
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel styryl-thiazole hybrids as potential anti-Alzheimer's agents†
Niki
Gouleni
a,
Annalisa
Di Rienzo
b,
Ahmet
Yılmaz
c,
Harun
Selvitopi
d,
Mehmet Enes
Arslan
c,
Adil
Mardinoglu
ef,
Hasan
Turkez
g,
Antonio
Di Stefano
b,
Stamatia
Vassiliou
*a and
Ivana
Cacciatore
 *b
*b
aLaboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Athens, Greece
bDepartment of Pharmacy, “G. D'Annunzio” University of Chieti-Pescara, 66100, Chieti Scalo, CH, Italy. E-mail: ivana.cacciatore@unich.it
cDepartment of Molecular Biology and Genetics, Faculty of Science, Erzurum Technical University, 25050, Erzurum, Turkey
dDepartment of Mathematics, Faculty of Sciences, Erzurum Technical University, 25050, Erzurum, Turkey
eScience for Life Laboratory, KTH-Royal Institute of Technology, SE-17121 Stockholm, Sweden
fCentre for Host-Microbiome Interactions, Faculty of Dentistry, Oral & Craniofacial Sciences, King's College London, London SE1 9RT, UK
gDepartment of Medical Biology, Faculty of Medicine, Atatürk University, Erzurum, Turkey
First published on 21st September 2023
Abstract
In this study, combining the thiazole and cinnamoyl groups into the styryl-thiazole scaffold, a series of novel styryl-thiazole hybrids (6a–p) was rationally designed, synthesized, and evaluated by the multi-target-directed ligands strategy as potential candidates for the treatment of Alzheimer's disease (AD). Hybrids 6e and 6i are the most promising among the synthesized hybrids since they are able to significantly increase cell viabilities in Aβ1–42-exposed-human neuroblastoma cell line (6i at the concentration of 50 μg mL−1 and 6e at the concentration of 25 μg mL−1 resulted in ∼34% and ∼30% increase in cell viabilities, respectively). Compounds 6e and 6i exhibit highly AChE inhibitory properties in the experimental AD model at 375.6 ± 18.425 mU mL−1 and 397.6 ± 32.152 mU mL−1, respectively. Moreover, these data were also confirmed by docking studies and in vitro enzyme inhibition assays. Compared to hybrid 6e and according to the results, 6i also has the highest potential against Aβ1–42 aggregation with over 80% preventive activity. The in silico prediction of the physicochemical properties confirms that 6i possesses a better profile compared to 6e. Therefore, compound 6i presents a promising multi-targeted active molecular profile for treating AD considering the multifactorial nature of AD, and it is reasonable to deepen its mechanisms of action in an in vivo experimental model of AD.
Introduction
In the last decades sulfur-containing therapeutics, such as glutathione, lipoic acid, and other thiol derivatives, have been widely investigated for the treatment of neurogenerative diseases such as Parkinson's (PD) or Alzheimer's diseases (AD).1–3 Notably, sulfur pharmacophores were conjugated or merged with other molecules endowed with anti-inflammatory, antioxidant, anti-Aβ-aggregation, anti-acetylcholinesterase, and anti-MAO activities in order to act on multiple targets involved in the onset and/or progression of these pathologies.4–6Heterocyclic rings are commonly used in the drug design of biologically active substances.7–9 Recently thiazole-based derivatives were proposed as potential lead compounds for the development of novel cholinesterase inhibitors or receptors for advanced glycation end-products (RAGE) antagonists.10–12 It was reported that the thiazole ring was able to create crucial interactions with the amino acid residues at the level of the active site of AChE.13 However, if on one hand the potential role of the thiazole scaffold as a cholinesterase inhibitor was widely explored, on the other hand, literature data about its potential as a multitarget AD therapeutic are very scarce.
Based on the above-mentioned evidence and considering the multifactorial nature of AD, it is reasonable to design novel styryl-thiazole hybrids able to interact with multiple targets involved in the pathogenesis of AD.14,15
This paper reports the synthesis of novel styryl-thiazole hybrids obtained by the fusion of two pharmacophoric groups capable of interacting with the active site of AChE (thiazole ring) and counteracting the inflammatory and antioxidant status of the cerebral areas of the brain affected by AD (cinnamic acid). Cinnamic acid and its derivatives display a broad spectrum of biological activities and they have been studied as anti-AD agents due to their anti-neuroinflammatory properties and their ability to inhibit Aβ aggregation by scavenging oxidants.16,17 However, the use of cinnamic acid is restricted by its poor water solubility, low oral bioavailability, and scarce capability to cross the blood–brain barrier (BBB).18
The incorporation of sulfur pharmacophore into the hybrid can enhance the pharmacokinetic properties of cinnamic acid along with the engagement of several targets involved in the multifaceted pathophysiology of AD resulting in an effective approach toward AD therapy. The thiazole group was merged with the styryl portion of cinnamic acid using the hybridization strategy schematically reported in Fig. 1. Starting from the basic scaffold – represented by (E)-2-styryl thiazole – a structure–activity relationships study was performed introducing a series of small substituents with different chemical–physical properties (steric hindrance, electron-donating or -withdrawing effects, hydrophilicity) on both thiazole and benzene rings. The synthesized hybrids 6a–6p were assessed by the neuroprotective effects on Aβ-neurotoxicity induced on SHSY-5Y neuroblastoma cells, AChEs inhibition, β-secretase inhibition, and genotoxic properties. ADME properties were also evaluated using Swiss ADME tool.
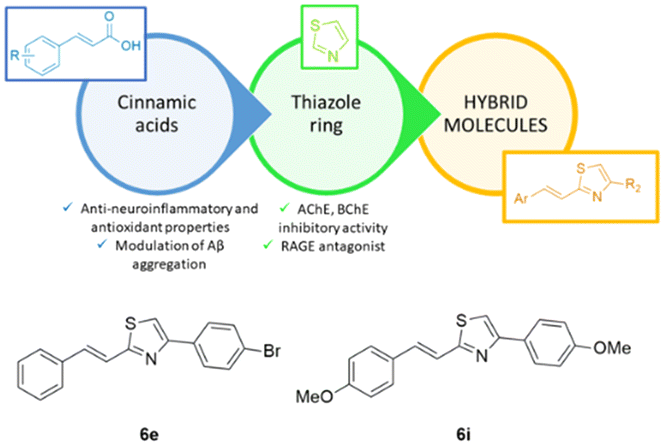 | ||
| Fig. 1 Design of styryl-thiazole hybrids. | ||
Results and discussion
Chemistry
The synthetic pathway toward the synthesis of E-styryl thiazoles 6a–p is outlined in Scheme 1. Cinnamamides 2a–h were efficiently prepared from cinnamic acids 1a–h through a two-step procedure.19–21 The acids were converted to their succinic esters using EDC·HCl and N-hydroxysuccinimide (NHS) in CH2Cl2 at room temperature. The crude active ester was subsequently converted to the corresponding amide after slow addition of 25% aq. NH3 in a biphasic system formed by a solvent mixture CHCl3/CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). Thionation of the amides with Lawesson's reagent resulted ineffective. On the other hand, heating amides 2a–h at 80 °C in dry dioxane in the presence of P2S5 (the reaction was followed by TLC) provided thioamides 3a–h in medium yields (17–60%) after column chromatography.22 The (E)-configuration of the double bond was confirmed by the large 1HNMR coupling constant (∼16 Hz). Additionally, the conversion of the amide to the corresponding thioamide was confirmed by the significant downfield shift (∼30 ppm) of the thiocarbonyl signal in 13CNMR to ∼200 ppm.
:2). Thionation of the amides with Lawesson's reagent resulted ineffective. On the other hand, heating amides 2a–h at 80 °C in dry dioxane in the presence of P2S5 (the reaction was followed by TLC) provided thioamides 3a–h in medium yields (17–60%) after column chromatography.22 The (E)-configuration of the double bond was confirmed by the large 1HNMR coupling constant (∼16 Hz). Additionally, the conversion of the amide to the corresponding thioamide was confirmed by the significant downfield shift (∼30 ppm) of the thiocarbonyl signal in 13CNMR to ∼200 ppm.
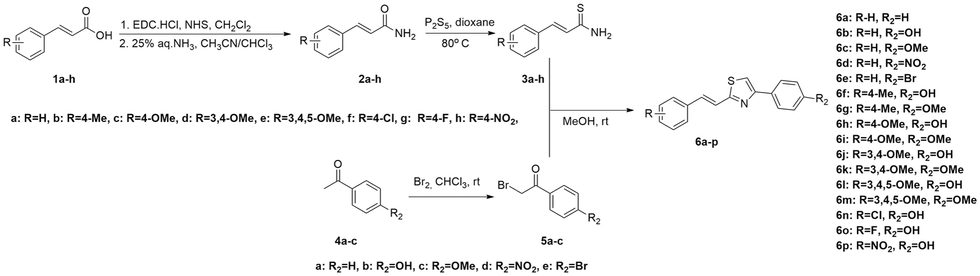 | ||
| Scheme 1 Synthesis of styryl-thiazole hybrids 6a–p. | ||
Compounds 5a–c were prepared from the corresponding acetophenones 4a–c23,24 while 5d–e were commercially available. Using the Hantzsch thiazole synthesis,25,26 α-bromo carbonyl compounds 5a–e were reacted with thioamides 3a–h to provide E-styryl thiazole hybrids 6a–p in medium to good yields (25–88%) after purification.
Biological studies
Human neuroblastoma SH-SY5Y cells provide a useful model for in vitro research, especially for neurodegenerative disorders such as Parkinson's and Alzheimer's diseases.27 To investigate the anti-AD potential of 6a–p, SHSY-5Y cells, treated with retinoic acid (RA)/brain-derived neurotrophic factor (BDNF), were differentiated into mature neuron-like cell cultures to obtain an AD in vitro experimental model.28 First, cytotoxicity studies on SHSY-5Y cells were performed for all the synthesized compounds 6a–p in the range of concentrations between 12.5 and 100 μg mL−1 (Fig. 2). Results showed that only 6e and 6i were found to have biocompatible properties showing no cytotoxic effect on the differentiated SHSY-5Y cells (Fig. 2) compared to the other compounds.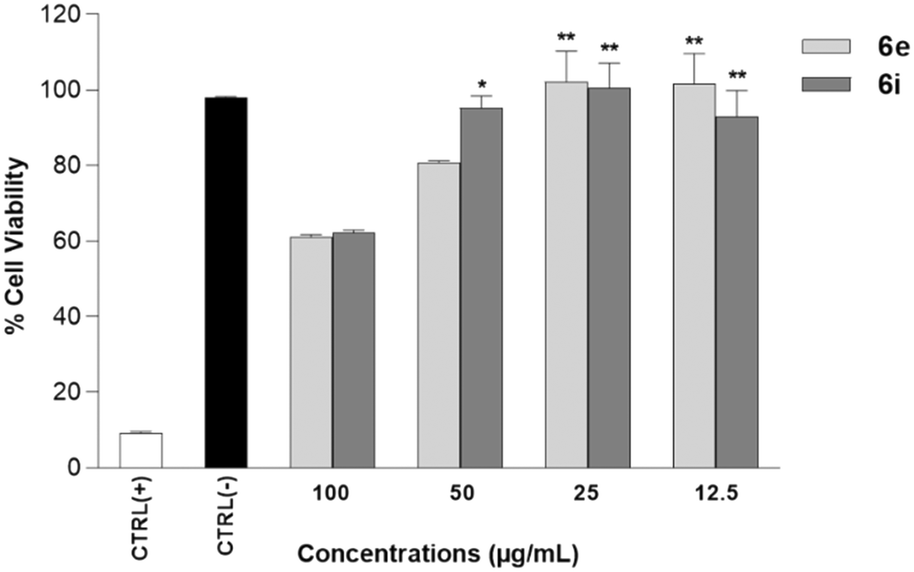 | ||
| Fig. 2 Cell viability assays of 6e and 6i on the human neuroblastoma RA/BDNF differentiated SH-SY-5Y cells. SH-SY-5Y cells were incubated for 24 h with 6e and 6i (12.5–100 μg mL−1) and then the % cell viability was assayed by MTT test. (−) Ctrl: untreated cells. (+) Ctrl: cells treated with 1.0% Triton X-100. Data are presented as the means ± SD for three independent experiments (n = 3). *p < 0.05 and **p < 0.01 significative vs. (+) Ctrl. | ||
Starting from these data, the genotoxic properties of 6e and 6i were also assessed on the differentiated SHSY-5Y cells. Genotoxic analysis of 6e and 6i by using Hoechst 33258 fluorescent nuclear staining showed that both 6e and 6i, tested at the concentration of 100 μg mL−1, did not enhance the genotoxic properties on the differentiated SHSY-5Y cells. In fact, in the negative control, 16 abnormal nuclear structures were observed (0.016 NA/1000 cells ± SD), comparable to the cells treated with 6e and 6i (0.017 NA/1000 cells ± SD) (Fig. 3 and Table 1).
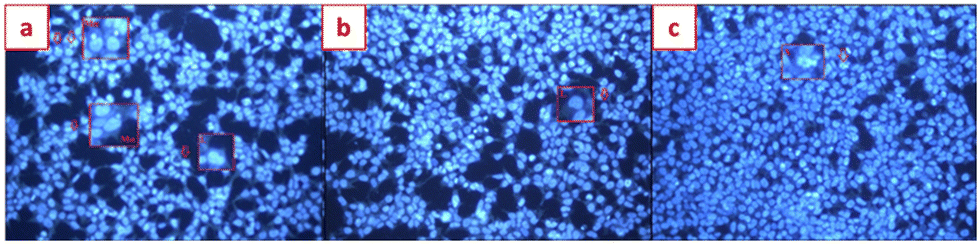 | ||
| Fig. 3 Hoechst 33258 fluorescent staining analysis for genotoxicity assessments. a) Negative control – 20× magnification, b) compound 6e – 20× magnification, and c) compound 6i – 20× magnification. MN, micronucleus; L, lobbed nuclei, N, notched nuclei, and A, apoptotic. | ||
| Treatment | Total MN | Total lobbed | Total notched | Mean NA/1000 cells ± SD |
|---|---|---|---|---|
| (−) Ctrl: cell culture plate. MN: micronucleus. Data are presented as the means ± SD for three independent experiments (n = 3).a p < 0.05 significative vs. (−) Ctrl. | ||||
| (−) Ctrl | 6 | 5 | 5 | 0.016 ± 0.007a |
| 6e | 4 | 5 | 9 | 0.017 ± 0.007a |
| 6i | 6 | 6 | 5 | 0.017 ± 0.008a |
The present investigation aimed to study the neuroprotective potential of 6e and 6i from degeneration associated with the Aβ1–42 in human neuroblastoma SH-SY5Y cells, chosen as an experimental in vitro AD model.
The AD in vitro cell model was generated by treating the SH-SY5Y cells with Aβ1–42 at the concentration of 20 μM for 24 h (data not shown). Results revealed that 6e and 6i were able to significantly increase cell viabilities compared to Aβ1–42-exposed cell cultures. 6i at the concentration of 50 μg mL−1 and 6e at the concentration of 25 μg mL−1 resulted in ∼34% and ∼30% increase in cell viabilities, respectively (Fig. 4).
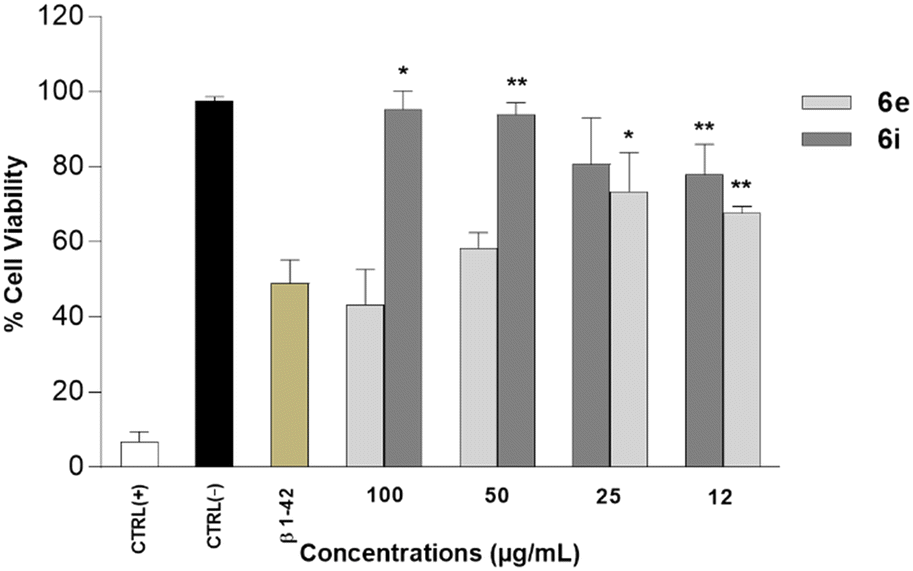 | ||
| Fig. 4 Neuroprotective properties of 6e and 6i (12.5–100 μg mL−1) against Aβ1–42-treated SH-SY-5Y cells assayed by MTT assay (−) Ctrl: untreated cells. (+) Ctrl: cells treated with 1.0% Triton X-100. Data are presented as the means ± SD for three independent experiments (n = 3). *p < 0.05 and **p < 0.01 significative vs. (+) Ctrl. | ||
Moreover, the neuroprotective mechanisms of 6e and 6i were investigated by using acetylcholinesterase (AChE), beta secretase (BACE) activity assays, and Aβ1–42 aggregation analyses. AChE activity results showed that experimental AD model cell cultures have higher AChE activity (459.6 ± 32.152 mU mL−1, 20 μM of Aβ1–42 exposure) compared to differentiated SHSY-5Y cell culture (363.6 ± 32.152 mU mL−1, negative control). Compounds 6e and 6i exhibited high AChE inhibitory properties in the experimental AD model at 375.6 ± 18.425 mU mL−1 and 397.6 ± 32.152 mU mL−1, respectively (Table 2). Furthermore, the BACE activity assay showed that Aβ1–42 application to the differentiated SHSY-5Y cell culture significantly increased BACE activity from 10.31 ± 0.82 (β-secretase activity, relative fluorescence units per mg protein) to 54.35 ± 4.34. On the other hand, 6e (17.41 ± 1.39) and compound 6i (14.13 ± 1.13) applications significantly ameliorated the negative impact of Aβ1–42 exposure on the BACE enzyme activity (Table 3).
| Compound | AChE activity (mU mL−1) |
|---|---|
| One milliunit (mU) of AChE activity was defined as the number of nmoles of acetylcholine hydrolyzed per min at 22 °C. (−) Ctrl: non-treated differentiated SH-SY5Y cells; (+) Ctrl: Aβ1–42-treated cells. Different letters (a, b) in the same column present statistical differences from each other. | |
| (−) Ctrl | 363.6 ± 32.152a |
| (+) Ctrl | 459.6 ± 32.152b |
| 6e | 375.6 ± 18.425a |
| 6i | 397.6 ± 32.152a |
| Compound | β-Secretase activity (relative fluorescence units per mg protein) |
|---|---|
| (−) Ctrl: non-treated differentiated SH-SY5Y cells; (+) Ctrl: Aβ1–42-treated cells. Different letters (a, b, c) in the same column present statistical differences from each other. | |
| (−) Ctrl | 10.31 ± 0.82a |
| (+) Ctrl | 54.35 ± 4.34b |
| 6e | 17.41 ± 1.39c |
| 6i | 14.13 ± 1.13c |
Both synthesized derivatives 6e and 6i were also tested to evaluate their intrinsic ability to inhibit both AChE and BACE1 enzymes (Fig. 5). The eserine, or physostigmine, is a well-characterized inhibitor of AChE – an enzyme responsible for the hydrolysis of the neurotransmitter acetylcholine in synaptic clefts – and was used as a recognized standard. Among the tested compounds, 6i showed the best ability to inhibit AChE with IC50 of 63.6 μg mL−1, while the weakest inhibitory activity was found in the case of 6e (IC50 = 78.5 μg mL−1). These data also confirmed a correlation between the increase in concentration and the percentage of inhibition for both derivatives.
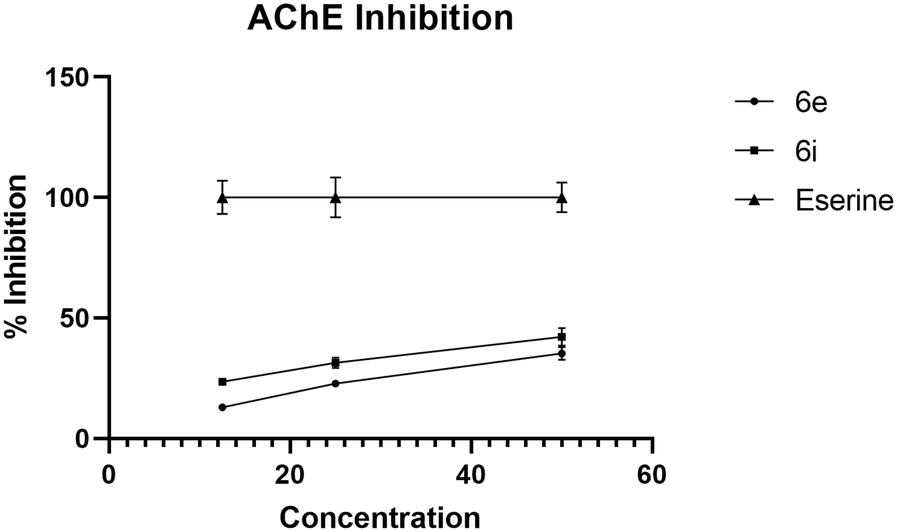 | ||
| Fig. 5 In vitro AChE inhibitory activity of 6e and 6i. | ||
We used OM99-2 – a well-recognized inhibitor of the BACE1 enzyme – as a positive control to validate the assay conditions, ensuring that they are conducive to BACE1 inhibition. The IC50 of 6e and 6i against BACE1 was 15.2 μg mL−1 and 25.3 μg mL−1, respectively (Fig. 6). The results indicated that the most active compound was 6e. Also, in this case, there was a dose-dependent effect: the percentage of inhibition increased with the enhancement of the concentration of compounds.
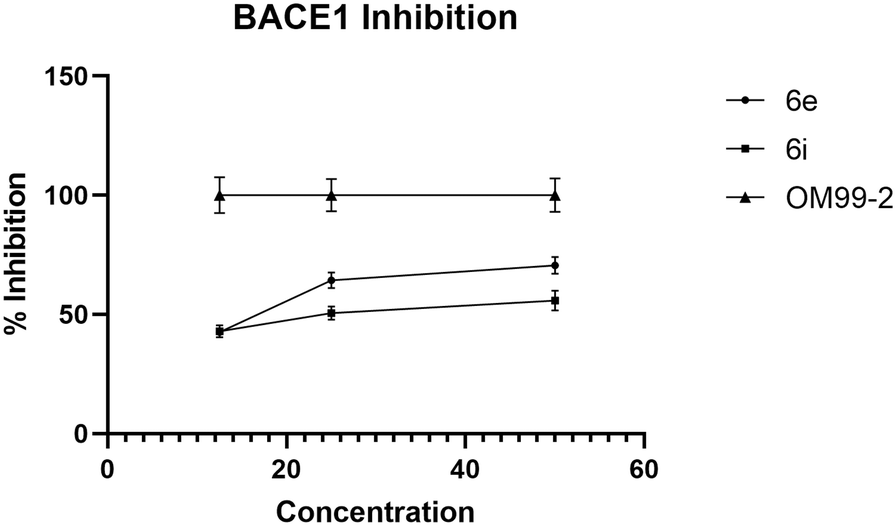 | ||
| Fig. 6 BACE1 inhibitory activity of 6e and 6i assessed by BACE1-FRET assay. | ||
Moreover, the impact of 6e and 6i on the Aβ1–42 aggregations was investigated by using the Congo red staining technique. Analyses showed that 6e and 6i have a positive impact on Aβ1–42 aggregation in the experimental AD model. According to the results, 6i has the highest potential against Aβ1–42 aggregation with over 80% preventive activity (Fig. 7).
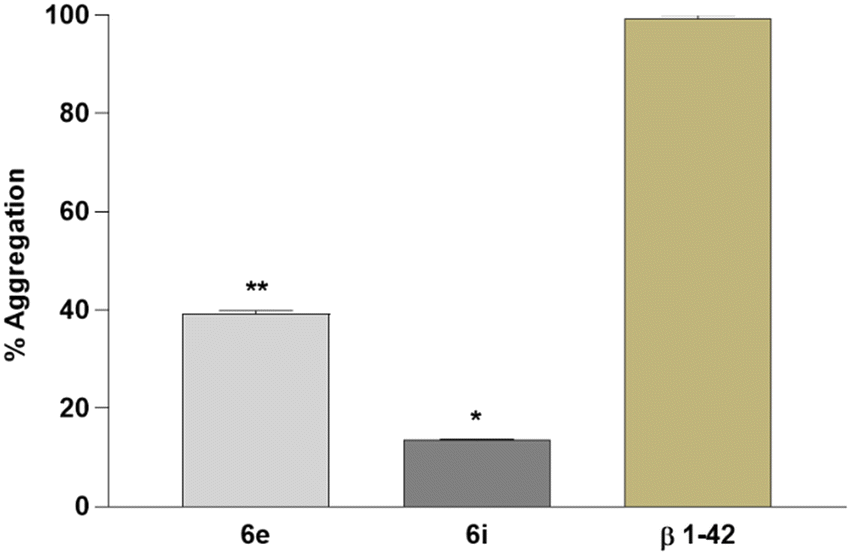 | ||
| Fig. 7 In vitro Aβ1–42 aggregation analysis in the experimental AD model. Data are presented as the means ± SD for three independent experiments (n = 3). *p < 0.05 and **p < 0.01. | ||
Flow cytometry analyses put forth that the Aβ1–42-induced cell death mechanism produced 36% apoptosis and 9% necrosis in the differentiated SHSY-5Y cell cultures (Fig. 8B). Hybrid 6i at the concentration of 50 μg mL−1 reduced the apoptotic cell death ratio by 30% and decreased necrotic cell deaths by 6% (Fig. 8C). On the contrary, 6e at lower concentrations (25 μg mL−1) resulted in the most effective treatment against Aβ1–42-induced apoptosis by reducing apoptosis by 20% and necrosis by 5%.
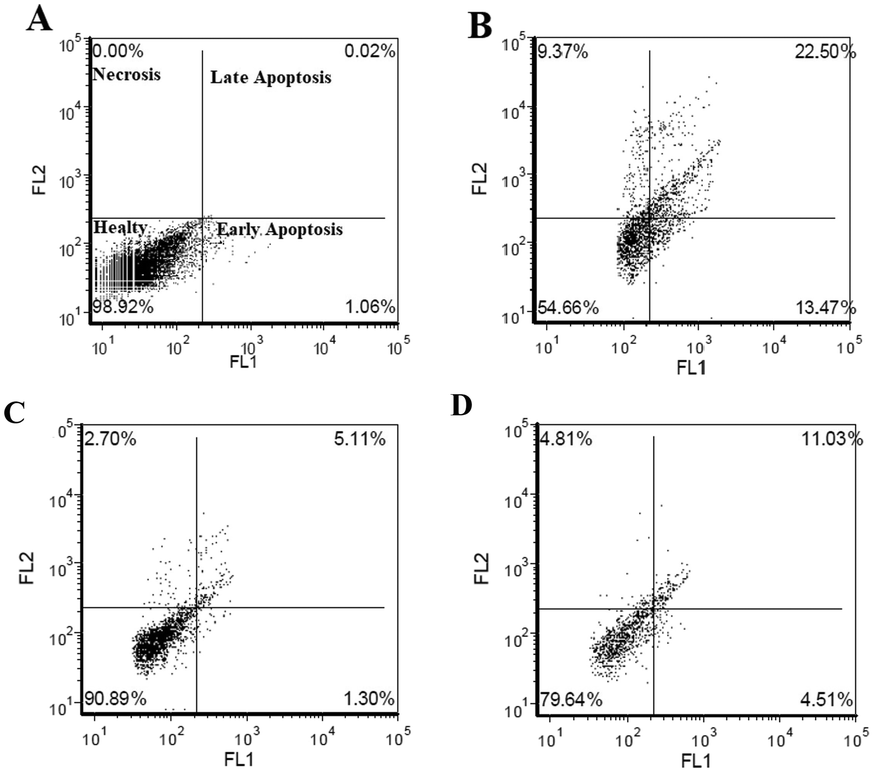 | ||
| Fig. 8 Flow cytometry analysis of 6e and 6i on the experimental AD model for 24 h. A) Negative control; B) Aβ1–42 treatment; C) compound 6i (50 μg mL−1); D) compound 6e (25 μg mL−1). | ||
Docking studies
To support the experimental results obtained by enzymatic inhibition, the interaction between AChE, BACE, and tested compounds 6e and 6i, molecular docking studies were performed.Fig. 7–10 show the protein–ligand interaction of the synthesized hybrids 6e and 6i with the human AChE (Fig. 9 and 10) and BACE enzymes (Fig. 11 and 12). It can be observed from investigations that the highest binding energy is −5.20 kcal mol−1 and the lowest binding energy is −9.58 kcal mol−1 between the different compounds and AChE enzyme. The docking analyses revealed that the binding energies between the different compounds considered in this study with the BACE are between −4.41 kcal mol−1 and −8.55 kcal mol−1. Moreover, molecular docking analyses showed that the lowest binding energy for AChE was found at −9.58 kcal mol−1 which was the interaction between compound 6e and the enzyme. The inhibition constant of compound 6e was investigated as 95.61 nM which was the lowest one among all the other compounds. The molecular interaction distance between 6e and AChE residue was calculated as 2.09 Å which was the closest interaction distance compared to other compounds (Fig. 7). Compound 6i also showed high interaction capabilities with 8.52 kcal mol−1 binding energies to the AChE enzyme (Fig. 9). When these results were compared to in vitro AChE activity assays, it was observed that compound 6e has the highest AChE enzyme inhibitory capacity and 6i has significant inhibitory potential (Table 2). All data, both theoretical and experimental AChE inhibition investigations, were correlated with each other.
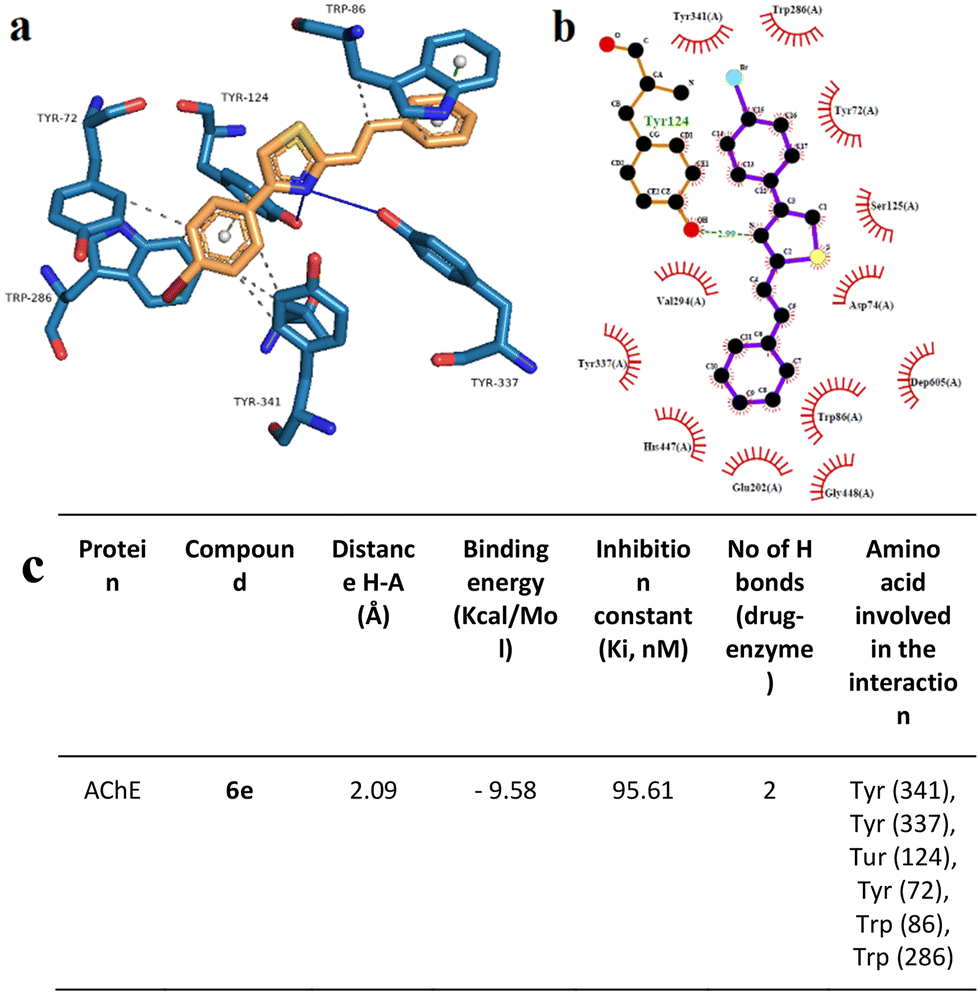 | ||
| Fig. 9 Binding mode of the best docking conformation of the binding mode of human AChE with 6e obtained by a) PyMol, b) LigPlot, and c) the table of hydrogen-bonds (H-bonds) (with distance Å), binding energy (kcal mol−1), inhibition constant, number of H-bonds (drug–enzyme) and amino acid involved in interaction obtained from Protein–Ligand Interaction Profiler. The blue line and dashed green show the H-bonds in PyMol and LigPlot, respectively. | ||
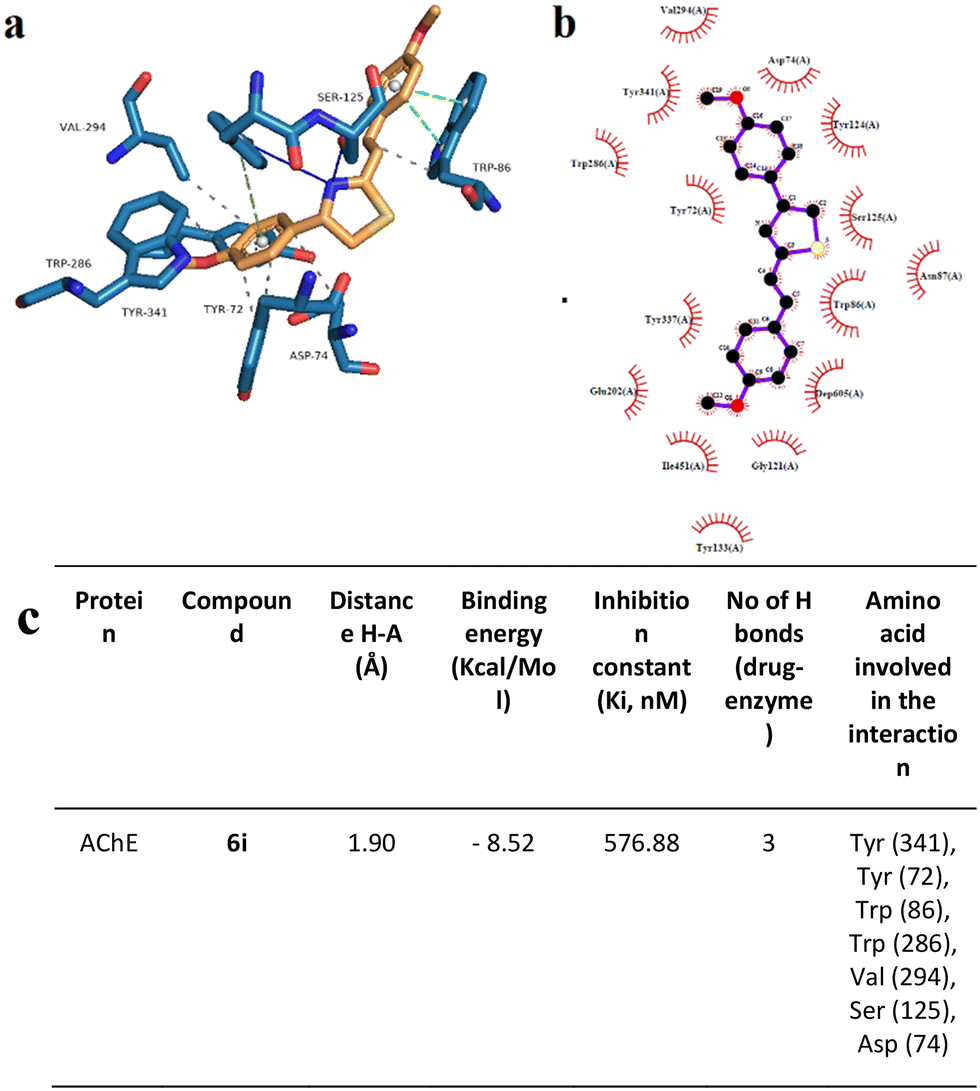 | ||
| Fig. 10 Binding mode of the best docking conformation of the binding mode of human AChE with 6i obtained by a) PyMol, b) LigPlot, and c) the table of hydrogen-bonds (H-bonds) (with distance Å), binding energy (kcal mol−1), inhibition constant, number of H-bonds (drug–enzyme) and amino acid involved in interaction obtained from Protein–Ligand Interaction Profiler. The blue line and dashed green show the H-bonds in Pymol and LigPlot, respectively. | ||
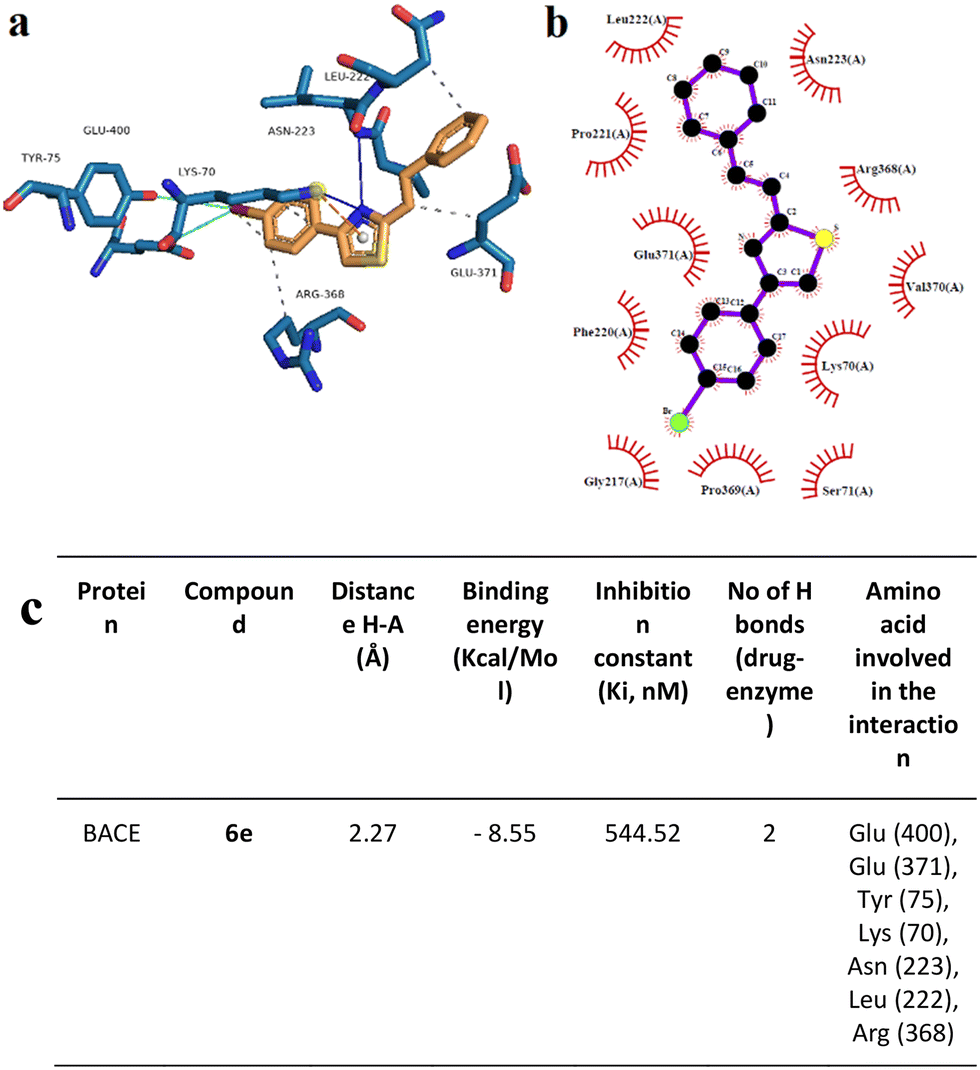 | ||
| Fig. 11 Interaction mode of the best docking conformation of the binding mode of human BACE with 6e obtained by a) PyMol, b) LigPlot, and c) the table of hydrogen-bonds (H-bonds) (with distance Å), binding energy (kcal mol−1), inhibition constant, number of H-bonds (drug–enzyme) and amino acid involved in interaction obtained from Protein–Ligand Interaction Profiler. The blue line and dashed green show the H-bonds in Pymol and LigPlot, respectively. | ||
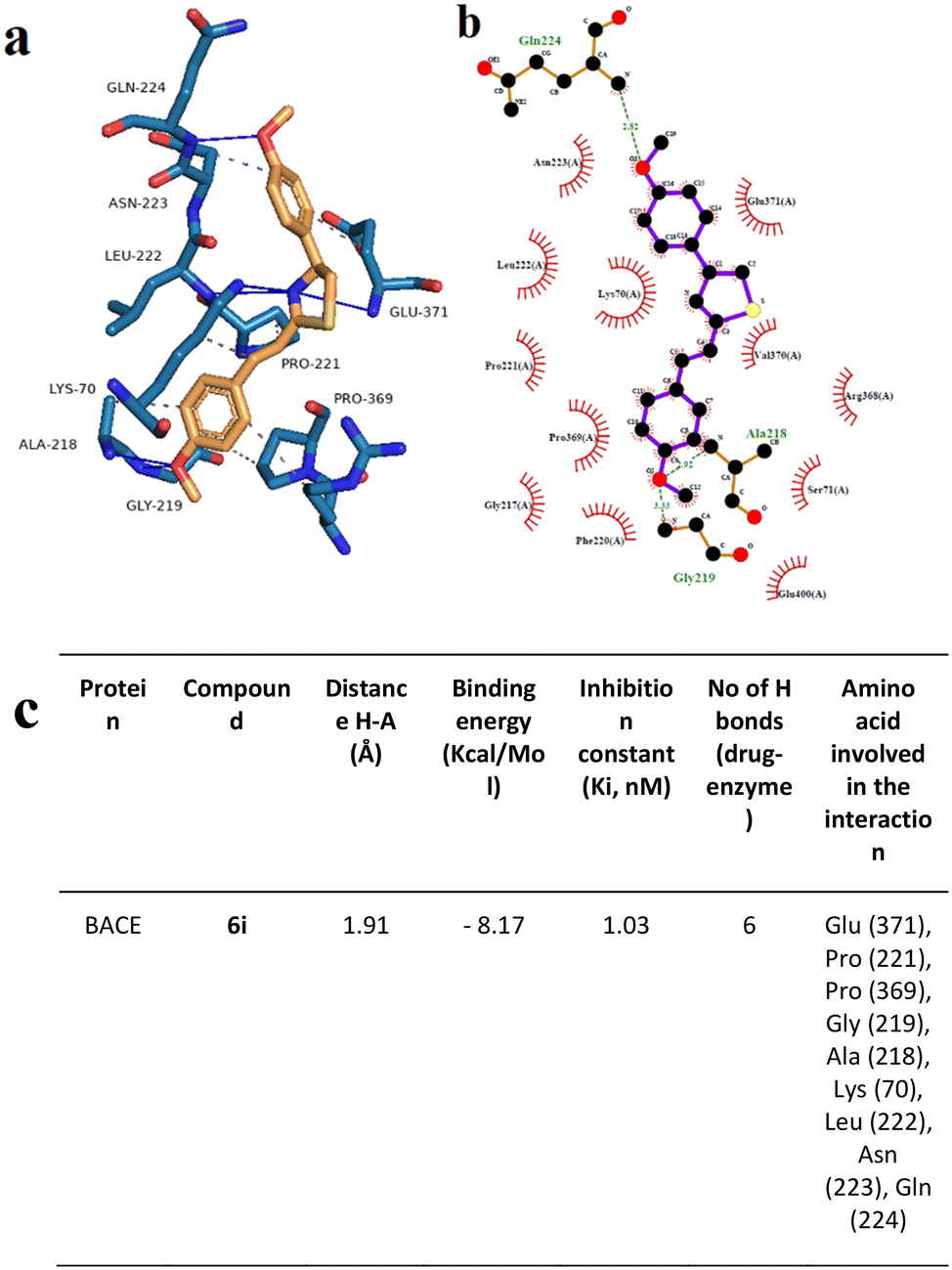 | ||
| Fig. 12 Interaction mode of the best docking conformation of the binding mode of human BACE with 6i obtained by a) PyMol, b) LigPlot, and c) the table of hydrogen-bonds (H-bonds) (with distance Å), binding energy (kcal mol−1), inhibition constant, number of H-bonds (drug–enzyme) and amino acid involved in interaction obtained from Protein–Ligand Interaction Profiler. The blue line and dashed green show the H-bonds in Pymol and LigPlot, respectively. | ||
Docking analysis on the BACE enzyme showed that the lowest binding energy belongs to compound 6e with −8.55 kcal mol−1. The inhibition constant of compound 6e was calculated as 544.52 nM and the closest interaction distance was found to be 2.27 Å (Fig. 11). According to the investigations, another strong interaction for the BACE enzyme was compound 6i with binding energies of −8.17 (Fig. 12). Furthermore, in vitro, BACE activity analysis showed that compounds 6e and 6i exhibited strong inhibitory potential against the BACE enzyme.
In silico evaluation of physicochemical properties
Inadequate efficacy and safety of a novel drug may be associated with its poor absorption, distribution, metabolism, and excretion (ADME).29 Properties such as molecular weight (MW), hydrogen bond receptor (HBA), hydrogen bond donor (HBD), number of rotatable bonds, lipid water partition coefficient (logPO/W), and topological polar surface area (TPSA) can be predicted by using SwissADME program (https://www.swissadme.ch/). The analysis of the data obtained by this method showed that (Tables 4 and 5).
| Hybrid | Water solubilitya (mg mL−1) | Lipinski's rule | Lipinski violationsa | TPSAa | MRa | RBa | |||
|---|---|---|---|---|---|---|---|---|---|
| MW (g mol−1) | logPa |
H-bond acceptorsa | H-bond donorsa | ||||||
|
a Prediction SwissADME platforms water solubility, Lipinski's rule parameters, topological polar surface area (TPSA), molar refractivity (MR), and rotatable bonds (RB) were predicted using the SwissADME software. Lipinski's rule of five determines the physicochemical properties of compounds as water/octanol partition coefficient (logP), molecular weight (MW), hydrogen bond acceptors (HBA), and hydrogen bond donors (HBD). A compound is characterized by good absorption and permeability when has a MW < 500, HBD < 5, logP value < 5, and HBA < 10.
|
|||||||||
| 6e | 1.99 × 10−5 | 342.25 | 5.05 | 1 | 0 | 1 | 41.13 | 90.62 | 3 |
| 6i | 7.24 × 10−5 | 323.41 | 4.38 | 3 | 0 | 0 | 59.59 | 95.90 | 5 |
| 6e | 6i | |
|---|---|---|
| a High absorption to CNS BBB penetration rate >2.0; middle absorption to CNS: BBB penetration rate <2.0–0.1; low absorption to CNS < 0.1. b Caco-2 permeability value <4: poorly permeable compound; Caco-2 permeability in the range 4–70 moderately permeable compound; Caco-2 permeability value >70: extremely permeable compound. c % HIA in the range 0–20: poor absorbance; % HIA in the range 20–70: moderate absorbance; % HIA in the range 70–100: good absorbance. d In vitro MDCK permeability value <25: poorly permeable compound; in vitro MDCK permeability value in the range 25–500: moderately permeable compound; in vitro MDCK permeability value >500: extremely permeable compound. e In vitro plasma protein binding value >90%: compound strongly bound; in vitro plasma protein binding value <90%: compound weakly bound. f Water solubility value <20 mg L−1: low; water solubility values in the range 10–1000 mg L−1: moderate; water solubility value >1000 mg L−1: high. | ||
| In vivo blood–brain barrier (BBB) penetrationa | 0.2 | 0.13 |
| In vitro Caco-2 cell permeability (nm s−1)b | 57.80 | 57.08 |
| Human intestinal absorption (HIA, %)c | 98.21 | 97.42 |
| In vitro MDCK cell permeability (nm s−1)d | 0.13 | 0.27 |
| In vitro plasma protein binding (%)e | 100 | 94.3 |
| Pure water solubility (mg L−1)f | 0.30 | 0.43 |
According to Table 4, only 6e violates Lipinski's rule due to a logP value slightly higher than 5, while 6i respects all parameters of the rule of five.
To confirm this prediction, water solubility and logP were also calculated experimentally using HPLC methods. Both hybrids 6e and 6i resulted from poor water solubility showing values equal to 1.75 × 10−5 and 7.19 × 10−5, respectively.
The logarithm of the ratio of the concentrations of the unionized hybrids in the solvents (logP) was calculated by the shake-flask method. Results confirmed the good lipophilicity of 6e (logP = 4.87) and 6i (logP = 4.18).
In silico pre-ADMET profile of both hybrids was predicted by using pre-ADMET software. As shown in Table 5, blood–brain barrier values (BBB) of 6e and 6i were in the range between 0.2 and 0.1 suggesting that both hybrids are scarcely permeable. Human gastrointestinal tract values (HIA) of these candidates are close to 98%, indicating these compounds can be well absorbed after oral administration.
Using Pre-ADMET software, both compounds showed good absorption properties (% human intestinal absorption, % HIA).30 However, in vitro MDCK cell permeability values are low, revealing that both compounds are poorly permeable for oral drug absorption.31 These values are in contrast with in vitro Caco-2 cell permeability values that suggested they are moderate parameters revealing that 6e, possessing Br substituent in the para position of the thiazole ring, showed a slightly higher BBB penetration rate (0.20) than 6i (0.13). Taking together all these data, 6e could be a slightly better candidate to cross BBB than 6i because the presence of Br in the para position could improve the BBB penetration rate of permeable compounds. Both compounds are strongly bound to plasma protein and are poorly soluble in water.
However, 6i was able to inhibit the AChE and to reduce at the same time the Aβ-aggregation more effectively than 6e. An important role was played by the nature and positions of substituents on both aromatic rings of hybrid molecules. The hybrid 6i bearing two methoxy groups at both aromatic rings emerged as the most effective inhibitor of AChE compared to 6e. On the other hand, 6e which had a Br in position 2 of the thiazole ring was found to be more effective than 6i in the inhibition of BACE. The docking results showed that both hybrids were in the active sites of both enzymes in the proper orientation. As suggested by previous papers, the thiazole group forms important interactions with the amino acid present at the active site of these enzymes. Moreover, 6i has been shown to have therapeutic potential by inhibiting the formation of Aβ aggregates. It could work by binding to Aβ and preventing its aggregation, as well as by stimulating the immune system to clear the aggregated protein.
Despite the effective pharmacological profile of 6i, physicochemical properties are not satisfactory. Its poor water solubility and scarce ability to cross the BBB, as evidenced by in silico predictive models, could hinder the efficacy on in vivo models. So, the development of new candidates that may be effective in AD can be followed rationally.
Experimental
General
All reagents used were purchased from commercial suppliers (Aldrich, Acros, TCI). Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm silica gel plates (silica gel 60F254) and components were envisioned by UV light absorbance. Purification of compounds by column chromatography was carried out on silica gel (70–230 mesh).1H and 13C spectra were recorded on Bruker Avance 400 spectrometer and Varian Mercury 200 MHz. ESI mass spectral analyses were performed on a mass spectrometer, using direct sample injection. Negative or positive ion ESI spectra were acquired by adjusting the needle and cone voltages accordingly. Microanalysis (C, H, N) was performed on a Carlo Erba instrument model E1110. Analyses indicated by the symbols of the elements were within ±0.4% of the theoretical values.
The purity of 6a–p was determined by chromatographic analyses. They were performed using an Agilent 1260 Infinity II HPLC (Agilent, Santa Clara, CA, USA) equipped with a 1260 Infinity II Quaternary Pump (model G7111A), 1260 Infinity II auto-sampler (model G7129A), a 1260 Infinity II Multicolumn Thermostat (model G7116A), and a 1260 Infinity II Diode Array Detector (model G7115A). Results were collected and integrated using Agilent OpenLAB CDS LC ChemStation software. The selected column was a Poroshell 120 EC-C18 (150 × 4.6 mm i.d., particle size 4 μm; Agilent, Santa Clara, USA), working at 20 °C. Samples were dissolved in acetonitrile (ACN) and analyzed using a mobile phase mixture of water (channel A) and acetonitrile (channel B), both containing 0.1% v/v of trifluoroacetic acid (TFA). The flow rate was 0.8 mL min−1 for a total run of 15 min. The UV detector was set at a length of 254 nm.
General method for the synthesis of styryl-thiazole hybrids 6a–p
To a stirred solution of thioamide 3a–h (1.0 mmol) in dry MeOH (7 mL) at room temperature, the corresponding phenacyl bromide 5a–e (1.0 mmol) was added, and the reaction mixture was stirred for 1 h. Then, the volatiles were removed in vacuo, ethyl acetate was added, and the organic phase was washed with 5% NaHCO3 and H2O, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography using an appropriate solvent system.:9). 1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 7.6 Hz, 2H), 7.57 (d, J = 7.3 Hz, 2H), 7.51–7.31 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 167.0, 156.3, 135.9, 134.7, 134.5, 129.1, 129.0, 128.9, 128.4, 127.3, 126.6, 121.8, 112.3. MS (ESI) m/z calculated for C17H14NS+ [M + H]+ 264.1, found 264.2. Calcd for C17H13NS: C, 77.53; H, 4.98; N, 5.32; S, 12.18. Found: C, 77.50; H, 5.00; N, 5.34; S, 12.17.
:6). 1H NMR (200 MHz, DMSO) δ 9.66 (s, 1H), 7.88–7.79 (m, 3H), 7.76–7.69 (m, 2H), 7.53 (s, 2H), 7.47–7.34 (m, 3H), 6.84 (d, J = 8.6 Hz, 2H). 13C NMR (50 MHz, DMSO) δ 165.6, 157.6, 155.4, 135.6, 133.7, 128.9, 127.6, 127.3, 125.4, 121.5, 115.5, 111.4. MS (ESI) m/z calculated for C17H14NOS+ [M + H]+ 280.1, found 279.9. Calcd for C17H13NOS: C, 73.09; H, 4.69; N, 5.01; O, 5.73; S, 11.48. Found: C, 73.11; H, 4.66; N, 5.01; O, 5.75; S, 11.47.
:9). 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 7.5 Hz, 2H), 7.50–7.29 (m, 6H), 6.97 (d, J = 8.3 Hz, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.7, 159.8, 156.1, 136.0, 134.5, 132.3, 129.0, 127.9, 127.4, 127.2, 121.8, 114.2, 110.6, 55.5. MS (ESI) m/z calculated for C18H16NOS+ [M + H]+ 294.1, found 294.1. Calcd for C18H15NOS: C, 73.69; H, 5.15; N, 4.77; O, 5.45; S, 10.93. Found: C, 73.71; H, 5.18; N, 4.75; O, 5.44; S, 10.91.
:6). 1H NMR (400 MHz, DMSO) δ 8.44 (s, 1H), 8.33 (d, J = 9.0 Hz, 2H), 8.27 (d, J = 9.0 Hz, 2H), 7.74 (d, J = 7.2 Hz, 2H), 7.59 (s, 2H), 7.43 (t, J = 7.3 Hz, 2H), 7.37 (t, J = 7.3 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 166.9, 152.7, 146.8, 139.9, 135.4, 134.9, 129.2, 128.9, 127.5, 127.0, 124.3, 121.1, 118.4. MS (ESI) m/z calculated for C17H13N2O2S+ [M + H]+ 309.1, found 309.1. Calcd for C17H12N2O2S: C, 66.22; H, 3.92; N, 9.08; O, 10.38; S, 10.40. Found: C, 66.23; H, 3.94; N, 9.10; O, 10.35; S, 10.38.
:9). 1H NMR (400 MHz, DMSO) δ 8.15 (s, 1H), 7.96 (d, J = 7.6 Hz, 2H), 7.73 (d, J = 7.5 Hz, 2H), 7.66 (d, J = 7.6 Hz, 2H), 7.56 (s, 2H), 7.42 (t, J = 7.3 Hz, 2H), 7.39–7.32 (m, 1H). 13C NMR (101 MHz, DMSO) δ 166.3, 153.8, 135.5, 134.3, 133.2, 131.8, 129.0, 128.9, 128.1, 127.4, 121.3, 121.2, 115.0. MS (ESI) m/z calculated for C17H13BrNS+ [M + H]+ 342.0, found 342.0. Calcd for C17H12BrNS: C, 59.66; H, 3.53; Br, 23.35; N, 4.09; S, 9.37. Found: C, 59.69; H, 3.54; Br, 23.37; N, 4.06; S, 9.34.
:7). 1H NMR (400 MHz, DMSO) δ 9.64 (s, 1H), 7.82 (s, 1H), 7.80 (d, J = 2.9 Hz, 2H), 7.61 (d, J = 8.1 Hz, 2H), 7.46 (d, J = 1.2 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 2.33 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.8, 157.6, 155.3, 138.6, 133.7, 132.8, 129.5, 127.6, 127.3, 125.4, 120.5, 115.5, 111.1, 21.0. MS (ESI) m/z calculated for C18H14NOS− [M–H]− 292.1, found 292.3. Calcd for C18H15NOS: C, 73.69; H, 5.15; N, 4.77; O, 5.45; S, 10.93. Found: C, 73.71; H, 5.17; N, 4.75; O, 5.46; S, 10.90.
:8). 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 8.9 Hz, 2H), 7.46 (d, J = 6.4 Hz, 1H), 7.43 (d, J = 14.3 Hz, 2H), 7.33 (d, J = 16.2 Hz, 1H), 7.25 (s, 1H), 7.20 (d, J = 8.0 Hz, 2H), 6.97 (d, J = 8.9 Hz, 2H), 3.85 (s, 3H), 2.38 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.1, 159.8, 155.9, 139.2, 134.6, 133.2, 132.4, 131.5, 129.7, 127.9, 127.2, 120.8, 114.2, 110.3, 55.5, 21.5. MS (ESI) m/z calculated for C19H18NOS+ [M + H]+ 308.1, found 308.3. Calcd for C19H17NOS: C, 74.23; H, 5.57; N, 4.56; O, 5.20; S, 10.43. Found: C, 74.26; H, 5.55; N, 4.59; O, 5.19; S, 10.40.
:1) 1H NMR (400 MHz, DMSO) δ 9.63 (s, 1H), 7.80 (d, J = 8.7 Hz, 2H), 7.76 (s, 1H), 7.67 (d, J = 8.8 Hz, 2H), 7.46 (d, J = 16.2 Hz, 1H), 7.37 (d, J = 16.2 Hz, 1H), 6.98 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 3.80 (s, 3H). 13C NMR (101 MHz, DMSO) δ 166.1, 160.0, 157.5, 155.2, 133.6, 128.8, 128.2, 127.5, 125.5, 119.3, 115.5, 114.4, 110.7, 55.3. MS (ESI) m/z calculated for C18H16NO2S+ [M + H]+ 310.1, found 310.1. Calcd for C18H15NO2S: C, 69.88; H, 4.89; N, 4.53; O, 10.34; S, 10.36. Found: C, 69.90; H, 4.91; N, 4.50; O, 10.32; S, 10.35.
:8) 1H NMR (200 MHz, DMSO) δ 7.92 (d, J = 8.7 Hz, 2H), 7.78 (d, J = 33.4 Hz, 3H), 7.65 (s, 1H), 7.43 (d, J = 6.8 Hz, 1H), 7.02 (d, J = 7.3 Hz, 2H), 6.97 (d, J = 7.4 Hz, 2H), 3.80 (s, 6H). 13C NMR (50 MHz, DMSO) δ 166.3, 160.0, 159.3, 154.8, 133.8, 128.9, 128.2, 127.5, 126.9, 119.2, 114.4, 114.2, 111.5, 55.2. MS (ESI) m/z calculated for C19H18NO2S+ [M + H]+ 324.1, found 324.1. Calcd for C19H17NO2S: C, 70.56; H, 5.30; N, 4.33; O, 9.89; S, 9.91. Found: C, 70.55; H, 5.32; N, 4.30; O, 9.88; S, 9.94.
:1). 1H NMR (400 MHz, DMSO) δ 9.63 (s, 1H), 7.81 (d, J = 8.6 Hz, 2H), 7.76 (s, 1H), 7.44 (s, 2H), 7.37 (s, 1H), 7.22 (d, J = 8.3 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 8.7 Hz, 2H), 3.83 (s, 3H), 3.79 (s, 3H). 13C NMR (101 MHz, DMSO) δ 166.1, 157.5, 155.2, 149.8, 149.1, 134.0, 128.5, 127.5, 125.5, 121.2, 119.5, 115.5, 111.7, 110.6, 109.7, 55.6, 55.6. MS (ESI) m/z calculated for C19H18NO3S+ [M + H]+ 340.1, found 340.1. Calcd for C19H17NO3S: C, 67.24; H, 5.05; N, 4.13; O, 14.14; S, 9.45. Found C, 67.26; H, 5.04; N, 4.15; O, 14.15; S, 9.41.
:6). 1H NMR (400 MHz, DMSO) δ 7.93 (d, J = 8.8 Hz, 2H), 7.86 (s, 1H), 7.46 (s, 2H), 7.38 (s, 1H), 7.22 (d, J = 8.3 Hz, 1H), 7.02 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 8.4 Hz, 1H), 3.84 (s, 3H), 3.80 (s, 3H), 3.79 (s, 3H). 13C NMR (101 MHz, DMSO) δ 166.3, 159.2, 154.8, 149.8, 149.1, 134.1, 128.4, 127.4, 126.9, 121.3, 119.4, 114.2, 111.7, 111.5, 109.7, 55.6, 55.6, 55.2. MS (ESI) m/z calculated for C20H20NO3S+ [M + H]+ 354.1, found 354.2. Calcd for: C20H19NO3S: C, 67.97; H, 5.42; N, 3.96; O, 13.58; S, 9.07. Found: C, 67.99; H, 5.43; N, 3.94; O, 13.57; S, 9.07.
:7). 1H NMR (400 MHz, DMSO) δ 9.63 (s, 1H), 7.82 (s, 1H), 7.80 (d, J = 2.2 Hz, 2H), 7.55 (d, J = 16.2 Hz, 1H), 7.45 (d, J = 16.1 Hz, 1H), 7.07 (s, 2H), 6.83 (d, J = 8.6 Hz, 2H), 3.84 (s, 6H), 3.69 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.8, 157.6, 155.4, 153.1, 138.3, 134.0, 131.2, 127.6, 125.4, 120.9, 115.5, 111.0, 104.8, 60.1, 59.8, 56.0. MS (ESI) m/z calculated for C20H20NO4S+ [M + H]+ 370.1, found 370.1. Calcd for: C20H19NO4S: C, 65.02; H, 5.18; N, 3.79; O, 17.32; S, 8.68. Found: C, 65.00; H, 5.21; N, 3.81; O, 17.31; S, 8.66.
:1). 1H NMR (400 MHz, DMSO) δ 7.94 (s, 1H), 7.91 (d, J = 9.1 Hz, 2H), 7.55 (s, 1H), 7.47 (d, J = 16.2 Hz, 1H), 7.08 (s, 2H), 7.02 (d, J = 8.8 Hz, 2H), 3.85 (s, 6H), 3.81 (s, 3H), 3.69 (s, 3H). 13C NMR (101 MHz, DMSO) δ 166.0, 159.3, 154.9, 153.1, 138.3, 134.1, 131.2, 127.4, 126.9, 120.9, 114.2, 111.9, 104.8, 60.1, 56.0, 55.2. MS (ESI) m/z calculated for C20H20NO4S+ [M + H]+ 384.1, found 384.1. Calcd for C21H21NO4S: C, 65.78; H, 5.52; N, 3.65; O, 16.69; S, 8.36. Found: C, 65.79; H, 5.54; N, 3.66; O, 16.67; S, 8.34.
:7). 1H NMR (400 MHz, DMSO) δ 9.65 (s, 1H), 7.84–7.81 (m, 3H), 7.75 (s, 2H), 7.54 (d, J = 6.5 Hz, 2H), 7.47 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 165.3, 157.6, 155.5, 134.6, 133.2, 132.3, 129.0, 128.9, 127.6, 125.3, 122.3, 115.5, 111.6. MS (ESI) m/z calculated for C17H13ClNOS+ [M + H]+ 314.0, found 314.0. Calcd for: C17H12ClNOS: C, 65.07; H, 3.85; Cl, 11.30; N, 4.46; O, 5.10; S, 10.22. Found: C, 65.06; H, 3.88; Cl, 11.33; N, 4.45; O, 5.09; S, 10.19.
:6). 1H NMR (400 MHz, DMSO) δ 9.62 (s, 1H), 7.85–7.79 (m, 5H), 7.50 (d, J = 4.0 Hz, 2H), 7.24 (t, J = 8.9 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 165.6, 162.4 (d, J = 246.7 Hz), 157.6, 155.4, 132.5, 132.25 (d, J = 3.0 Hz), 129.4 (d, J = 8.2 Hz), 127.6, 125.4, 121.4, 115.8 (d, J = 21.7 Hz), 115.5, 111.3. MS (ESI) m/z calculated for C17H13FNOS+ [M + H]+ 298.1, found 298.2. Calcd for: C17H12FNOS: C, 68.67; H, 4.07; F, 6.39; N, 4.71; O, 5.38; S, 10.78. Found: C, 68.69; H, 4.08; F, 6.41; N, 4.69; O, 5.37; S, 10.76.
:6). 1H NMR (400 MHz, DMSO) δ 9.66 (bs, 1H), 8.25 (d, J = 8.8 Hz, 2H), 8.00 (d, J = 8.9 Hz, 2H), 7.93 (s, 1H), 7.83–7.79 (m, 3H), 7.66 (d, J = 16.2 Hz, 1H), 6.85 (d, J = 8.7 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 164.6, 157.7, 155.9, 146.9, 142.5, 131.1, 128.2, 127.6, 125.6, 125.2, 124.1, 115.6, 112.8. MS (ESI) m/z calculated for C17H13N2O3S+ [M + H]+ 325.1, found 324.9. Calcd for: C17H12N2O3S: C, 62.95; H, 3.73; N, 8.64; O, 14.80; S, 9.89. Found C, 62.97; H, 3.72; N, 8.66; O, 14.79; S, 9.87.
Biological assays
:F12 (1:1, Gibco®) with 10% fetal bovine serum (FBS, Sigma-Aldrich®) and 1% penicillin/streptomycin (Sigma-Aldrich®) at 37 °C and 5% CO2. After the cultures reached confluence, the cells were harvested by using trypsin/EDTA (Sigma-Aldrich®) and transferred into 48 well plates. To differentiate neuroblastoma cell lines to mature neuron-like cell cultures, 10 μM retinoic acid (RA) (Sigma-Aldrich®) was added to cell cultures, and cell medium (DMEM:F12 + 3% fetal bovine serum + 1% penicillin/streptomycin) was refreshed every 2 days until 8th day. Then, 25 nM brain-derived neurotrophic factor (BDNF) (Promega®) was integrated into the cultures for another 5 days to complete cellular differentiation. Differentiated cells were determined under the inverted microscope and cell cycle analysis was performed by using flow cytometry/cell cycle analysis for further confirmation of differentiation.32
Biocompatibility investigation by cytotoxicity and genotoxicity assays
Genotoxicity analysis by Hoechst 33258 fluorescent nuclear staining
Possible genotoxic properties of 6e and 6i were investigated by using Hoechst 33258 fluorescent nuclear staining. 6e and 6i were applied to the differentiated SHSY-5Y cell cultures for 24 h and cell culture mediums were removed from plates and cells were washed with PBS 3 times. Then, cell cultures were fixed by using 4% paraformaldehyde at 4 °C for 30 minutes. After the fixation procedure, cell cultures were washed by using PBS 3 times, and 200 μL of Hoechst 33258 fluorescent dye (1 mM) was added to cell cultures and incubated for 5 minutes at room temperature. Nuclear abnormalities (MN: micro nucleus, L: lobbed, and N: notched mutations) were observed under a fluorescent microscope. For each compound application, a total of 1000 cells were counted for triple replicates, and nuclear abnormality rations were investigated visually.34Experimental Alzheimer's disease model
In vitro Alzheimer's disease model was constituted via the differentiation of SHSY-5Y cell culture and the integration of Aβ1–42 into cell cultures. 20 μM of Aβ1–42 was incubated in a cell culture medium for 24 h to produce amyloid fibrilization. After cellular differentiation, 20 μM of fibrilized Aβ1–42 was added to cell cultures and incubated for 24 h to reach a 50% cytotoxic effect. To investigate the neuroprotective properties of 6e and 6i, 20 μM of fibrilized Aβ1–42 and 6e and 6i were integrated into differentiated cell cultures for 24 h incubation period. The neuroprotective effect of 6e and 6i was observed by using an MTT cell viability assay.35Biochemical investigations
In vitro β-amyloid aggregation analysis by using Congo red staining
After 6e and 6i applications to the experimental disease model, cells were fixed by using 4% paraformaldehyde in phosphate-buffered saline at 4 °C for 30 min. Cultures were washed with PBS and 100 μL of Congo red (1 μM, C.I.22120, Direct Red 28; Aldrich, Milwaukee, WI) was added to each well. Samples were incubated for 1 h in the dark and washed with PBS 3 times. To extract Congo Red dye from beta-amyloid fibrils, 100 μL of DMSO was added to each well. Color intensities were measured by using a microplate reader at 497 nm.36Flow cytometry analysis
Apoptotic and necrotic status in the experimental Alzheimer's disease model against 6e and 6i applications were determined by using Annexin V-FITC Apoptosis Kit (Abcam®, BioVision, UK) according to the manufacturer's instructions. Briefly, 105 cells were seeded into a 48-well plate, and compounds were applied to cultures for 24 hours. Experimental analyses were divided into six groups; 20 μM of fibrilized Aβ1–42 applied AD model, 6i (50 μg mL−1), and 6e (25 μg mL−1) applied AD model for 24 h. Cell cultures were transferred into a fresh Eppendorf tube by using trypsin incubation for 3 minutes at 37 °C. Cells were centrifuged at 500 × g for 5 minutes and supernatants were discarded. Cells were resuspended in 500 μL of binding buffer and 5 μL of Annexin V-FITC/5 μL of propidium iodide was added to cell cultures. After 5 min incubation at room temperature, cells were analyzed by flow cytometry (CyFlow® Cube 6, Germany).Molecular docking
Docking investigations were performed using Auto Dock software (https://autodock.scripps.edu) in accordance with the previously published protocol.37 AChE and BACE protein structures were obtained from the Protein Data Bank (RCSB PDB, https://www.rcsb.org/). Structures of 6e and 6i in PDB (Protein database) format were constituted by using ChemDraw (Chem3D) software. Then, the Auto Dock molecular modeling software was used to acquire the protein–ligand interactions. In the process, the final docking score for each molecule was calculated by taking the average score of the protein–ligand docking scores. Subsequently, the resulting average scores were ranked from highest to lowest. The obtained docking results using Auto Dock were analyzed by the Protein–Ligand Interaction Profiler (https://plip-tool.biotec.tu-dresden.de) online software in order to monitor the distance between ligand and amino acid residues in detail. Finally, PyMol software (https://pymol.org) was utilized to obtain 3-dimensional residual interaction plots, and also the LigPlot software (Version 4.5.3, https://www.ebi.ac.uk/thornton-srv/software/LIGPLOT) was performed to obtain data related to interactions between protein and ligand in 2-dimensional sketches.38,39HPLC-UV assays
The analytical HPLC apparatus consisted of a Waters 600 HPLC pump (Waters Corporation, Milford, MA, USA). The mobile phase was a mixture of H2O and ACN + 0.1% TFA flushing using isocratic conditions with a flow of 1 mL min−1, and a Hypersil GOLD C18 column (250 × 4.6 mm) was used. Solubility was determined as previously reported.40 The logP values were determined by “shake-flask” method.41 Approximately 5 mg of compound was dissolved in 1 mL of n-octanol and mixed with an equal volume of phosphate buffer solution (PBS, pH 7.4). The mixture was subjected to repeated inversions (200 times, for 5 min) and then allowed to stand for 30 min for the complete separation of the two phases. Then, each phase was analyzed by HPLC.
Conclusions
In this paper, sixteen styryl thiazole hybrids were designed, synthesized, characterized, and tested as potential anti-Alzheimer drug candidates.Docking studies confirmed the capability of the most active hybrids 6e and 6i to inhibit AChE and BACE, two important enzymes involved in the pathogenesis of AD. Moreover, 6i has been shown to have therapeutic potential by inhibiting the formation of Aβ aggregates. This hybrid could be considered a potential lead compound for the development of multi-target drugs with AChE, BACE, and Aβ-aggregation inhibitory properties. All these findings encourage medicinal chemists to improve the pharmacokinetic profile of this hybrid since its water solubility and capability to cross the BBB are very low. Further research is needed to fully understand the mechanism of action and optimize its pharmacokinetic properties for clinical use.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research project was supported by the Italian Ministry of Education, University and Research (University “G. d'Annunzio” of Chieti-Pescara) FAR 2020 and by NKUA-ELKE K.E.16672.Notes and references
- H. Zhu, V. Dronamraju, W. Xie and S. S. More, Med. Chem. Res., 2021, 30, 305–352 CrossRef CAS PubMed.
- C. Cornacchia, L. Marinelli, A. Di Rienzo, M. P. Dimmito, F. Serra, G. Di Biase, B. De Filippis, H. Turkez, A. Mardinoglu, I. Bellezza, A. Di Stefano and I. Cacciatore, Eur. J. Med. Chem., 2022, 243, 114746 CrossRef CAS PubMed.
- Y. H. Ham, K. K. Jason Chan and W. Chan, Chem. Res. Toxicol., 2020, 33, 1815–1821 Search PubMed.
- I. Cacciatore, H. Turkez, A. Di Rienzo, M. Ciulla, A. Mardinoglu and A. Di Stefano, RSC Med. Chem., 2021, 12, 1944–1949 RSC.
- P. K. Mandal, S. Saharan, M. Tripathi and G. Murari, Biol. Psychiatry, 2015, 78, 702–710 CrossRef CAS PubMed.
- M. Rosini, E. Simoni, M. Bartolini, A. Tarozzi, R. Matera, A. Milelli, P. Hrelia, V. Andrisano, M. L. Bolognesi and C. Melchiorre, Eur. J. Med. Chem., 2011, 46, 5435–5442 CrossRef CAS PubMed.
- T. B. Callis, T. R. Garrett, A. P. Montgomery, J. J. Danon and M. Kassiou, J. Med. Chem., 2022, 65, 13483–13504 CrossRef CAS PubMed.
- H. Wang, Z. Cai, S. Zheng, H. Ma, H. Lin and X. Zheng, Lett. Drug Des. Discovery, 2018, 15, 388–397 CrossRef CAS.
- B. Sever, M. D. Altintop, H. K. Gençer, H. A. Kapkaç, O. Atli, M. Baysal and A. Özdemir, Lett. Drug Des. Discovery, 2018, 15, 744–756 CrossRef CAS.
- R. Hussain, H. Ullah, F. Rahim, M. Sarfraz, M. Taha, R. Iqbal, W. Rehman, S. Khan, S. A. A. Shah, S. Hyder, M. Alhomrani, A. S. Alamri, O. Abdulaziz and M. A. Abdelaziz, Molecules, 2022, 27, 6087 CrossRef CAS PubMed.
- G. Ghotbi, M. Hamzeh-Mivehroud, A. Taghvimi, S. Davaran and S. Dastmalchi, Pharm. Sci., 2021, 27, 366–377 CrossRef CAS.
- K. Luo, J. Chen, H. Li, D. Wu, Y. Du, S. Zhao and X. Fu, Bioorg. Chem., 2023, 138, 106596 CrossRef CAS PubMed.
- V. K. Nuthakki, S. Choudhary, C. N. Reddy, S. Bhatt, A. Jamwal, A. Jotshi and S. B. Bharate, ACS Chem. Neurosci., 2023, 14, 1193–1219 CrossRef CAS PubMed.
- Y. S. Lee, H. Kim, Y. H. Kim, E. J. Roh, H. Han and K. J. Shin, Bioorg. Med. Chem. Lett., 2012, 22, 7555–7561 CrossRef CAS PubMed.
- A. Y. Hemaida, G. S. Hassan, A. R. Maarouf, J. Joubert and A. A. El-Emam, ACS Omega, 2021, 6, 19202–19211 CrossRef CAS PubMed.
- K. Wang, J. Shi, Y. Zhou, Y. He, J. Mi, J. Yang, S. Liu, X. Tang, W. Liu, Z. Tan and Z. Sang, Bioorg. Chem., 2021, 112, 104879 CrossRef CAS PubMed.
- N. Ruwizhi and B. A. Aderibigbe, Int. J. Mol. Sci., 2020, 21, 5712 CrossRef CAS PubMed.
- Y. Chen, Y. Ma and W. Ma, Eur. J. Drug Metab. Pharmacokinet., 2009, 34, 51–56 CrossRef CAS PubMed.
- Y. Yan, X. Xu, X. Jie, J. Cheng, R. Bai, Q. Shuai and Y. Xie, Tetrahedron Lett., 2018, 59, 2793–2796 CrossRef CAS.
- R. Maity, S. Naskar and I. Das, J. Org. Chem., 2018, 83, 2114–2124 CrossRef CAS PubMed.
- J. Petrova, S. Momchilova and N. G. Vassilev, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 164, 87–94 CrossRef CAS.
- D. Orr, A. Tolfrey, J. M. Percy, J. Frieman, Z. A. Harrison, M. Campbell-Crawford and V. K. Patel, Chem. – Eur. J., 2013, 19, 9655–9662 CrossRef CAS PubMed.
- L. Huang, T. Su, W. Shan, Z. Luo, Y. Sun and F. He, Bioorg. Med. Chem., 2012, 20, 3038–3048 CrossRef CAS PubMed.
- Q. Jiang, W. Sheng and C. Guo, Green Chem., 2013, 15, 2175 RSC.
- V. Facchinetti, M. M. Avellar, A. C. S. Nery, C. R. B. Gomes, T. R. A. Vasconcelos and M. V. N. de Souza, Synthesis, 2015, 48, 437–440 CrossRef.
- Y. B. Yu, H. L. Chen, L. Y. Wang, X. Z. Chen and B. Fu, Molecules, 2009, 14, 4858–4865 CrossRef CAS PubMed.
- L. F. Hoffmann, A. Martins, F. Majolo, V. Contini, S. Laufer and M. I. Goettert, Neural Regener. Res., 2023, 18, 1265–1266 CrossRef PubMed.
- H. Turkez, I. Cacciatore, M. E. Arslan, E. Fornasari, L. Marinelli, A. Di Stefano and A. Mardinoglu, Biomolecules, 2020, 10, 737 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 23, 3–26 CrossRef.
- S. Yee, Pharm. Res., 1997, 14, 763 CrossRef CAS PubMed.
- S. Yamashita, T. Furubayashi, M. Kataoka, T. Sakane, H. Sezaki and H. Tokuda, Eur. J. Pharm. Sci., 2000, 10, 195–204 CrossRef CAS PubMed.
- H. Turkez, M. E. Arslan, A. Yilmaz, F. Doru, O. Caglar, E. Arslan, A. Tatar, A. Hacımuftuoglu and A. M. Abd El-Aty, J. Food Biochem., 2021, 45, e13990 CrossRef CAS PubMed.
- A. Pagoni, L. Marinelli, A. Di Stefano, M. Ciulla, H. Turkez, A. Mardinoglu, S. Vassiliou and I. Cacciatore, Eur. J. Med. Chem., 2020, 186, 111880 CrossRef CAS PubMed.
- R. Küçükdoğru, H. Türkez, M. E. Arslan, Ö. Ö. Tozlu, E. Sönmez, A. Mardinoğlu, I. Cacciatore and A. Di Stefano, Metab. Brain Dis., 2020, 35, 947–957 CrossRef PubMed.
- L. Marinelli, E. Fornasari, A. Di Stefano, H. Turkez, M. E. Arslan, P. Eusepi, M. Ciulla and I. Cacciatore, Neuropeptides, 2017, 66, 52–58 CrossRef PubMed.
- X. Li, X. Zhang, R. Xing, F. Qi, J. Dong, D. Li, X. Tian, B. Yu, M. Huang, L. Zhang, X. Yuan, Y. Yang, H. Wu, L. Zang, X. Mao and R. Sui, Int. J. Biol. Macromol., 2021, 192, 491–497 CrossRef CAS PubMed.
- D. Seeliger and B. L. de Groot, J. Comput.-Aided Mol. Des., 2010, 24, 417–422 CrossRef CAS PubMed.
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS PubMed.
- S. Yuan, H. C. S. Chan and H. Zhenquan, WIREs Comput. Mol. Sci., 2017, 7, e1298 CrossRef.
- I. Cacciatore, L. Marinelli, E. Fornasari, L. S. Cerasa, P. Eusepi, H. Türkez, C. Pomilio, M. Reale, C. D'Angelo, E. Costantini and A. Di Stefano, Int. J. Mol. Sci., 2016, 17, 1035 CrossRef PubMed.
- P. Sozio, L. S. Cerasa, S. Laserra, I. Cacciatore, C. Cornacchia, E. S. Di Filippo, S. Fulle, A. Fontana, A. Di Crescenzo, M. Grilli, M. Marchi and A. Di Stefano, Eur. J. Pharm. Sci., 2013, 41, 187–198 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and HPLC chromatograms of 6a–p. See DOI: https://doi.org/10.1039/d3md00308f |
| This journal is © The Royal Society of Chemistry 2023 |