
Discovery of N-sulfonylated aminosalicylic acids as dual MCL-1/BCL-xL inhibitors†
Lijia
Chen
a,
Jay
Chauhan
a,
Jeremy L.
Yap
a,
Christopher C.
Goodis
a,
Paul T.
Wilder
bc and
Steven
Fletcher
 *ac
*ac
aDepartment of Pharmaceutical Sciences, University of Maryland School of Pharmacy, 20 N. Pine St., Baltimore, MD 21201, USA. E-mail: steven.fletcher@rx.umaryland.edu
bUniversity of Maryland School of Medicine, 20 S. Greene St., Baltimore, MD 21201, USA
cUniversity of Maryland Greenebaum Cancer Center, 20 S. Greene St., Baltimore, MD 21201, USA
First published on 27th October 2022
Abstract
The anti-apoptotic protein MCL-1, which is overexpressed in multiple cancers, is presently a focus for the development of targeted drugs in oncology. We previously discovered inhibitors of MCL-1 based on 1-sulfonylated 1,2,3,4-tetrahydroquinoline-6-carboxylic acids (“1,6-THQs”). However, with the nitrogen atom constrained in the bicyclic ring, we were unable to modify the alkyl portion of the tertiary sulfonamide functionality. Moreover, the introduction of additional functional groups onto the benzene ring portion of the THQ bicycle would not be trivial. Therefore, we elected to deconstruct the piperidine-type ring of the 6-carboxy-THQ lead to create a new 4-aminobenzoic acid scaffold. Given its simplicity, this permitted us to introduce diversity at the sulfonamide nitrogen, as well as vary the positions and substituents of the benzene ring. One of our most potent MCL-1 inhibitors, 6e-OH, exhibited a Ki of 0.778 μM. Heteronuclear single quantum coherence experiments suggested 6e-OH bound in the canonical BH3-binding groove, with significant perturbations of R263, which forms a salt bridge with MCL-1's pro-apoptotic binding partners, as well as residues in the p2 pocket. Selectivity studies indicated that our compounds are dual inhibitors of MCL-1 and BCL-xL, with 17cd the most potent dual inhibitor: Ki = 0.629 μM (MCL-1), 1.67 μM (BCL-xL). Whilst selective inhibitors may be more desirable in certain instances, polypharmacological agents whose additional target(s) address other pathways associated with the disease state, or serve to counter resistance mechanisms to the primary target, may prove particularly effective therapeutics. Since selective MCL-1 inhibition may be thwarted by overexpression of sister anti-apoptotic proteins, including BCL-xL and BCL-2, we believe our work lays a solid foundation towards the development of multi-targeting anti-cancer drugs.
Introduction
Apoptosis is a mode of programmed cell death that is regulated by the BCL-2 proteins.1,2 Accordingly, more than three decades of research have been dedicated towards this family in the development of targeted antineoplastics.3–6 The family comprises two sub-populations: the anti-apoptotic proteins, which include MCL-1, BCL-xL and BCL-2, and the pro-apoptotic proteins, which include the multi-domain BH proteins, such as PUMA and NOXA, and the BH3-only proteins BAX and BAK.1,2 The balance of these two sub-populations is tightly regulated to direct the cell towards survival or death as required to maintain a healthy population.1,2 Ultimately, this fate is dictated by a protein–protein interaction (PPI) between the anti-apoptotic and pro-apoptotic proteins that serves to neutralize each other's function;1,2 whichever protein class is present in excess will determine the cell's destiny. Many cancers hijack this process, over-expressing anti-apoptotic proteins, which leads to cellular immortality.1,2 Since the BH3 α-helical domains of pro-apoptotic proteins recognize a shallow groove on the surface of anti-apoptotic proteins, the strategy to inhibit over-expressed anti-apoptotic proteins to treat cancer has centered around the deployment of synthetic “BH3 mimetics” to deceive the anti-apoptotic proteins, thereby enabling the BH3-only proteins BAX and BAK to homodimerize in the outer mitochondrial membrane, and initiate the apoptosis cascade.1,2 Particularly, the BH3 α-helix projects key residues along one face into pockets denoted p1–p4 in the BH3-binding groove on its partner protein.1,2,7 In addition, a conserved aspartate on the opposing face of the helix forms a salt bridge with a conserved arginine (R263) on the side of the groove. Synthetic BH3 mimetics typically target one or more of these pockets as well as R263.1,2,4–8Initial BCL-2 inhibitors were pan-inhibitors, exhibiting similar binding affinities to multiple anti-apoptotic BCL-2 members, such as the pyrogallol derivative TW37.9 Intense drug discovery programs have guided the design of selective inhibitors of these proteins,5,6 including the FDA-approved BCL-2 inhibitor venetoclax,10 the BCL-xL inhibitor A-1331852 (ref. 11) and, more recently MCL-1 inhibitors, such as the indole-2-carboxylic acids12,13 and -2-acylsulfonamides14 (Fig. 1), typified by 1a and 1b, a family of sulfonylated aminonaphthalenes,15e.g. UMI-77 (2), and the macrocyclic compound AZD5991 (3),16 which is currently in clinical trials for haematological malignancies, although one trial remains suspended owing to an adverse cardiac event.17 Our laboratory has also discovered MCL-1 inhibitors based on, among others,18–21,22 a racemic 1,2,3,4-tetrahydroquinoline-3-carboxylic acid (3-THQ) scaffold, such as (±)-4.23 A significant majority of reported MCL-1 inhibitors present a carboxylic acid to bind R263, as well as hydrophobic groups to bind the p2 pocket (and sometimes additional pockets).4–6,8 With selective inhibitors in hand, it becomes possible to interrogate the biological implications of the inhibitions of specific targets. However, ultimately a therapeutic regimen may comprise a drug “cocktail” in which multiple drugs bind multiple targets that together result in a more efficacious treatment of the disease.24 The major determinants of resistance to the BCL-2 drug venetoclax are overexpression of MCL-1 and/or BCL-xL.25 Accordingly, combination therapies of venetoclax with MCL-1 or BCL-xL inhibitors are being investigated.25 An alternative strategy to combination therapy, or polypharmacy, is the development of single agents that are purposely designed to bind multiple targets, i.e. polypharmacology. Polypharmacy and polypharmacology have been reviewed extensively elsewhere, and each carries its own advantages and disadvantages.26 The most significant concern in polypharmacology is the inability to alter the dosing of one pharmacophore over the other, yet that might be outweighed by enhanced additive or synergistic effects. Herein, we describe our continued efforts into the development of inhibitors of the BCL-2 anti-apoptotic proteins, and report the discovery of dual MCL-1/BCL-xL inhibitors based on sulfonylated aminosalicylic acids.
 | ||
| Fig. 1 Some MCL-1 inhibitors spanning a range of chemotypes. | ||
Previously, we reported the 3-THQ derivative (±)-4 as an inhibitor of MCL-1 (Ki = 0.12 μM).23 This compound presented a carboxylic acid to engage R263 through a salt bridge, and a 4-chloro-3,5-dimethylphenoxyphenyl group projected from the nitrogen atom of a sulfonamide linker, which was predicted to probe deep into the p2 pocket. The 3-THQ scaffold was then isomerized into a 1,2,3,4-tetrahydroquinoline-6-carboxylic acid (6-THQ) scaffold to remove the chiral center, as exemplified by compound 5 (Fig. 2), although this led to a significant decrease in MCL-1 binding affinity (Ki = 2.73 μM).27 In the structure of the 6-THQ scaffold, the nitrogen atom is fused in the THQ ring, restricting the ability to alter the N-alkyl component, and modification of the phenyl ring is not trivial. To overcome these barriers to further optimization of the 6-THQ series, we decided to re-engineer the 6-THQ scaffold by deconstructing the piperidine-type ring, thereby transforming the core into a 4-aminobenzoic acid scaffold (e.g.6e) as shown in Fig. 2. This particular scaffold offers an acyclic nitrogen atom at the same para position compared to the 6-THQ scaffold, but now it may be readily doubly-functionalized. Although this modification may be deleterious owing to a loss of rigidity, benefits towards structure–activity relationship studies include (i) a wide range of commercially available 4-aminobenzoic acids, (ii) the symmetry renders the introduction of additional functionality more controlled, and (iii) the location of the nitrogen atom may be easily shifted.
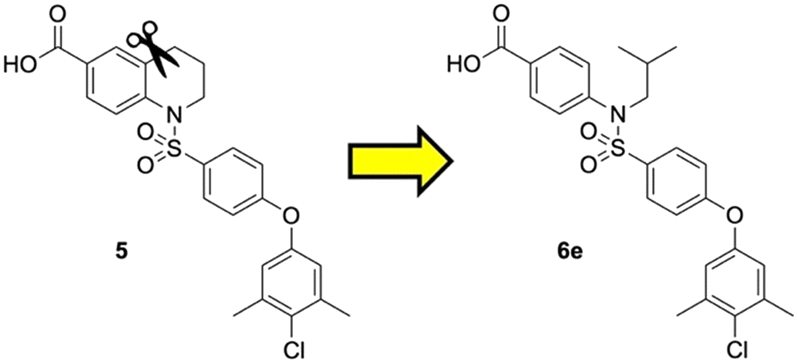 | ||
| Fig. 2 Dismantling the 6-THQ-based MCL-1 inhibitor 5 into a more flexible and more optimizable ligand. | ||
A GOLD docking experiment was performed to investigate the possibility of compound 6e binding to the MCL-1 protein. The small-molecule was first MM2 energy minimized, whilst the MCL-1 protein crystal structure (4HW2) was processed to remove the ligand and water molecules. The binding site was defined as a 10 Å radius sphere around residue M231, which is located at the entrance to the p2 pocket. No further constraints were used. One high-scoring result is shown in Fig. 3. As illustrated, the 4-chloro-3,5-dimethylphenyl moiety of compound 6e successfully docked into the p2 binding pocket of MCL-1, there are potential electrostatic interactions and hydrogen bonds between the carboxylic acid and sulfonamide of 6e with R263, and there appears to be a hydrophobic interaction between the isobutyl group of 6e and a shallow hydrophobic pocket formed by A227, F228, M231 and F270, as seen elsewhere.12
 | ||
| Fig. 3 Predicted binding mode of 4-sulfonamidobenzoic acid derivative 6e with MCL-1 (PDB ID: 4HW2) using GOLD software: (a) the MCL-1 protein is shown in grey helical structure with key residues in the p2 pocket shown in sticks (carbon: gray; nitrogen: blue; oxygen: red; sulfur: yellow), 6e is shown in stick structure (carbon: green; nitrogen: blue; oxygen: red; sulfur: yellow); hydrogen bonds are shown in black dashed lines; (b) as in (a) but with the MCL-1 protein shown with a solvent-accessible surface (hydrophobic: grey; basic: blue; acidic: red; sulfur: yellow), and the locations of the pockets p1–p4 are indicated. | ||
Encouraged by the docking result, a focused library of compounds was synthesized according to Scheme 1. Briefly, 4-nitrobenzoic acid (7) was esterified with methanol and sulfuric acid to yield methyl ester 8. The nitro group of 8 was reduced with tin(II) chloride to deliver aniline 9, which underwent reductive amination with isobutyraldehyde to afford compound 10. Subsequently, sulfonylations of 10 generated the library of compounds 11a–11d, which were then saponified with lithium hydroxide to yield the 4-sulfonamidobenzoic acid final molecules 6a–6d. Alternatively, compound 11d was further elaborated in an SNAr reaction with 4-chloro-3,5-dimethylphenol, followed by saponification to give final compound 6e.
 | ||
Scheme 1 Synthesis of 4-sulfonamidobenzoic acid analogues. Reagents and conditions: (a) H2SO4, MeOH, 80 °C, overnight, 98%; (b) SnCl2·2H2O, EtOAc, 50 °C, overnight, 85%; (c) isobutyraldehyde, NaBH(OAc)3, DCE, RT, overnight, 86%; (d) RSO2Cl, DIPEA, DMAP, CHCl3, 60 °C, overnight, 80–92%; (e) LiOH·H2O, THF–MeOH–H2O, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, RT, overnight, 90–98%; (f) 4-chloro-3,5-dimethylphenol, K2CO3, DMSO, 100 °C, overnight, 86%. :1:1, RT, overnight, 90–98%; (f) 4-chloro-3,5-dimethylphenol, K2CO3, DMSO, 100 °C, overnight, 86%. | ||
The binding affinities of the final molecules were evaluated using a well-established fluorescence polarization competition assay (FPCA) where the compounds were titrated into a mixture of MCL-1172-327 and FITC-BAK BH3 peptide to monitor their abilities to disrupt this interaction, as described previously.18–20,22,23,28,29 Although the unsubstituted phenyl analogue 6a and the bioisostere 6d demonstrated no inhibitory activity, building off the para-position as in biphenyl analogue 6c resulted in moderate inhibition of MCL-1 with a Ki of 16.7 μM. In contrast to 6c, the naphthyl analogue 6b was inactive (Ki > 500 μM). The data indicate that the geometrical shape of rigid hydrophobic moieties may have significant influence on binding affinities. Analogous to our discovery in the 3-THQ and 6-THQ SAR library studies,23 further extension of the R group with the 4-chloro-3,5-dimethylphenoxy moiety yielded compound 6e as the most potent analogue in this batch, whose binding affinity (Ki = 2.06 μM) was ≥10-fold stronger than 6c. Taken together, these findings suggest that although the binding domain for the sulfonamide R group – possibly the p2 pocket – is not wide, it is deep, which is consistent with previous reports.12
With further investigation of the docking result in Fig. 3, we suspected that installing extra polar functional groups ortho to the carboxylic acid may yield additional interactions with either R263 and/or T266. Accordingly, we transformed 4-aminobenzoic acid derivative 6e into its corresponding 4-aminosalicylic acid derivative “6e-OH” and performed another GOLD docking experiment; one high scoring result is depicted in Fig. 4. In this particular predicted conformation, the carboxylic acid forms a salt bridge with R263, as well as a hydrogen bond with T266. Additionally, the hydroxyl successfully recognized and hydrogen bonded to R263. Therefore, we hypothesized that 4-sulfonamidosalicylic acids, such as 6e, would be stronger binders compared to the corresponding 4-sulfonamidobenzoic acids (Table 1).
 | ||
| Fig. 4 Predicted binding mode of 4-sulfonamidosalicylic acid 6e-OH with MCL-1 (PDB ID: 4HW2) using GOLD: the MCL-1 protein is shown with a solvent-accessible surface (hydrophobic: grey; basic: blue; acidic: red; sulfur: yellow), and the locations of the pockets p1–p4 are indicated; 6e-OH is shown in stick structure (carbon: green; nitrogen: blue; oxygen: red; sulfur: yellow). | ||
|
|
||
|---|---|---|
| Compound | R | MCL-1 Ki (μM) |
| 6a |

|
>500 |
| 6b |
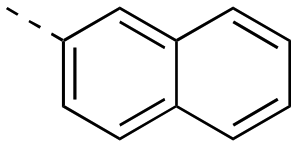
|
>500 |
| 6c |
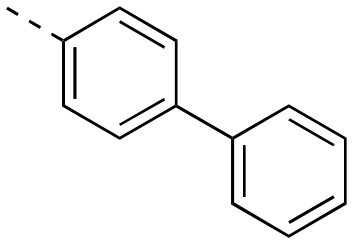
|
16.7 ± 2.3 |
| 6d |
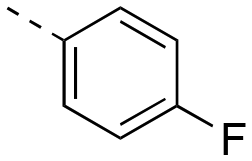
|
>500 |
| 6e |

|
2.06 ± 0.12 |

To test this hypothesis, we designed and synthesized a library of 4-sulfonamidosalicylic acid analogues according to Scheme 2. Esterification of 4-aminobenzoic acid (12) yielded methyl ester 13, whose phenolic hydroxyl was protected to afford compound 14. Reductive aminations of aniline 14 with isobutyraldehyde, benzaldehyde or cyclopentanone installed the R1 group, furnishing compounds 15a–15c. Sulfonylations (introduction of the R2 group) of the secondary amines 15 delivered sulfonamides 16, which were then saponified followed by TFA-mediated debenzylation to generate the target molecules 17.30 Alternatively, compounds 16ad, 16bc and 16cc were further elaborated with different substituted phenols R3OH through SNAr chemistry to afford biphenyl ethers 16ah–16ak, 16bd and 16cd. Subsequently, these compounds were subjected to the same deprotection protocol as before to yield the target molecules 17ah–17aj, 6e-OH, 17bd and 17cd. Additionally, compound 14 was directly sulfonylated by 4-phenoxybenzenesulfonyl chloride to yield, after saponification, compound 17da′, which was subsequently debenzylated to furnish target molecule 17da.
 | ||
| Scheme 2 Synthesis of 4-sulfonamidosalicylic acid analogues. Reagents and conditions: (a) H2SO4, MeOH, reflux, overnight, 95%; (b) BnBr, KOtBu, DMF, RT, overnight, 56–70%; (c) RCHO or cyclopentanone, NaBH(OAc)3, DCE, RT, overnight, 64–86%; (d) R2–SO2Cl, DIPEA, DMAP, CHCl3, 60 °C, overnight, 13–92%; (e) LiOH·H2O, THF–MeOH–H2O, 3:1:1, RT, overnight, 80–99%; (f) TFA, toluene, RT, overnight 64–83%; (g) R3-phenol, K2CO3, DMSO, 100 °C, overnight, 59–87%. | ||
Using the same FPCA experiment as described earlier, the MCL-1 binding affinities of the 4-sulfonamidosalicylic acid derivatives were determined (Table 2). Excitingly, all of these compounds exhibited stronger MCL-1 binding affinities compared to their corresponding 4-sulfonamidobenzoic acid counterparts in Table 1, demonstrating the significant contribution from the addition of the OH group, as predicted by the molecular modeling (Fig. 4). Furthermore, increasing the R2 size and hydrophobicity yielded stronger inhibitors, illustrated by the decreasing Kis of almost 50-fold from the phenyl 17aa (Ki = 192 μM), 4-flurophenyl 17ad, 4-methylphenyl 17af, 4-bromophenyl 17ae, 2-naphthyl 17ab to 4-biphenyl 17ac (Ki = 4.11 μM). Further expansion of the R2 moiety from the para position of the benzene sulfonamide to deliver biphenyl ethers produced the group of tighter binders 17ag–17aj and 6e-OH, indicating a larger and kinked R2 group is desirable, affording greater hydrophobic interactions with the BH3-bindng groove, likely enabling deeper access to the p2 pocket. Again, the most potent analogue 6e-OH was equipped with the 4-chloro-3,5-dimethylphenoxy group, inhibiting MCL-1 with a Ki of 778 nM.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | MCL-1 Ki (μM) | Compound | R1 | R2 | MCL-1 Ki (μM) |
| 17aa |
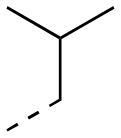
|
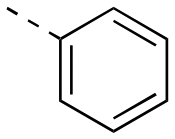
|
192 ± 37.5 | 6e-OH |

|

|
0.778 ± 0.050 |
| 17ab |
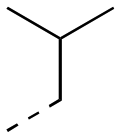
|

|
13.8 ± 1.3 | ||||
| 17ac |
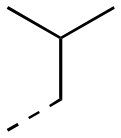
|
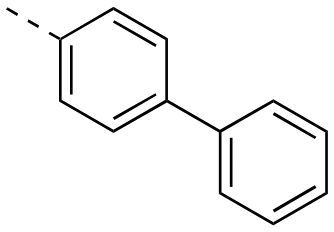
|
4.11 ± 0.25 | 17ba |

|
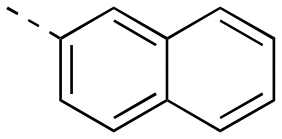
|
24.7 ± 9.4 |
| 17ad |

|

|
99.4 ± 9.4 | 17bb |
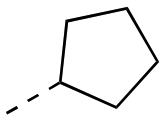
|

|
5.24 ± 0.69 |
| 17ae |
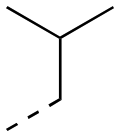
|
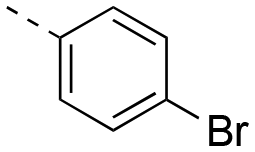
|
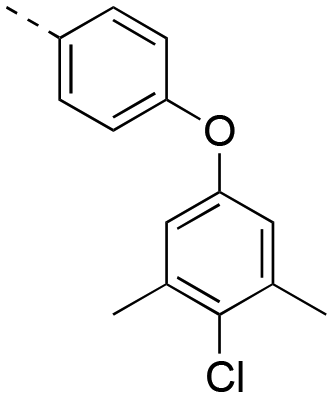
16.9 ± 1.5 | 17bd |

|
|
1.76 ± 0.21 |
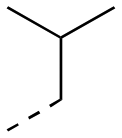
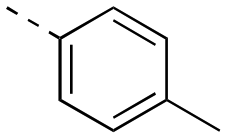
| 17af |
|
|
61.2 ± 4.5 | ||||
| 17ag |
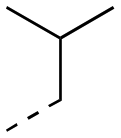
|

|
5.77 ± 0.46 | 17ca |
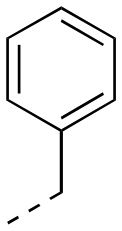
|
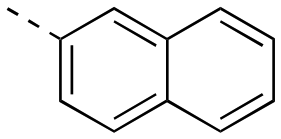
|
11.4 ± 1.6 |
| 17ah |
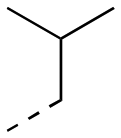
|
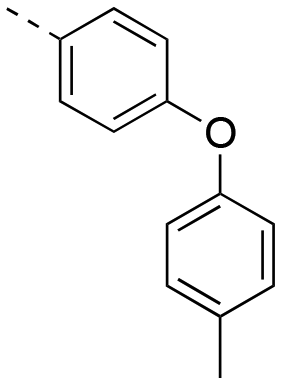
|
2.26 ± 0.17 | 17cb |

|
|
1.71 ± 0.14 |
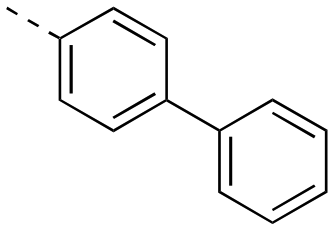
| 17ai |

|
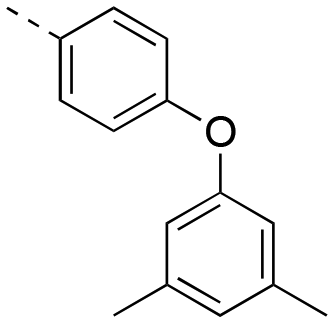
|
2.96 ± 0.29 | 17cd |
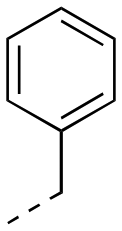
|
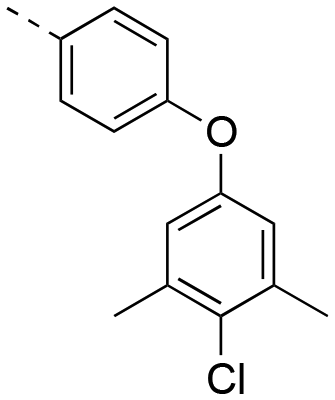
|
0.659 ± 0.040 |
| 17aj |
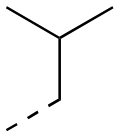
|
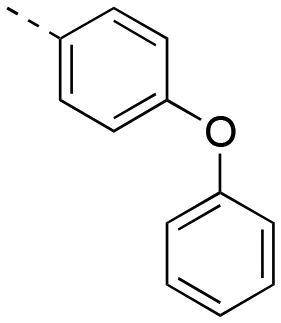
|
2.74 ± 0.26 | 17da | H |
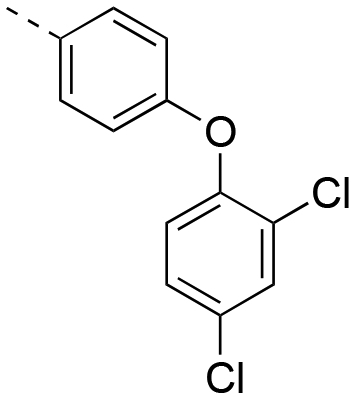
|
13.7 ± 1.1 |

In addition to varying the R2 sulfonyl moiety, we modified the R1 group to determine its impact on binding to MCL-1. In each case, consistent increases in binding affinities from naphthyl to 4-biphenyl to 4-chloro-3,5-dimethylphenoxyphenyl were observed within a family of R1 compounds, with R1 = cyclopentyl the worst, and R1 = benzyl the best. Lastly, it is noteworthy that completely eliminating the hydrophobic R1 group confirmed its significance: 17da (R1 = H), Ki = 13.7 μM; 17ag (R1 = isobutyl), Ki = 5.77 μM.
Upon dismantling the 6-THQ ring, as well as relinquishing the constraint on the identity of the nitrogen alkyl appendage, there was also now no restriction on the relative position of the nitrogen atom. Inspection of previous docking results suggested that the optimal relative locations of the carboxylic acid and the sulfonamido groups were para and meta. Thus, we designed 5-sulfonamidosalicylic acids, such as 6e-OH-5, to ascertain which substitution pattern was optimal. As illustrated in Fig. 5, 6e-OH-5 favorably fitted into the MCL-1 binding pocket and developed similar polar interactions with R263 and T266 compared to its predecessor 4-sulfonamidosalicylic acid 6e-OH (Fig. 4). Additionally, the hydrophobic 4-chloro-3,5-dimethylphenyl moiety probed into the p2 pocket of MCL-1. Following the docking study, a library of 5-aminosalicylic acids was designed and synthesized according to Scheme 3 to validate the in silico model. In addition, the function of the hydroxyl group was re-assessed in this new scaffold by deleting it to generate 3-sulfonamidobenzoic acid derivatives.
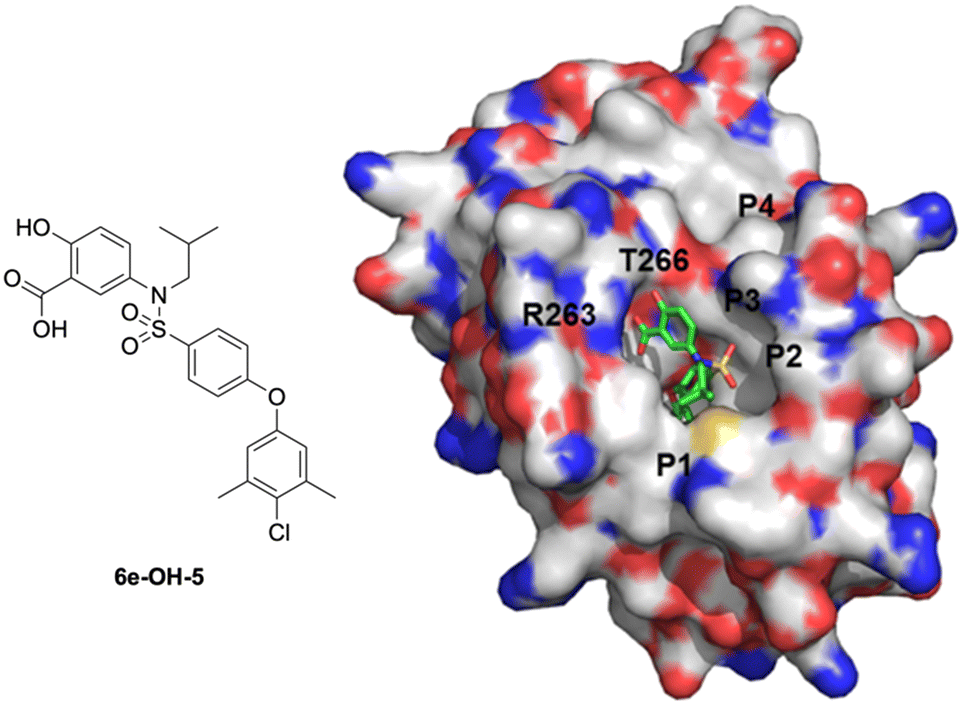 | ||
| Fig. 5 Predicted binding mode of 5-sulfonamidosalicylic acid 6e-OH-5 with MCL-1 (PDB ID: 4HW2) using GOLD: the MCL-1 protein is shown with a solvent-accessible surface (hydrophobic: grey; basic: blue; acidic: red; sulfur: yellow), and the locations of the pockets p1–p4 are indicated; 6e-OH-5 is shown in stick structure (carbon: green; nitrogen: blue; oxygen: red; sulfur: yellow). | ||
 | ||
| Scheme 3 Synthesis of 3-sulfonamidobenzoic acid and 5-sulfonamidosalicylic acid analogues. Reagents and conditions: (a) H2SO4, MeOH, 80 °C, overnight, 68–90%; (b) isobutyraldehyde, NaBH(OAc)3, DCE, RT, overnight, 63–73%; (c) RSO2Cl, DIPEA, DMAP, CHCl3, 60 °C, overnight, 24–98%; (d) LiOH·H2O, THF–MeOH–H2O, 3:1:1, RT, overnight, 45–98%; (e) BnBr, KOtBu, DMF, RT, overnight, 70%; (f) 4-chloro-3,5-dimethylphenol, K2CO3, DMSO, 100 °C, overnight, 30–60%; (g) TFA, toluene, RT, overnight, 98%. | ||
The target molecules were synthesized similarly to in Schemes 1 and 2, with the R1 group constrained as isobutyl. Briefly, esterifications of 3-aminobenzoic acid (18a) and 5-aminosalicylic acid (18b) afforded compounds 19a and 19b, respectively. Reductive aminations with isobutyraldehyde furnished compounds 20a and 20b, which were further functionalized with different sulfonyl chlorides (RSO2Cl) to yield sulfonamides 21aa–21ad, and 21ba–21bc. These compounds were then subjected to saponifications to yield the final molecules 22aa–22ad, and 22ba–22bc. Also, compound 21ad of the 3-sulfonamidobenzoic acid family was further elaborated by an SNAr reaction with 4-chloro-3,5-dimethylphenol, followed by saponification to afford compound 22af. In the case of the 5-sulfonamidosalicylic acid family, compound 21bd was first O-benzylated to give compound 21bd′ before undergoing an SNAr with 4-chloro-3,5-dimethylphenol to afford 21be, and was then subjected to saponification followed by TFA-mediated debenzylation to yield final compound 6e-OH-5.30
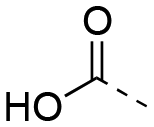
The SAR data for both 3-sulfonamidobenzoic acid and 5-sulfonamidosalicylic acid derivatives are reported in Table 3. The pronounced differences of Kis between the benzoic acids (left) and the salicylic acids (right) demonstrate the compelling benefits generated by the hydroxyl group as seen earlier with the 4-aminobenzoic/salicylic acids by comparing Tables 1 and 2. For example, 4-biphenyl 22ac was inactive, yet conversion of the scaffold into the corresponding salicylic acid 22bc resulted in a moderate inhibitor of MCL-1 (Ki = 8.70 μM). Intriguingly, in the benzoic acid class, the 2-naphthyl analogue 22ab (Ki = 61.7 μM) exhibited a stronger binding affinity than the 4-biphenyl counterpart 22ac (Ki > 500 μM), which is in stark contrast to the relationships between the corresponding 4-aminobenzoic acid derivatives 6b and 6c, respectively (Table 1). This suggests that the shifted nitrogen may have changed the binding orientation of the scaffold, potentially now directing the 4-biphenyl moiety at an unfavorable angle towards MCL-1, while guiding the 2-naphthyl group in a favorable direction towards the BH3-binding groove, possibly the p2 pocket. The most potent compounds 22af and 6e-OH-5 carry the 4-chloro-3,5-dimethyl-4-chlorophenoxy group, with Ki = 1.45 μM and 586 nM, respectively, as with other series of compounds. Once again, in light of the data for biphenyls 22ac, in particular, and 22bc, it appears that the ether oxygens of 22af and 6e-OH-5 are pivotal in realizing more effective inhibition of MCL-1, probably through the inherent kink of the ether functional group that promotes better interactions with the terminal (and now more substituted) aryl ring. Although the library of 5-sulfonamidosalicylic acids is limited, the data suggest that the 4-sulfonamidosalicylic acids and the 5-sulfonamidosalicylic acids isomers are comparable in MCL-1 inhibitory activity.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | X | R | MCL-1 Ki (μM) | Compound | X | R | MCL-1 Ki (μM) |
| 22aa | H |
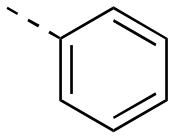
|
>500 | 22ba | OH |

|
46.5 ± 9.8 |
| 22ab | H |

|
61.7 ± 4.7 | ||||
| 22ac | H |

|
>500 | 22bb | OH |
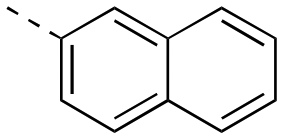
|
7.51 ± 0.84 |
| 22ad | H |

|
>500 | 22bc | OH |
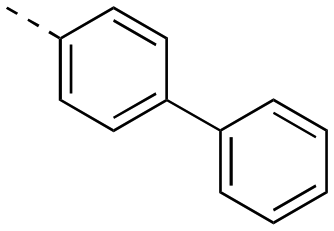
|
8.70 ± 2.05 |
| 22ae | H |

|
155 ± 14 | ||||
| 22af | H |

|
1.45 ± 0.89 | 6e-OH-5 | OH |
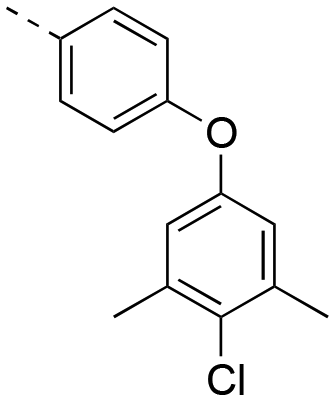
|
0.586 ± 0.041 |

HSQC-NMR studies of the binding pattern of 4-aminosalicylic acid with MCL-1
To gain a better understanding of the likely binding site of our compounds, one representative compound (6e-OH) was examined by heteronuclear single quantum coherence (HSQC)-NMR studies in complex with 15N-MCL-1. The 2D 1H–15N HSQC spectra of MCL-1 were collected with 6e-OH (red) and without 6e-OH (black), and they are overlaid in Fig. 6. The chemical shift (δ) perturbations have been plotted in Fig. 7, with significant perturbations (Δδ > 0.3 ppm) shown in red. These figures clearly show that R263 and T266 were both significantly perturbed upon the binding of 6e-OH. Moreover, the majority of hydrophobic residues in the p2 binding pocket, which are primarily located at MCL-1 protein helixes α2 to α6, were heavily perturbed. The residues whose chemical shifts were not significantly perturbed are shown in grey, the bulk of which are located far from the BH3-binding groove. Subsequently, we mapped the significant perturbations (Δδ > 0.3 ppm) onto the MCL-1 crystal structure (PDB ID: 4HW3), colouring those residues in red (Fig. 8): as illustrated, the bulk of those residues are located in and around the BH3-binding groove, providing support to our proposed binding mode. | ||
| Fig. 6 1H–15N HSQC spectra overlay of apo-MCL-1 (black) and 6e-OH bound MCL-1 (red). | ||
 | ||
| Fig. 7 Plot of chemical shift of MCL-1 protein upon 6e-OH binding, red columns are the amino acid residues that were significantly perturbed (Δ chemical shift >0.3 ppm). | ||
 | ||
| Fig. 8 NMR chemical shift perturbations of the MCL-1/6e-OH complex mapped onto MCL-1 crystal coordinates (PDB ID: 4HW3) in PyMOL. Residues experiencing chemical shifts of at least 0.3 ppm are shaded red. | ||
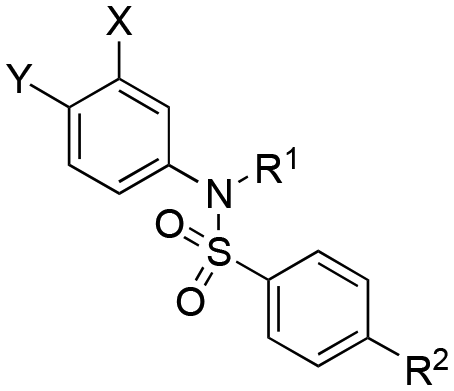
MCL-1/BCL-xL binding selectivity of 4-aminosalicylic acids and 5-aminosalicylic acids
Selective MCL-1 inhibitors first appeared over a decade ago.12,14,15,31–34 We were curious to determine if our compounds were also selective for MCL-1, so we evaluated the BCL-xL binding affinities of our more potent MCL-1 inhibitors, and the data are displayed in Table 4. Whilst our compounds present many similarities to other published, selective MCL-1 inhibitors, such as an aryl carboxylic acid and p2-binding group that is hydrophobic and connected to the scaffold with a good degree of flexibility to support a re-modeling of the p2 pocket,4,6,12,15 we observed little-to-no-selectivity. In fact, the most selective was 17cd at just under 3-fold selectivity for MCL-1 over BCL-xL. The most striking difference between our non-selective inhibitors and several of the more potent and selective MCL-1 inhibitors is that ours are devoid of a bicyclic scaffold harboring the R263-targeting carboxylic acid, in contrast to the reported bicyclic (or even tricyclic32) indole12,14 and naphthalene15 derivatives. Indeed, we revisited our original THQ compounds and discovered that (±)-4 exhibited a selectivity for MCL-1 over BCL-xL of around 42-fold, comparable with other selectivities with bicyclic scaffolds,12,15 ((±)-4: Ki = 0.12 μM (MCL-1), 5.01 μM (BCL-xL)). To the contrary, THQ 5 offered no selectivity (Ki = 2.73 μM (MCL-1) and 3.56 μM (BCL-xL)). Taken together, these findings suggest that the structure of the scaffold, as well as the relative positionings of the acidic function and the p2-binding group are all significant determinants of selectivity.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | X | Y | R1 | R2 | MCL-1 Ki (μM) | BCL-xL Ki (μM) |
K
i (MCL-1):Ki (BCL-xL) |
| 17aa | OH |
|

|
|
192 ± 37 | 154 ± 28 | 1:0.80 |
| 17ab | OH |
|

|
|
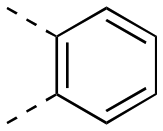
13.8 ± 1.3 | 43.1 ± 14.7 | 1:3.12 |
| 17ac | OH |

|

|

|
4.11 ± 0.25 | 10.8 ± 2.6 | 1:2.63 |
| 17ag | OH |

|
|

|
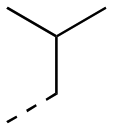
5.77 ± 0.46 | 17.9 ± 4.6 | 1:3.12 |
| 6e-OH | OH |
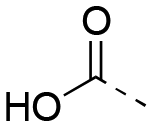
|
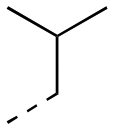
|
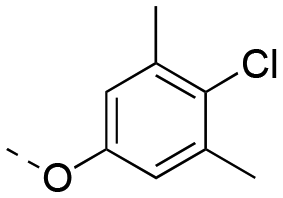
|
0.778 ± 0.050 | 1.63 ± 0.18 | 1:2.09 |
| 17bd | OH |
|
|
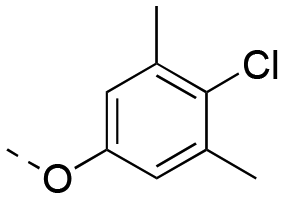
|
1.76 ± 0.21 | 1.91 ± 0.12 | 1:1.08 |
| 17cd | OH |
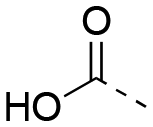
|
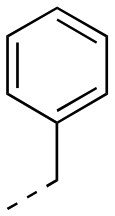
|
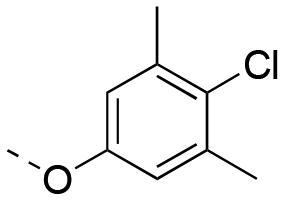
|
0.629 ± 0.040 | 1.67 ± 0.35 | 1:2.65 |
| 22ba |
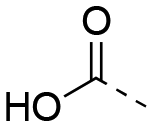
|
OH |
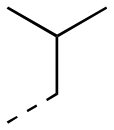
|
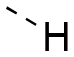
|
46.5 ± 9.8 | 49.3 ± 5.0 | 1:1.06 |
| 22bb |
|
OH |

|
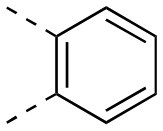
|
7.51 ± 0.84 | 44.6 ± 5.9 | 1:5.94 |
| 22bc |
|
OH |

|
|
8.70 ± 2.05 | 13.9 ± 3.3 | 1:1.59 |
| 6e-OH-5 |

|
OH |

|

|
0.586 ± 0.041 | 2.91 ± 0.19 | 1:4.96 |
Conclusions
We previously reported a class of MCL-1 inhibitors – herein discovered to be selective for MCL-1 over BCL-xL – based on chiral 1-sulfonamido-THQ-3-carboxylic acids. Subsequent isomerization to the achiral 1-sulfonamido-THQ-6-carboxylic acid counterparts27 followed by deconstructing the piperidine-type ring to afford 4-sulfonamidobenzoic acid derivatives resulted in moderately potent MCL-1 inhibitors, but none bound MCL-1 as tightly as the original THQ (±)-4. Nevertheless, the simpler 4-sulfonamidobenzoic acid scaffold permitted more facile diversification with greater commercial availabilities of precursors. The introduction of a hydroxyl group ortho to the carboxylic acid enhanced MCL-1 binding in every case, and the isomeric 5-sulfonamidosalicylic acids exhibited comparable inhibitory activities. At the same time, the MCL-1 selectivity observed by (±)-4 was lost. We surmise this may be related to the loss of a bicyclic scaffold, as previously reported indoles and naphthalenes exhibit excellent selectivity profiles for MCL-1 over other BCL-2 anti-apoptotic protein,12,15 and/or the substitution pattern of the bicycle. Future work will include continued SAR of our aminosalicylic acid scaffolds, as well as enlargement of inhibitors by converting the carboxylic acid of lead compounds into acylsulfonamides to reach into the p4 pocket,14 guided by X-ray co-crystal studies that will also shed light on selectivity profiles. Two of the more common methods by which MCL-1 evades targeted drugs is the concomitant upregulation of BCL-2 and/or BCL-xL.35 As dual inhibitors of MCL-1 and BCL-xL, our lead inhibitors offer a platform for the development of novel drugs that may thwart the cancer cell's ability to overcome inhibition of MCL-1 with selective drugs. Furthermore, the continued development of more potent inhibitors is directly facilitated by our novel and simple aminosalicylic acid scaffolds.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We thank the University of Maryland School of Pharmacy, the University of Maryland School of Medicine Chemical Biology Therapeutics program and the National Institutes of Health (NIH/NIGMS T32 GM066706) for financial support of this research.Notes and references
- J. Kale, E. J. Osterlund and D. W. Andrews, Cell Death Differ., 2018, 25, 65–80 CrossRef CAS PubMed
.
- J. M. Adams and S. Cory, Cell Death Differ., 2018, 25, 27–36 CrossRef CAS PubMed
- A. R. D. Delbridge, S. Grabow, A. Strasser and D. L. Vaux, Nat. Rev. Cancer, 2016, 16, 99–109 CrossRef CAS PubMed
- S. Fletcher, Expert Opin. Ther. Pat., 2019, 29, 909–919 CrossRef CAS
- J. L. Yap, L. Chen, M. E. Lanning and S. Fletcher, J. Med. Chem., 2017, 60, 821–838 CrossRef CAS
- S. T. Diepstraten, M. A. Anderson, P. E. Czabotar, G. Lessene, A. Strasser and G. L. Kelly, Nat. Rev. Cancer, 2022, 22, 45–64 CrossRef CAS
- M. Sattler,
et al.
, Science, 1997, 275, 983–986 CrossRef CAS PubMed
- A. Negi and P. V. Murphy, Eur. J. Med. Chem., 2021, 210, 113038 CrossRef CAS PubMed
- C. Billard, Mol. Cancer Ther., 2013, 12, 1691–1700 CrossRef CAS
- A. J. Souers,
et al.
, Nat. Med., 2013, 19, 202–208 CrossRef CAS PubMed
- L. Wang,
et al.
, ACS Med. Chem. Lett., 2020, 11, 1829–1836 CrossRef CAS
- A. Friberg,
et al.
, J. Med. Chem., 2013, 56, 15–30 CrossRef CAS PubMed
- T. Lee,
et al.
, J. Med. Chem., 2019, 62, 3971–3988 CrossRef CAS PubMed
- N. F. Pelz,
et al.
, J. Med. Chem., 2016, 59, 2054–2066 CrossRef CAS
- F. A. Abulwerdi,
et al.
, J. Med. Chem., 2014, 57, 4111–4433 CrossRef CAS
- A. E. Tron,
et al.
, Nat. Commun., 2018, 9, 5341 CrossRef CAS PubMed
- AstraZeneca, “A Phase 1/1b/2a, 3-Part, Open-Label, Multicentre Study to Assess the Safety, Tolerability, Pharmacokinetics and Preliminary Antitumor Activity of Ascending Doses of AZD5991 Monotherapy and in Combination With Venetoclax in Subjects With Relapsed or Refractory Haematologic Malignancies”, https://clinicaltrials.gov, Clinical trial registration NCT03218683, Jan. 2022. Accessed: Feb. 14, 2022, Available: https://clinicaltrials.gov/ct2/show/NCT03218683.
- I. L. Conlon,
et al.
, Bioorg. Med. Chem. Lett., 2018, 28, 1949–1953 CrossRef CAS
- M. E. Lanning,
et al.
, Eur. J. Med. Chem., 2016, 113, 273–292 CrossRef CAS PubMed
- M. E. Lanning,
et al.
, Org. Biomol. Chem., 2015, 13, 8642–8646 RSC
- E. Whiting,
et al.
, Bioorg. Med. Chem. Lett., 2018, 28, 523–528 CrossRef CAS PubMed
- B. Drennen,
et al.
, RSC Med. Chem., 2022, 13, 963–969 RSC
- L. Chen,
et al.
, Org. Biomol. Chem., 2016, 14, 5505–5510 RSC
- Y. Gilad, G. Gellerman, D. M. Lonard and B. W. O'Malley, Cancers, 2021, 13, 669 CrossRef CAS
- P. Bose, V. V. Gandhi and M. Y. Konopleva, Leuk. Lymphoma, 2017, 58, 2026–2039 CrossRef CAS
- E. Proschak, H. Stark and D. Merk, J. Med. Chem., 2019, 62, 420–444 CrossRef CAS
- L. Chen,
et al., 1-Sulfonylated 1,2,3,4-Tetrahydroquinoline-6-carboxylic Acids as Simple, Readily-Accessible MCL-1 Inhibitors, Drug Dev. Res., 2022 DOI:10.1002/ddr.22004
- B. Drennen,
et al.
, ChemMedChem, 2016, 11, 827–833 CrossRef CAS PubMed
- I. L. Conlon,
et al.
, ChemMedChem, 2020, 15, 1691–1698 CrossRef CAS
- S. Fletcher and P. T. Gunning, Tetrahedron Lett., 2008, 49, 4817–4819 CrossRef CAS
- A. M. Petros,
et al.
, Bioorg. Med. Chem. Lett., 2014, 24, 1484–1488 CrossRef CAS PubMed
- J. P. Burke,
et al.
, J. Med. Chem., 2015, 58, 3794–3805 CrossRef CAS PubMed
- J. D. Leverson,
et al.
, Cell Death Discovery, 2015, 6, e1590 CrossRef CAS
- M. Bruncko,
et al.
, J. Med. Chem., 2015, 58, 2180–2194 CrossRef CAS
- A. Bolomsky,
et al.
, Blood Adv., 2021, 5, 4125–4139 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details, and 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d2md00277a |
| This journal is © The Royal Society of Chemistry 2023 |