
The Mycobacterium tuberculosis mycothiol S-transferase is divalent metal-dependent for mycothiol binding and transfer†
Yahani P.
Jayasinghe
 a,
Michael T.
Banco‡
b,
Jared J.
Lindenberger§
b,
Lorenza
Favrot¶
b,
Zuzana
Palčeková
c,
Mary
Jackson
c,
Shino
Manabe
de and
Donald R.
Ronning
*a
a,
Michael T.
Banco‡
b,
Jared J.
Lindenberger§
b,
Lorenza
Favrot¶
b,
Zuzana
Palčeková
c,
Mary
Jackson
c,
Shino
Manabe
de and
Donald R.
Ronning
*a
aDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska, USA. E-mail: don.ronning@unmc.edu
bDepartment of Chemistry and Biochemistry, University of Toledo, Toledo, Ohio, USA
cMycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, Colorado, USA
dLaboratory of Functional Molecule Chemistry, Pharmaceutical Department and Institute of Medicinal Chemistry, Hoshi University, Tokyo, Japan
eResearch Center for Pharmaceutical Development, Graduate School of Pharmaceutical Sciences & Faculty of Pharmaceutical Sciences, Tohoku University, Miyagi, Japan
First published on 26th January 2023
Abstract
Mycothiol S-transferase (MST) (encoded by the rv0443 gene) was previously identified as the enzyme responsible for the transfer of Mycothiol (MSH) to xenobiotic acceptors in Mycobacterium tuberculosis (M.tb) during xenobiotic stress. To further characterize the functionality of MST in vitro and the possible roles in vivo, X-ray crystallographic, metal-dependent enzyme kinetics, thermal denaturation studies, and antibiotic MIC determination in rv0433 knockout strain were performed. The binding of MSH and Zn2+ increases the melting temperature by 12.9 °C as a consequence of the cooperative stabilization of MST by both MSH and metal. The co-crystal structure of MST in complex with MSH and Zn2+ to 1.45 Å resolution supports the specific utilization of MSH as a substrate as well as affording insights into the structural requirements of MSH binding and the metal-assisted catalytic mechanism of MST. Contrary to the well-defined role of MSH in mycobacterial xenobiotic responses and the ability of MST to bind MSH, cell-based studies with an M.tb rv0443 knockout strain failed to provide evidence for a role of MST in processing of rifampicin or isoniazid. These studies suggest the necessity of a new direction to identify acceptors of the enzyme and better define the biological role of MST in mycobacteria.
Introduction
Mycothiol (MSH) is a low molecular weight thiol produced by Mycobacterium tuberculosis (M.tb) and many other actinomycetes.1 It protects the bacterium during oxidative stress, supports electrophilic toxin elimination, and xenobiotic detoxification.2 Additionally, MSH is the major cytosolic thiol-containing reductant used to maintain redox homeostasis within mycobacterial cells.3 MSH is thereby functionally analogous to low molecular weight thiols in other organisms such as glutathione (GSH), the primary thiol molecule produced by eukaryotes and many Gram-negative bacteria, and bacillithiol produced by various Bacillus species.4,5In Mycobacteria, MSH is produced by a series of enzymes consisting of MshA, MshA2, MshB, MshC, and MshD.6 Previous gene knockout studies suggest that the defects of these enzymes make the bacteria more vulnerable to oxidative stress and most antibiotics.7 Defects in mshA and mshC genes significantly decrease MSH levels to nondetectable levels.6 Importantly, the mshC knockout strains exhibit an increase in susceptibility to some antibiotics including rifampicin, erythromycin, azithromycin, and vancomycin.2 In contrast, these same gene knockouts exhibit increased resistance to isoniazid and ethionamide suggesting a possible role for MSH in activation of these prodrugs.8–10 The decrease in MSH levels in these strains shows a concomitant increase in the production of ergothioneine (EGT), which is another low molecular weight thiol secreted by mycobacteria.11 Additionally, the lower production of MSH in the knockout strains is associated with increasing levels of production of hydroperoxide reductase enzyme, which presumptively reduces oxidative damage caused by accumulation of reactive oxygen species within the M.tb bacillus.8,12 These data suggest that MSH is an important molecule for normal redox homeostasis as well as maintaining mycobacterial viability during times of oxidative or xenobiotic stress.
With emphasis on understanding the role of MSH in xenobiotic detoxification, researchers have identified one M.tb-encoded enzyme to be a functional analog to glutathione S-transferase (GST). Fahey et al. identified the enzyme encoded by the rv0443 gene in M.tb strain H37Rv and named it mycothiol-S-transferase (MST).4 As the proposed mechanism is suggested to be a simple nucleophilic substitution reaction, MST is expected to transfer MSH to any electrophilic acceptor recognized by the MST enzyme (Fig. 1). Based on protein sequence similarity, MST was identified as a member of the DinB superfamily of proteins belonging to the Domain of Unknown Function 664 family.4 Other proteins in the same superfamily of proteins include Bacillus subtillus YfiT (PDB: 1RXQ) and Mycobacterium thermoresistibile EgtB (PDB: 4X8E), which suggests a metal-assisted catalytic mechanism for MST.13,14 Monochlorobimane (mClB) was identified as a surrogate MSH acceptor substrate for the enzyme that affords facile enzymatic evaluation of MST and the proposal that MSH can be transferred to a variety of acceptors that include first and second-line TB drugs.4 However, the S–C bond forming reaction catalyzed by EgtB requires Fe2+ and proceeds through the formation of an oxygen radical, so the presumed electrophilic attack by an MSH thiolate may not be fully representative of the biologically relevant enzymatic reaction.14 Additionally, the broad structural and chemical scope of the proposed MSH acceptors suggests that further characterization of the catalyzed reaction and evaluation of the possible MSH transfer mechanism is necessary to assess the potential role of MST in M.tb xenobiotic modification.
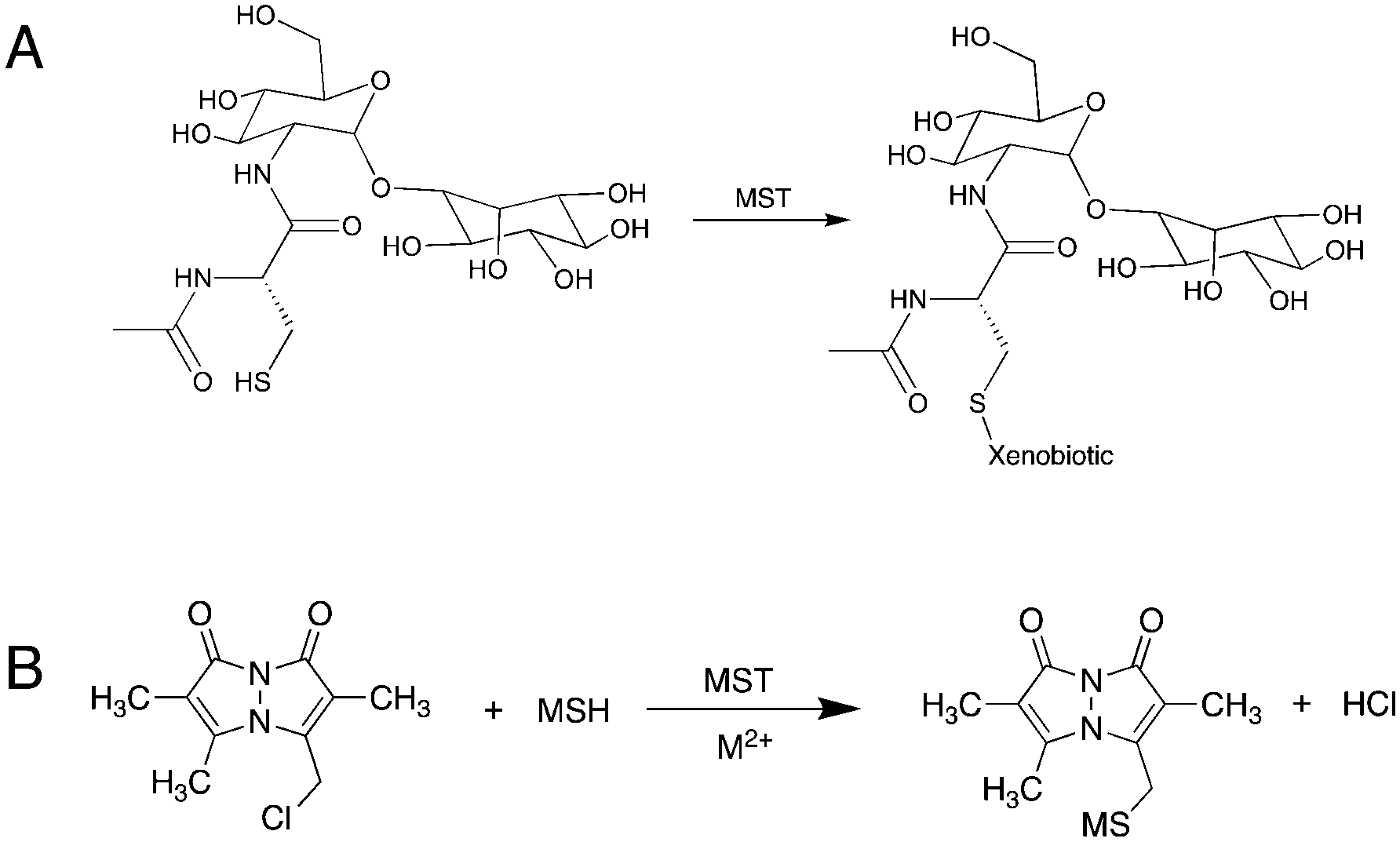 | ||
| Fig. 1 (A) Proposed MSH transfer reaction by MST to xenobiotics. (B) The reaction used in the MST enzyme kinetic assay. Monochlorobimane is used as a surrogate substrate that reacts with MSH with the enzyme assists catalytic reaction to produce the conjugated products, which give an elevated florescence signal to detect the reaction progress. | ||
This study presents the crystal structure of MST alone and in complex with the MSH substrate and divalent metal ions to provide structural details of the protein architecture and the substrate binding sites. The structure confirms that MST is a member of the DinB superfamily of proteins. These structural details also illustrate unexpected structural changes in response to MSH and metal binding as well as highlighting possible roles for other conserved active site residues. Additionally, the in vitro and cell-based assays with current TB drugs, rifamycin and isoniazid, provide novel perceptions about the previous hypotheses of the role of MST in M.tb drug metabolism.
Results
Metal dependency of MST
As most of the DinB superfamily of proteins use a metal ion for their catalytic mechanisms, differential scanning fluorometry (DSF) studies were performed to assess the ability of MST to selectively bind various metals with and without MSH (Fig. 2). The melting temperature of the wild-type MST protein lacking any metal or other ligands was shown to be 59.0 °C in DSF studies. The addition of divalent metal cations to the reaction mixture exhibits slight shifts to higher melting temperatures that ranged from 2.2 °C with the addition of Fe2+ to 11.6 °C using Zn2+ (Table 1). The magnitude of these changes in MST melting temperature are commensurate with observed changes in other metal binding enzymes and proteins.15 The melting temperature was shifted further when MSH was added along with the metal cation. The addition of different metal cations to the reaction mixture causes different degrees of temperature shifts. The metal dependency of the protein was further confirmed by the increased catalytic rates of the protein when added a metal ion to the reaction mixture (Fig. 2B).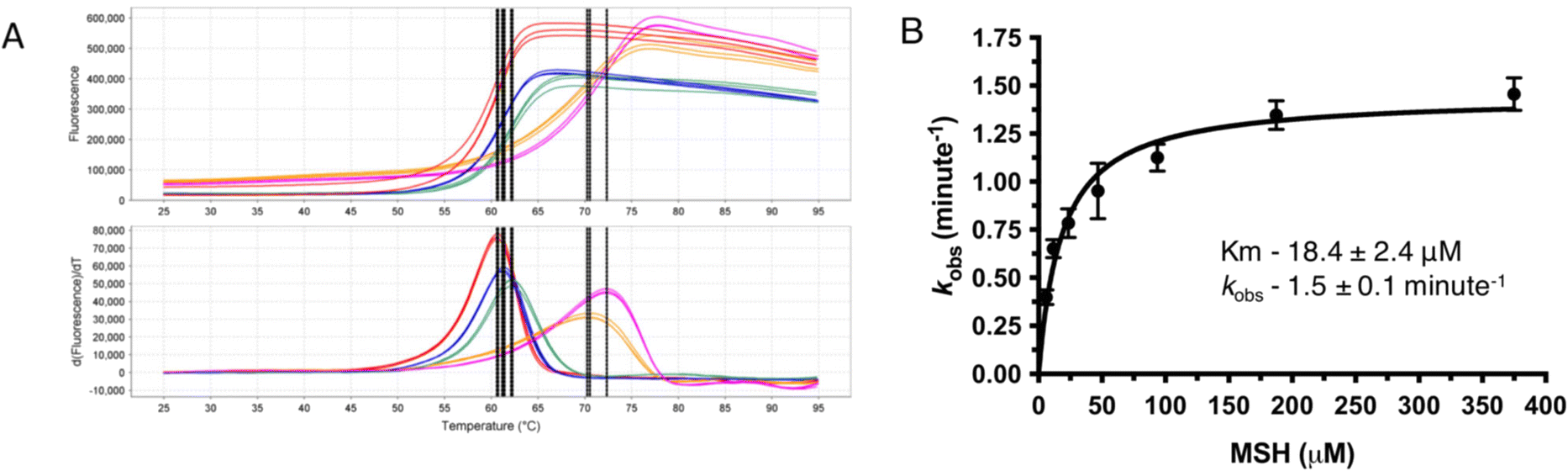 | ||
| Fig. 2 (A) Differential scanning fluorimetry of MST with varying metals. Red represents EDTA, blue represents ferrous, orange represents zinc, green represents ferrous with MSH, and the pink represents zinc with MSH. (B) Steady state kinetics for MST in the presence of Zn2+ as the catalytic metal. | ||
| Sample | Determined Tm (°C) | ΔTm (°C) |
|---|---|---|
| MST alone | 59.0 ± 0.1 | — |
| + MSH | 59.0 ± 0.2 | 0 |
| + Zinc | 70.6 ± 0.2 | 11.6 |
| + Cobalt | 64.5 ± 0.1 | 5.5 |
| + Nickle | 68.5 ± 0.2 | 9.5 |
| + Ferrous | 61.3 ± 0.1 | 2.3 |
| + Zinc and MSH | 71.9 ± 0.1 | 12.9 |
| + Cobalt and MSH | 65.1 ± 0.1 | 6.1 |
| + Nickle and MSH | 68.9 ± 0.1 | 9.9 |
| + Ferrous and MSH | 62.2 ± 0.1 | 3.2 |
Steady-state kinetic experiments further confirm the metal dependency of MST
MST shows only residual activity when performed the enzymatic reaction without adding any metal to catalyze the reaction. There is a detectable enzyme activity when a divalent metal ion is added to the reaction mixture. As Zn2+ is stable in the reaction mixture giving consistent data enzyme kinetic parameters of MST were determined using Zn2+ as the catalytic metal. The Km of MSH in the presence of Zn2+ was determined as 9.8 ± 1.0 μM and Vmax as 1484 RFU min−1 (Table S1†). The rates of MST-catalyzed reactions using other metal ions, Co2+ and Fe2+ were also determined. The mean initial velocity values with Co2+, Zn2+, and Fe2+ are 2.7 ± 0.1, 0.7 ± 0.0 and 0.6 ± 0.0 μM min−1, respectively (Table S1†). While the reaction rates with Fe2+ and Zn2+were similar, the enzymatic rate using Co2+ exhibits a greater than 4-fold higher mean velocity when compared to Zn2+ and Fe2+.MST catalyzes MSH transfer to Nitrocefin
The colorimetric β-lactamase substrate nitrocefin was tested to use as an alternative to monitor the MST transfer reaction and begin to evaluate the MST substrate breadth. In this case, determining if MST can catalyze MSH transfer and open the β-lactam ring. The data suggest the transfer of MSH to Nitrocefin in MST catalyzed reaction. The reaction was performed by measuring the absorbance at 500 nm and the β-lactam ring-opening reaction of Nitrocefin, rather than elimination of a nitro group from the bis-dinitro-aryl moiety, was observed in an enzyme-dependent manner.16 The reactions using 500 nM MST while varying nitrocefin concentration are consistent with saturation kinetics and analysis of the reaction products by HPLC separation on a reverse phase column and mass spectrometry experiments show a 1002.19 m/z ion corresponding to nitrocefin–MSH conjugate and a 1024.98 ion corresponding to the sodiated form (Fig. 3).16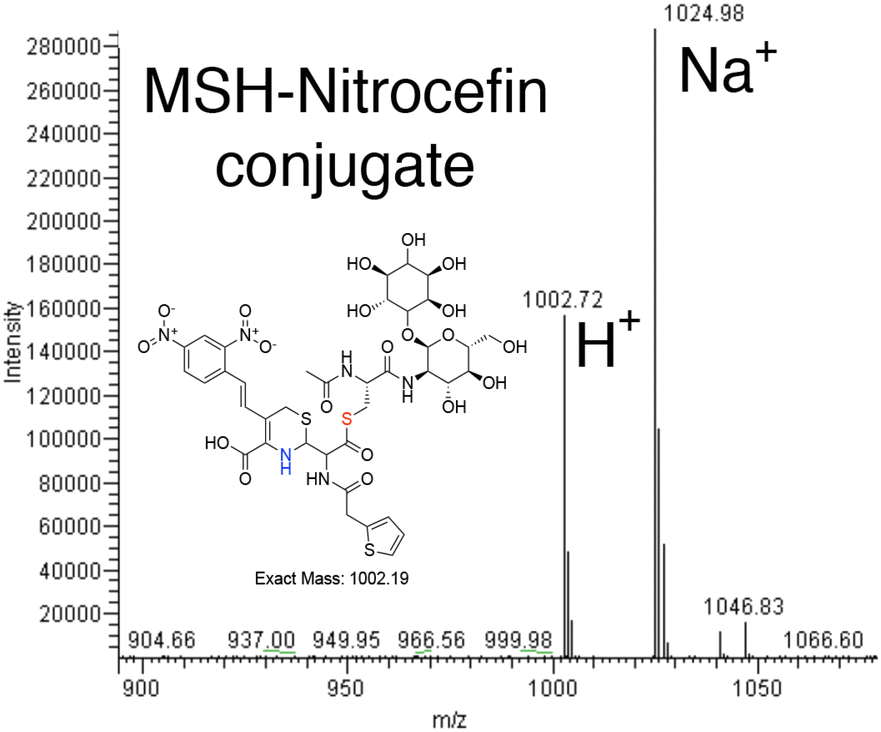 | ||
| Fig. 3 Formation of a MSH–nitrocefin conjugate. | ||
MST shows structural characteristics of the DinB superfamily of proteins
The DSF studies and enzyme kinetic assays suggest higher stability of the protein when it binds with the substrate, MSH. The crystallographic experiments were performed to observe the binding of MSH to the protein and to identify the catalytic site residues of the protein. In an effort to obtain this information, we first solved the crystal structure of ligand-free M.tb MST.The crystal diffracted to 1.36 Å resolution (Table 1). The solved crystal structure (PDB: 8FX9) consists of one molecule in the asymmetric unit (Fig. 4A). The overall fold of the MST protein is similar to other proteins in the DinB superfamily of proteins, which consists of conserved four α-helical bundles giving an up-down-up topology with is found in all DinB family proteins (Fig. 4B). Additionally, helices most of the DinB superfamily enzymes are homodimers.4 The native ESI-MS experiments with MST confirmed this is a homodimer. This is also reflected in the crystal packing arrangement where the dyad of the homodimer aligns with a crystallographic 2-fold axis of symmetry. The MST ternary complex structure gives further evidence to the homodimeric character of MST. The α1 and α6 helices of one molecule interact with the same helices of the neighbouring chain in the asymmetric unit. In DinB proteins, the second and fourth helices of the four helical bundle, helices 2 and 8 in the case of MTS, harbor three conserved metal-binding residues. In MST, these correspond to His52, Asp155, and His159. In other DinB proteins, the aspartate residue is commonly replaced by another histidine residue.17 However, difference maps within the proposed MST metal binding site lacked density corresponding to any metal ions. Due to the modularity of the 4-helical bundle and the ability to bind catalytic metals, the activity of proteins in the DinB family are expected to be enzymatically versatile by employing the homodimer interface as a substrate recognition region or by fusing the DinB domain with an accessory protein possessing affinity to a substrate. This second example seems to be the case with bacterial EgtB enzymes that catalyse one step of the ergothioneine biosynthetic pathway.18
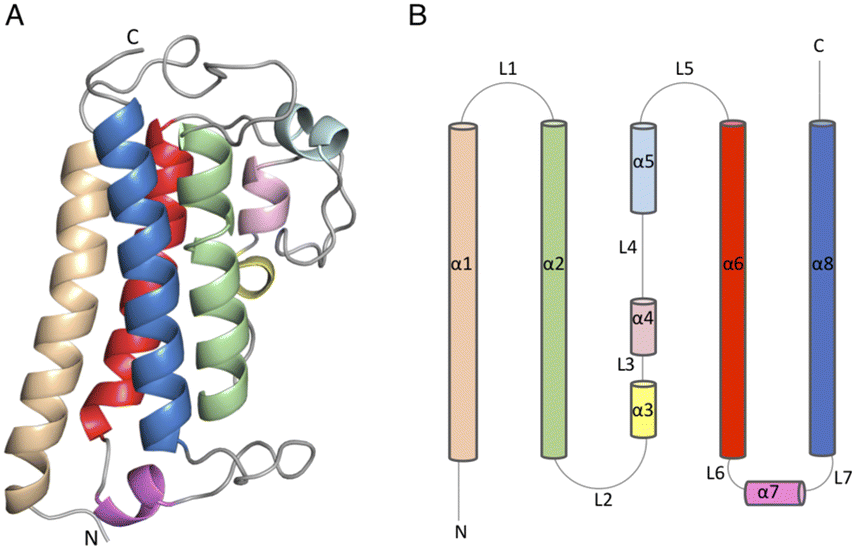 | ||
| Fig. 4 (4) (A) Apo structure of MST consists of a four-helical bundle (B) topology diagram of MST apo structure which has a similar topology as other DinB superfamily of proteins. | ||
To obtain a better understanding of the interactions between MST and MSH, co-crystallization experiments were performed. Unfortunately, the only difference density observed in the solved structure corresponded to a covalent attachment of MST at residue C36 by MSH instead of binding in the catalytic site (Fig. S1†). While this complies with the suggested modification of surface cysteine residues with MSH to reversibly protect proteins during times of oxidative stress, this structure lacked insight regarding the MSH binding mode within the MST active site and what residues are involved in the catalytic mechanism.
Structure of MST with bound MSH and Zn2+
As the DSF experiments exhibit significant enhancement of MST when binding both MSH and metal ions, co-crystallization experiments were performed for MST with MSH and various metals. The crystal structure of MST with MSH and Zn2+ was solved to 1.45 Å resolution in the P21212 space group (PDB: 8F5V). The crystallographic statistics are provided in Table 2. Consistent with the apo and covalent MSH structures, the asymmetric unit contains two MST molecules, but each MST monomer possesses a bound Zn2+ ion and one molecule of MSH (Fig. 5A). The difference density representing MSH affords accurate modelling of the interactions necessary for recognition and metal-dependent binding by MST (Fig. 5B).| MST Apo structure (PDB: 8FX9) | MST/MSH/Zn2+ complex (PDB: 8F5V) | |
|---|---|---|
| Crystallographic data | ||
| Beamline/facility | 21-ID-F-APS | 21-ID-D-APS |
| Wavelength | 1.00 | 1.00 |
| Resolution range | 29.43–1.36 (1.41–1.36) | 30.67–1.45 (1.50–1.45) |
| Space group | C2221 | P21212 |
| Unit cell dimensions | ||
| a, b, c (Å) | 65.377 98.891 49.97 | 75.027 76.732 53.26 |
| α, β, γ (°) | 90 90 90 | 90 90 90 |
| Total reflections | 424![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 289 (7504) 289 (7504) |
661127 (33581) |
| Unique reflections | 31576 (2669) |
53633 (4383) |
| Multiplicity | 11.8 (4.2) | 12.3 (7.7) |
| Completeness (%) | 99.89 (99.93) | 97.25 (80.50) |
| Mean I/sigma(I) | 19.61 (3.10) | 16.19 (2.55) |
| Wilson B-factor (Å2) | 13.21 | 16.51 |
| R-merge | 0.074 (0.49) | 0.085 (0.73) |
| R-meas | 0.913 (0.82) | 0.088 (0.78) |
| R-pim | 0.025 (0.39) | 0.024 (0.28) |
| CC1/2 | 0.99 (0.94) | 0.999 (0.84) |
| CC* | 1 (0.961) | 1 (0.957) |
| Refinement | ||
| Reflections used in refinement | 40358 (3999) |
53621 (4383) |
| Reflections used for R-free | 1605 (159) | 1998 (171) |
| R-work | 0.1912 (0.40) | 0.18 (0.22) |
| R-free | 0.2229 (0.40) | 0.20 (0.23) |
| CC (work) | 0.925 (0.666) | 0.966 (0.904) |
| CC (free) | 0.940 (0.749) | 0.949 (0.860) |
| Number of non-hydrogen atoms | 1596 | 3135 |
| Macromolecules | 1368 | 2693 |
| Ligands | — | 68 |
| Solvent | 228 | 374 |
| Protein residues | 171 | 342 |
| RMS (bonds) (Å) | 0.009 | 0.005 |
| RMS (angles) (°) | 1.11 | 0.81 |
| Ramachandran favored (%) | 100 | 99.41 |
| Ramachandran allowed (%) | 0.00 | 0.59 |
| Ramachandran outliers (%) | 0.00 | 0.00 |
| Rotamer outliers (%) | 0.00 | 0.71 |
| Clashscore | 2.97 | 1.87 |
| Average B-factor (Å2) | 18.80 | 22.51 |
| Macromolecules | 17.30 | 21.39 |
| Ligands | — | 21.09 |
| Solvent | 27.70 | 30.85 |
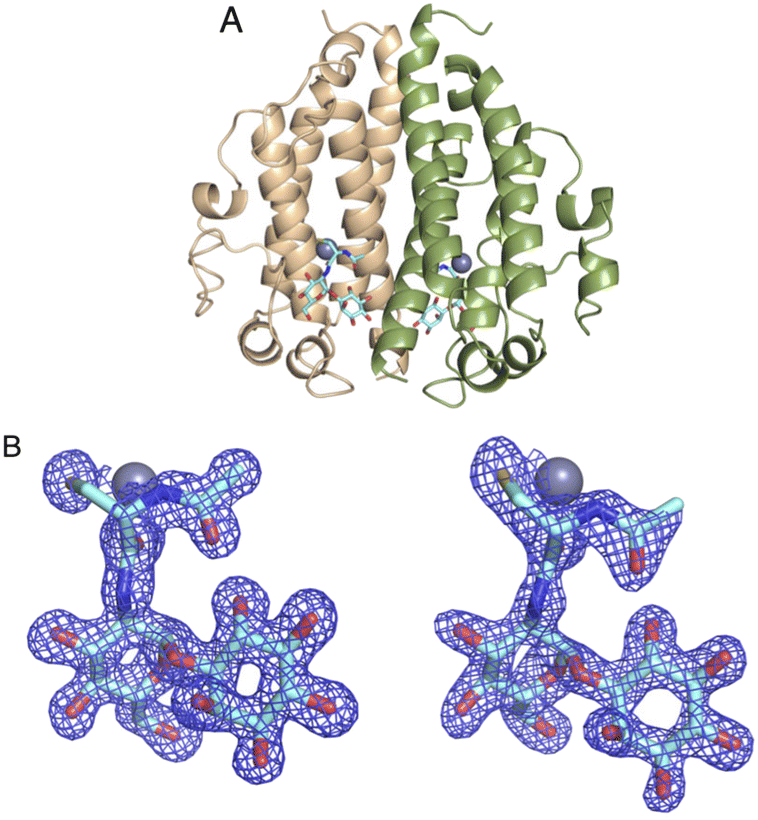 | ||
| Fig. 5 (A) The ternary structure of MST in complex with MSH and Zn2+. Two chains of the dimer are colored in peach and green (B) calculated Fo − Fc maps for MSH at the active sites of the two chains in the asymmetric unit (chain A and B respectively). The map is contoured at 3σ in blue mesh. Atom color: carbon is light blue in MSH, nitrogen blue, oxygen red, and sulfur yellow. The zinc ion is represented by the slate-colored sphere. | ||
The Zn2+ is coordinated mainly by two active site histidine residues, His52 and His159, with each histidine residue forming a monodentate interaction with the zinc ion. These residues are conserved and form similar interactions to coordinate the metal ion in other DinB superfamily homologs and further strengthen the hypothesis that metal-binding is central to the biological function of enzymes in that superfamily.19,20 The sequence alignment of MST with the 600 DinB superfamily proteins shows the residues involved in metal and MSH binding are conserved (Fig. 6A). The third protein ligand for Zn2+ is represented by an ionic interaction with the side chain of the conserved Asp155 residue. Additionally, the zinc ion forms two interactions with MSH to fill the typical penta-coordinate complex of a catalytically active zinc. Specifically, the carbonyl oxygen of the amide linkage between the N-acetyl cysteine and glucosamine moieties forms a very short 2.2 Å polar interaction (Fig. 6B), and the sulfur of MSH, presumably in a thiolate form, forms a likely ionic interaction.
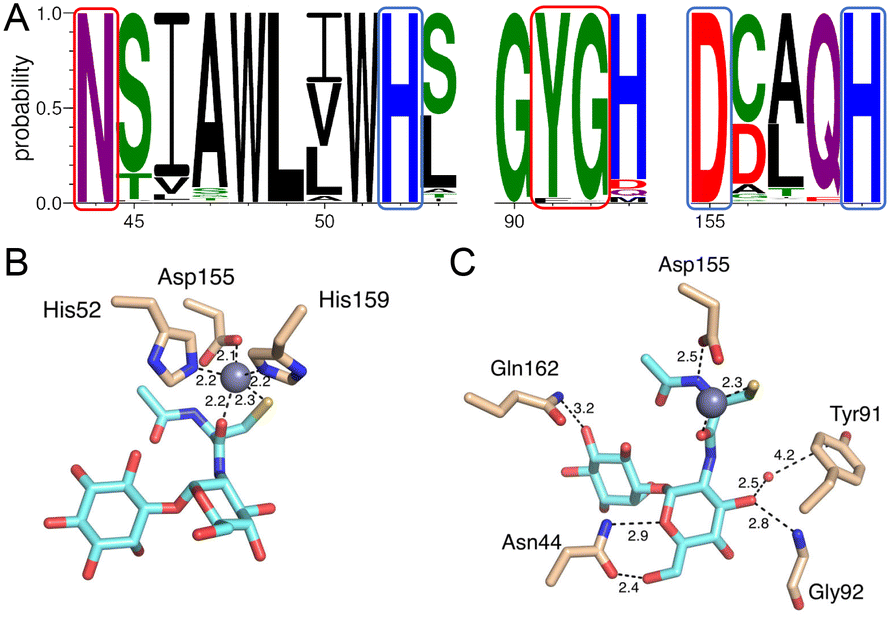 | ||
| Fig. 6 (A) Selected portions of the sequence alignment of 600 actinobacteria MST homologs highlighting conserved active site residues involved in metal and MSH binding. Residue numbering is consistent with the M.tb MST enzyme. Blue and red boxes indicate metal binding and MSH binding residues, respectively. (B) The interactions of Zn2+ with active site residues (His52, Asp55, and His159) of MST and MSH thiolate and carbonyl oxygen atoms. (C) The interactions of MSH with MST protein residues. Atom color: carbon is cyan in MSH, tan in protein residues. In both B and C, nitrogen atoms are blue, oxygen atoms are red and sulfur atoms are yellow. | ||
Mycothiol binding to the MST active site coordinates by backbone residues of the protein (Fig. 6C). The backbone nitrogen of Gly92 and Gln162 forms hydrogen bonding interactions with MSH. In addition to that the carbonyl oxygen of Asn44 forms an interaction with a hydroxyl group of N-acetyl glucosamine moiety and the backbone nitrogen of Asn44 forms another hydrogen bonding interaction with MSH to form two hydrogen-bonding interactions. The conserved Asp155, in addition to forming an aforementioned ionic interaction with the zinc ion, forms a hydrogen bond with the N-acetyl cysteine amide.
The implications for second substrate-binding
Comparison of the ligand-free MST structure and the MSH/Zn2+-bound MST complex, highlights structural changes in the active site upon binding MSH and Zn2+. The largest magnitude structural change is a dynamic loop region that includes the conserved Trp139 residue (Fig. 7A). Additionally, the conserved residue Tyr91 undergoes a rotamer change. It is intriguing that the repositioning of these two residues due to MSH/Zn2+ binding, decreases the distance between Tyr91 and Trp139 from 10.4 Å to only 5.4 Å (Fig. 7B).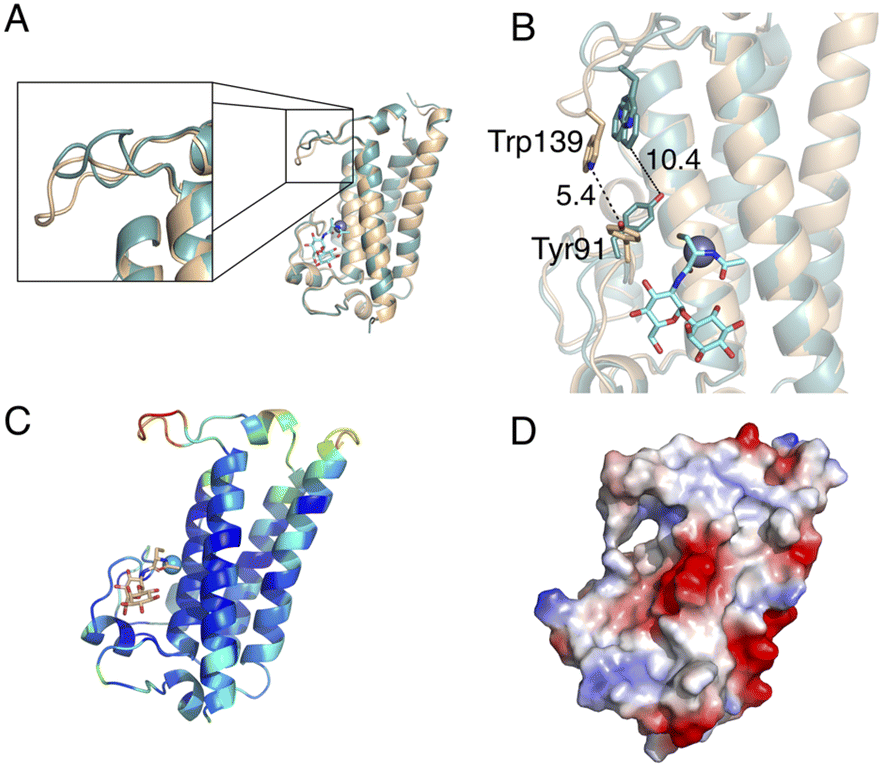 | ||
| Fig. 7 The conformational changes of protein upon MSH binding. The superimposed crystal structures of MST apoprotein (teal) and MST–MSH–Zn2+ ternary structure (pale orange). (A) Significant conformational change of the loop involves in second substrate binding (B) conformational changes in Tyr91 and Trp139 to in response to MSH/Zn2+ binding. (C) MST ternary structure colored according to the B-factors indicating elevated B-factors for the loop responsible for second substrate recognition (D) surface of MST colored by electrostatic potential illustrates the slight positive potential in the MSH-acceptor substrate-binding site. | ||
The location of the ligands in the MST/MSH/Zn2+ ternary complex also offers insights into the likely second substrate-binding site of MST. The residues identified as the second substrate-binding domain are highly conserved among MST homologs. Necessitated by the expected direct attack of the MSH thiol on the MSH acceptor, the second substrate-binding site must be in proximity to the MSH binding site allowing the transfer of MSH to the acceptor. Additionally, the observed structural changes to the loop harbouring Trp139 exhibits elevated B-factors suggesting that the induced structural changes may support the binding of the second substrate via an induced-fit mechanism as the increased dynamics in the complex may be due to lack of filling the second substrate binding site (Fig. 7C and D and S2†).
The electrostatic surface potential indicates that the predicted second substrate binding site possesses a relatively neutral-charge and is less positively charged when compared to the charged active site surface near the metal ion, indicating the possibility of binding a moderately amphipathic acceptor molecule to the binding site (Fig. 7D). More specifically, the electrostatics and the hydrophobicity of the surface generated according to the Kyte-Doolittle scale suggest that the binding site accommodating the MSH acceptor possesses both hydrophobic and hydrophilic characteristics (Fig. S3†).21,22
Possible catalysis by Fe2+
Other DinB superfamily enzymes, such as the M.tb-encoded EgtB enzyme that conjugates the thiol of γ-glutamyl cysteine to the ε-carbon of the L-hercyanine side chain, use Fe2+ as the catalytic metal using a radical reaction mechanism.14 This suggested the possibility that in vivo, MST uses Fe2+ and molecular oxygen to activate the MSH thiol via radical formation to afford C–S bond formation with the second substrate similar to that proposed for the EgtB-catalyzed C–S bond formation between γ-glutamyl cysteine and L-hercynine.14 When performing the MST enzymatic assays where Zn2+ is replaced by Fe2+, the kinetic results exhibit a similar reaction rate, suggesting possible catalysis by Fe2+. The ascorbic acid is meant to afford redox cycling during the reaction. The purified MST showed residual activity when the protein is not dialyzed with EDTA and that suggested the possibility of MST co-purifying with a metal cation. The ferrozine assay was performed to identify whether the confirm the bound metal is Fe2+. The ferrozine assay shows that the co-purified metal is Fe2+ as suggested by the absorbance maximum at 550 nm similar to the FeSO positive control. This suggests Fe2+ may be the biologically relevant metal for catalysis (Fig. S4†).Metalloenzymes that employ a non-heme iron(II) ion and molecular oxygen in a radical transfer reaction mechanism typically possess conserved active site Tyr and Trp residues that afford electron hopping and radical resonance stabilization. The conserved Tyr91 is positioned near the metal binding site but does not interact directly with MSH or the catalytic metal. Instead, it exhibits a rotamer change upon MSH binding that repositions the Tyr91 side chain 5 Å closer to the conserved Trp139 residue. This suggests the possibility that residue Tyr91, along with the conserved Trp139, plays a similar electron transfer role in the MST-catalyzed reaction. Evaluating the MST crystal structures to identify electron transfer active pathways shows that the potential electron transfer pathway through Tyr91 and Trp 139 is the shortest pathway to transfer electrons (Fig. S5†).23 For this modelling, the ternary structure of MST with bound Zn2+ binding site was used, but Fe2+ was modelled in place of the Zn2+.
MST does not impact the efficacy of first-line TB drugs, rifampicin and isoniazid
To further evaluate the importance of MST in catalyzing the transfer of MSH to xenobiotics, growth studies were performed on M.tb and an rv0443 knockout strain. The M.tb cell-based studies show that the first-line TB drugs rifampicin and isoniazid exhibit equivalent MIC values for both the wild-type M.tb strain and rv0443 knockout strain (Table 3). In contrast to published results using strains producing undetectable levels of MSH where rifampicin sensitivity increases and isoniazid sensitivity decreases, the results presented here suggests that MST has little, if any, effect on modulating the sensitivity to rifampicin or isoniazid in M.tb (Table 3).12,24 As MST was shown to tightly bind MSH in mycobacterial extracts, these new results suggest that the primary function of MST in M.tb may be distinct from xenobiotic modification.4| Genotype | RIF MIC | INH MIC |
|---|---|---|
| CDC1551 WT | 0.0025 μg ml−1 | 0.02 μg ml−1 |
| rv0443 KO strain | 0.0025 μg ml−1 | 0.02 μg ml−1 |
Discussion
As suggested by sequence similarity in previous studies, the X-ray crystal structure of MST presented here places it in the DinB superfamily of proteins. The canonical four-helical bundle creates a metal binding site using a combination of three coordinating residues. In the case of MST and its close homologs, these conserved residues are H52, D155, and H159. The crystal structure of the MST/MSH/Zn2+ complex clearly defines the cooperative polar interactions between MST and Zn2+ necessary to bind MSH. The identity of the bound metal in the biological context and the specific chemical role of this metal is still not clear. Indeed, the metal preference may differ between the members of this family. The DSF studies with MST and various divalent metals suggest that each metal enhances MST thermal stability and supports MSH binding. Experiments determined that residual Fe2+ co-purifies with MST suggesting that MST uses Fe2+ as the catalytic metal in vivo. Considering that data as well as the structural similarities to other DinB superfamily members, functional parallels to the ergothioneine biosynthetic enzyme, EgtB, are drawn resulting in a hypothesis that MST catalyzes MSH transfer using an Fe2+-assisted radical reaction mechanism where the thiol of MSH is oxidatively activated.14 Additional kinetic experiments under appropriate hypoxic or anoxic conditions may give additional insights regarding the required metal and the MST catalytic mechanism.Structural differences between the ligand-free MST and the MST/MSH/Zn2+ complex exhibit clear impacts on second substrate binding. Complex formation with MSH requires structural changes in and around the MSH binding site as well as promoting structural changes in the L4 loop. The apparent increase in L4 loop dynamics as indicated by elevated B-factors, along with the proximity to the metal binding site, suggest this loop of conserved residues is responsible for binding the second MST substrate.
The other important aspect of the observed structural changes relates to the conserved aromatic residues, Tyr91 and Trp139, near the catalytic metal, which also supports an electron transfer role in an MST mechanism involving an oxygen radical. Again, when considering the potential role of MST Tyr91 when compared to the available EgtB/substrate complexes, another function of Tyr91 may be that of a general acid/base. The C–S bond formation catalyzed by EgtB was proposed to be a radical reaction through the formation of a complex of Fe3+, thiyl radical of γ-glutamyl cysteine, and a peroxide formed by oxygen and a proton donated from Tyr377 of EgtB. The change in rotamer conformation of MST Tyr91 upon binding of MSH may correlate with a similar role to that of Tyr377 in EgtB, but the probability of a phenylalanine residue at this position in roughly 3% of the MST homologs slightly weakens this hypothesis (Fig. 6).
Another outstanding question relates to the MSH acceptor in the MST-catalysed reaction. While many of the residues in L4 loop are conserved among actinobacteria suggesting the importance of those residues for the enzyme function, the lack of unambiguous results from the MST assays performed in vitro support a retrograde inspection of MST function in M.tb. The results of M.tb knockout studies show that lack of MST does not alter the sensitivity of M.tb to either rifampicin or isoniazid. This contrasts with previous studies clearly showing that the lack of MSH production in mycobacteria alters sensitivity to isoniazid and rifampicin. Further experiments are needed to clarify the impact of MST function on those two first-line TB drugs under different oxidative conditions and to identify alternative MSH acceptors or an alternative role for MST. Alternatively, this enzyme may play a role in MSH transfer to as yet uncharacterized acceptors and maybe involved in a novel biosynthetic pathway.
Experimental
Mycothiol synthesis
Mycothiol was synthesized according to the previous paper by using an endocyclic cleavage-recyclization process.25Molecular cloning and purification of MST
A codon-optimized gene encoding MST of Mycobacterium tuberculosis was synthesized by Integrated DNA Technology and was cloned into a pET32 plasmid (EMD Biosciences) using the Gibson assembly (New England BioLabs). The C36S mutant of MST was constructed for crystallization experiments, by site-directed mutagenesis. The mutant gene was inserted into the same pET32-based plasmid followed by the Gibson assembly (New England BioLabs). Nucleotide sequences of constructs were confirmed by gene sequencing (Eurofins MWG Operon). The E. coli BL-21 (DE3) cells (New England BioLabs) were transformed with MST constructs for expression. The cells were grown at 37 °C in Luria Broth (Research Products International) using Carbenicillin (Gold Biotechnology) as the antibiotic until an OD600 nm reached 0.6. Protein production was induced by adding 1 mM isopropyl β-D-1-thiogalactopyranoside (Gold Biotechnology) and the cultures were incubated at 16 °C for 24 hours. The E. coli cells were harvested by centrifugation at 3724 × g and cells were resuspended in a buffer containing 20 mM Tris pH 8.0, 0.2 M sodium chloride, and 5 mM β-mercaptoethanol, 5 mM imidazole.The resuspended cells were lysed with lysozyme (Hampton Research) and DNase (Roche) in ice for thirty minutes, followed by sonication (Sonicator 3000, Misonix). The crude lysate was separated by centrifugation at 10000 g (fixed-angle rotor, 5810-R Centrifuge, Eppendorf), and the supernatant was applied to a 5 mL HisTrap™ TALON® crude cobalt column (Cytiva). The column was washed with 15 column volumes of resuspension buffer (20 mM Tris pH 8.0, 0.2 M sodium chloride, and 5 mM β-mercaptoethanol, 5 mM imidazole), followed by an isocratic elution. The thioredoxin and poly-histidine fusion tag was cleaved by adding recombinant human rhinovirus 3C protease to the eluted protein and dialyzed overnight against the resuspension buffer. The fusion tag and the protease tag-removed enzymes were removed by subjecting the sample again through a cobalt column. The enzyme was then dialyzed in a buffer containing 20 mM Tris pH 8.0 and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP). The protein concentration was measured using Biotek Synergy H4 Hybrid Reader by obtaining the absorbance at 280 nm. The extinction coefficient of the protein (36440 M−1 cm−1) was calculated by the ProtParam function from the ExPASY proteomics server.26
Differential scanning fluorimetry
To examine the metal dependency, any bound metal of the purified protein was chelated by extensively dialyzing the protein in a buffer containing ethylenediaminetetraacetic acid (EDTA) for 5 days. The residual EDTA was removed by dialyzing the protein in 25 mM TRIS at pH 8.0 buffer. DSF experiments were done using a QuantStudio 3 real-time PCR thermocycler (ThermoFisher). The amount of 50 μM enzyme, 0.5 mM TCEP, 1 X SYPRO™ Orange dye (Thermofisher), and 25 mM TRIS at pH 8 were added to the DSF reaction mixture. The 100 μM of different metals (zinc acetate, cobalt chloride, nickel chloride, and ferrous sulfate) was added to the reaction mixture to analyze the metal dependency. The protein was subjected to denaturation to determine the melting temperature by monitoring the increase in the fluorescence corresponding to the temperature increase from 22 to 95 °C by continuously increasing the temperature at a rate of 0.5 °C per minute. The Tm was obtained by calculating the derivative of the data.Enzyme kinetic assays
Conjugation of MSH to monochlorobimane (mBCl) was monitored by measuring fluorescent signal at 394 nm excitation and 490 nm emission using a Biotek Synergy H4 Hybrid Reader at 37 °C.3 The reaction conditions were 500 nM MST, 200 μM MSH, 100 μM zinc acetate, 500 μM TCEP, and 25 mM HEPES at pH 7.5. The reaction was initiated by addition of 1 mM mBCl. After background subtraction, the fluorescence values were converted into product concentration using cysteine-bimane as a standard curve. For the enzymatic assay results triplicate data were plotted using Prism 9 software and non-linear regression fitting using the equation, Y = Vmax × X/(Km + X). The kcat value was determined by dividing the Vmax by the enzyme concentration.Crystallization and structure determination of MST–MSH–Zn2+ ternary complex
Crystals of MST and MST-C36S were grown using the hanging-drop vapor diffusion technique. To get the structure of the ternary complex 1.3 mM MSH and 500 μM Zinc acetate were added to a 500 μM MST-C36S sample. The mixture was incubated for 15 min on ice. Co-crystals of MST-C36S were grown using the hanging drop vapor diffusion method. Protein crystals were obtained in a well solution containing 0.1 M Bis-Tris pH 6.5, 0.2 M Magnesium chloride hexahydrate, 16% Polyethylene glycol 3350, and 0.1 M Ammonium sulfate.Diffraction datasets for all the crystals were collected at 100 K at the LS-CAT beam line at the Advanced Photon Source Argonne National Laboratory (APS-ANL, IL). The collected diffraction datasets were processed using autoProc or HKL2000.27–32 Initially, a molecular replacement was performed using the Enterococcus faecalis EF_3021 (PDB: 3CEX) as a search model with the apo MST dataset, whereas subsequent datasets were phased by isomorphous replacement when appropriate. The apo MST structure was used as the search model for the MST–MSH–Zn2+ ternary complex. Rigid body refinement, simulated annealing, and positional and B-factor refinements were carried out using PHENIX.33 Manual refinement of the structure was performed using COOT.34 Structural validation was performed using Molprobity.35
Conclusions
This study provides the first structural details of M.tb MST confirming that it belongs to the DinB superfamily of proteins. The X-ray crystal structure of the MST–MSH–Zn2+ ternary complex provides detailed interactions supporting of MSH binding within the enzyme active site as well as the cooperative function of divalent metals in MSH binding. Further characterization of the protein suggests Fe2+ as the active site metal in vivo and its function in a radical-based enzymatic reaction. Further experiments are needed to fully understand how MST catalyzes MSH transfer as well as the identity of biologically relevant acceptors. M.tb cell-based study shows that this enzyme does not significantly contribute to increasing the resistance of M.tb to rifampicin or the sensitivity to the prodrug isoniazid. These data support additional studies MST to identify authentic MST substrates and to fully understand the MST mechanism of catalysis for MSH transfer to those acceptors. Finally, more studies are advised to identify the M.tb enzyme that plays a more prominent role in antitubercular drug modification using MSH.Author contributions
D. R. R. conceptualized the study. D. R. R., S. M. and M. J. provided resources, supervision, and validation. L. F., J. J. L., M. T. B., Y. P. J., and Z. P. performed the experiments and formal analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research was supported in part by NIH grant R21AI151924 to DRR. The following reagents were obtained through BEI Resources, NIAID, NIH: Mycobacterium tuberculosis, Strain CDC1551, Transposon Mutant 3271 (MT0459, Rv0443), NR-18896. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).References
- V. K. Jothivasan and C. J. Hamilton, Mycothiol: synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes, Nat. Prod. Rep., 2008, 25(6), 1091–1117 RSC.
- G. L. Newton, N. Buchmeier and R. C. Fahey, Biosynthesis and Functions of Mycothiol, the Unique Protective Thiol of Actinobacteria, Microbiol. Mol. Biol. Rev., 2008, 72(3), 471 CrossRef CAS PubMed.
- R. C. Fahey, Glutathione analogs in prokaryotes, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830(5), 3182–3198 CrossRef CAS PubMed.
- G. L. Newton, S. S. Leung, J. I. Wakabayashi, M. Rawat and R. C. Fahey, The DinB Superfamily Includes Novel Mycothiol, Bacillithiol, and Glutathione S-Transferases, Biochemistry, 2011, 50(49), 10751–10760 CrossRef CAS PubMed.
- G. L. Newton, K. Arnold, M. S. Price, C. Sherrill, S. B. Delcardayre, Y. Aharonowitz, G. Cohen, J. Davies, R. C. Fahey and C. Davis, Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes, J. Bacteriol., 1996, 178(7), 1990 CrossRef CAS PubMed.
- F. Fan, M. W. Vetting, P. A. Frantom and J. S. Blanchard, Structures and mechanisms of the mycothiol biosynthetic enzymes, Curr. Opin. Chem. Biol., 2009, 13(4), 451–459 CrossRef CAS PubMed.
- G. L. Newton, M. D. Unson, S. J. Anderberg, J. A. Aguilera, N. N. Oh, S. B. delCardayre, Y. Av-Gay and R. C. Fahey, Characterization of Mycobacterium smegmatis Mutants Defective in 1-d-myo-Inosityl-2-amino-2-deoxy-α-d-glucopyranoside and Mycothiol Biosynthesis, Biochem. Biophys. Res. Commun., 1999, 255(2), 239–244 CrossRef CAS PubMed.
- M. Rawat, G. L. Newton, M. Ko, G. J. Martinez, R. C. Fahey and Y. Av-Gay, Mycothiol-Deficient Mycobacterium smegmatis Mutants Are Hypersensitive to Alkylating Agents, Free Radicals, and Antibiotics, Antimicrob. Agents Chemother., 2002, 46(11), 3348 CrossRef CAS PubMed.
- X. Xu, C. Vilchèze, Y. Av-Gay, A. Gómez-Velasco and W. R. Jacobs, Jr, Precise null deletion mutations of the mycothiol synthesis genes reveal their role in isoniazid and ethionamide resistance in Mycobacterium smegmatis, Antimicrob. Agents Chemother., 2011, 55(7), 3133–3139 CrossRef CAS PubMed.
- C. Vilchèze, Y. Av-Gay, S. W. Barnes, M. H. Larsen, J. R. Walker, R. J. Glynne and W. R. Jacobs, Jr, Coresistance to isoniazid and ethionamide maps to mycothiol biosynthetic genes in Mycobacterium bovis, Antimicrob. Agents Chemother., 2011, 55(9), 4422–4423 CrossRef PubMed.
- C. Sao Emani, J. Williams Monique, J. Wiid Ian, F. Hiten Nicholas, J. Viljoen Albertus, D. Pietersen Ray-Dean, D. van Helden Paul and B. Baker, Ergothioneine Is a Secreted Antioxidant in Mycobacterium smegmatis, Antimicrob. Agents Chemother., 2013, 57(7), 3202–3207 CrossRef PubMed.
- N. A. Buchmeier, G. L. Newton, T. Koledin and R. C. Fahey, Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics, Mol. Microbiol., 2003, 47(6), 1723–1732 CrossRef CAS PubMed.
- S. S. Rajan, X. Yang, L. Shuvalova, F. Collart and W. F. Anderson, YfiT from Bacillus subtilis is a probable metal-dependent hydrolase with an unusual four-helix bundle topology, Biochemistry, 2004, 43(49), 15472–15479 CrossRef CAS PubMed.
- K. V. Goncharenko, A. Vit, W. Blankenfeldt and F. P. Seebeck, Structure of the sulfoxide synthase EgtB from the ergothioneine biosynthetic pathway, Angew. Chem., Int. Ed., 2015, 54(9), 2821–2824 CrossRef CAS PubMed.
- S. Alquethamy, K. Ganio, Z. Luo, S. I. Hossain, A. J. Hayes, T. Ve, M. R. Davies, E. Deplazes, B. Kobe and C. A. McDevitt, Structural and biochemical characterization of Acinetobacter baumannii ZnuA, J. Inorg. Biochem., 2022, 231, 111787 CrossRef CAS PubMed.
- A. M. Rice, A. Faig, D. E. Wolff and S. B. King, Sodium borohydride and thiol mediated nitrite release from nitroaromatic antibiotics, Bioorg. Med. Chem. Lett., 2021, 48, 128245 CrossRef CAS PubMed.
- D. R. Cooper, K. Grelewska, C.-Y. Kim, A. Joachimiak and Z. S. Derewenda, The structure of DinB from Geobacillus stearothermophilus: a representative of a unique four-helix-bundle superfamily, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66(3), 219–224 CrossRef CAS PubMed.
- K. V. Goncharenko, A. Vit, W. Blankenfeldt and F. P. Seebeck, Structure of the Sulfoxide Synthase EgtB from the Ergothioneine Biosynthetic Pathway, Angew. Chem., Int. Ed., 2015, 54(9), 2821–2824 CrossRef CAS PubMed.
- S. S. Rajan, X. Yang, L. Shuvalova, F. Collart and W. F. Anderson, YfiT from Bacillus subtilis Is a Probable Metal-Dependent Hydrolase with an Unusual Four-Helix Bundle Topology, Biochemistry, 2004, 43(49), 15472–15479 CrossRef CAS PubMed.
- D. R. Cooper, K. Grelewska, C. Y. Kim, A. Joachimiak and Z. S. Derewenda, The structure of DinB from Geobacillus stearothermophilus: a representative of a unique four-helix-bundle superfamily, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66(Pt 3), 219–224 CrossRef CAS PubMed.
- E. C. Meng, E. F. Pettersen, G. S. Couch, C. C. Huang and T. E. Ferrin, Tools for integrated sequence-structure analysis with UCSF Chimera, BMC Bioinf., 2006, 7(1), 339 CrossRef PubMed.
- J. Kyte and R. F. Doolittle, A simple method for displaying the hydropathic character of a protein, J. Mol. Biol., 1982, 157(1), 105–132 CrossRef CAS PubMed.
- R. N. Tazhigulov, J. R. Gayvert, M. Wei and K. B. Bravaya, eMap: A Web Application for Identifying and Visualizing Electron or Hole Hopping Pathways in Proteins, J. Phys. Chem. B, 2019, 123(32), 6946–6951 CrossRef CAS PubMed.
- M. Hernick, Mycothiol: a target for potentiation of rifampin and other antibiotics against Mycobacterium tuberculosis, Expert Rev. Anti-infect. Ther., 2013, 11(1), 49–67 CrossRef CAS PubMed.
- S. Manabe and Y. Ito, Mycothiol synthesis by an anomerization reaction through endocyclic cleavage, Beilstein J. Org. Chem., 2016, 12, 328–333 CrossRef CAS PubMed.
- E. H. C. Gasteiger, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, Protein identification and analysis tools on the ExPASy server, in The proteomics protocols handbook, ed. J. M. Walker, Humana Press Inc, Totowa, NJ, 2005, pp. 571–607 Search PubMed.
- Z. Otwinowski and W. Minor, Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol., 1997, 276, 307–326 CAS.
- W. Kabsch, XDS, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(2), 125–132 CrossRef CAS PubMed.
- P. Evans, Scaling and assessment of data quality, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62(1), 72–82 CrossRef PubMed.
- P. R. Evans and G. N. Murshudov, How good are my data and what is the resolution?, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69(7), 1204–1214 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Overview of the CCP4 suite and current developments, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67(4), 235–242 CrossRef CAS PubMed.
- C. Vonrhein, I. J. Tickle, C. Flensburg, P. Keller, W. Paciorek, A. Sharff and G. Bricogne, Advances in automated data analysis and processing within autoPROC, combined with improved characterisation, mitigation and visualisation of the anisotropy of diffraction limits using STARANISO, Acta Crystallogr., Sect. A: Found. Adv., 2018, 74(a1), a360 Search PubMed.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Towards automated crystallographic structure refinement with phenix. refine, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68(4), 352–367 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(Pt 4), 486–501 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall, 3rd, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(Pt 1), 12–21 CrossRef CAS PubMed.
- A. Martin, M. Camacho, F. Portaels and C. Palomino Juan, Resazurin Microtiter Assay Plate Testing of Mycobacterium tuberculosis Susceptibilities to Second-Line Drugs: Rapid, Simple, and Inexpensive Method, Antimicrob. Agents Chemother., 2003, 47(11), 3616–3619 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00401a |
| ‡ Current address: Biochemistry and Biophysics Center, National Heart, Lung, and Blood Institute, Bethesda, Maryland, USA. |
| § Current address: Duke Human Vaccine Institute, Durham, NC, USA. |
| ¶ Current address: Irving Institute for Cancer Dynamics, Columbia University, New York, NY, USA. |
| This journal is © The Royal Society of Chemistry 2023 |