as new antiparasitic drug candidates†
Renzo
Carlucci‡
 a,
Gabriel
Di Gresia‡
a,
María Gabriela
Mediavilla
b,
Julia A.
Cricco
b,
Babu L.
Tekwani
e,
Shabana I.
Khan
c and
Guillermo R.
Labadie
*ad
a,
Gabriel
Di Gresia‡
a,
María Gabriela
Mediavilla
b,
Julia A.
Cricco
b,
Babu L.
Tekwani
e,
Shabana I.
Khan
c and
Guillermo R.
Labadie
*ad
aInstituto de Química Rosario (IQUIR) UNR, CONICET, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, S2002LRK, Rosario, Argentina. E-mail: labadie@iquir-conicet.gov.ar; Fax: +54 341 4370477; Tel: +54 341 4370477
bInstituto de Biología Molecular y Celular de Rosario (IBR), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) – Universidad Nacional de Rosario (UNR), Suipacha 531, S2002LRK, Rosario, Argentina
cNational Center for Natural Products Research & Department of Biomolecular Sciences, School of Pharmacy, University of Mississippi, University, MS 38677, USA
dDepartamento de Química Orgánica, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, S2002LRK, Rosario, Argentina
eDepartment of Infectious Diseases, Division of Scientific Platforms, Southern Research, Birmingham, AL 35205, USA
First published on 31st October 2022
Abstract
We have previously shown that prenyl and aliphatic triazoles are interesting motifs to prepare new chemical entities for antiparasitic and antituberculosis drug development. In this opportunity a new series of prenyl-1,2,3-triazoles were prepared from isoprenyl azides and different alkynes looking for new antimalarial drug candidates. The compounds were prepared by copper(I) catalyzed dipolar cycloaddition of the isoprenyl azide equilibrium mixture providing exclusively 1,4-disubstituted 1,2,3-triazoles in a regiospecific fashion. The complete collection of 64 compounds was tested on chloroquine-sensitive (CQ sensitive), Sierra Leone (D6), and the chloroquine-resistant, Indochina (W2), strains of Plasmodium falciparum and those compounds which were not previously reported were also tested against Leishmania donovani, the causative agent for visceral leishmaniasis. Thirteen analogs displayed antimalarial activity with IC50 below 10 μM, while the antileishmanial activity of the newly reported analogs could not improve upon those previously reported. Compounds 1o and 1r were identified as the most promising antimalarial drug leads with IC50 below 3.0 μM for both CQ-sensitive and resistant P. falciparum strains with high selectivity index. Finally, a chemoinformatic in silico analysis was performed to evaluate physicochemical parameters, cytotoxicity risk and drug score. The validation of a bifunctional farnesyl/geranylgeranyl diphosphate synthase PfFPPS/GGPPS as the potential target of the antimalarial activity of selected analogs should be further investigated.
1. Introduction
Neglected tropical diseases (NTDs) are a group of infectious pathologies prevalent in intertropical and subtropical countries with deficient health, socioeconomic and environmental conditions.1 These diseases are considered neglected because they are widespread among poor and outcast populations that live in rural areas of developing countries. Those conditions are favourable for propagating the vectors that transmit most NTDs. Malaria2 and leishmaniasis3 are two infectious diseases that affect an enormous part of the tropical and subtropical population in the World.Malaria is a parasitic disease transmitted by the female mosquito from Anopheles genus. Plasmodium spp. are the etiological agents of the disease with five species that can infect humans. P. falciparum has the highest prevalence and severity of the infection and is mostly found in sub-Saharan Africa. In Africa, the infections by this parasite are responsible for most of mortality in children. P. vivax, which is mostly endemic of Asia4 and South America5 is now recognized as fatal. With up to 15 million infections every year, and up to 2500 million people at risk, P. vivax is a significant threat for all developing countries in tropical and subtropical regions.6 The strengthening of prevention and control measures has led to a 29% reduction in mortality rates by malaria around the world since 2010. Sub-Saharan Africa keeps having a highly unequal share of the World's malaria cases and deaths.2
Leishmaniasis is a parasitic disease caused by Leishmania spp., with over 20 species that causes different clinical manifestations. The infection has three different forms known as cutaneous (CL), mucocutaneous (ML) and visceral (VL), damaging the skin, mucous or organs, respectively. The parasites are transmitted to the host by the bite of female mosquito of Phlebotomus genus (Africa and Asia) or Lutzomyia genus (Latin America). This endemic disease affects the population of more than 98 countries with a global prevalence of 12 million people. CL is mainly distributed along Middle Eastern and Latin American regions, whereas VL is mostly found in India, East Africa and Brazil.7 Over the last 10 years there were a world epidemic expansion in VL which has a high probability of death in untreated cases within the first 2 years.8,9
There are areas where visceral leishmaniasis (VL) is co-endemic with malaria, making coinfections with both diseases possible. There are different reports with cases of co-infection of these parasites in humans in different regions of Africa.10,11 Therefore, there is a need to identify optimal and cost-effective strategies to control co-infections to reduce the risk of spreading these diseases.12
Chloroquine has been the first line drug for malaria treatment, but due to emergence of resistance, new treatments, like the artemisinin combination therapy (ACT) are recommended by the WHO.13–15 (Fig. 1) For leishmaniasis, Sb(V) derivatives are still first line treatment, together with amphotericin B, miltefosine, pentamidine, and paromomycin. The first two are the preferred chemotherapy despite the high toxicity and development of resistance.16 (Fig. 1) Therefore, more effective antiparasitic agents with novel mechanism of action and no side-effects are needed.
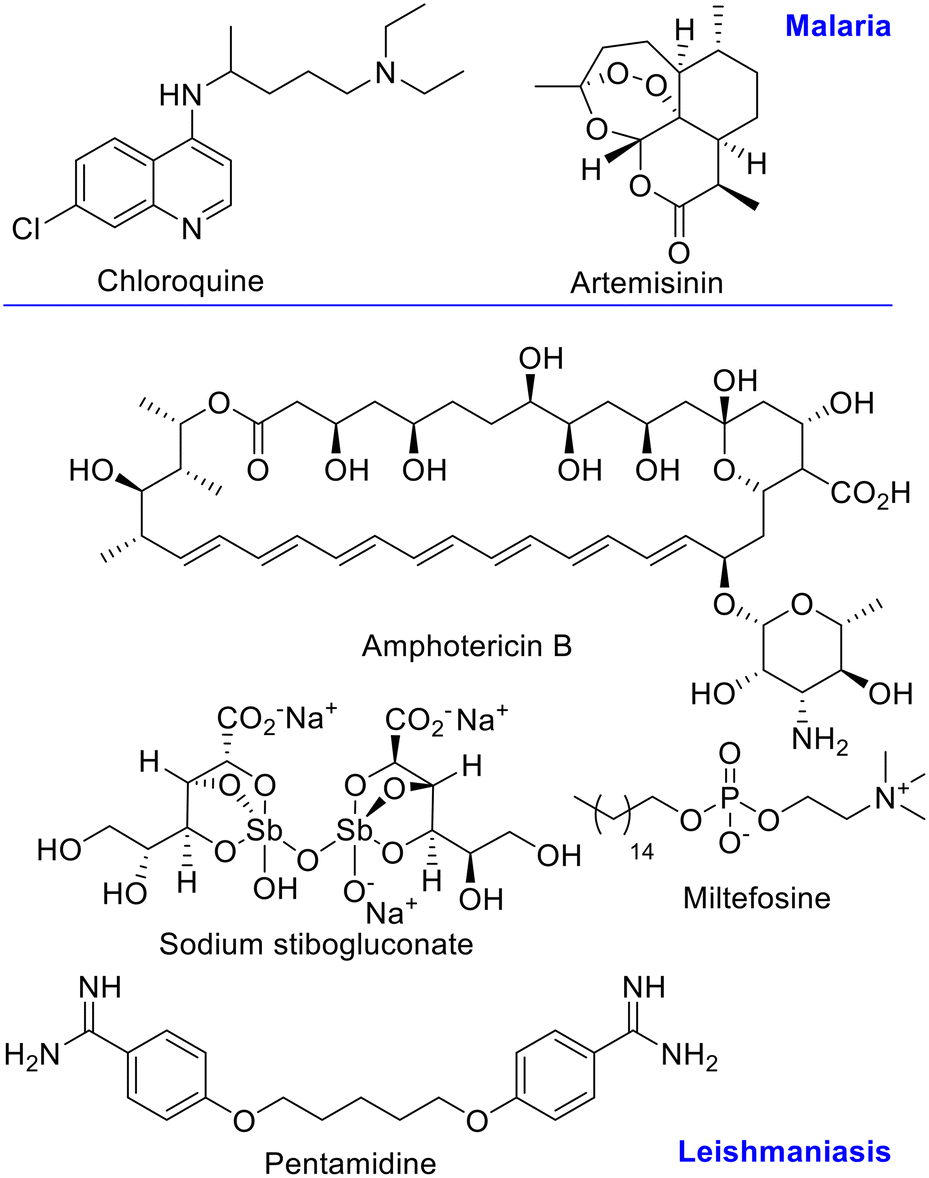 | ||
| Fig. 1 Available drugs for malaria and leishmaniasis treatment. | ||
The isoprene biosynthesis is linked to metabolites that are required for the cellular viability, like dolichol, ubiquinone and heme A. Isoprenes are also involved in the post-translational modification of proteins required for anchoring to the membrane.17P. falciparum and L. donovani, like most of eukaryotes, possess prenyltransferases that incorporate farnesyl or geranylgeranyl on substrate proteins.18–20
The inhibition of the isoprenoid biosynthesis blocks the proteins prenylation and is lethal for P. falciparum and L. donovani. One important enzyme of that essential pathway is the farnesyl pyrophosphate synthase (FPPS). FPPS has been pointed out as a drug target for parasitic diseases, including malaria and leishmaniasis.21 Bisphosphonates, that were initially introduced as bone disorder drug candidates, lately have become the preferred scaffold to develop new inhibitors of FPPS.22 In fact, over the last years different biphosphonates have been reported as FPPS inhibitors displaying activity on P. vivax,23 both on the liver stage assays and in vivo.24 Interestingly, the compounds also inhibit geranylgeranyl pyrophosphate synthase (GGPPS), which has emerged as a new target for antimalarial drug development.25,26 Other bisphosphonate derivatives have displayed activity against Trypanosoma cruzi and Toxoplasma gondii,27 of which the fatty acids substituted analogs have shown significant inhibition of the FPPS of different neglected diseases related parasites.28–30
P. falciparum possess an enzyme responsible for the synthesis of farnesyl pyrophosphate, which is different to the FPPS from humans, Leishmania genus, and other organisms. This parasite is believed to have a bifunctional enzyme that synthesizes both farnesyl pyrophosphate and geranylgeranyl pyrophosphate, called bifunctional farnesyl/geranylgeranyl diphosphate synthase (PfFPPS/GGPPS, TDR Target database code: PF3D7_1128400). This enzyme is a validated target in malaria, although the classic inhibitors of FPPS have little bioavailability and low inhibitory activities.31 Thereby, more studies related to the inhibition of the enzyme are needed.
The synthetic accessibility of 1,2,3-triazoles by CuAAC, the flagship reaction of the “Click chemistry”, have broken into the medicinal chemistry arena being extensively used to prepare new collections of putative active compounds.32,33 Drug discovery for parasitic diseases, including malaria34,35 and leishmaniasis,36 has not been an exception where examples of active 1,2,3-triazoles have been reported.37 Some examples of antimalarial 1,2,3 triazoles38 including naphtyl derivatives,39 are shown in Fig. 2.
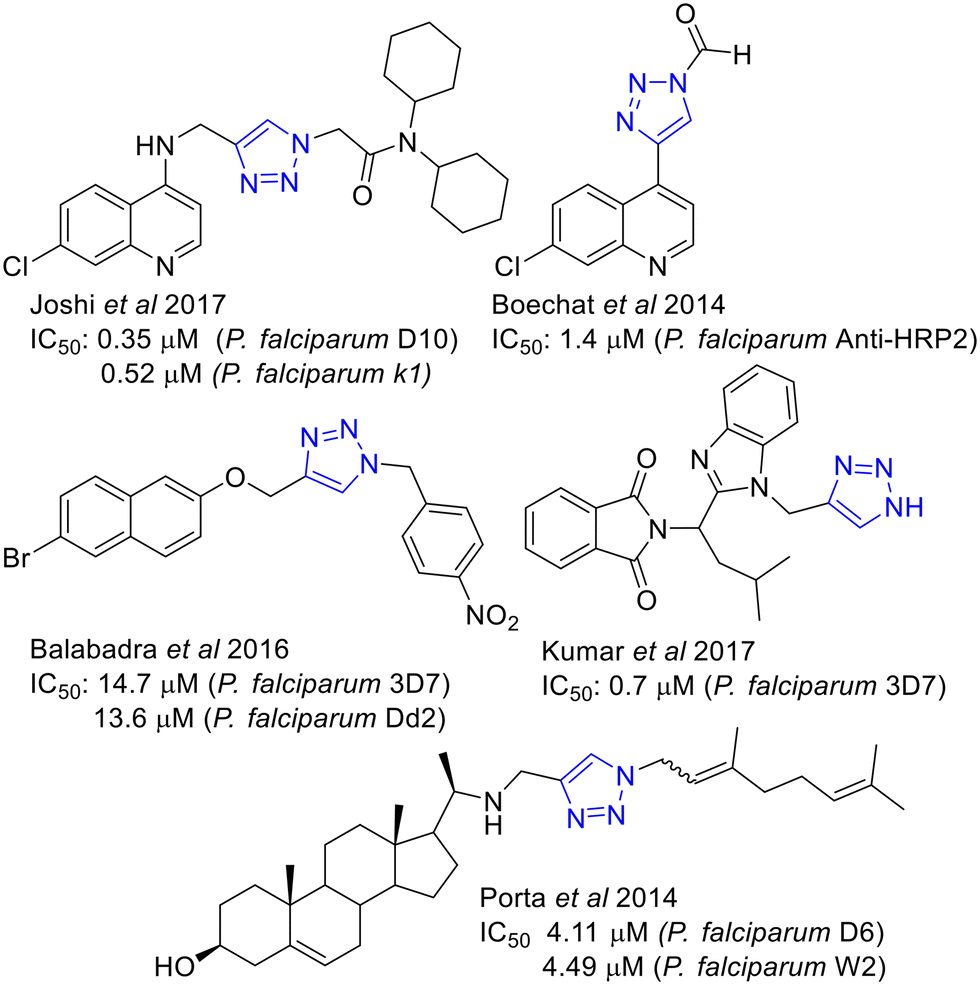 | ||
| Fig. 2 Reported antimalarial 1,2,3-triazolyl derivatives. | ||
Prenyl 1,2,3-triazoles, recently reported by our group, are an interesting example or those heterocycles. Those compounds displayed activity against T. cruzi and L. donovani at low micromolar concentrations,40 being promising antiparasitic candidates.
2. Results and discussion
2.1. Design and synthesis
Looking to find new antiparasitic 1,2,3-triazoles we decided to expand the previously reported collection of our group. (Fig. 3) As can be seen, those compounds are mostly N1-prenylated-1,2,3-triazoles holding non-polar substituents on C4.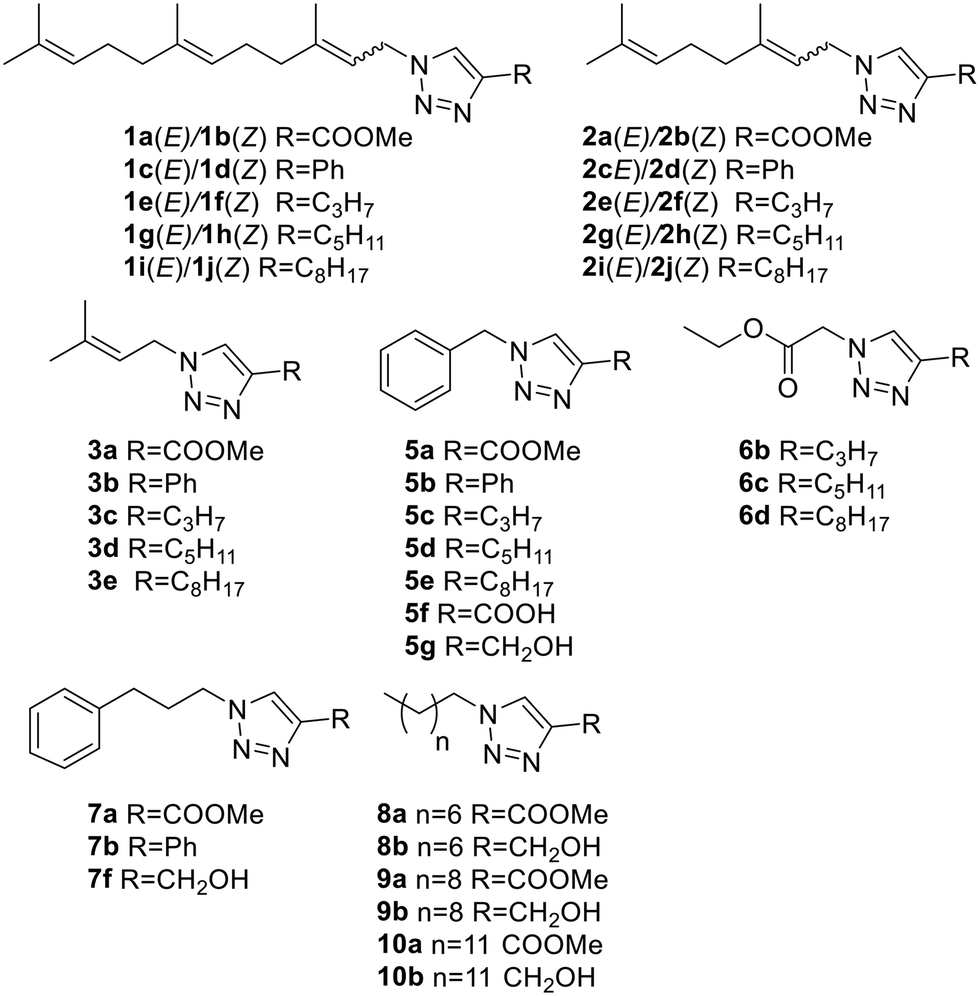 | ||
| Fig. 3 Previously reported 1,2,3-triazoles. | ||
To target FPPS/GGPPS, a substrate mimic should have an isoprenyl chain and a polar group that will fill the pyrophosphate pocket. To that purpose the new analog series will include hydroxy-groups on the C4-substituent. (Fig. 4) Additionally, to expand the collection covering a wide spectrum of steric and electronic demand, phenyl-alkylated substituents were also introduced.
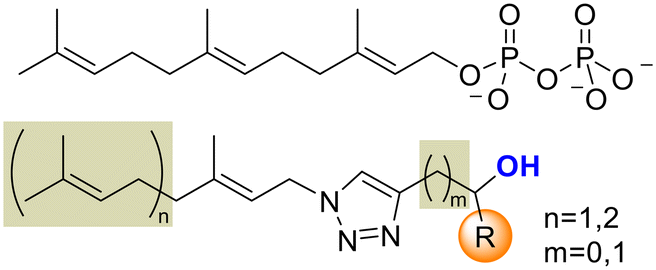 | ||
| Fig. 4 Potential substrate mimic structures. | ||
The necessary azides were prepared following previously reported procedures.41 Cinnamyl azide and prenyl azides were prepared from the corresponding alcohols by the Thompson's reaction.42 Farnesyl and geranyl azides were obtained as a mixture of regioisomers that interconvert rapidly at room temperature, due to a [3,3]-sigmatropic Winstein's rearrangement.40 On the contrary, cinnamyl azide is not in equilibrium with its tertiary isomer as has been previously reported.43 Once the necessary building blocks were synthetized, CuAAC conditions were used to prepare the 1,4-disubstituted 1,2,3-triazoles.
The reactions were conducted using CuSO4 as the copper source, sodium ascorbate as a reductant in a tBuOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O mixture.44 Eight terminal alkynes were used as a synthetic counterpart of the allylic azides (Scheme 1).
:H2O mixture.44 Eight terminal alkynes were used as a synthetic counterpart of the allylic azides (Scheme 1).
 | ||
| Scheme 1 Synthesis of 1,2,3 triazoles collection. | ||
Following that procedure, four farnesyl derivatives were obtained as E:Z (1:2) mixtures and were separated by column chromatography. (Fig. 5) Additionally, five cinnamyl and five phenyl propyl analogs were also obtained. (Fig. 5) Finally, the C10 derivative of the 5 series (compound 5h) and the diester 6a were also prepared. Together the compounds added to the previous collection (Fig. 4) were obtained with a 77% average yield after purification.
 | ||
| Fig. 5 1,2,3-triazoles synthesized in this work. | ||
2.2. Biology
| Cmpd | Azide | R | E/Z | P. f. D6b IC50 μM | SI | P. f. W2c IC50 μM | SI | Cytotoxicity Vero cells IC50 μM |
|---|---|---|---|---|---|---|---|---|
| a Analogs not displayed on this table have IC50 > 25 μM on both strains. b P. falciparum chloroquine sensitive-Sierra Leone clone (D6). c P. falciparum chloroquine resistant-Indochina clone (W2), CHQ = chloroquine, ART = artemisinin, SI = selectivity index calculated as IC50 Vero cells/IC50P. falciparum. | ||||||||
| 1g | Farnesyl | C5H11 | E | 3.49 | >3.96 | 6.69 | >2.07 | >13.8 |
| 1o | Farnesyl | C(OH)(CH3)2 | E | 2.35 | >6.09 | 1.99 | >7.20 | >14.3 |
| 1r | Farnesyl | CH2CH2OH | Z | 2.74 | >5.46 | 2.05 | >7.31 | >15.0 |
| 3a | Prenyl | COOMe | — | 24.3 | >1 | 23.1 | 1.06 | >24.3 |
| 4a | Cinnamyl | Ph | — | 11.5 | >1.58 | 13.0 | >1.40 | >18.2 |
| 4b | Cinnamyl | n-C3H7 | — | 20.9 | <1 | 22.0 | <1 | >20.9 |
| 4d | Cinnamyl | n-C8H17 | — | 10.8 | >1.48 | 10.1 | >1.58 | >16.0 |
| 5h | Benzyl | n-C10H21 | — | 11.0 | >1.44 | 10.7 | >1.48 | >15.9 |
| 6a | CH2COOEt | COOMe | — | 12.3 | >1.70 | 12.3 | >1.70 | >20.9 |
| 7c | (CH2)3Ph | n-C3H7 | — | 15.3 | >1.36 | 11.3 | >1.83 | >20.7 |
| 7d | (CH2)3Ph | n-C5H11 | — | 16.3 | >1.13 | 10.9 | >1.70 | >18.5 |
| 8b | n-C8H17 | CH2OH | — | 13.3 | >1.70 | 6.63 | >3.39 | >22.5 |
| 9a | n-C10H21 | COOMe | — | 11.2 | >1.58 | 6.73 | >2.64 | >17.8 |
| CHQ | 0.083 | >179 | 0.422 | >35.2 | ||||
| ART | 0.094 | >179 | 0.094 | >179 | ||||
 | ||
| Fig. 6 Compounds active against P. falciparum. | ||
The most active compounds present IC50 below 25 μM, with those of the most active members in the low micromolar range. Interestingly, there is no significant difference between the antimalarial activity on both strains and over half of the active compounds exhibited a selectivity index greater than 1.5. (Table 1).
| Cmpd | Azide | R | E/Z | L. d. promastigotesb IC50 μM |
|---|---|---|---|---|
| a Analogs not displayed on this table have IC50 > 100 μM. Pen = pentamidine, Amp = amphotericin. b All the analogs were tested at least at six different concentrations, each in duplicate. The mean values were used to generate the growth inhibition curves and determination of IC50 values. | ||||
| 1k | Farnesyl | CH2OH | E | 56.0 |
| 1l | Farnesyl | CH2OH | Z | 59.3 |
| 1m | Farnesyl | CH(OH)CH3 | E | 56.7 |
| 1n | Farnesyl | CH(OH)CH3 | Z | 59.9 |
| 1o | Farnesyl | C(OH)(CH3)2 | E | 48.3 |
| 1p | Farnesyl | C(OH)(CH3)2 | Z | 63.4 |
| 1q | Farnesyl | CH2CH2OH | E | 63.0 |
| 1r | Farnesyl | CH2CH2OH | Z | 63.0 |
| 4c | Cinnamyl | n-C5H11 | — | 74.4 |
| 5a | Benzyl | COOMe | — | 92.1 |
| 5b | Benzyl | Ph | — | 85.0 |
| 5e | Benzyl | n-C8H17 | — | 22.1 |
| 5f | Benzyl | COOH | — | 18.7 |
| 7b | (CH2)3Ph | Ph | — | 68.4 |
| 9a | n-C10H21 | COOMe | — | 78.5 |
| Pen | 6.17 | |||
| Amp | 0.35 | |||
The cytotoxicity against Vero cells of the compounds presented on Table 2 was, in all cases, considerably lower than the antileishmanial activity. However, the selectivity index was <1 and, in that case, no compound could be considered a hit.45
 | ||
| Fig. 7 P. falciparum and L. donovani activities heat map. | ||
Finally, the compounds with farnesyl chains were those displaying the best antimalarial activity. Those compounds have IC50s below 4 μM being 3 to 5 times more potent than the rest of the analogs that display antimalarial activity. The analog 1g has an IC50 of 3.49 μM on CQ-sensitive P. falciparum (D6) but was considerably less active on the CQ-resistant strain W2 (6.69 μM).
The other farnesylated 1,2,3-triazoles derivatives are compounds 1o and 1r which are the two most potent antimalarial compounds of the whole collection. Compound 1o has an IC50 of 2.35 μM and 1r has an IC50 of 2.74 μM, for the CQ-sensitive strain D6. Interestingly, they were slightly more active on the CQ-resistant strain W2 with IC50 of 1.99 and 2.05 μM, respectively, being therefore very promising candidates. Another interesting outcome of this series is the regiochemistry of the farnesyl chain. On the one side, 1g and 1o were Z and on the other side 1r was E, while their regioisomers were inactive. In summary, the combination of substituents with alcohols and farnesyl chains seems to enhance the antimalarial activity being promising candidates for future studies. A more detailed look at the antileishmanial activity revealed that farnesyl derivatives 1k–1r displayed IC50 between 48.27 to 63.00 μM. The activity of the previously reported prenylated 1,2,3-triazoles was highly dependent on the size of the isoprenyl units, where the farnesyl derivatives were the most active. The antileishmanial activity of the farnesyl series 1a–1j, that range from 11.0 to 52.0 μM, was considerably more potent than 1k–1r. The main difference between both series is the hydroxyl group on the alkyl substituent present on analogs 1k–1r that seems to be detrimental for the activity. As an example, 1b (Z,E-farnesyl R = COOMe) has an IC50 of 11 μM that increased to 59 μM on 1l (Z,E-farnesyl R = CH2OH).
The other series contain aromatic substituents, including benzyl, cinnamyl and 3-phenyl propyl that have IC50s ranging from 18.7 to 92.7 μM. The most active member of the newly reported compounds belongs to the benzyl series. Analog 5e, a non-polar derivative holding a pentyl substituent, has an IC50 of 22 μM and 5f, a very simple carboxylic acid, is the most active member of the new collection having an IC50 of 18.7 μM. Also, that analog is the only new derivative with no explicit toxicity (SI = 1.25).
In summary, the newly introduced analogs did not improve the activity of the previously reported derivatives 1b and 1h. Nevertheless, the structurally simple derivative 5f seems to be an interesting structure to further develop an antileishmanial agent.
It has been recently reported that MMV019313, a non-bisphosphonate FPPS inhibitor, is capable of inhibiting Pf FPPS/GGPPS in a non-conventional way.46 Indeed, MMV019313 interact with the enzyme in a non-catalytic site being 100-times more selective to Pf FPPS/GGPPS than hFPPS. Gisselberg et al. have proposed that Pf FPPS/GGPPS could have an allosteric inhibition site like the human enzyme.31 Unfortunately, the site has not been characterized in the plasmodial enzyme yet and molecular docking in the apo Pv FPPS/GGPPS structure was inconclusive.
However, we can hypothesize that the antimalarial activity of these farnesylated derivatives may be related to the inhibition of Pf FPPS/GGPPS, or another isoprene biosynthesis enzyme, considering the essentiality of these important metabolites. Such as is the case for protein farnesyl transferase, which is required for the viability of the parasite.
In L. donovani there are two different enzymes that synthesize FPP and GGPP. In that way, it is possible to assume that it should be more difficult to identify compounds that inhibit both Pf FPPS/GGPPS and the leishmanial enzyme. The fact that most active compounds on the kinetoplastid parasite did not have hydroxy groups on their structure and were 5 times less active than the most potent (1b and 1hL. donovani IC50 = 11 μM vs.1k and 1h IC50s of 56.0 and 48.3 μM, respectively) does not support isoprene enzymes as possible targets. Nevertheless, a complete study of the mechanism of action should be required to confirm those hypotheses.
Finally, to identify candidates with wide-spectrum activity, they should have similar potency as antimalarial and antileishmanial drugs. As is clearly seen on Fig. 7 heatmap, in general the activity hot spots are different for each parasite. Nevertheless, there are 4 analogs (1g,1o, 1r and 9a) that seem to be the most adequate starting point for the development of malaria-visceral leishmaniasis co-infection treatments.
2.3. Cheminformatics analysis
As it was mentioned, FPPS is an important target for numerous pathologies and infectious diseases drug design, for which many inhibitors have been previously reported. Taking advantage of the ChEMBL database, a chemical database of bioactive molecules maintained by the European Bioinformatics Institute (EMBL-EBI),47 a complete search was performed that provided 470 compounds with reported inhibitory activity on the enzyme. To determine the chemical space explored by the reported FPPS inhibitors, the collection was first manually curated, leading to 235 compounds. Then, the similarity of those compounds was analysed on the web-based platform Chemmine Tools.48 (Tanimoto coefficient >0.7) The results were plotted in a 3D scatter plot and were grouped according to the different clusters found. The 1,2,3-triazoles were also included in the same analysis to identify their similarity with the reported inhibitors. Fig. 8 shows the clusters as distinct colours dots. The white, light blue, and blue clusters belong to previously reported inhibitors found on ChemBL, while the orange are 1,2,3-triazole derivatives. The analysis performed demonstrates that the prepared collection has low similarity with previously reported compounds and is a new chemotype that constitutes an unexplored section of the chemical space of the FPPS inhibitors. | ||
| Fig. 8 Analysis of molecular similarity of the FPPS inhibitors stored on ChemBL and the 1,2,3-triazoles reported here (circled on dotted green line). The high similarity areas are clearly delimited. | ||
2.4. Physicochemical parameters
There is consensus that oral administration is the most convenient, cost-effective, and commonly used medication administration route. Therefore, it is critical over the development process of new chemical entities to consider their pharmacokinetic properties, particularly their administration, distribution, metabolization, excretion and potential toxicity properties (ADME/Tox). Compounds that have promissory activities but have poor bioavailability are neglected. It has been reported a 90% attrition rate of drug candidates on the transition from preclinical trials to marketing surveillance trials because of their poor physicochemical properties.49There are some rules or numerical cut-offs to label a molecule as a good candidate in terms of its physicochemical properties. Considering those proposed by Lipinski (1997),52 named the rules of five and other contribution like Veber's (2002),53 we explored our full collection of 64 synthesized molecules, and predicted their physicochemical properties computationally. To do that, we used the free web services Osiris,50 and Molinspiration.51
Computational predictions of the physicochemical properties of the collection using DataWarrior showed few exceptions of the Lipinski's rules. (Table 3) The library satisfies the restriction of MW (179–386 Da), number of H-bond donors (0 or 1) and acceptors (3 to 7). However, 22 of the 64 compounds had clogP > 5 (−0.39 to 8.77). Analysing the parameters for the antimalarial active compounds we could identify narrower intervals. While the number of bond donors and acceptors remain the same, the MW ranged from 195–344 Da. In contrast, the clogP of the active compounds was narrower but still have a broad window (−0.39–7.40). Interestingly the 3 more active compounds (1g, 1o and 1r) were also the more lipophilic, being active even violating one of the rules of five.
Osiris platform provided the toxicity risk, drug likeness and a drug score. The results revealed that all the compounds do not have any toxicity risk, undesired effects, tumorigenic features, or effect on the reproductive system (Table 4). The solubility is a critical property which aids in the circulation of a drug after the administration and into the bloodstream. More than 80% of marketed drugs have logS > 4.
| Toxicity riska | Drug-score | |||||
|---|---|---|---|---|---|---|
| Mb | Tc | Id | Ree | Drug likeness | Drug-score | |
| a Ranked according to: (−) not toxic, (±) slightly toxic, (+) highly toxic. b M = mutagenic. c T = tumorigenic. d I = irritant. e Re = reproductive effective. | ||||||
| Library | — | — | — | — | −25.40–14.35 | 0.17–0.63 |
| Active compounds | — | — | — | — | −21.86–14.35 | 0.22–0.49 |
| Chloroquine | + | — | + | — | 7.39 | 0.25 |
| Artemisinin | — | + | + | — | −1.97 | 0.17 |
Osiris platform estimates the solubility in mol L−1, where our collection displayed a promising range of −4.91 to −0.72. In terms of drug likeness, the prepared collection showed a poor profile (−25.4 to 14.35) compared to commercial drugs like chloroquine or artemisinin (7.39 and −1.975, respectively). That poor behaviour is balanced with the rest of the parameters used to calculate the drug score (solubility, logP, MW and toxicity risk) delivering moderate to good values (0.17 to 0.63) compared with standard drugs (0.17 to 0.25). A detailed analysis of compounds 1o and 1r showed that they have a very good drug score (0.33 and 0.30) compared with chloroquine (0.25) and share very similar physicochemical properties. (Table 5) Considering those parameters and the antiplasmodial potency on the sensitive and the resistant strains, 1o and 1r are the most promising candidates for further antimalarial drug development.
3. Experimental
3.1. General
All reactions progress was monitored on GP TLC plates. Column chromatography was performed with silica gel 60 (230–400 mesh) under a low pressure of nitrogen, using increasing EtOAc–hexane gradients as a solvent. All the solvents (hexane, ethyl acetate) were distilled before use. Chemical reagents were purchased from commercial suppliers and used without further purification, unless otherwise noted. Solvents were analytical grade or were purified by standard procedures prior to use. 1H and 13C NMR spectra were measured on a 300 MHz Bruker Avance II using CDCl3 as a solvent. Chemical shifts were reported in ppm downfield from tetramethylsilane (δ) as the internal standards and coupling constants are in hertz (Hz). Assignments of proton resonances were confirmed by correlated spectroscopy (heteronuclear single quantum coherence, HSQC). The following abbreviations are used to indicate NMR signal multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet. High-resolution mass spectra (HRMS) were recorded on a Bruker MicroTOF II spray source. All the melting points were determined in open Pyrex capillaries with an Electrothermal 9000 melting point apparatus.3.2. Synthesis
:H2O (1:1) and then 1 M CuSO4 solution and finally 1 M sodium ascorbate solution were added, and the mixture was stirred overnight at room temperature. Brine was added, and the solution was extracted with dichloromethane. Combined organic extracts were dried over sodium sulphate and evaporated. Products were purified by column chromatography in silica gel with increasing hexane/ethyl acetate or ethyl acetate/ethanol gradients.
3.3. Biology
In vitro Antileishmanial assays. Antileishmanial activity of the compounds was tested in vitro on a culture of L. donovani promastigotes. In a 96-well microplate the appropriate dilutions of the compounds were added to the promastigote cultures (2 × 106 cell per mL) to get the final concentrations of 40, 8 and 1.6 μg mL−1. The plates were incubated at 26 °C for 72 hours and growth was determined by Alamar blue assay.58 Pentamidine and amphotericin B were tested as the standard antileishmanial agents and DMSO was used as negative control.
Statistics and data analysis. IC50 values were obtained by non-linear regression of dose response logistic functions, using the Microsoft Excel-based plug-in XLfit. All experiments were performed in triplicate.
a) Screening at a maximum concentration of 4.75 μg mL−1. Vero cells (African green monkey kidney cells) were cultured in RPMI medium supplemented with 10% heat inactivated FCS, 0.15% (w/v) NaHCO3, 100 U mL−1 penicillin and 100 U mL−1 streptomycin at 37 °C in an atmosphere of 5% CO2 and 95% humidity.
Cytotoxicity assays: Neutral Red assay was used to evaluate the cytotoxicity of analogs. Vero cells were plated on 96-well plate and, upon confluency, incubated with various concentrations of each analog up to a maximum concentration of 4.75 μg mL−1, for 48 hours at 37 °C in a cell culture incubator. DMSO was used as negative control. Cell viability was measured and IC50 value for each compound was computed from the concentration response curve.40,59
b) Cytotoxicity assay on Vero cells for hit candidates. Vero cells (African green monkey kidney cells) were cultured in DMEM high glucose medium supplemented with 10% heat inactivated FCS at 37 °C in a 5% CO2 and 95% relative humidity atmosphere.
Cytotoxicity assays: MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to evaluate the cytotoxicity of analogs. Vero cells were seeded on 96-well plate at a density of 15.6 × 103 cell per cm2 and, 24 h later, incubated with up to 100 μM of each analog for 48 hours in the same conditions of growth as described above. Incubation with DMSO was used as negative control. Cell viability was measured incubating 1 h with 0.5 mg mL−1 MTT, solubilizing the violet insoluble formazan produced with DMSO and reading absorbance at 540 nm. IC50 value for each compound was computed from the logarithm of concentration response curve.
4. Conclusions
We have previously shown that prenyl and aliphatic triazoles are interesting motifs that provided new chemical entities for antiparasitic and antitubercular drug development.40,44 In this opportunity the previously reported collections were expanded with 20 new analogs. The complete collection of 64 compounds was tested on two different strains of the malaria parasite (P. falciparum) and the antileishmanial activity was evaluated on promastigotes of L. donovani. Only 13 analogs displayed antimalarial activity. The antileishmanial activity of newly tested analogs was less potent than the previously reported compounds.The isomeric hydroxy derivatives 1o and 1r were selected as the most promising antimalarial drug candidates, based on their activity and selectivity. (Fig. 9) Their structural and selectivity profile prompt us to propose a possible mechanism of action by interfering the isoprene metabolism. The farnesyl pyrophosphate synthase could be involved as the main molecular target, nevertheless the participation of other validated enzymes like farnesyl transferase could not be excluded. Those hypotheses will be further validated, and analogs 1g, 1o, 1r and 9a will be modified to develop malaria-visceral leishmaniasis co-infection treatments.
 | ||
| Fig. 9 Most promising antimalarial compounds of the synthesized chemical library. | ||
Author contributions
Renzo Carlucci: investigation, formal analysis, visualization; writing original draft. Gabriel Di Gresia: investigation, formal analysis, visualization; writing original draft. María Gabriela Mediavilla: investigation; formal analysis; validation. Julia A. Cricco: conceptualization; methodology; resources; writing – review & editing. Babu Tekwani: conceptualization; methodology; resources; writing – review & editing. Shabana I. Khan: conceptualization; methodology; resources; writing – review & editing. Guillermo R. Labadie: conceptualization; data curation; funding acquisition; methodology; project administration; resources; supervision; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by grants from UNR (Universidad Nacional de Rosario 80020190300101UR), CONICET (PIP 2021-2023 11220200101045CO), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT PICT-2017-2096 awarded to G. R. L.) The research leading to these results has, in part, received funding from UK Research and Innovation via the Global Challenges Research Fund under grant agreement ‘A Global Network for Neglected Tropical Diseases’ grant number MR/P027989/1. GRL, MGM and JAC are members of the Research Career of the Consejo Nacional de Investigaciones Científicas y Técnicas of Argentina (CONICET). RC thanks CONICET for the award of a fellowship, GDG has a fellowship of Agencia I+D+i associated to grant no. PICT 2017-2096.Notes and references
- M. N. Malecela and C. Ducker, Trans. R. Soc. Trop. Med. Hyg., 2021, 115, 121–123 CrossRef.
- WHO, World malaria report, 2017 Search PubMed.
- H. L. Choi, S. Jain, J. A. Ruiz Postigo, B. Borisch and D. A. Dagne, PLoS Neglected Trop. Dis., 2021, 15, e0009181 CrossRef.
- E. D. Benavente, E. Manko, J. Phelan, M. Campos, D. Nolder, D. Fernandez, G. Velez-Tobon, A. T. Castaño, J. G. Dombrowski, C. R. F. Marinho, A. C. C. Aguiar, D. B. Pereira, K. Sriprawat, F. Nosten, R. Moon, C. J. Sutherland, S. Campino and T. G. Clark, Nat. Commun., 2021, 12, 3160 CrossRef CAS.
- K. E. Battle and J. K. Baird, PLoS Med., 2021, 18, e1003799 CrossRef CAS.
- R. N. Price, R. J. Commons, K. E. Battle, K. Thriemer and K. Mendis, Trends Parasitol., 2020, 36, 560–570 CrossRef PubMed.
- N. N. H. Valero and M. Uriarte, Parasitol. Res., 2020, 119, 365–384 CrossRef.
- M. Shrestha, M. Khatri-Chhetri, R. C. Poudel, J. Maharjan, S. P. Dumre, K. D. Manandhar, B. D. Pandey, S. B. Pun and K. Pandey, BMC Infect. Dis., 2019, 19, 444 CrossRef.
- L. H. C. Sangenis, S. R. D. A. Lima, C. X. D. Mello, D. T. Cardoso, J. N. Mello, M. C. C. do Espírito Santo and W. Tavares, Rev. Inst. Med. Trop. Sao Paulo, 2014, 56, 271–274 CrossRef.
- Y. Aschale, A. Ayehu, L. Worku, H. Tesfa, M. Birhanie and W. Lemma, BMC Infect. Dis., 2019, 19, 239 CrossRef.
- E. van den Bogaart, M. M. Z. Berkhout, A. B. Y. M. Nour, P. F. Mens, A.-B. A. Talha, E. R. Adams, H. B. M. Ahmed, S. H. Abdelrahman, K. Ritmeijer, B. Y. M. Nour and H. D. F. H. Schallig, BMC Public Health, 2013, 13, 332 CrossRef PubMed.
- F. B. Agusto and I. M. ELmojtaba, PLoS One, 2017, 12, e0171102 CrossRef PubMed.
- R. T. Eastman and D. A. Fidock, Nat. Rev. Microbiol., 2009, 7, 864–874 CrossRef CAS.
- G. R. Pereira, G. C. Brandão, L. M. Arantes, H. A. De Oliveira Jr, R. C. De Paula, M. F. A. Do Nascimento, F. M. Dos Santos, R. K. Da Rocha, J. C. D. Lopes and A. B. De Oliveira, Eur. J. Med. Chem., 2014, 73, 295–309 CrossRef CAS PubMed.
- K. F. Suazo, C. Schaber, C. C. Palsuledesai, A. R. Odom John and M. D. Distefano, Sci. Rep., 2016, 6, 38615 CrossRef CAS.
- S. R. B. Uliana, C. T. Trinconi and A. C. Coelho, Parasitology, 2018, 145, 464–480 CrossRef.
- M. H. Gelb, J. D. Scholten and J. S. Sebolt-Leopold, Curr. Opin. Chem. Biol., 1998, 2, 40–48 CrossRef CAS.
- D. Chakrabarti, T. Da Silva, J. Barger, S. Paquette, H. Patel, S. Patterson and C. M. Allen, J. Biol. Chem., 2002, 277, 42066–42073 CrossRef CAS PubMed.
- D. Chakrabarti, T. Azam, C. DelVecchio, L. Qiu, Y.-I. Park and C. M. Allen, Mol. Biochem. Parasitol., 1998, 94, 175–184 CrossRef CAS.
- M. Brioschi, A. Martinez Fernandez and C. Banfi, Expert Rev. Proteomics, 2017, 14, 515–528 CrossRef CAS.
- A. Srivastava, P. Mukherjee, P. V. Desai, M. A. Avery and B. L. Tekwani, Infect. Disord.: Drug Targets, 2008, 8, 16–30 CAS.
- J. B. Rodriguez, B. N. Falcone and S. H. Szajnman, Expert Opin. Drug Discovery, 2016, 11, 307–320 CrossRef CAS.
- D. Mukkamala, J. H. No, L. M. Cass, T.-K. Chang and E. Oldfield, J. Med. Chem., 2008, 51, 7827–7833 CrossRef CAS PubMed.
- A. P. Singh, Y. Zhang, J.-H. No, R. Docampo, V. Nussenzweig and E. Oldfield, Antimicrob. Agents Chemother., 2010, 54, 2987–2993 CrossRef CAS PubMed.
- J. H. No, F. de Macedo Dossin, Y. Zhang, Y. L. Liu, W. Zhu, X. Feng, J. A. Yoo, E. Lee, K. Wang, R. Hui, L. H. Freitas-Junior and E. Oldfield, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4058–4063 CrossRef CAS.
- J. D. Artz, A. K. Wernimont, J. E. Dunford, M. Schapira, A. Dong, Y. Zhao, J. Lew, R. G. Russell, F. H. Ebetino, U. Oppermann and R. Hui, J. Biol. Chem., 2011, 286, 3315–3322 CrossRef CAS PubMed.
- S. H. Szajnman, A. Montalvetti, Y. Wang, R. Docampo and J. B. Rodriguez, Bioorg. Med. Chem. Lett., 2003, 13, 3231–3235 CrossRef CAS PubMed.
- S. H. Szajnman, G. E. Garcia Linares, Z. H. Li, C. Jiang, M. Galizzi, E. J. Bontempi, M. Ferella, S. N. Moreno, R. Docampo and J. B. Rodriguez, Bioorg. Med. Chem., 2008, 16, 3283–3290 CrossRef CAS PubMed.
- V. S. Rosso, S. H. Szajnman, L. Malayil, M. Galizzi, S. N. Moreno, R. Docampo and J. B. Rodriguez, Bioorg. Med. Chem., 2011, 19, 2211–2217 CrossRef CAS PubMed.
- M. Recher, A. P. Barboza, Z. H. Li, M. Galizzi, M. Ferrer-Casal, S. H. Szajnman, R. Docampo, S. N. Moreno and J. B. Rodriguez, Eur. J. Med. Chem., 2013, 60, 431–440 CrossRef CAS.
- J. E. Gisselberg, Z. Herrera, L. M. Orchard, M. Llinás and E. Yeh, Cell Chem. Biol., 2018, 25, 185–193 CrossRef CAS PubMed.
- A. Rani, G. Singh, A. Singh, U. Maqbool, G. Kaur and J. Singh, RSC Adv., 2020, 10, 5610–5635 RSC.
- K. Bozorov, J. Zhao and H. A. Aisa, Bioorg. Med. Chem., 2019, 27, 3511–3531 CrossRef CAS.
- M. C. Joshi, K. J. Wicht, D. Taylor, R. Hunter, P. J. Smith and T. J. Egan, Eur. J. Med. Chem., 2013, 69, 338–347 CrossRef CAS.
- N. Boechat, L. Ferreira Mde, L. C. Pinheiro, A. M. Jesus, M. M. Leite, C. C. Junior, A. C. Aguiar, I. M. de Andrade and A. U. Krettli, Chem. Biol. Drug Des., 2014, 84, 325–332 CrossRef CAS.
- E. O. Porta, P. B. Carvalho, M. A. Avery, B. L. Tekwani and G. R. Labadie, Steroids, 2014, 79, 28–36 CrossRef CAS PubMed.
- X. M. Chu, C. Wang, W. L. Wang, L. L. Liang, W. Liu, K. K. Gong and K. L. Sun, Eur. J. Med. Chem., 2019, 166, 206–223 CrossRef CAS PubMed.
- P. Kumar, A. O. Achieng, V. Rajendran, P. C. Ghosh, B. K. Singh, M. Rawat, D. J. Perkins, P. Kempaiah and B. Rathi, Sci. Rep., 2017, 7, 6724 CrossRef.
- S. Balabadra, M. Kotni, V. Manga, A. D. Allanki, R. Prasad and P. S. Sijwali, Bioorg. Med. Chem., 2017, 25, 221–232 CrossRef CAS PubMed.
- E. O. J. Porta, S. N. Jäger, I. Nocito, G. I. Lepesheva, E. C. Serra, B. L. Tekwani and G. R. Labadie, MedChemComm, 2017, 8, 1015–1021 RSC.
- G. R. Labadie, A. de la Iglesia and H. R. Morbidoni, Mol. Diversity, 2011, 15, 1017–1024 CrossRef CAS PubMed.
- A. S. Thompson, G. R. Humphrey, A. M. DeMarco, D. J. Mathre and E. J. J. Grabowski, J. Org. Chem., 1993, 58, 5886–5888 CrossRef CAS.
- E. O. J. Porta, M. M. Vallejos, A. B. J. Bracca and G. R. Labadie, RSC Adv., 2017, 7, 47527–47538 RSC.
- D. G. Ghiano, A. de la Iglesia, N. Liu, P. J. Tonge, H. R. Morbidoni and G. R. Labadie, Eur. J. Med. Chem., 2017, 125, 842–852 CrossRef CAS PubMed.
- K. Katsuno, J. N. Burrows, K. Duncan, R. Hooft van Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. T. Slingsby, Nat. Rev. Drug Discovery, 2015, 14, 751–758 CrossRef CAS PubMed.
- S. Kabeche, J. Aida, T. Akther, T. Ichikawa, A. Ochida, M. J. Pulkoski-Gross, M. Smith, P. S. Humphries and E. Yeh, Bioorg. Med. Chem. Lett., 2021, 41, 127978 CrossRef CAS.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis and E. Cibrián-Uhalte, Nucleic Acids Res., 2017, 45, D945–D954 CrossRef CAS PubMed.
- https://chemminetools.ucr.edu/ .
- S. Kar and J. Leszczynski, Expert Opin. Drug Discovery, 2020, 15, 1473–1487 CrossRef CAS PubMed.
- https://www.organic-chemistry.org/prog/peo/ .
- https://www.molinspiration.com, v2014.11, 20-08- 2015.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- L. S. Campbell-Verduyn, L. Mirfeizi, R. A. Dierckx, P. H. Elsinga and B. L. Feringa, Chem. Commun., 2009, 2139–2141 RSC.
- C. Girard, E. Önen, M. Aufort, S. Beauvière, E. Samson and J. Herscovici, Org. Lett., 2006, 8, 1689–1692 CrossRef CAS PubMed.
- S. S. Maurya, A. Bahuguna, S. I. Khan, D. Kumar, R. Kholiya and D. S. Rawat, Eur. J. Med. Chem., 2019, 162, 277–289 CrossRef CAS.
- E. A. Panozzo-Zénere, E. O. J. Porta, G. Arrizabalaga, L. Fargnoli, S. I. Khan, B. L. Tekwani and G. R. Labadie, Eur. J. Med. Chem., 2018, 143, 866–880 CrossRef PubMed.
- J. Mikus and D. Steverding, Parasitol. Int., 2000, 48, 265–269 CrossRef CAS PubMed.
- J. Fernández de Luco, A. I. Recio-Balsells, D. G. Ghiano, A. Bortolotti, J. M. Belardinelli, N. Liu, P. Hoffmann, C. Lherbet, P. J. Tonge, B. Tekwani, H. R. Morbidoni and G. R. Labadie, RSC Med. Chem., 2021, 12, 120–128 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00324d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |