
Structural rationalization of GSPT1 and IKZF1 degradation by thalidomide molecular glue derivatives†
Radosław P.
Nowak
 ab,
Jianwei
Che
ab,
Silas
Ferrao
a,
Nikki R.
Kong
ab,
Hu
Liu
ab,
Breanna L.
Zerfas
ab and
Lyn H.
Jones
*ab
ab,
Jianwei
Che
ab,
Silas
Ferrao
a,
Nikki R.
Kong
ab,
Hu
Liu
ab,
Breanna L.
Zerfas
ab and
Lyn H.
Jones
*ab
aCenter for Protein Degradation, Dana-Farber Cancer Institute, 360 Longwood Avenue, Boston, MA, USA
bDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA, USA. E-mail: lyn_jones@dfci.harvard.edu
First published on 26th January 2023
Abstract
Thalidomide and its derivatives are molecular glues that bind cereblon (CRBN), a component of an E3 ubiquitin ligase complex, and mediate protein interactions with neosubstrates resulting in their polyubiquitination and proteasomal degradation. The structural features of neosubstrate binding have been elucidated that highlight key interactions with a β-hairpin degron containing a glycine, which is present in a wide-range of proteins, including zinc-finger transcription factors such as IKZF1, and the translation termination factor GSPT1. Here, we profile 14 closely-related thalidomide derivatives in CRBN occupancy, and IKZF1 and GSPT1 degradation cell-based assays, and use crystal structures, computational docking and molecular dynamics to delineate subtle structure–activity relationships. Our findings will enable the rational design of CRBN modulators in the future, and help avoid the degradation of GSPT1 which is broadly cytotoxic.
Introduction
Immunomodulatory drugs thalidomide, lenalidomide and pomalidomide known as IMiDs bind cereblon (CRBN),1 a component of a ubiquitin E3 ligase complex (CRL4CRBN), and induce the recruitment, polyubiquitination and proteasomal degradation of neosubstrates.2,3 The drugs feature a glutarimide motif that binds in a tri-tryptophan cage in the thalidomide binding site at the surface of CRBN.4,5 The isoindolinone or phthalimide portion of the IMiD then mediates new protein interactions with a key recognition motif of the neosubstrate called a structural degron. The degron is a β-hairpin loop that contains an essential glycine residue – larger amino acid side chains at this position would clash with the CRBN-IMiD complex. Many neosubstrates are zinc-finger transcription factors, which possess the G-loop degron (a CxxCG motif), which have been difficult to drug using traditional small molecule modalities because they lack a suitable binding pocket.6 The proteins Ikaros (IKZF1) and Aiolos (IKZF3) are physiologically relevant neosubstrates of the IMiDs because they are essential transcription factors in multiple myeloma.3 Another important target is the embryonic transcription factor SALL4 because its degradation has been linked to the potential cause of teratogenicity of the IMiDs.7–9 Other neosubstrates are not zinc-finger transcription factors but still contain the G-loop degron. One such example is GSPT1 (G1 to S phase transition 1) which is degraded by certain thalidomide derivatives.10–16 GSPT1, although currently being targeted for the treatment of acute myeloid leukaemia (AML),17,18 is a translation termination factor and its depletion is broadly cytotoxic due to its essential cellular function,13,19 as reflected in the GSPT1 profile in the Cancer Dependency Map (DepMap, https://www.depmap.org).Considerable efforts have been made to structurally diversify ‘molecular glue’20,21 IMiD degraders to expand the repertoire of therapeutically relevant degradable proteins. However, a lack of understanding of the structural features that drive target and off-target degradation has hindered rational degrader design and medicinal chemistry optimization. Additionally, CRBN binding motifs have been incorporated into heterobifunctional proteolysis targeting chimeras (PROTACs) that hijack the E3 ubiquitin machinery to degrade bound targets.22,23 Certain PROTACs also possess the IMiD molecular glue mechanism and degrade targets such as GSPT1.19,24–29 It is important that drug discovery efforts to expand the utility of CRBN molecular glues and PROTACs consider the consequences of modulating targets that possess potential toxicities.30 GSPT1 off-target degradation may narrow therapeutic indices, that could be particularly important for non-oncology indications, or hinder the pharmacological validation of new degradable targets.
For these reasons, we decided to screen a variety of thalidomide derivatives to help establish GSPT1 and IKZF1 structure–activity relationships (SARs). Our study explores not only the features that affect CRBN binding but also the molecular interactions within the ternary complexes that are required to mediate subsequent degradation.
Results and discussion
To facilitate the generation of useful and transferrable SARs that elucidate key features of neosubstrate binding, we screened relatively simple IMiD derivatives. The 14-member glutarimide-containing compound set included the unsubstituted phthalimide (thalidomide) and isoindolinone (EM12) derivatives, the amino-substituted drugs lenalidomide and pomalidomide and their regioisomers, and their hydroxy congeners (Fig. 1). All compounds were acquired from commercial sources, or synthetically prepared using reported procedures (see ESI†).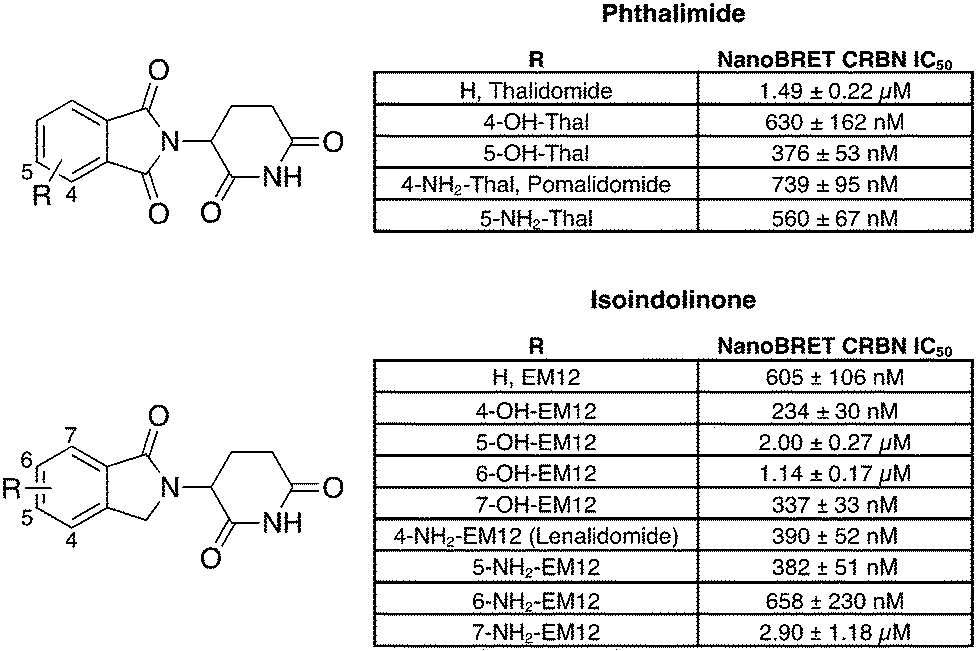 | ||
| Fig. 1 Cell-based cereblon binding potency of molecular glue degraders profiled in this work. The data are presented as IC50 ± Std. error of two replicates (N = 2). | ||
Our group recently developed a NanoBRET CRBN occupancy assay to help understand the cell-based pharmacology of CRBN-based degraders.31 The assay measures the dose-dependent reduction in BRET signal following displacement of a fluorescent CRBN tracer from NanoLuc-tagged CRBN in live cells. Noticeably, the range of potencies observed for these compounds was relatively narrow, all within a 12-fold span (Fig. 1). This likely reflects the predominant contribution to the binding energy from the recognition of the glutarimide motif within the tri-tryptophan cage, and the overall similar physicochemical nature of the ligands.
Development of a HiBiT IKZF1 cell-based degradation assay was also reported by us recently, which uses CRISPR-mediated tagging of the endogenous protein with a HiBiT peptide to enable quantitative measurement of IKZF1 degradation by the compound set using luminescence (Fig. 2A and D).32 For this study we employed a GSPT1Δ GFP/RFP degradation assay to assess the potency and efficacy of the derivatives (Fig. 2B and E).33 We utilized the GFP/RFP reporter system rather than HiBiT as it provides a very sensitive readout of GSPT1 stability based on a truncated GSPT1 (aa 389-499) construct.
 | ||
| Fig. 2 Cell-based HiBiT IKZF1 and GSPT1 GFP/mCherry degradation data of phthalimides (A–C) and isoindolinones (D–F). Compounds were incubated for 24 h for IKZF1 and 5 h for GSPT1 assay and the data are presented as two replicates (N = 2). Dmax, presented as mean ± std. error, corresponds to the maximal depth of degradation. The DC50 reflects the concentration where 50% degradation was observed. | ||
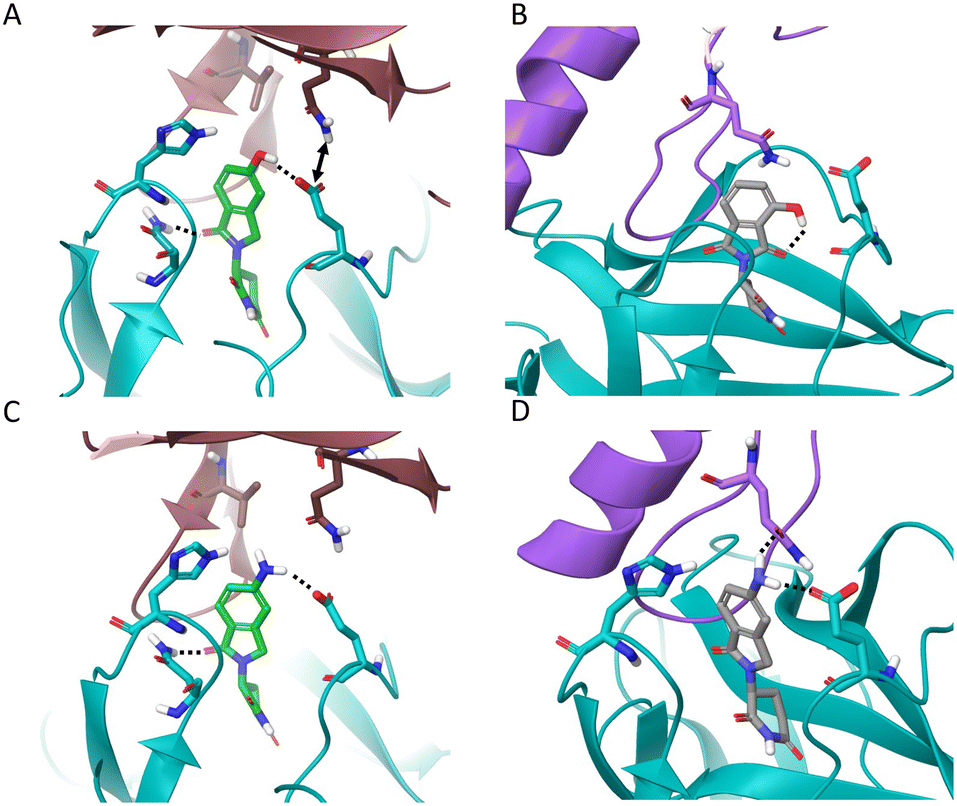
The reported structures of pomalidomide with CRBN/DDB1ΔB/IKZF1ZnF2 (PDB 6H0F),6 the thalidomide metabolite 5-OH-Thal with CRBN/SALL4,34 and the GSPT1 molecular glue degraders CC-885 and CC-90009 with CRBN/DDB1/GSPT1 (PDB 5HXB and 6XK9 respectively)12,17 were used to help rationalize the observed trends in CRBN occupancy and degradation potency.
We found that the isoindolinone EM12 is a moderately active degrader of IKZF1 (DC50 1.7 μM, Dmax 69 ± 6%), but it does not degrade GSPT1. 4-OH-EM12 more effectively degrades IKZF1 (DC50 28 nM, Dmax 82 ± 1%) because it likely binds in a similar manner to pomalidomide (Fig. 3A) by making a hydrogen bond (H-bond) with CRBN Glu377, holding the side chain in a conformation that enables an interaction through water to the Gln146 side chain in the IKZF1 G-loop. Additionally, 4-OH-EM12 more potently engages CRBN than EM12, likely due to the Glu377 H-bond, that could also translate to greater degradation potency.
 | ||
| Fig. 3 CRBN co-crystal structures of A) 4-NH2-Thal (pomalidomide) with IKZF1 ZnF2 (PDB: 6H0F), B) 5-OH-Thal with SALL4 ZnF2 (PDB: 7BQV), C) CC-885 with GSPT1 (PDB: 5HXB), and D) CC-90009 with GSPT1 (PDB: 6XK9). Dashed lines indicate hydrogen bonding. The residue numbers are based on the corresponding PDB structures – note residue index shift between the GSPT1 structures. | ||
5-OH-EM12 does not degrade IKZF1. Substitution at the 5-position may induce conformational changes in the proximal side chain of His353 in CRBN that prevent favorable interactions with Asn148 in the IKZF1 G-loop that are visible in PDB 6H0F (i.e., π–π stacking, and H-bonding through water) (Fig. 3A). Also, since the –NH2 motif of the Gln146 amide side chain acts as an H-bond donor to the backbone carbonyl of Gly151 across the β-hairpin, the carbonyl oxygen atom of the amide is likely placed in proximity to, and potentially clashes with the oxygen atom of the phenol present in 5-OH-EM12 (see Fig. 3A).
Surprisingly, 5-OH-EM12 degrades GSPT1 (Dmax 47 ± 9%) (Fig. 2E). The GSPT1 degraders CC-885 and CC-90009 also possess H-bonding motifs connected to the 5-position of the IMiD scaffold. An inspection of the crystal structures of CC-885 and CC-90009 clearly show H-bonding with CRBN Glu377 (Fig. 3C and D). The putative binding mode of 5-OH-EM12 based on computational docking is illustrated in Fig. 4A. Using molecular dynamics simulations (methods described in ESI†), we observe that the 5-OH group forms a direct H-bond with Glu377 72% of the time and a water-bridged H-bond 11% of the time. These interactions would position the Glu377 carboxylate side chain in a conformation that forms a salt bridge with Lys628 in GSPT1, as observed for CC-885 and CC-90009. However, the salt bridge interaction may not be as stable for 5-OH-EM12 because it is observed 64% of the time, which is consistent with its weaker degradation of GSPT1 compared to CC-885 and CC-90009.
 | ||
| Fig. 4 Docking models of A) 5-OH-EM12 in GSPT1/CRBN, B) 4-OH-Thal in IKZF1/CRBN, C) 5-NH2-EM12 in GSPT1/CRBN, and D) 5-NH2-EM12 in IKZF1/CRBN. The dotted lines indicate key potential hydrogen bonds that describe the data. | ||
The simulation positions the Glu377 residue to form a direct H-bond 70% of the time, and additionally form a water-mediated H-bond 22% of the time, with the Gln534 side chain in GSPT1. Effectively, these two residues interact almost all the time in the simulation (illustrated by an arrow in Fig. 4A), and this is likely essential to the recruitment and subsequent degradation of GSPT1.
These SARs also appear to hold for 5-OH-Thal, which degrades GSPT1 even more effectively (DC50 130 nM, Dmax 72 ± 2%) but not IKZF1 (the latter result was also reported recently) (Fig. 2D and E).35 The enhanced GSPT1 degradation efficacy of 5-OH-Thal versus 5-OH-EM12 may be driven by its more potent engagement of CRBN (Fig. 1). The crystal structure of 5-OH-Thal with CRBN and the neosubstrate SALL4ZnF2 was recently published (Fig. 3B, PDB 7BQV).34 It shows a H-bond through water between the phenol and His353 and the binding mode appears to be commensurate with our interpretation of the SAR at the 5-position above. Additionally, the equivalent residue to IKZF1 Gln146 is Val411 in SALL4 which allows enough room in the pocket for the phenol moiety to be accommodated, unlike IKZF1 (Fig. 3B).34 The corresponding GSPT1 amino acid is also a valine (Val570), suggesting 5-OH-Thal binds in a similar manner to that in SALL4 enabling its degradation (shown in our docking model).
4-OH-Thal does not significantly degrade either protein. We hypothesize that this may be the result of an intramolecular H-bond between the 4-hydroxy group and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the phthalimide ring, negating the gain of an H-bond donor interaction with Glu377 that could significantly affect its position in CRBN, potentially preventing favorable interactions with Gln146 in IKZF1 and Lys628/Gln534 in GSPT1 (see the docking model Fig. 4B). In the MD simulation, the 4-OH group indeed forms an intramolecular H-bond 45% of the time (which competes with H-bonding to water). The motif formed an H-bond to Glu377 only 15% of the time, significantly less than the 5-OH-Thal derivative. Therefore, the simulation results confirm our hypothesis and are consistent with the docking model.
O in the phthalimide ring, negating the gain of an H-bond donor interaction with Glu377 that could significantly affect its position in CRBN, potentially preventing favorable interactions with Gln146 in IKZF1 and Lys628/Gln534 in GSPT1 (see the docking model Fig. 4B). In the MD simulation, the 4-OH group indeed forms an intramolecular H-bond 45% of the time (which competes with H-bonding to water). The motif formed an H-bond to Glu377 only 15% of the time, significantly less than the 5-OH-Thal derivative. Therefore, the simulation results confirm our hypothesis and are consistent with the docking model.
In contrast to 5-OH-EM12 and 5-OH-Thal, the amino congeners 5-NH2-EM12 and 5-NH2-Thal do not degrade GSPT1, and only very weakly degrade IKZF1. The amino group may bind His353 through water, as for 5-OH-Thal in PDB 7BQV, but in a manner that likely requires significant rotamer change that negatively affects interactions with the G-loop (observed in the docking model Fig. 4C). In the simulation with GSPT1, there is no stable direct H-bond with His353, and the water mediated H-bond only exists 9% of the time. Also, the –NH2 group is not able to properly engage Gln534 in GSPT1 directly, but it does through water 11% of the time (Fig. 4C). Conversely, it may form weak H-bond interactions due to improper angular arrangements with Gln146 in IKZF1 (Fig. 4D), that are observed 11% of the time directly, and 12% of the time through a water bridge in the simulation, which is consistent with the observed, albeit weak, degradation of IKZF1.
Substitutions at the 6- and 7-positions of the isoindolinone scaffold cause a loss of degradation capacity, even though the derivatives retain CRBN engagement in cells. These findings are not surprising because substitutions at these vectors would be expected to clash with the G-loop degron based on the crystal structure information.
Conclusions
GSPT1 is a translation termination factor that possesses a G-loop degron enabling its recruitment to CRBN by certain molecular glues and PROTACs that mediate its polyubiquitination and proteasomal degradation. GSPT1 is an essential gene for normal cell function, and protein depletion is broadly cytotoxic.13,19 Hydrogen bonding motifs connected to the 5-position of the IMiD scaffolds appear to be important for GSPT1 degradation activity, as observed for the degraders CC-885 and CC-90009, and here shown for the simpler derivatives 5-OH-EM12 and 5-OH-Thal. Hydrogen bonding between these groups and CRBN Glu377 positions the carboxylate side chain to optimally form a salt bridge with GSPT1 Lys628, and bonding to CRBN His353 may help additional interactions with GSPT1 Lys572 in the G-loop. Glu377 is likely positioned through interactions with the IMiD phenol in such a way as to form a H-bond with the Gln534. Although the 5-OH substituted IMiDs do not appear to directly interact with the Gln534 side chain, weak water mediated H-bonding is a possibility based on our model and simulation. Protein interactions mediated by molecular glues in this region of the pocket clearly play important roles in the recruitment of neosubstrates beyond the G-loop degron. Additionally, GSPT1 Val570 within the β-hairpin accommodates substitutions in the 5-position, as seen in the 5-OH-Thal CRBN/SALL4 structure.It should therefore be possible to use the insights from our work to design degraders that do not possess GSPT1 activity and so avoid potentially broad cytotoxicity, or conversely, enhance GSPT1 degradation for certain cancer indications. In support of this, PROTACs tethered to kinase ligands via an NH-alkyl linker connected to the 5-position of the phthalimide scaffold were found to serendipitously degrade GSPT1, in agreement with the SAR found in our study.19 Similarly, a BTK PROTAC was recently designed to incorporate potent GSPT1 degradation through the use of an NH-alkyl tether attached to the 5-position of thalidomide (the 4-position linked PROTAC was a considerably weaker GSPT1 degrader).26 Additionally, a JAK2 PROTAC that was linked through the phthalimide 5-position to avoid an H-bond donor in this region and thus spare GSPT1 degradation.27
In the non-PROTAC molecular glue area, several examples beyond CC-885 and CC-90009 have reported the use of H-bond donors at the 5-position (or its equivalent in alternative CRBN degrader scaffolds) to achieve potent GSPT1 degradation,11,13,14,36 and our structural analyses provide a clear rationale for these results. Our data also show that substitutions at the 6- and 7-positions of the IMiD rings ablate neosubstrate binding and degradation, which could be incorporated into PROTAC design to completely avoid off-target effects driven by molecular glue activity.
Thalidomide and lenalidomide are known to be hydroxylated at the 5-position of the IMiD scaffold. Our results have safety implications for IMiD-based molecular glue and PROTAC degrader development due to the potential downregulation of SALL4 and GSPT1 by 5-OH metabolites, as seen for 5-OH-Thal. Blocking the 5-position of the phthalimide and isoindolinone scaffolds from metabolism could be an important design strategy to avoid the formation of toxic CRBN modulators in vivo.
Author contributions
SF, NRK, RPN and BLZ developed the assays and screened compounds; JC performed computational docking; LHJ conceived and directed the project; LHJ wrote the manuscript with contributions from all authors.Conflicts of interest
L. H. J. serves on the scientific advisory boards for, and holds equity in, Interline Therapeutics, Rapafusyn Pharmaceuticals, Ananke Therapeutics and Umbra Therapeutics, consults for Matchpoint Therapeutics, and holds equity in Jnana Therapeutics. The Center for Protein Degradation at DFCI receives research funding from Deerfield. J. C. is a consultant for Soltego, Allorion and Matchpoint Therapeutics, and holds equity in Soltego, Allorion, Matchpoint and M3 Bioinformatics & Technology Inc.Acknowledgements
We thank Wuxi for compound synthesis, and for running the CRBN NanoBRET and degradation assays, and Eric Fischer, Nathanael Gray and all members of the CPD past and present for useful discussions. The Center for Protein Degradation at DFCI receives research funding from Deerfield.References
- T. Ito, H. Ando, T. Suzuki, T. Ogura, K. Hotta, Y. Imamura, Y. Yamaguchi and H. Handa, Identification of a primary target of thalidomide teratogenicity, Science, 2010, 327(5971), 1345–1350, DOI:10.1126/science.1177319.
- P. P. Chamberlain and L. G. Hamann, Development of targeted protein degradation therapeutics, Nat. Chem. Biol., 2019, 15(10), 937–944, DOI:10.1038/s41589-019-0362-y.
- J. Krönke, N. D. Udeshi, A. Narla, P. Grauman, S. N. Hurst, M. McConkey, T. Svinkina, D. Heckl, E. Comer and X. Li, et al., Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells, Science, 2014, 343(6168), 301–305, DOI:10.1126/science.1244851.
- E. S. Fischer, K. Böhm, J. R. Lydeard, H. Yang, M. B. Stadler, S. Cavadini, J. Nagel, F. Serluca, V. Acker and G. M. Lingaraju, et al., Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide, Nature, 2014, 512(7512), 49–53, DOI:10.1038/nature13527.
- P. P. Chamberlain, A. Lopez-Girona, K. Miller, G. Carmel, B. Pagarigan, B. Chie-Leon, E. Rychak, L. G. Corral, Y. J. Ren and M. Wang, et al., Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs, Nat. Struct. Mol. Biol., 2014, 21(9), 803–809, DOI:10.1038/nsmb.2874.
- Q. L. Sievers, G. Petzold, R. D. Bunker, A. Renneville, M. Słabicki, B. J. Liddicoat, W. Abdulrahman, T. Mikkelsen, B. L. Ebert and N. H. Thomä, Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN, Science, 2018, 362(6414), eaat0572, DOI:10.1126/science.aat0572.
- K. A. Donovan, J. An, R. P. Nowak, J. C. Yuan, E. C. Fink, B. C. Berry, B. L. Ebert and E. S. Fischer, Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome, eLife, 2018, 7, e38430, DOI:10.7554/eLife.38430.
- M. E. Matyskiela, S. Couto, X. Zheng, G. Lu, J. Hui, K. Stamp, C. Drew, Y. Ren, M. Wang and A. Carpenter, et al., SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate, Nat. Chem. Biol., 2018, 14(10), 981–987, DOI:10.1038/s41589-018-0129-x.
- M. E. Matyskiela, T. Clayton, X. Zheng, C. Mayne, E. Tran, A. Carpenter, B. Pagarigan, J. McDonald, M. Rolfe and L. G. Hamann, et al., Crystal structure of the SALL4-pomalidomide-cereblon-DDB1 complex, Nat. Struct. Mol. Biol., 2020, 27(4), 319–322, DOI:10.1038/s41594-020-0405-9.
- J. D. Hansen, M. Correa, M. Alexander, M. Nagy, D. Huang, J. Sapienza, G. Lu, L. A. LeBrun, B. E. Cathers and W. Zhang, et al., CC-90009: A Cereblon E3 Ligase Modulating Drug That Promotes Selective Degradation of GSPT1 for the Treatment of Acute Myeloid Leukemia, J. Med. Chem., 2021, 64(4), 1835–1843, DOI:10.1021/acs.jmedchem.0c01489.
- J. D. Hansen, K. Condroski, M. Correa, G. Muller, H. W. Man, A. Ruchelman, W. Zhang, F. Vocanson, T. Crea and W. Liu, et al., Protein Degradation via CRL4, J. Med. Chem., 2018, 61(2), 492–503, DOI:10.1021/acs.jmedchem.6b01911.
- M. E. Matyskiela, G. Lu, T. Ito, B. Pagarigan, C. C. Lu, K. Miller, W. Fang, N. Y. Wang, D. Nguyen and J. Houston, et al., A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase, Nature, 2016, 535(7611), 252–257, DOI:10.1038/nature18611.
- G. Nishiguchi, F. Keramatnia, J. Min, Y. Chang, B. Jonchere, S. Das, M. Actis, J. Price, D. Chepyala and B. Young, et al., Identification of Potent, Selective, and Orally Bioavailable Small-Molecule GSPT1/2 Degraders from a Focused Library of Cereblon Modulators, J. Med. Chem., 2021, 64(11), 7296–7311, DOI:10.1021/acs.jmedchem.0c01313.
- A. D. Takwale, E. Y. Kim, Y. Jang, D. H. Lee, S. Kim, Y. Choi, J. H. Kim, D. Y. Lee, Y. Kim and S. M. Lee, et al., Structure-activity relationship analysis of novel GSPT1 degraders based on benzotriazinone scaffold and its antitumor effect on xenograft mouse model, Bioorg. Chem., 2022, 127, 105923, DOI:10.1016/j.bioorg.2022.105923.
- F. Xiong, L. Kong, L. Chen, M. Xue, F. Cao, S. Zhang, H. Li, H. Yan, Y. Li and Z. Zuo, Discovery of potential novel CRBN modulators by virtual screening and bioassay, Eur. J. Med. Chem., 2022, 236, 114355, DOI:10.1016/j.ejmech.2022.114355.
- C. E. Powell, G. Du, J. Che, Z. He, K. A. Donovan, H. Yue, E. S. Wang, R. P. Nowak, T. Zhang and E. S. Fischer, et al., Selective Degradation of GSPT1 by Cereblon Modulators Identified via a Focused Combinatorial Library, ACS Chem. Biol., 2020, 15(10), 2722–2730, DOI:10.1021/acschembio.0c00520.
- C. Surka, L. Jin, N. Mbong, C. C. Lu, I. S. Jang, E. Rychak, D. Mendy, T. Clayton, E. Tindall and C. Hsu, et al., CC-90009, a novel cereblon E3 ligase modulator, targets acute myeloid leukemia blasts and leukemia stem cells, Blood, 2021, 137(5), 661–677, DOI:10.1182/blood.2020008676.
- R. S. Sellar, A. S. Sperling, M. Słabicki, J. A. Gasser, M. E. McConkey, K. A. Donovan, N. Mageed, D. N. Adams, C. Zou and P. G. Miller, et al., Degradation of GSPT1 causes TP53-independent cell death in leukemia whilst sparing normal hematopoietic stem cells, J. Clin. Invest., 2022, 132(16), e153514, DOI:10.1172/JCI153514.
- M. Ishoey, S. Chorn, N. Singh, M. G. Jaeger, M. Brand, J. Paulk, S. Bauer, M. A. Erb, K. Parapatics and A. C. Müller, et al., Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders, ACS Chem. Biol., 2018, 13(3), 553–560, DOI:10.1021/acschembio.7b00969.
- X. Tan, L. I. Calderon-Villalobos, M. Sharon, C. Zheng, C. V. Robinson, M. Estelle and N. Zheng, Mechanism of auxin perception by the TIR1 ubiquitin ligase, Nature, 2007, 446(7136), 640–645, DOI:10.1038/nature05731.
- S. L. Schreiber, The Rise of Molecular Glues, Cell, 2021, 184(1), 3–9, DOI:10.1016/j.cell.2020.12.020.
- J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew and K. Coleman, et al., Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4, Chem. Biol., 2015, 22(6), 755–763, DOI:10.1016/j.chembiol.2015.05.009.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation, Science, 2015, 348(6241), 1376–1381, DOI:10.1126/science.aab1433.
- L. J. Alcock, Y. Chang, J. A. Jarusiewicz, M. Actis, S. Nithianantham, A. Mayasundari, J. Min, D. Maxwell, J. Hunt and B. Smart, et al., Development of Potent and Selective Janus Kinase 2/3 Directing PG-PROTACs, ACS Med. Chem. Lett., 2022, 13(3), 475–482, DOI:10.1021/acsmedchemlett.1c00650.
- S. Lier, A. Sellmer, F. Orben, S. Heinzlmeir, L. Krauß, C. Schneeweis, Z. Hassan, C. Schneider, A. Patricia Gloria Schäfer and H. Pongratz, et al., A novel Cereblon E3 ligase modulator with antitumor activity in gastrointestinal cancer, Bioorg. Chem., 2022, 119, 105505, DOI:10.1016/j.bioorg.2021.105505.
- Z. Yang, Y. Sun, Z. Ni, C. Yang, Y. Tong, Y. Liu, H. Li and Y. Rao, Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently, Cell Res., 2021, 31(12), 1315–1318, DOI:10.1038/s41422-021-00533-6.
- Y. Chang, J. Min, J. A. Jarusiewicz, M. Actis, S. Yu-Chen Bradford, A. Mayasundari, L. Yang, D. Chepyala, L. J. Alcock and K. G. Roberts, et al., Degradation of Janus kinases in CRLF2-rearranged acute lymphoblastic leukemia, Blood, 2021, 138(23), 2313–2326, DOI:10.1182/blood.2020006846.
- B. Wang, J. Liu, I. Tandon, S. Wu, P. Teng, J. Liao and W. Tang, Development of MDM2 degraders based on ligands derived from Ugi reactions: Lessons and discoveries, Eur. J. Med. Chem., 2021, 219, 113425, DOI:10.1016/j.ejmech.2021.113425.
- J. Yang, Y. Li, A. Aguilar, Z. Liu, C. Y. Yang and S. Wang, Simple Structural Modifications Converting a Bona fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders, J. Med. Chem., 2019, 62(21), 9471–9487, DOI:10.1021/acs.jmedchem.9b00846.
- L. H. Jones, C. A. Mitchell, L. Loberg, M. Pavkovic, M. Rao, R. Roberts, K. Stamp, L. Volak, M. B. Wittwer and S. Pettit, Targeted protein degraders: a call for collective action to advance safety assessment, Nat. Rev. Drug Discovery, 2022, 21(6), 401–402, DOI:10.1038/d41573-022-00055-9.
- J. T. Cruite, G. P. Dann, J. Che, K. A. Donovan, S. Ferrao, E. S. Fischer, N. S. Gray, F. Huerta, N. R. Kong and H. Liu, et al., Cereblon covalent modulation through structure-based design of histidine targeting chemical probes, RSC Chem. Biol., 2022, 3, 1105–1110 RSC.
- N. R. Kong, H. Liu, J. Che and L. H. Jones, Physicochemistry of Cereblon Modulating Drugs Determines Pharmacokinetics and Disposition, ACS Med. Chem. Lett., 2021, 12(11), 1861–1865, DOI:10.1021/acsmedchemlett.1c00475.
- E. S. Wang, A. L. Verano, R. P. Nowak, J. C. Yuan, K. A. Donovan, N. A. Eleuteri, H. Yue, K. H. Ngo, P. H. Lizotte and P. C. Gokhale, et al., Acute pharmacological degradation of Helios destabilizes regulatory T cells, Nat. Chem. Biol., 2021, 17(6), 711–717, DOI:10.1038/s41589-021-00802-w.
- H. Furihata, S. Yamanaka, T. Honda, Y. Miyauchi, A. Asano, N. Shibata, M. Tanokura, T. Sawasaki and T. Miyakawa, Structural bases of IMiD selectivity that emerges by 5-hydroxythalidomide, Nat. Commun., 2020, 11(1), 4578, DOI:10.1038/s41467-020-18488-4.
- S. Yamanaka, H. Murai, D. Saito, G. Abe, E. Tokunaga, T. Iwasaki, H. Takahashi, H. Takeda, T. Suzuki and N. Shibata, et al., Thalidomide and its metabolite 5-hydroxythalidomide induce teratogenicity via the cereblon neosubstrate PLZF, EMBO J., 2021, 40(4), e105375, DOI:10.15252/embj.2020105375.
- J. Zou, R. J. Jones, H. Wang, I. Kuiatse, F. Shirazi, E. E. Manasanch, H. C. Lee, R. Sullivan, L. Fung and N. Richard, et al., The novel protein homeostatic modulator BTX306 is active in myeloma and overcomes bortezomib and lenalidomide resistance, J. Mol. Med., 2020, 98(8), 1161–1173, DOI:10.1007/s00109-020-01943-6.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00347c |
| This journal is © The Royal Society of Chemistry 2023 |