
Design, synthesis and evaluation of halogenated phenazine antibacterial prodrugs targeting nitroreductase enzymes for activation†
Ke
Liu
 ,
Tao
Xiao
,
Hongfen
Yang
,
Manyun
Chen
,
Qiwen
Gao
,
Beau R.
Brummel
,
Yousong
Ding
and
Robert W.
Huigens
III
*
,
Tao
Xiao
,
Hongfen
Yang
,
Manyun
Chen
,
Qiwen
Gao
,
Beau R.
Brummel
,
Yousong
Ding
and
Robert W.
Huigens
III
*
Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, USA. E-mail: rhuigens@cop.ufl.edu
First published on 6th June 2023
Abstract
It is of great importance to develop new strategies to combat antibiotic resistance. Our lab has discovered halogenated phenazine (HP) analogues that are highly active against multidrug-resistant bacterial pathogens. Here, we report the design, synthesis, and study of a new series of nitroarene-based HP prodrugs that leverage intracellular nitroreductase (NTR) enzymes for activation and subsequent release of active HP agents. Our goals of developing HP prodrugs are to (1) mitigate off-target metal chelation (potential toxicity), (2) possess motifs to facilitate intracellular, bacterial-specific HP release, (3) improve water solubility, and (4) prevent undesirable metabolism (e.g., glucuronidation of HP's phenol). Following the synthesis of HP-nitroarene prodrugs bearing a sulfonate ester linker, NTR-promoted release experiments demonstrated prodrug HP-1-N released 70.1% of parent HP-1 after 16 hours (with only 6.8% HP-1 release without NTR). In analogous in vitro experiments, no HP release was observed for control sulfonate ester compounds lacking the critical nitro group. When compared to parent HP compounds, nitroarene prodrugs evaluated during these studies demonstrate similar antibacterial activities in MIC and zone of inhibition assays (against lab strains and clinical isolates). In conclusion, HP-nitroarene prodrugs could provide a future avenue to develop potent agents that target antibiotic resistant bacteria.
Introduction
Since the end of the antibiotic “golden age” in the late 1960s, very few new classes of antibiotics have entered the clinic.1,2 In addition to the limited antibiotic discovery pipeline over the last 50 years, numerous problems associated with antibiotic resistance have significantly increased the need for new antibacterial agents with novel modes of action.3,4 Currently in the United States, more than 2.8 million bacterial infections occur and result in > 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths each year.5 Bacteria are notorious for their ability to develop resistance and evade conventional antibiotic treatments using well-characterized mechanisms, which include: 1) target mutation resulting in a reduction/loss of antibiotic binding, 2) target overproduction, 3) drug modification/degradation, 4) alterations in membrane chemistry to reduce drug penetration, and 5) efflux pump action to reduce antibiotic accumulation within the cell.3,6 If unanswered, antibiotic resistant infections are expected to be the leading cause of death in 2050 with 10 million deaths per year.7
000 deaths each year.5 Bacteria are notorious for their ability to develop resistance and evade conventional antibiotic treatments using well-characterized mechanisms, which include: 1) target mutation resulting in a reduction/loss of antibiotic binding, 2) target overproduction, 3) drug modification/degradation, 4) alterations in membrane chemistry to reduce drug penetration, and 5) efflux pump action to reduce antibiotic accumulation within the cell.3,6 If unanswered, antibiotic resistant infections are expected to be the leading cause of death in 2050 with 10 million deaths per year.7
Our lab has discovered and developed a series of halogenated phenazine (HP) antibacterial agents that target Gram-positive human pathogens and M. tuberculosis.8–18 The discovery of HP agents was initially inspired by a microbial competition interaction within the lungs of cystic fibrosis (CF) patients. Many young CF patients initially experience chronic Staphylococcus aureus infections within their lungs; however, as these patient age, Pseudomonas aeruginosa co-infects the lungs and eradicates the established S. aureus infection by secreting redox-active phenazine antibiotics (e.g. pyocyanin, 1-hydroxyphenazine).19–21
During preliminary investigations, we have reported the chemical synthesis, microbiological studies, and structure–activity relationships (SAR) of > 100 HP analogues. Potent HP analogues have demonstrated excellent activities against planktonic bacteria (minimum inhibitory concentration, MIC ≤ 0.1 μM) and established biofilms (minimum biofilm eradication concentration, MBEC ≤ 10 μM), including methicillin-resistant S. aureus (MRSA), methicillin-resistant S. epidermidis (MRSE), and vancomycin-resistant Enterococcus faecium (VRE).9–11,13,15,18 Unlike redox-active phenazine antibiotics (e.g., pyocyanin), HPs operate through a unique iron-starvation mechanism utilizing the critical iron chelation moiety (1-hydroxyl group and the adjacent nitrogen) in the HP scaffold.12,15,16,18 In transcript profiling (RNA-seq) studies, we demonstrated HP-14 rapidly induces the transcription of multiple iron uptake gene clusters in MRSA biofilms.12 In follow-up work, we have shown that other potent HP analogues have induced the transcription of iron-uptake biomarkers (e.g., sbnC, staphyloferrin B, siderophore used for iron uptake from local environment; isdB, iron-regulated surface determinant, involved in iron acquisition from heme).15,16,18
In separate work, HP-29 demonstrated in vivo efficacy in dorsal wound infections against S. aureus UAMS-1 (0.82-log10 reduction in CFU/lesion, 3 day treatment in mice) and E. faecium OG1RF (1.73-log10 reduction in CFU/lesion, 3 day treatment).15 Currently, we are working to expand HP treatments to address systemic infections through the development of prodrugs of lead HPs. To do this, we are synthetically functionalizing the phenolic hydroxyl group of active HPs to afford prodrug molecules that (1) mitigate off-target metal chelation (addressing toxicity issues), (2) possess trigger motifs that allow intracellular, bacterial-specific release of HP molecules, (3) improve water solubility, and (4) prevent undesirable metabolism (e.g., glucuronidation of the phenolic group in HP agents).11,13,14,16,17 We have reported several types of HP prodrugs that include carbonate-based PEGs,11,13 antibiotic conjugates,14,16 and quinone triggers (QuAOCOM- and ether-linked).13,17
In this study, we describe a new HP prodrug strategy that targets nitroreductase (NTR) enzymes found within most bacteria.22–24 NTR enzymes are known to reduce nitroarenes to the corresponding aniline products. Here, we aim to leverage NTR's ability to reduce nitroarenes and implement Chopra and Chakrapani's design that utilizes a sulfonate ester linker to develop HP prodrugs.22 The sulfonate ester linker (1) allows a straightforward synthesis of HP-nitroarene prodrug analogues, and (2) facilitates the release of the active HP warhead following reduction of the nitroarene to aniline (which includes loss of SO2 gas; see Fig. 1). For this work, we selected 6,8-CF3-HP (compound 8, a new potent HP analogue; Fig. 2), and three potent HP analogues (HP-1: 9, 7-Cl-HP: 10, HP-29: 11; Fig. 2B)8,13,15 to synthesize and investigate nitroarene prodrugs. These HP prodrugs were further evaluated in enzymatic release experiments and microbiological assays.
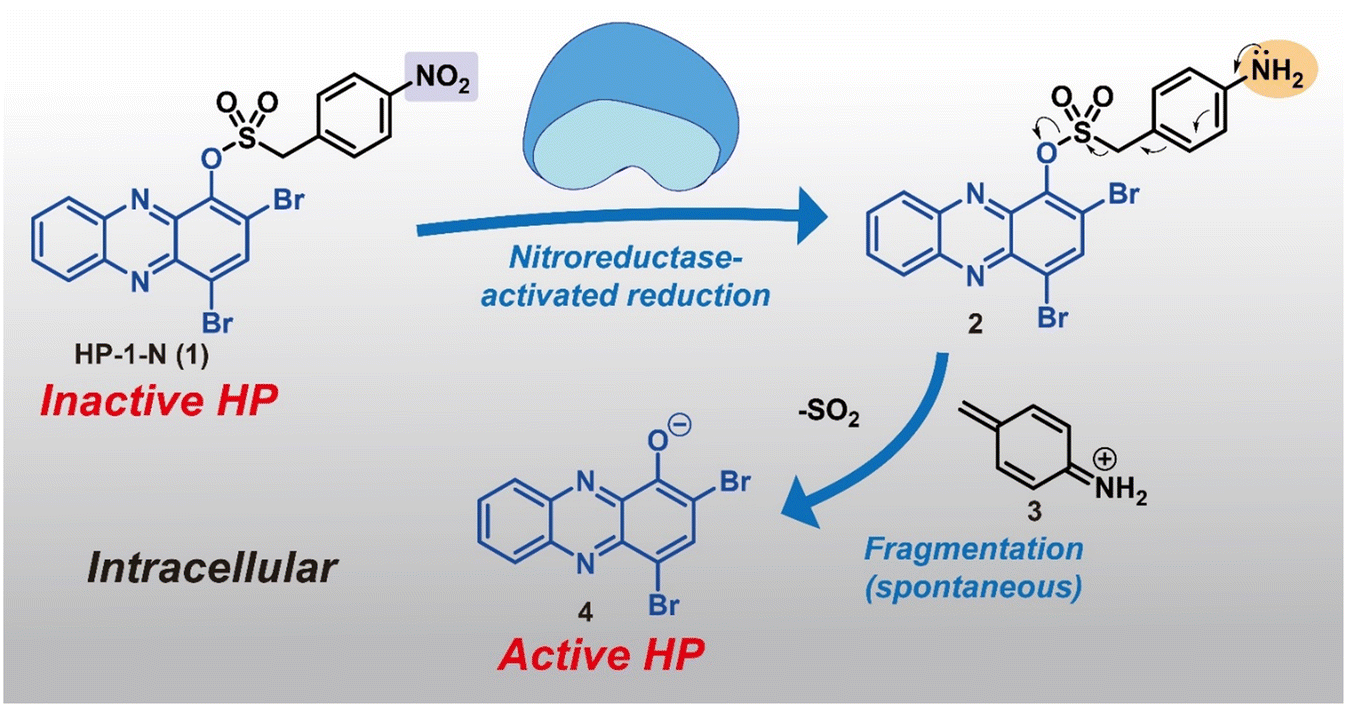 | ||
| Fig. 1 Within the bacterial cell, HP-nitroarene prodrugs (e.g., HP-1-N) are designed to release active HP 4 following NTR-promoted reduction of the nitroarene to aniline, and subsequent release of sulfur dioxide (SO2). | ||
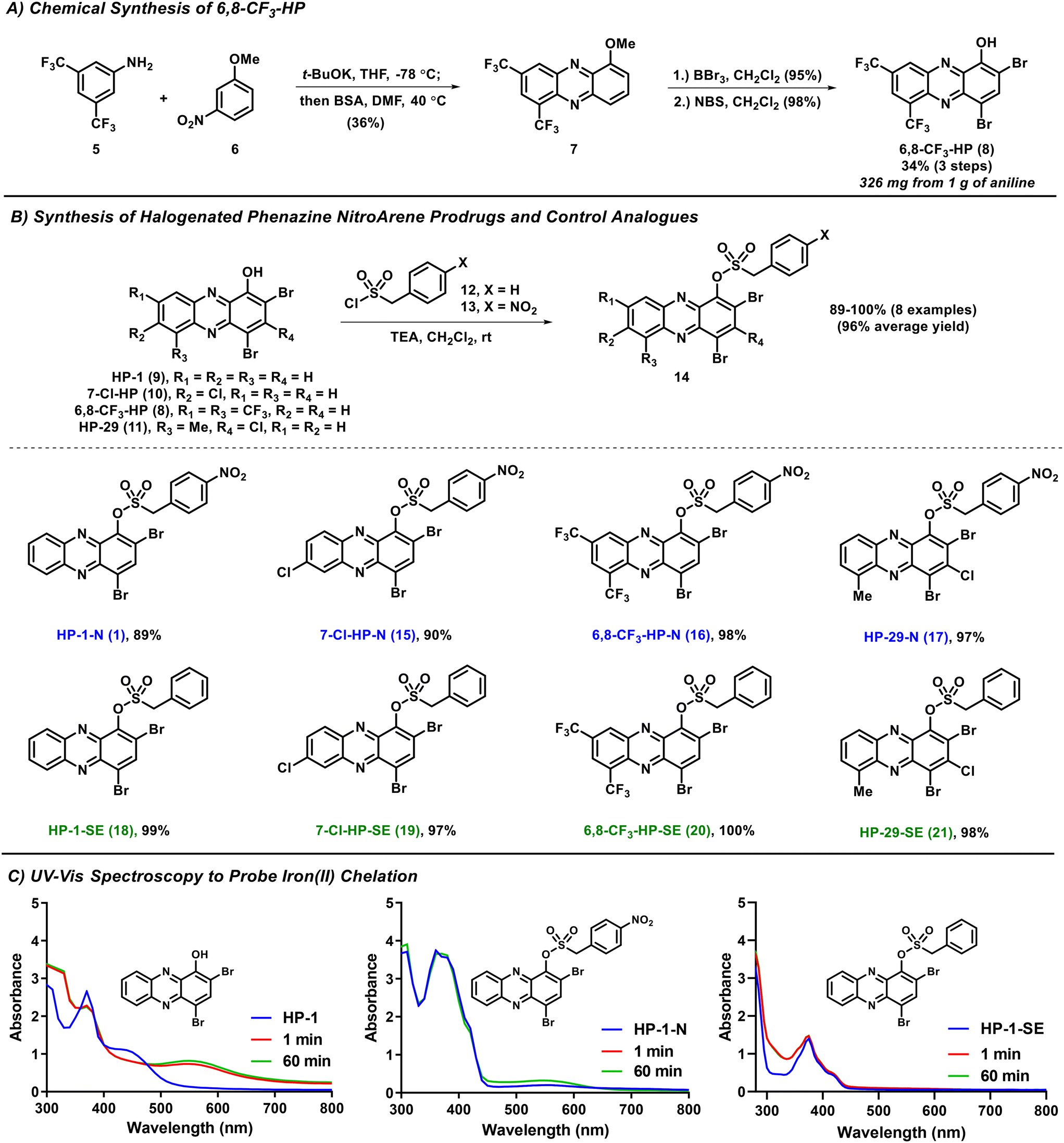 | ||
| Fig. 2 Synthetic schemes of 6,8-CF3-HP, HP-nitroarene prodrugs and sulfonate ester control compounds. UV-vis spectroscopy showing HP-nitroarene prodrugs and control compounds unable to bind iron(II). | ||
Results and discussion
The synthesis of 6,8-CF3-HP was initiated with a potassium tert-butoxide-promoted condensation between 3,5-bis(trifluoromethyl)aniline 5 and 3-nitroanisole 6 to form a nitroso intermediate (not shown) that was immediately taken forward and reacted with N,O-bis(trimethylsilyl)acetamide (BSA) to produce 1-methoxy-6,8-bis(trifluoromethyl)phenazine 7 with a 39% yield (Fig. 2A). Our synthesis of phenazine 7 was inspired by Wróbel and co-workers.25–28 1-Methoxyphenazine 7 was then subjected to (1) demethylation with boron tribromide (BBr3, 95% yield), and (2) bromination using N-bromosuccinimide (NBS) to synthesize 6,8-CF3-HP (98% yield). We found this route to be scalable, producing 326 milligrams of 6,8-CF3-HP from 1 gram of starting 3,5-bis(trifluoromethyl)aniline 5.The synthesis of initial nitroarene prodrugs involved a single reaction between four potent HP agents and (4-nitrophenyl)methanesulfonyl chloride in the presence of triethylamine to afford target HP-nitroarene prodrugs HP-1-N (1; 89% yield), 7-Cl-HP-N (15; 90%), 6,8-CF3-HP-N (16; 98%) and HP-29-N (17; 97%) in good to excellent yields (Fig. 2B). In addition to HP-nitroarene prodrugs, we synthesized the analogous sulfonate ester analogues without the key nitro functional group to probe this particular molecular trigger in this prodrug design. To this end, the same four HP molecules were reacted with 4-phenylmethanesulfonyl chloride to yield sulfonate ester comparator compounds HP-1-SE (18; 99%), 7-Cl-HP-SE (19; 97%), 6,8-CF3-HP-SE (20; 100%), and HP-29-SE (21, 98%).
Following the synthesis of HP nitroarene prodrugs and sulfonate ester control compounds, we used UV-vis spectroscopy to determine if our new analogues are able to directly chelate iron(II) or not.15,18 This is a critical design element to HP prodrugs as we aim to make analogues that are unable to bind iron(II), and other metal cations, until the molecular trigger is activated and HP is released within the bacterial cell. For all parent HPs, we observed direct iron(II) binding, as expected, based on a new peak formed in the UV-vis spectrum at ∼550 nm (see HP-1, Fig. 2C). In UV-vis experiments with HP nitroarenes and sulfonate ester control compounds, we observed no direct iron(II) binding, as designed (Fig. 2C; ESI†).
Next, we subjected the HP-nitroarenes and sulfonate esters to in vitro prodrug release experiments22 in the presence of nitroreductase (NTR from Escherichia coli; Fig. 3). Experimentally, all test compounds were incubated at 37 °C in Tris buffer with or without NTR for 2 and 16 hours (compound concentration: 1 mM; enzyme concentration: 0.1 mg mL−1). The percent HP release from HP-nitroarene and sulfonate ester control compounds was quantified using LC-MS (see Supporting Information for details). After 2 hours incubation with NTR, 30.3% of HP-1 was released from prodrug HP-1-N, whereas only 3.5% HP-1 was released in the absence of the enzyme (likely caused by sulfonate hydrolysis). After 16 hours, NTR promoted 70.1% release of HP-1 from HP-1-N, whereas only 6.8% HP-1 release was observed in the absence of NTR. As expected, no HP-1 was released from the corresponding sulfonate ester control compound HP-1-SE following incubation with NTR. HP-nitroarene compounds 7-Cl-HP-N and 6,8-CF3-HP-N (and their corresponding sulfonate ester control compounds) demonstrated similar HP-release profiles in experiments with NTR (Fig. 3); however, HP-29-N gave highly inconsistent results in these experiments (data not shown), likely due to solubility issues.
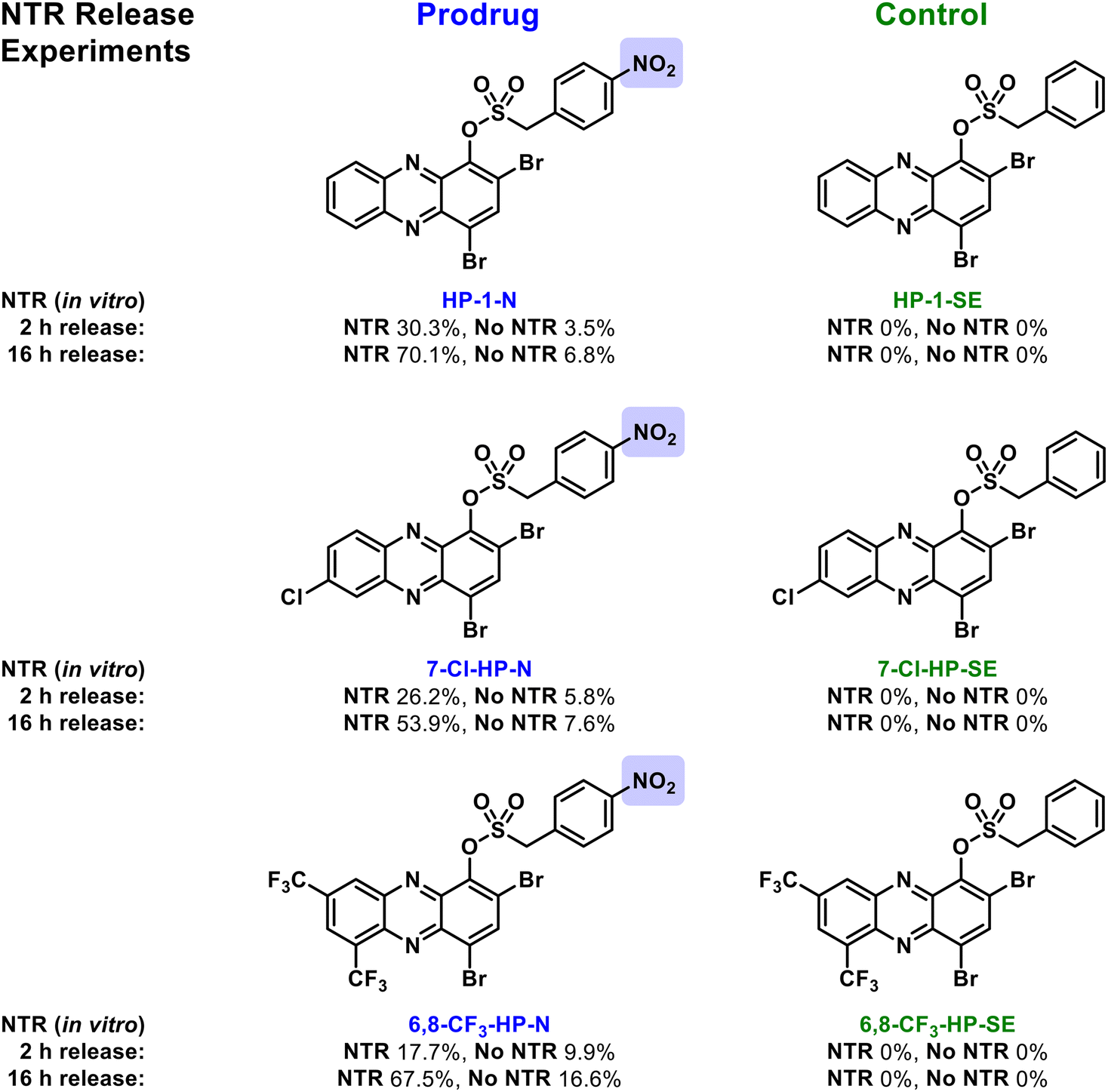 | ||
| Fig. 3 In vitro findings with nitroreductase-promoted release of HP analogues from nitroarene prodrugs (e.g., HP-1-N) alongside sulfonate ester control compounds (e.g., HP-1-SE). | ||
Following in vitro assessment of bacterial nitroreductase, we evaluated HP-nitroarene prodrugs against MRSA 1707 using Petri dishes to determine relative antibacterial activity of analogues via the relative zone of inhibition in agar diffusion assays (Fig. 4). Our experimental approach was to evaluate HP-1, prodrug HP-1-N and control HP-1-SE alongside on a single Petri dish (minimum of three independent replicates; other focused HP prodrug series were evaluated using an analogues approach, see Supporting Information). In these experiments, HP-1 demonstrated the most potent activity with a zone of inhibition at 5.08 ± 0.52 cm2 against MRSA 1707, while nitroarene prodrug HP-1-N gave a zone of inhibition of 1.13 ± 0.24 cm2. We believe the overall reduction in zone of inhibition results for HP-1-N compared to HP-1 is indicative of the time needed for the processing of the nitroarene prodrug moiety to release the active HP warhead. As designed, HP-1-SE showed no inhibition activity against MRSA 1707, demonstrating the (1) requirement of the nitro-group functionality of HP-1-N, and (2) stability of the sulfonate ester linker for HP-1-SE. Similar activity profiles were observed for the 7-Cl-HP and HP-29 series in the Petri dish growth inhibition experiments against MRSA 1707; however, control compound 6,8-CF3-HP-SE (designed to be inactive against bacteria) demonstrated some antibacterial activities in these experiments, likely due to some non-specific linker cleavage (Table 1).
| Compound | Iron binding (UV-vis) | 2 h release (%) | 16 h release (%) | MRSA 1707 zone of inhibition (cm2) |
|---|---|---|---|---|
| NTR/no NTR | NTR/no NTR | |||
| HP-1 | Yes | — | — | 5.08 ± 0.52 |
| HP-1-N | No | 30.3 ± 5.6/3.5 ± 1.4 | 70.1 ± 15.9/6.8 ± 3.0 | 1.13 ± 0.24 |
| HP-1-SE | No | 0/0 | 0/0 | 0 |
| 7-Cl-HP | Yes | — | — | 1.70 ± 0.07 |
| 7-Cl-HP-N | No | 26.3 ± 11.9/5.8 ± 2.9 | 53.9 ± 17.4/7.6 ± 3.3 | 0.66 ± 0.07 |
| 7-Cl-HP-SE | No | 0/0 | 0/0 | 0 |
| 6,8-CF 3 -HP | Yes | — | — | 4.58 ± 0.17 |
| 6,8-CF 3 -HP-N | No | 17.7 ± 7.3/9.9 ± 4.3 | 67.5 ± 32.7/16.6 ± 9.4 | 2.57 ± 0.19 |
| 6,8-CF 3 -HP-SE | No | 0/0 | 0/0 | 1.07 ± 0.08 |
| HP-29 | Yes | — | — | 3.85 ± 0.18 |
| HP-29-N | No | — | — | 0.87 ± 0.09 |
| HP-29-SE | No | — | — | 0 |
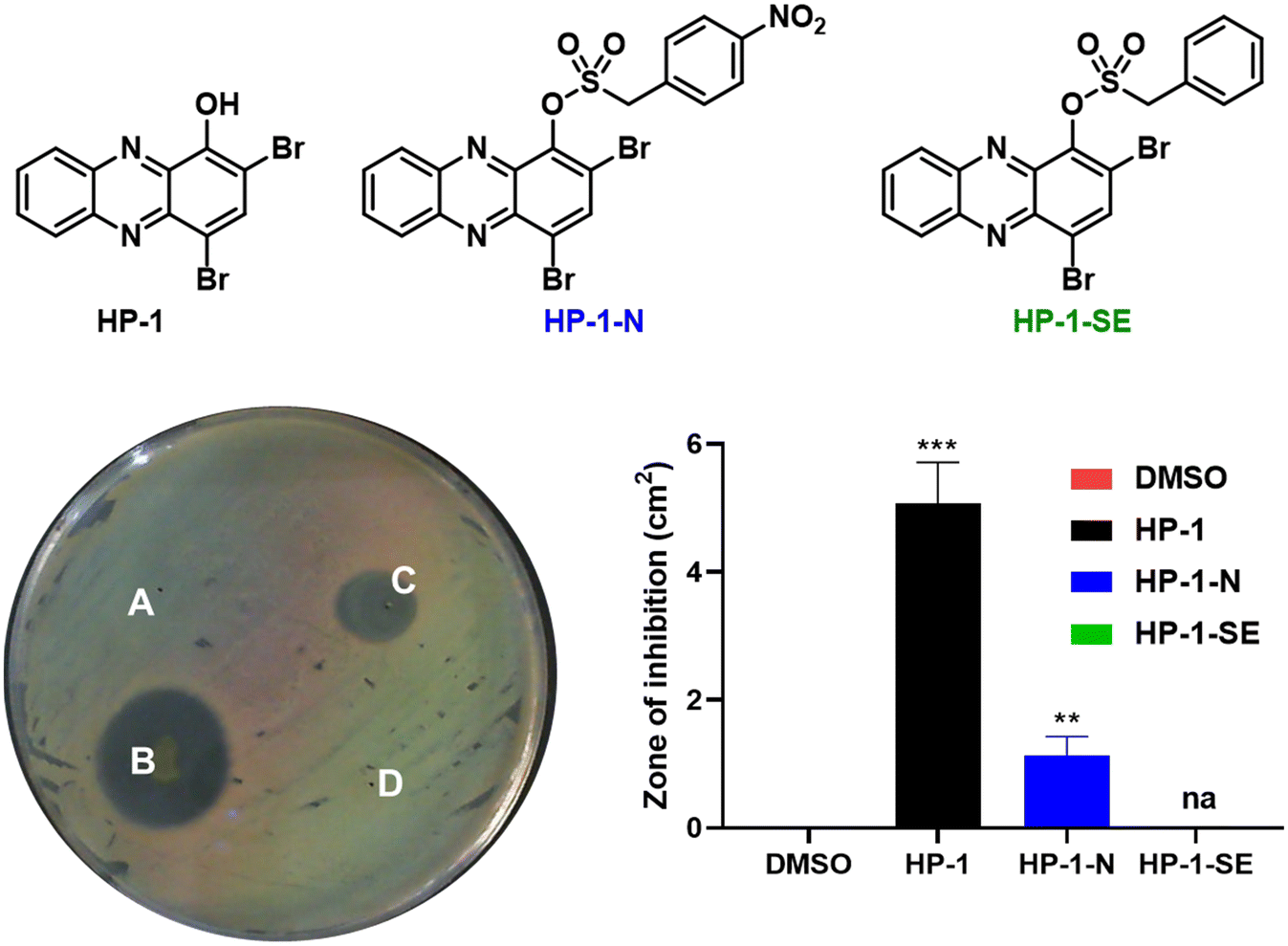 | ||
| Fig. 4 Agar diffusion assay to determine the zone of inhibition against MRSA 1707. (A.) DMSO, negative control, (B.) HP-1, zone of inhibition = 5.08 ± 0.52 cm2, (C.) HP-1-N, zone of inhibition = 1.13 ± 0.24 cm2, and (D.) HP-1-SE, zone of inhibition = 0 cm2. **P-Value <0.01, ***P-value <0.001, na: not applicable (Students' T-test). | ||
Following agar diffusion experiments, we assessed antibacterial activities of HP prodrugs alongside their corresponding HP parent and non-nitro containing sulfonate ester analogue in minimum inhibition concentration (MIC) assays (Fig. 5; Table 2). During the course of these studies, prodrug HP-1-N demonstrated good-to-moderate antibacterial activities against MRSA, MRSE, VRE, E. faecalis and S. pneumoniae strains (MIC profiles can be viewed in Table 2), similar to the activity profile of parent HP-1 (≤4-fold difference in MIC values). In addition, the sulfonate ester control HP-1-SE was found to be inactive across all strains (MIC > 100 μM), demonstrating ideal linker stability in these experiments. In addition, the HP-29 series demonstrated ideal MIC profiles with prodrug HP-29-N (MIC = 0.10–0.59 μM against MRSA, MRSE, VRE, S. pneumoniae; MIC = 4.69 μM against E. faecalis OG1RF) demonstrating near equipotent activity compared to HP-29 (MIC = 0.10–0.39 μM) across most strains while HP-29-SE reported no activity (MIC > 100 μM) in these assays.
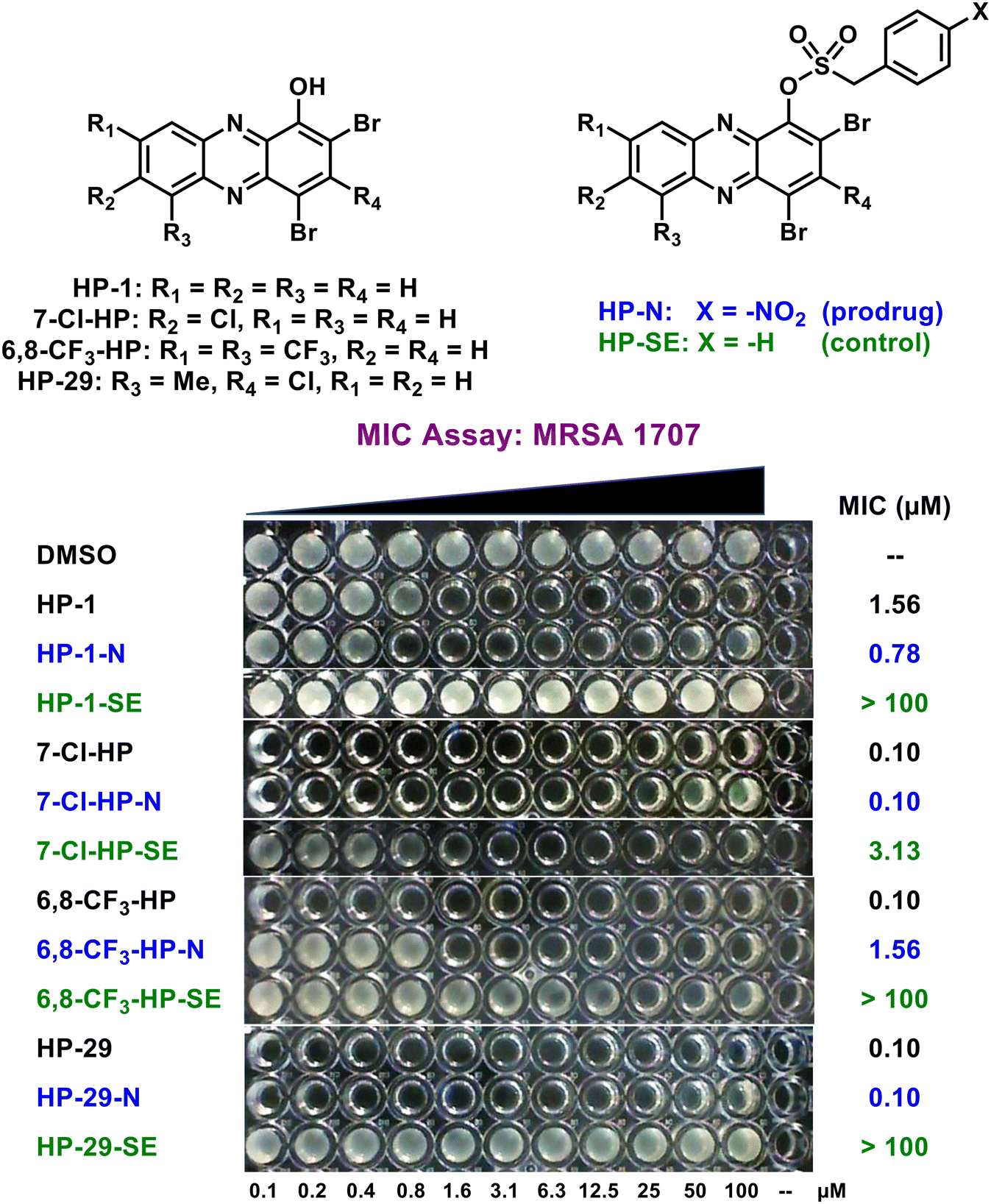 | ||
| Fig. 5 MIC assay of HP prodrug analogues against MRSA 1707. | ||
| Compound | MRSA BAA-1707 | MRSA BAA-44 | S. epi 12228 | MRSE 35984 | VRE 700221 | E. faecalis OG1RF | S. pneu. 6303 | % hemolysis (200 μM) | HEK-293 (CC50) |
|---|---|---|---|---|---|---|---|---|---|
| a Midpoint value for 2-fold range in MIC values observed. b Lowest concentration tested. “Hemolysis” refers to percent lysis of red blood cells observed at 200 μM. Each data point is the result of three independent experiments. | |||||||||
| HP-1 | 1.17a | 1.56 | 2.35a | 1.56 | 4.69a | 18.8a | 1.17a | <1 | — |
| HP-1-N | 1.17a | 1.56 | 4.69a | 4.69a | 9.38a | 37.5a | 4.69a | <1 | 51.7 |
| HP-1-SE | >100 | >100 | >100 | >100 | >100 | >100 | >100 | — | >100 |
| 7-Cl-HP | 0.075a | 0.15a | 0.30a | 0.39 | 0.59a | 4.69a | 0.30a | <1 | — |
| 7-Cl-HP-N | 0.10b | 0.15a | 0.15a | 0.39 | 0.59a | 4.69a | 0.15a | <1 | 6.4 |
| 7-Cl-HP-SE | 2.35a | 1.17a | 3.13 | 4.69a | >100 | >100 | 1.56 | — | 30.3 |
| 6,8-CF 3 -HP | 0.15a | 0.15a | 0.10b | 0.05b | 0.20 | 0.10b | 0.10b | 16.7 | — |
| 6,8-CF 3 -HP-N | 1.56 | 4.69a | 0.15a | 0.78 | 1.56 | 0.78 | 0.20 | 10.4 | 2.9 |
| 6,8-CF 3 -HP-SE | >100 | 18.8a | 0.15a | 1.56 | 0.78 | 0.39 | 0.20 | — | 3.6 |
| HP-29 | 0.10b | 0.10b | 0.10b | 0.10b | 0.15a | 0.39 | 0.10b | <1 | 18.3 |
| HP-29-N | 0.10b | 0.30a | 0.59a | 0.15a | 0.59a | 4.69a | 0.15a | <1 | 34.1 |
| HP-29-SE | >100 | >100 | >100 | >100 | >100 | >100 | >100 | — | 49.6 |
| Vancomycin | 0.78 | 0.30a | 1.56 | 0.59a | >100 | 1.56 | 0.59a | <1 | — |
| QAC-10 | 4.69a | — | — | 2.35a | 2.35a | — | — | >99 | — |
Despite the HP-1-N and HP-29-N series demonstrating ideal MIC profiles, the 7-Cl-HP-N and 6,8-CF3-HP-N series demonstrated less-than-ideal sulfonate ester linker stabilities as evident from the SE (control) analogues (Table 2). All SE analogues in this study were designed to be inactive in antibacterial assays (expecting MIC values > 100 μM) and synthesized to probe general linker stability; however, 7-Cl-HP-SE reported MIC values of 1.17–4.69 μM against 5 of the 7 strains used in our panel, while 6,8-CF3-HP-SE was found to demonstrate antibacterial activities against 6 of 7 strains (MIC = 0.15–18.8 μM). Evaluating the 7-Cl-HP-N and 6,8-CF3-HP-N analogue series were insightful and their linker stability concerns became the primary reason we moved forward with the HP-1-N and HP-29-N series.
Following our initial antibacterial assessment, HP analogues were evaluated in hemolysis and cytotoxicity assays (Table 2). HP and nitroarene prodrug analogues demonstrated negligible (< 1%) hemolysis activity against red blood cells (RBC) at 200 μM with the exception of 6,8-CF3-HP and 6,8-CF3-HP-N reporting 16.7 and 10.4% hemolysis at this concentration (note: QAC-10 is a known membrane-lysing antibacterial agent29 and was used as a comparator in these experiments, demonstrating >99% hemolysis during these studies). Several HP analogues were evaluated against HEK-293 cells with HP prodrugs HP-1-N (CC50 = 51.7 μM) and HP-29-N (CC50 = 34.1 μM) demonstrated good cytotoxicity profiles (note: when comparing the CC50 values in HEK-293 cells to MIC values against MRSA, a selectivity index of ∼200 can be quantified for prodrug HP-29-N). Finally, we note improved cytotoxicity against HEK-293 cells regarding prodrug HP-29-N (CC50 = 34.1 μM) compared to parent HP-29 (CC50 = 18.3 μM).
After we determined the HP-1 and HP-29 prodrug series demonstrated good antibacterial targeting profiles, we evaluated these focused collections against a panel of multidrug-resistant MRSA and MRSE clinical isolates that were obtained from patients treated at Shands Hospital (Gainesville, FL; Table 3). In addition to evaluating select HP prodrug series, we profiled the clinical isolate strains with six conventional antibiotics, including vancomycin, methicillin, ciprofloxacin, tetracycline, erythromycin, and tobramycin. All clinical isolates demonstrated resistance to methicillin (MIC values = 6.25 to > 100 μM) and at least one other antibiotic tested. Isolate S. aureus 129 was found to be resistant to four of the six antibiotics tested (methicillin, MIC = 37.5 μM; ciprofloxacin, erythromycin, tobramycin, MIC > 100 μM); however, most of the MRSA and MRSE isolates in our panel demonstrated resistance to three or more conventional antibiotics. Prodrug HP-1-N demonstrated 2- to 4-fold reductions in antibacterial potency (MIC = 3.13–6.25 μM) compared to parent HP-1 (MIC = 0.78–2.35 μM) against the clinical isolates, which we believe is indicative of prodrug processing by NTR. As designed, HP-1-SE was completely inactive against the clinical isolates (MIC > 100 μM), demonstrating the requirement of the nitro-group in HP-1-N for activity. In addition, the HP-29 series demonstrated similar activity profiles against the clinical isolates with increased overall potency as HP-29-N reported elevated MIC values (0.30–2.35 μM) compared to HP-29 (MIC = 0.10–0.39 μM); however, HP-29-SE was inactive (MIC > 100 μM) as designed.
| Compound | MRSA 2 | MRSA 1 | S. aureus 129 | S. aureus 138 | S. aureus 147 | S. aureus 156 | S. epidermidis CL-1 |
|---|---|---|---|---|---|---|---|
| a Midpoint value for 2-fold range in MIC values observed. b Midpoint value for 4-fold range in MIC values observed. c Lowest concentration tested. Each data point is the result of three independent experiments. | |||||||
| HP-1 | 2.35a | 0.78 | 1.56 | 1.56 | 1.56 | 1.56 | 1.56 |
| HP-1-N | 3.13b | 3.13 | 6.25 | 4.69a | 6.25 | 6.25 | 6.25 |
| HP-1-SE | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| HP-29 | 0.15a | 0.10c | 0.10c | 0.10c | 0.10c | 0.10c | 0.39 |
| HP-29-N | 0.30a | 0.30a | 1.17a | 0.39 | 0.59a | 0.59a | 2.35a |
| HP-29-SE | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| Vancomycin | 0.39 | 0.39 | 0.39 | 0.39 | 0.59a | 0.39 | 0.59a |
| Methicillin | >100 | 37.5a | 37.5a | 37.5a | 6.25 | 25 | 12.5 |
| Ciprofloxacin | >100 | 0.78 | >100 | >100 | 1.17a | 0.78 | 0.30a |
| Tetracycline | 0.10c | 0.10c | 0.10c | 0.15a | 0.10c | 0.15a | 25 |
| Erythromycin | 18.8a | 100 | >100 | >100 | 37.5a | >100 | >100 |
| Tobramycin | 6.25 | 6.25 | >100 | 12.5 | 6.25 | 12.5 | 1.56 |
Collectively, our findings show that select halogenated phenazine analogues can be successfully advanced as nitroreductase-targeting prodrug molecules. We focused these initial investigations on determining the potential for this prodrug strategy, which included: (1) investigating a focused series of HP antibacterial agents as starting points for prodrug synthesis, (2) demonstrating target prodrugs do not bind iron(II), (3) characterizing in vitro NTR-promoted release of prodrugs, and (4) evaluating antibacterial activity profiles of HP-NTR prodrugs. From a focused series of four HP-NTR prodrug collections, we found a spectrum of ideal to non-ideal prodrug profiles to rank and guide future studies in this area (Fig. 6). Each prodrug was rapidly synthesized in good yields and were shown to not bind iron(II) directly, which was a critical starting point for further investigations; however, additional examination of these compounds revealed insights critical to the development of HP-NTR prodrugs.
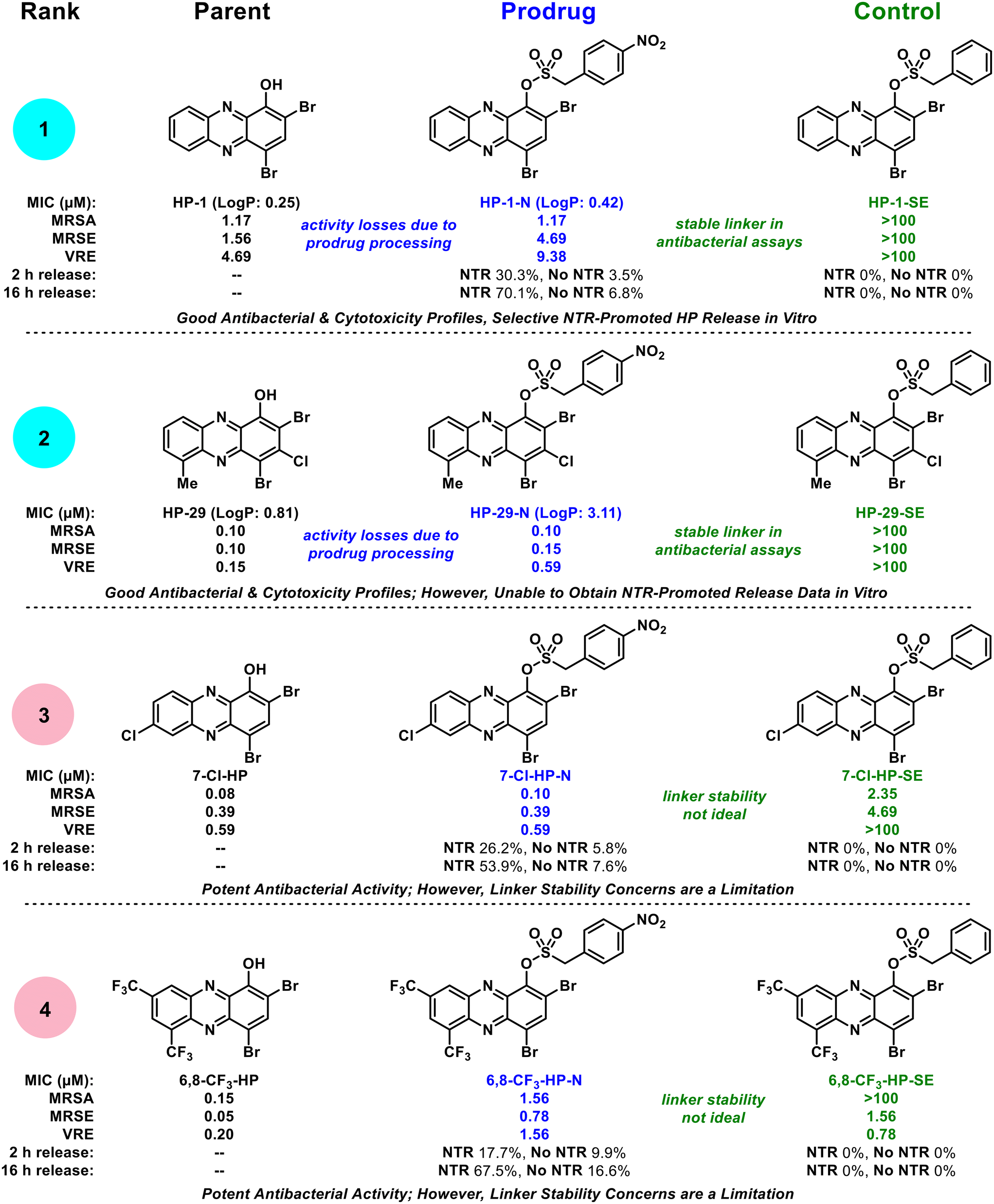 | ||
| Fig. 6 Biological activity profile overview of HP-nitroarene prodrug analogues. The HP-nitroarenes are ranked in order from most ideal (rank 1) to least ideal (rank 4) prodrugs for future studies. | ||
During the course of these studies, we felt our most ideal prodrug was HP-1-N (Fig. 6) as it demonstrated: (1) the most significant NTR-promoted HP release in vitro (with minimal hydrolysis in analogous experiments without NTR present), (2) ideal antibacterial activity profiles in Petri dish experiments and MIC assays (against lab strains and clinical isolates), and (3) the control HP-1-SE proved inactive against all strains demonstrating a stable sulfonate ester linkage and the absolute requirement for the nitro group in HP-1-N to possess antibacterial activity. In addition, we used a shake method30–33 protocol to show HP-1-N possesses acceptable water solubility with an experimental logP of 0.42 (parent HP-1, logP = 0.25). The prodrug we ranked as second best in this study is HP-29-N, which demonstrated ideal antibacterial profiles (significantly more potent than HP-1-N, as expected, and control HP-29-SE was inactive in all antibacterial assays); however, we were unable to obtain in vitro data to show NTR-release despite multiple attempts, likely due to solubility issues (logP = 3.11). We ranked prodrug 7-Cl-HP-N as third (and unacceptable) as unexpected antibacterial activity was observed for control compound 7-Cl-HP-SE demonstrating this molecule has an unstable linker that is likely susceptible to hydrolysis liberating the 7-Cl-HP warhead that can subsequently bind iron and inhibit bacterial growth. We ranked 6,8-CF3-HP-N as our lowest performing prodrug for linker stability concerns, which were apparent in the in vitro experiments without NTR present, Petri dish experiments and MIC assays.
During the course of these studies, we synthesized and evaluated a new series of HP prodrugs designed to utilize a nitroarene trigger to be reduced by nitroreductase enzymes in bacteria. Following synthesis of a focused series of HP-N prodrugs and control molecules (designed to assess linker stability), we showed these compounds were unable to bind iron(II), which is critical for their mode of action and required for mitigating off-target cytotoxicity. Following chemical synthesis, in vitro nitroreductase-promoted release assays and antibacterial assessments were used to characterize the action of HP-N prodrug molecules that led to the identification of HP-1-N as an ideal candidate due to a combination of targeted release by nitroreductase, antibacterial activities, linker stability, and a good cytotoxicity profile. Future optimization efforts will focus on the development of the nitroarene motif and a modified stable linker. Overall, we believe the development of HP prodrug antibacterial agents can lead to groundbreaking discoveries and provide new treatment options for antibiotic resistant infections.
Author contributions
R. H. designed and directed this study. K. L. performed all nitroreductase experiments, microbiological assessments (zone of inhibition and MIC assays), and UV-vis spectroscopy experiments for metal(II) binding. T. X. preformed the majority of the chemical synthesis experiments and characterized new compounds. H. Y. synthesized and characterized HP 8. M. C. performed cytotoxicity experiments with select compounds in HEK-293 cells. Q. G. determined logP values for select analogues. B. B. performed some chemical synthesis to support biological studies. Y. D. oversaw the cytotoxicity experiments in HEK-293 cells. R. H. and K. L. wrote the manuscript with input from each of the authors. All authors have reviewed the data, agree with the findings, and approve the final manuscript.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the University of Florida and the National Institute of General Medical Sciences for providing generous support for this work (R35GM128621 to R. W. H.). B. R. B. was supported by the NIH-NIDCR training grant T90 DE021990 “Comprehensive Training Program in Oral Biology” at the University of Florida. Y. D. is partially supported by NIH and acknowledges grant R35GM128742. High resolution mass spectra were obtained for novel synthesized compounds from the University of Florida's Mass Spectrometry Research and Education Center and supported by NIH S10 OD021758-01A1. We obtained MRSA and MRSE clinical isolates from the Emerging Pathogens Institute (EPI) at the University of Florida. These isolates were obtained from patients treated at Shands Hospital (Gainesville, FL).References
- K. Lewis, The Science of Antibiotic Discovery, Cell, 2020, 181, 29–45 CrossRef CAS PubMed.
- P. S. Hoffman, Antibacterial Discovery: 21st Century Challenges, Antibiotics, 2020, 9, 213–213 CrossRef PubMed.
- Y. Abouelhassan, A. T. Garrison, H. Yang, A. Chávez-Riveros, G. M. Burch and R. W. Huigens III, Recent Progress in Natural-Product-Inspired Programs Aimed at Addressing Antibiotic Resistance and Tolerance, J. Med. Chem., 2019, 62, 7618–7642 CrossRef CAS PubMed.
- K. Liu and R. W. Huigens III, Instructive Advances in Chemical Microbiology Inspired by Nature's Diverse Inventory of Molecules, ACS Infect. Dis., 2020, 6, 541–562 CrossRef CAS PubMed.
- M. A. Fitzpatrick, Real-World Antibiotic Needs for Resistant Gram-Negative Infections, Lancet Infect. Dis., 2020, 20, 1108–1109 CrossRef PubMed.
- J. M. Munita and C. A. Arias, Mechanisms of Antibiotic Resistance, Microbiol. Spectrum, 2016, 4, VMBF-0016-2015 Search PubMed.
- Antimicrobial Resistance Collaborators, Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis, Lancet, 2022, 399, 629–655 CrossRef PubMed.
- N. V. Borrero, F. Bai, C. Perez, B. Q. Duong, J. R. Rocca, S. Jin and R. W. Huigens III, Phenazine Antibiotic Inspired Discovery of Potent Bromophenazine Antibacterial Agents against Staphylococcus aureus and Staphylococcus epidermidis, Org. Biomol. Chem., 2014, 12, 881–886 RSC.
- A. T. Garrison, Y. Abouelhassan, D. Kallifidas, F. Bai, M. Ukhanova, V. Mai, S. Jin, H. Luesch and R. W. Huigens III, Halogenated Phenazines that Potently Eradicate Biofilms, MRSA Persister Cells in Non-biofilm Cultures, and Mycobacterium tuberculosis, Angew. Chem., Int. Ed., 2015, 54, 14819–14823 CrossRef CAS PubMed.
- A. T. Garrison, Y. Abouelhassan, V. M. Norwood IV, D. Kallifidas, F. Bai, M. Nguyen, M. Rolfe, G. M. Burch, S. Jin, H. Luesch and R. W. Huigens III, Structure-Activity Relationships of a Diverse Class of Halogenated Phenazines that Targets Persistent, Antibiotic-Tolerant Bacterial Biofilms and Mycobacterium tuberculosis, J. Med. Chem., 2016, 59, 3808–3825 CrossRef CAS PubMed.
- H. Yang, Y. Abouelhassan, G. M. Burch, D. Kallifidas, G. Huang, H. Yousaf, S. Jin, H. Luesch and R. W. Huigens III, A Highly Potent Class of Halogenated Phenazine Antibacterial and Biofilm-Eradicating Agents Accessed Through a Modular Wohl-Aue Synthesis, Sci. Rep., 2017, 7, 2003 CrossRef PubMed.
- Y. Abouelhassan, Y. Zhang, S. Jin and R. W. Huigens III, Transcript Profiling of MRSA Biofilms Treated With a Halogenated Phenazine Eradicating Agent: A Platform for Defining Cellular Targets and Pathways Critical to Biofilm Survival, Angew. Chem., Int. Ed., 2018, 57, 15523–15528 CrossRef CAS PubMed.
- A. T. Garrison, Y. Abouelhassan, D. Kallifidas, H. Tan, Y. S. Kim, S. Jin, H. Luesch and R. W. Huigens III, An Efficient Buchwald-Hartwig/Reductive Cyclization for the Scaffold Diversification of Halogenated Phenazines: Potent Antibacterial Targeting, Biofilm Eradication and Prodrug Exploration, J. Med. Chem., 2018, 61, 3962–3983 CrossRef CAS PubMed.
- T. Xiao, K. Liu and R. W. Huigens III, Progress Towards a Stable Cephalosporin-Halogenated Phenazine Conjugate for Antibacterial Prodrug Applications, Bioorg. Med. Chem. Lett., 2020, 30, 127515 CrossRef CAS PubMed.
- H. Yang, S. Kundra, M. Chojnacki, K. Liu, M. A. Fuse, Y. Abouelhassan, D. Kallifidas, P. Zhang, G. Huang, S. Jin, Y. Ding, H. Luesch, K. H. Rohde, P. M. Dunman, J. A. Lemos and R. W. Huigens III, A Modular Synthetic Route Involving N-Aryl-2-Nitrosoaniline Intermediates Leads to a New Series of 3-Substituted Halogenated Phenazine Antibacterial Agents, J. Med. Chem., 2021, 64, 7275–7295 CrossRef CAS PubMed.
- H. Yang, K. Liu, S. Jin and R. W. Huigens III, Design, Synthesis and Biological Evaluation of a Halogenated Phenazine-Erythromycin Conjugate Prodrug for Antibacterial Applications, Org. Biomol. Chem., 2021, 19, 1483–1487 RSC.
- R. W. Huigens III, H. Yang, K. Liu, Y. S. Kim and S. Jin, An Ether-Linked Halogenated Phenazine-Quinone Prodrug Model for Antibacterial Applications, Org. Biomol. Chem., 2021, 19, 6603–6608 RSC.
- K. Liu, M. Brivio, T. Xiao, V. M. Norwood IV, Y. S. Kim, S. Jin, A. Papagni, L. Vaghi and R. W. Huigens III, Modular Synthetic Routes to Fluorine-Containing Halogenated Phenazine and Acridine Agents that Induce Rapid Iron Starvation in MRSA Biofilms, ACS Infect. Dis., 2022, 8, 280–295 CrossRef CAS PubMed.
- Z. A. Machan, T. L. Pitt, W. White, D. Watson, G. W. Taylor, P. J. Cole and R. Wilson, Interaction Between Pseudomonas aeruginosa and Staphylococcus aureus: Description of an Antistaphylococcal Substance, J. Med. Microbiol., 1991, 34, 213–217 CrossRef CAS PubMed.
- J. B. Laursen and J. Nielsen, Phenazine Natural Products: Biosynthesis, Synthetic Analogues, and Biological Activity, Chem. Rev., 2004, 104, 1663–1685 CrossRef CAS PubMed.
- A. Price-Whelan, L. E. P. Dietrich and D. K. Newman, Rethinking “Secondary” Metabolism: Physiological Roles for Phenazine Antibiotics, Nat. Chem. Biol., 2006, 2, 71–78 CrossRef CAS PubMed.
- K. A. Pardeshi, T. A. Kumar, G. Ravikumar, M. Shukla, G. Kaul, S. Chopra and H. Chakrapani, Targeted Antibacterial Activity Guided by Bacteria-Specific Nitroreductase Catalytic Activation to Produce Ciprofloxacin, Bioconjugate Chem., 2019, 30, 751–759 CrossRef CAS PubMed.
- S. Xu, Q. Wang, Q. Zhang, L. Zhang, L. Zuo, J. D. Jiang and H. Y. Hu, Real Time Detection of ESKAPE Pathogens by a Nitroreductase-Triggered Fluorescence Turn-on Probe, Chem. Commun., 2017, 53, 11177–11180 RSC.
- H. A. J. Hibbard and M. M. Reynolds, Synthesis of Novel Nitroreductase Enzyme-Activated Nitric Oxide Prodrugs to Site-Specifically Kill Bacteria, Bioorg. Chem., 2019, 93, 103318 CrossRef CAS PubMed.
- Z. Wróbel and A. Kwast, 2-Nitroso-N-Arylanilines: Products of Acid-Promoted Transformation of σH Adducts of Arylamines and Nitroarenes, Synlett, 2007, 1525–1528 CrossRef.
- Z. Wróbel and A. Kwast, Simple Synthesis of N-Aryl-2-Nitrosoanilines in the Reaction of Nitroarenes with Aniline Anion Derivatives, Synthesis, 2010, 3865–3872 CrossRef.
- A. Kwast, K. Stachowska, A. Trawczyński and Z. Wróbel, N-Aryl-2-Nitrosoanilines as Intermediates in the Synthesis of Substituted Phenazines from Nitroarenes, Tetrahedron Lett., 2011, 52, 6484–6488 CrossRef CAS.
- Z. Wróbel, K. Plichta and A. Kwast, Reactivity and Substituent Effects in the Cyclization of N-Aryl-2-Nitrosoanilines to Phenazines, Tetrahedron, 2017, 73, 3147–3152 CrossRef.
- M. C. Jennings, L. E. Ator, T. J. Paniak, K. P. C. Minbiole and W. M. Wuest, Biofilm-Eradicating Properties of Quaternary Ammonium Amphiphiles: Simple Mimetics of Antimicrobial Peptides, ChemBioChem, 2014, 15, 2211–2215 CrossRef CAS PubMed.
- C. D. Schönsee and T. D. Bucheli, Experimental Determination of Octanol–Water Partition Coefficients of Selected Natural Toxins, J. Chem. Eng. Data, 2020, 65, 1946–1953 CrossRef.
- S. K. Dubey, G. Singhvi, A. Tyagi, H. Agarwal and K. V. Krishna, Spectrophotometric Determination of pKa and LogP of Risperidone, J. Appl. Pharm. Sci., 2017, 7, 155–158 CAS.
- M. F. Harris and J. L. Logan, Determination of Log Kow Values for Four Drugs, J. Chem. Educ., 2014, 91, 915–918 CrossRef CAS.
- N. Bodor, Z. Gabanyi and C. K. Wong, A New Method for the Estimation of Partition Coefficient, J. Am. Chem. Soc., 1989, 111, 3783–3786 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00204g |
| This journal is © The Royal Society of Chemistry 2023 |