
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 2-aminopropyl benzopyran derivatives as potential agents against triple-negative breast cancer†
Ainhoa
García
 ab,
Sandra
Torres-Ruiz
b,
Laura
Vila
b,
Carlos
Villarroel-Vicente
ab,
Álvaro
Bernabeu
a,
Pilar
Eroles
*bcd,
Nuria
Cabedo
*ab and
Diego
Cortes
a
ab,
Sandra
Torres-Ruiz
b,
Laura
Vila
b,
Carlos
Villarroel-Vicente
ab,
Álvaro
Bernabeu
a,
Pilar
Eroles
*bcd,
Nuria
Cabedo
*ab and
Diego
Cortes
a
aDepartment of Pharmacology, University of Valencia, 46100 Valencia, Spain. E-mail: ncabedo@uv.es
bInstitute of Health Research-INCLIVA, University Clinic Hospital of Valencia, 46010 Valencia, Spain
cDepartment of Physiology, University of Valencia, 46010 Valencia, Spain. E-mail: pilar.eroles@uv.es
dCenter for Biomedical Network Research on Cancer (CIBERONC), 28019 Madrid, Spain
First published on 4th October 2023
Abstract
Synthesis of three series of 2-aminopropyl derivatives containing a benzopyran nucleus was performed to evaluate their performance against triple-negative breast cancer cell lines (MDA-MB-231 and MDA-MB-436) and normal breast epithelial cells (MCF10A). For the three series, the cytotoxic activity was as follows: N-methylated derivatives (tertiary amines) 5b, 6b, and 7b > secondary amine benzopyrans 5, 6, and 7 > quaternary amine salts 5c, 6c, and 7c > free phenolic derivatives 5a, 6a, and 7a. The structure–activity relationship showed the importance of the presence of an amine group and a p-fluorobenzyloxy substituent in the chromanol ring (IC50 values from 1.5 μM to 58.4 μM). In addition, 5a, 5b, 6a, and 7b displayed slight selectivity towards tumor cells. Compounds 5, 5a, 5b, 6, 6a, 6c, 7, and 7b showed apoptotic/necrotic effects due to, at least in part, an increase in reactive oxygen species generation, whereas 5b, 5c, 6b, 7a, and 7c caused cell cycle arrest in the G1 phase. Further cell-based mechanistic studies revealed that 5a, 6a, and 7b, which were the most promising compounds, downregulated the expression of Bcl-2, while 5b downregulated the expression of cyclins CCND1 and CCND2. Therefore, 2-aminopropyl benzopyran derivatives emerge as new hits and potential leads for developing useful agents against breast cancer.
1. Introduction
Breast cancer (BC) is a complex and heterogeneous disease, and the most frequently diagnosed and leading cause of cancer-related deaths in women, with increasing mortality rates affecting more than 1 million women worldwide.1–4 The selection of drug therapy for patients depends on the cancer subtype: hormone receptor-positive, human epidermal growth factor receptor 2 (HER-2), and triple-negative BC (TNBC).TNBC is characterized as lacking estrogen receptors (ERs), progesterone receptors (PRs), and HER-2.5–9 This is the most aggressive subtype, and represents 15% of all breast cancers.10 Consequently, TNBC patients do not respond to hormonal and anti-HER2 monoclonal antibody trastuzumab-based targeted therapies. An affordable therapeutic strategy includes cytotoxic chemotherapy with taxane- and tetracycline-based combination treatment and platinum-based agents, but drug resistance is one of the major challenges that is currently being addressed by TNBC research.10,11 The discovery of new compounds that decrease the survival of tumor cells in this particular subtype of breast cancer is highly significant for enhancing treatment of this disease.10
Several natural products containing a benzopyran skeleton (Fig. 1) have exhibited anti-tumor properties against BC through pleiotropic mechanisms involving apoptosis and cell cycle arrest, among others. Genistein, an estrogenic soy-derived compound belonging to the isoflavone class, induces apoptosis in TNBC cells.12–15 Wogonin, a natural flavone isolated from the roots of Scutellaria baicalensis Georgi, suppresses tumor angiogenesis in BC.16,17 Moreover, hesperitin derivatives also present anti-tumor activity against BC.18,19 Therefore, the synthesis of the benzopyran nucleus has been performed by numerous medicinal chemistry groups in their search for new anti-tumor agents.20,21
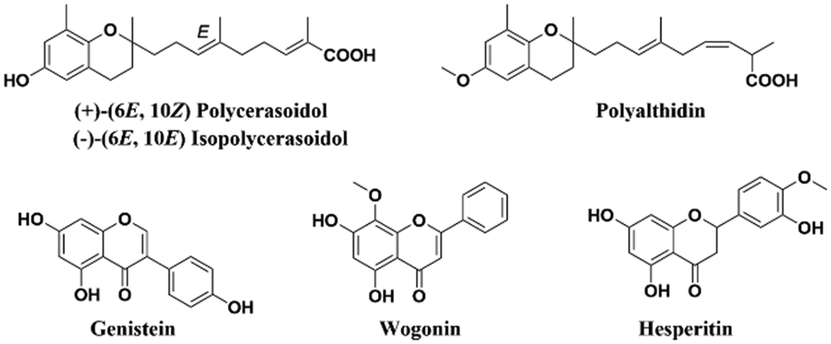 | ||
| Fig. 1 Natural products containing a benzopyran skeleton. | ||
Our research group previously isolated polycerasoidol and polyalthidin (Fig. 1) from the stem bark of Polyalthia cerasoides (Annonaceae).22,23 Polycerasoidol and synthetic analogs displayed PPAR-activating properties, anti-inflammatory effects, and ameliorated metabolic derangements,24,25 while it has been shown that polyalthidin is an inhibitor of the mammalian mitochondrial respiratory chain.23 In addition, it has been reported that isopolycerasoidol induces mitochondrial-mediated apoptosis in human BC cell lines.26
Based on these pieces of evidence, our goal was to discover new molecules for future TNBC treatment. Therefore, we synthesised 2-aminopropyl benzopyran derivatives bearing a p-methoxyphenylethylamine (series 1), diphenylethylamine (series 2), or isoquinoline moiety (series 3) (Scheme 1). The 15 new compounds were tested against human TNBC cell lines (MDA-MB-231 and MDA-MB-436) and normal breast epithelial cells (MCF10A) for cytotoxic activity. Then, we studied their impact on the apoptosis/necrosis effect or cell cycle arrest, as well as their reactive oxygen species (ROS) production and the gene expression of some promising compounds.
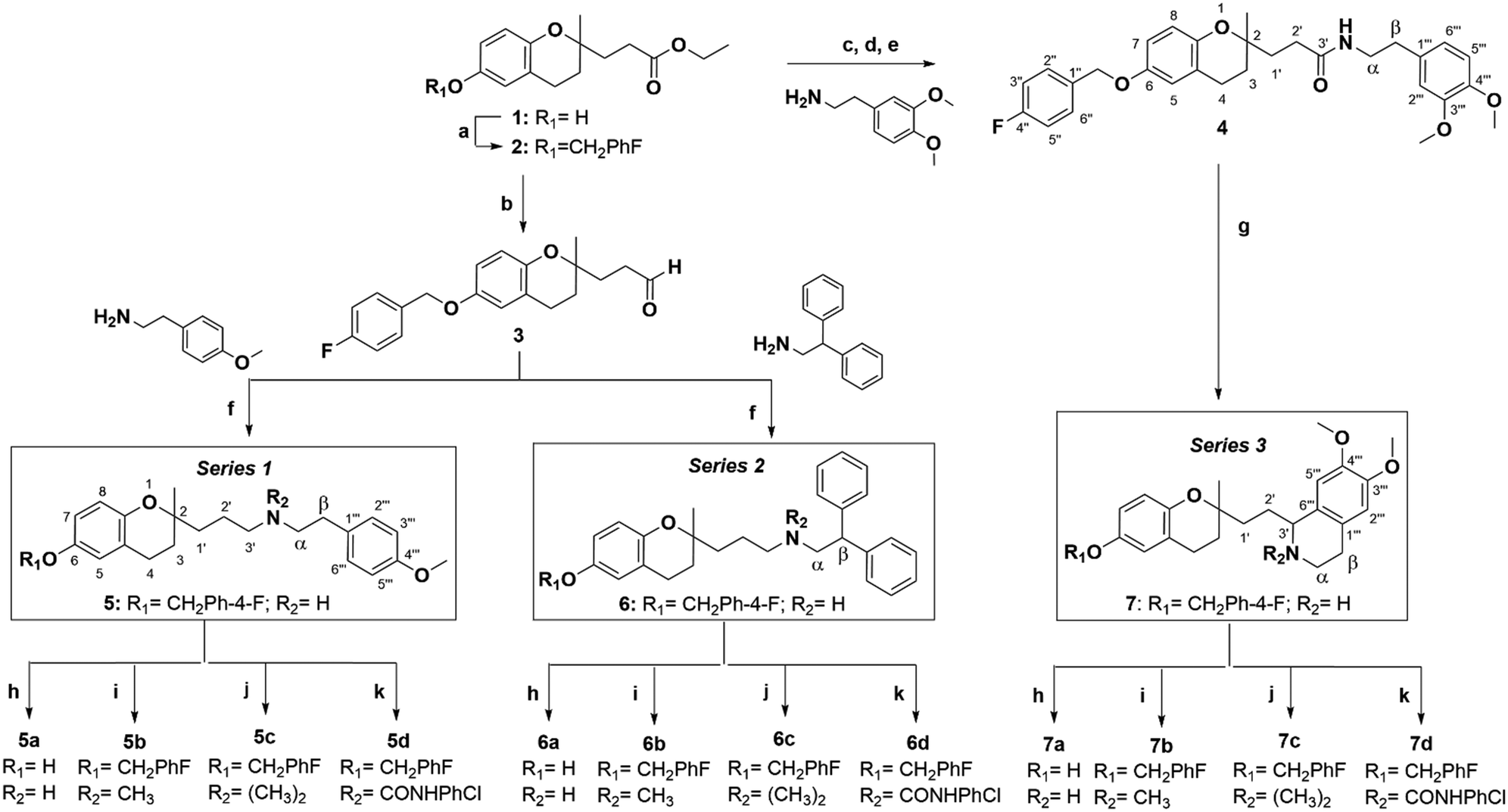 | ||
Scheme 1 Reagents and conditions: (a) p-fluorobenzyl chloride, K2CO3, EtOH, reflux, N2, 4 h, 79%, (b) DIBAL-H, DCM, −78 °C, N2, 15 min, 92%, (c) MeOH, KOH 20%, reflux, N2, 4 h, (d) SOCl2, DCM, reflux, N2, 3 h, (e) 4-DMAP, Et3N, DCM, rt., N2, overnight, 63%, (f) NaBH(OAc)3, AcOH, (CH2Cl)2, rt., N2, 1 h, 72% for 5 and 54% for 6, (g) POCl3, CH2Cl2, reflux, N2, overnight, NaBH4, MeOH, rt., N2, 2 h, 53%, (h) HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH 1:1, reflux, N2, 3 h, 92–96%, (i) HCHO, HCO2H, MeOH, reflux, N2, 1 h, NaBH4, rt., N2, 45 min, 76–86%, (j) MeI, DMF, reflux, N2, 30 min–1 h, 79–96%, (k) 4-chlorophenyl isocyanate, Et3N, DCM, rt., N2, overnight, 68–92%. :EtOH 1:1, reflux, N2, 3 h, 92–96%, (i) HCHO, HCO2H, MeOH, reflux, N2, 1 h, NaBH4, rt., N2, 45 min, 76–86%, (j) MeI, DMF, reflux, N2, 30 min–1 h, 79–96%, (k) 4-chlorophenyl isocyanate, Et3N, DCM, rt., N2, overnight, 68–92%. | ||
2. Results and discussion
2.1. Chemistry
Using different methodologies, we synthesized three series of compounds with a benzopyran skeleton. 2-Aminopropyl benzopyran derivatives were synthesised via a chroman-4-one scaffold. These derivatives were obtained by aldol condensation between o-hydroxyacetophenones and methyl-ketones in the presence of a secondary amine by Michael addition, as previously reported.27–30 The free phenolic group of precursor benzopyran 1 was protected using p-fluorobenzyl chloride in the presence of potassium carbonate to afford benzopyran ester 2 (Scheme 1).To obtain 2-aminopropyl benzopyran derivatives, two different synthetic pathways were followed from benzopyran ester 2. The first was to synthesise p-methoxyphenylethylamine derivatives (series 1) and diphenylethylamine derivatives (series 2), and the second was used to synthesise isoquinoline derivatives (series 3). For the first pathway, controlled reduction by diisobutylaluminum hydride (DIBAL-H) of the ester group was carried out to obtain aldehyde intermediate 3, from which compounds 5 (series 1) and 6 (series 2) were obtained by reductive amination.
The condensation of 3 with 4-methoxyphenethylamine and 2,2-diphenylethylamine resulted in secondary amines 5 and 6, respectively, via a Schiff base, followed by reduction with sodium triacetoxyborohydride under acid conditions. The second pathway was based on the formation of the amide intermediate 4 from an acid chloride. The ester group of 2 was submitted to basic hydrolysis, followed by a reaction with thionyl chloride. Then, acid chloride was reacted with 3,4-dimethoxyphenethylamine to yield amide 4, from which compound 7 was obtained by Bischler–Napieralski cyclodehydration.
In each series, four derivatives were obtained from amines 5, 6, and 7. The subsequent acid hydrolysis of the p-fluorobenzyl group yielded amines 5a, 6a, and 7a. N-Methylated derivatives 5b, 6b, and 7b were obtained with formic acid and formaldehyde, followed by reduction with sodium borohydride, while N-dimethylated salts 5c, 6c, and 7c were obtained with methyl iodide. The functionalization of the amine group in the urea group was achieved by reacting it with 4-chlorophenyl isocyanate to obtain urea derivatives 5d, 6d, and 7d (Scheme 1).
2.2. Pharmacology
| Structure | MDA-MB-231 (μM) | MDA-MB-436 (μM) | MCF10A (μM) | SI MCF10A/MDA-MB-231 | SI MCF10A/MDA-MB-436 | |
|---|---|---|---|---|---|---|
| The data are presented as the mean ± SD of three independent experiments performed in triplicate.a P < 0.05.b P < 0.01.c P < 0.001 vs.5b, 6b, and 7b for each family. Selectivity index (SI): ratio of cytotoxicity in MCF10A cells compared to that in cancer cells. | ||||||
| 5 |
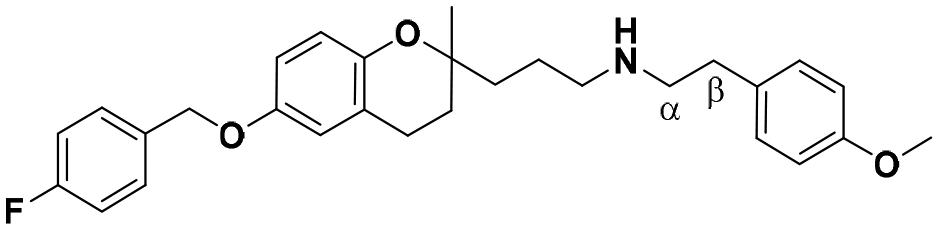
|
13.2 ± 1.6a | 13.0 ± 2.0 | 6.3 ± 0.9 | 0.48 | 0.48 |
| 5a |
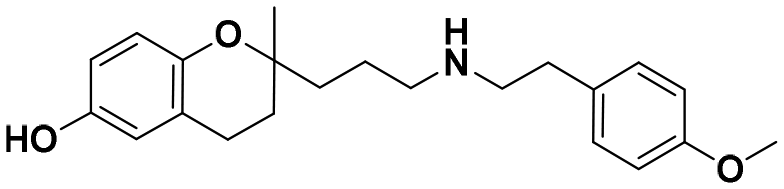
|
51.0 ± 5.8a | 30.9 ± 3.5a | 76.8 ± 0.5b | 1.51 | 2.50 |
| 5b |
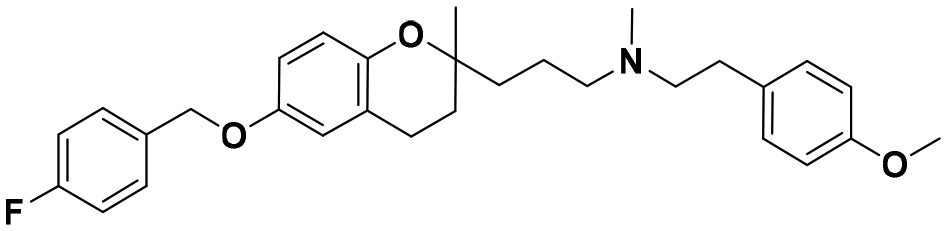
|
7.3 ± 0.7 | 12.6 ± 1.5 | 12.1 ± 2.8 | 1.65 | 0.96 |
| 5c |
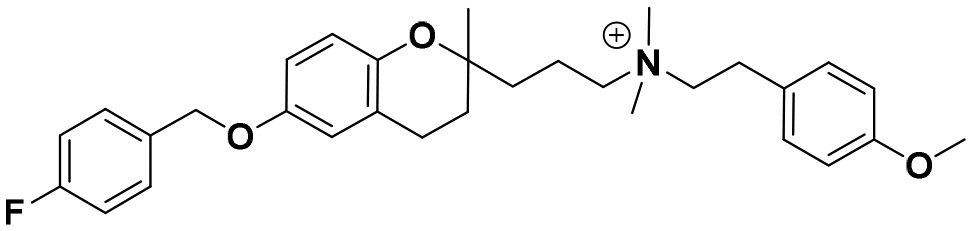
|
25.0 ± 1.1c | 24.0 ± 1.2b | 25.0 ± 2.0a | 1.0 | 1.0 |
| 5d |
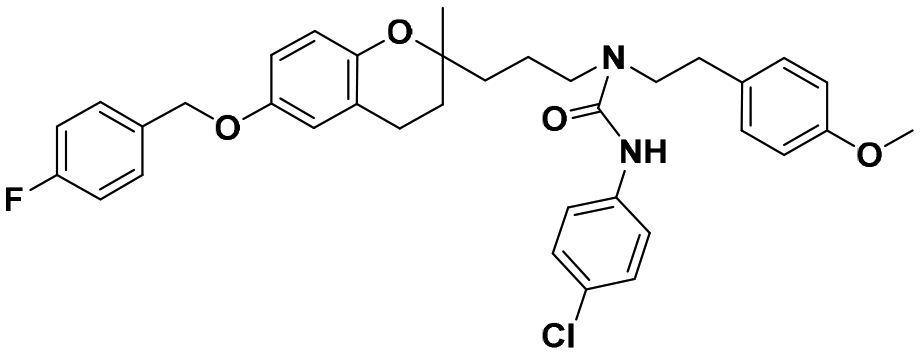
|
>100 | >100 | >100 | — | — |
| 6 |
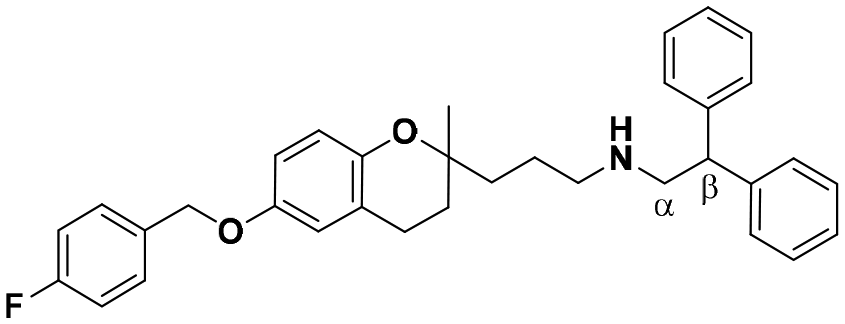
|
11.0 ± 2.6a | 19.3 ± 0.8b | 12.9 ± 1.8a | 1.17 | 0.67 |
| 6a |
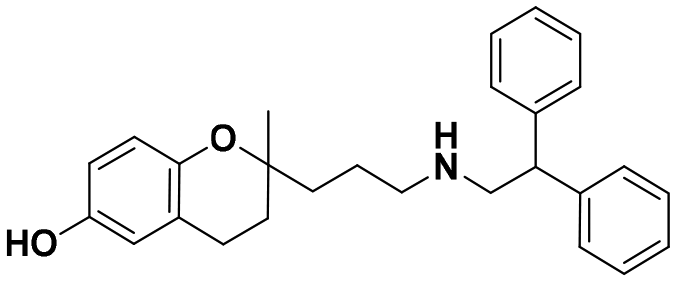
|
48.3 ± 4.2a | 31.1 ± 5.5c | 46.6 ± 3.6b | 0.96 | 1.5 |
| 6b |
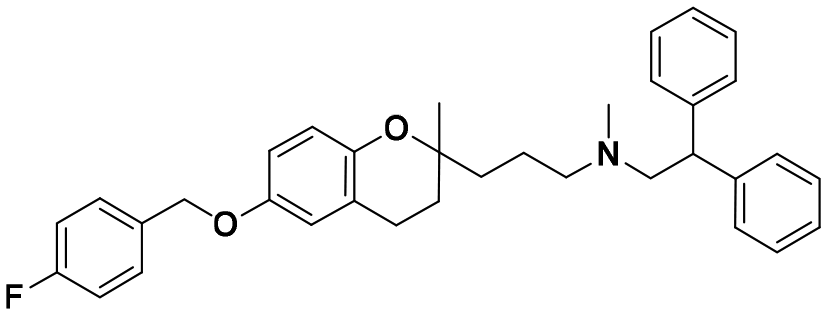
|
1.5 ± 0.1 | 2.0 ± 1.7 | 1.4 ± 0.7 | 0.94 | 0.70 |
| 6c |
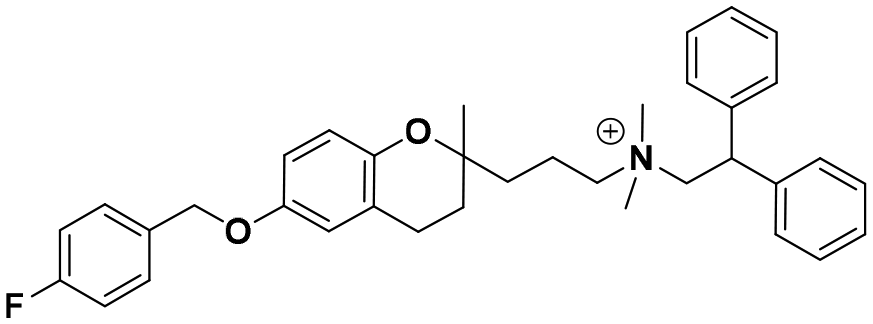
|
19.2 ± 2.9b | 24.6 ± 1.1b | 20.8 ± 4.0 | 1.09 | 0.84 |
| 6d |
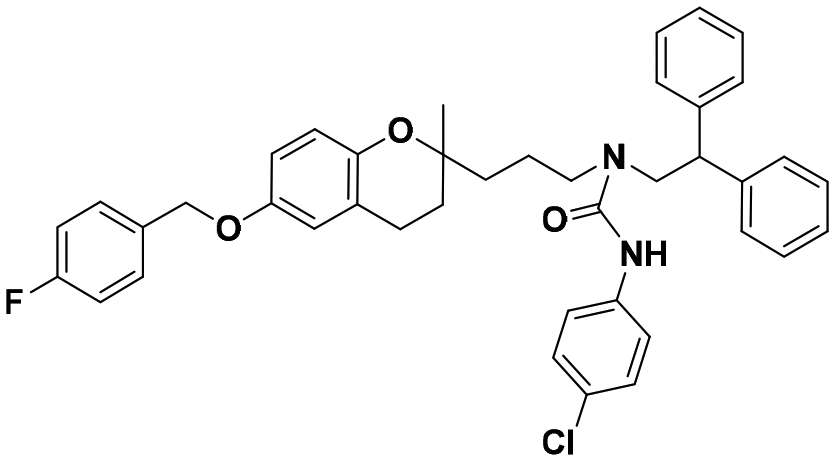
|
>100 | >100 | >100 | — | — |
| 7 |
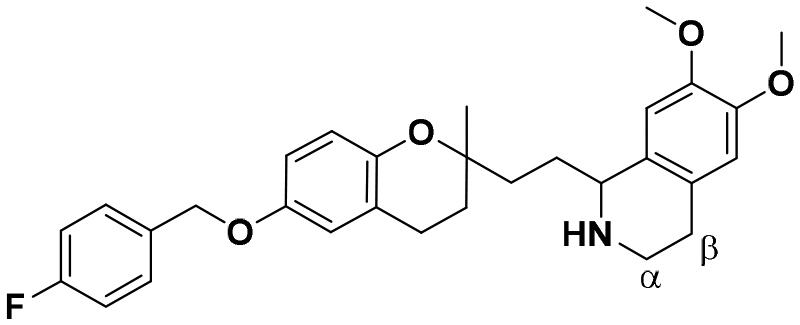
|
22.2 ± 1.8a | 22.0 ± 0.8 | 21.9 ± 3.8 | 0.98 | 0.99 |
| 7a |
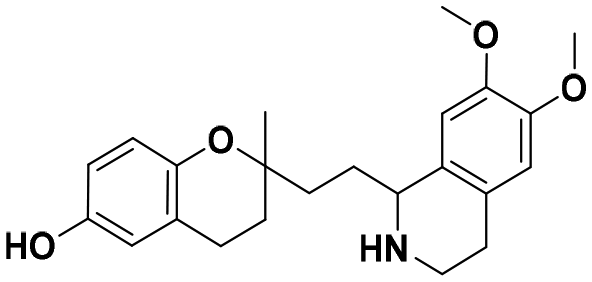
|
58.4 ± 4.0a | 55.2 ± 5.6a | 54.2 ± 3.7b | 0.93 | 0.98 |
| 7b |
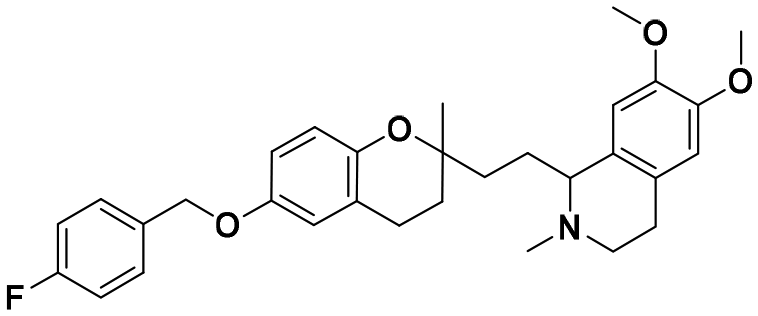
|
14.0 ± 0.8 | 21.8 ± 0.6 | 20.1 ± 1.9 | 1.44 | 0.92 |
| 7c |
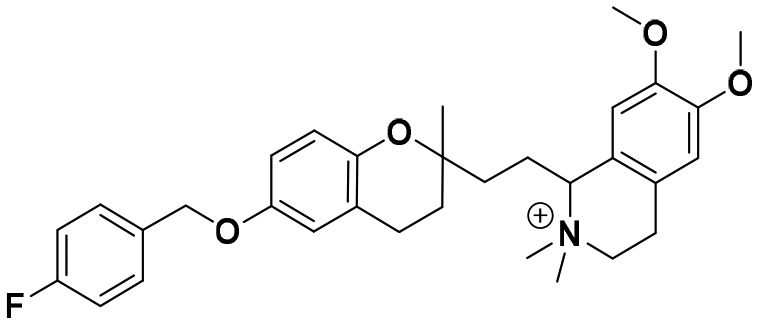
|
42.2 ± 4.9a | 53.5 ± 1.1c | 41.4 ± 2.1b | 0.98 | 0.77 |
| 7d |
|
>100 | >100 | >100 | — | — |
| TMX | 25.9 ± 0.1 | 15.80 ± 0.7 | 26.8 ± 0.5 | 1.03 | 1.70 | |
For the three series, it was observed that the compounds were equally cytotoxic to cancer and normal cells. In the MDA-MB-231 cell line, the cytotoxic activity of the N-methylated compounds 5b, 6b, and 7b (IC50 = 1.5–14.0 μM) was more potent than that of secondary amines 5, 6, and 7 (IC50 = 11.0–22.0 μM), followed by N-dimethylated derivatives as quaternary ammonium salts 5c, 6c, and 7c (IC50 = 19.2–42.2 μM), as well as secondary amines with a free phenolic group 5a, 6a, and 7a (IC50 = 58.4–48.3 μM).
In the MDA-MB-436 cells, the N-methylated compounds 5b, 6b, and 7b (IC50 = 2.0–21.8 μM) were also more potent than secondary amines 5, 6, and 7 (IC50 = 13.0–22.0 μM), followed by N-dimethylated derivatives 5c, 6c, and 7c (IC50 = 24.0–53.5 μM), and secondary amines with a free phenolic group 5a, 6a, and 7a (IC50 = 30.9–55.2 μM).
In the MCF10A cells, N-methylated compounds were more potent than secondary amines, followed by N-dimethylated derivatives, and secondary amines with a free phenolic group. However, urea derivatives (5d, 6d, and 7d) did not produce any cellular cytotoxicity in either cell line.
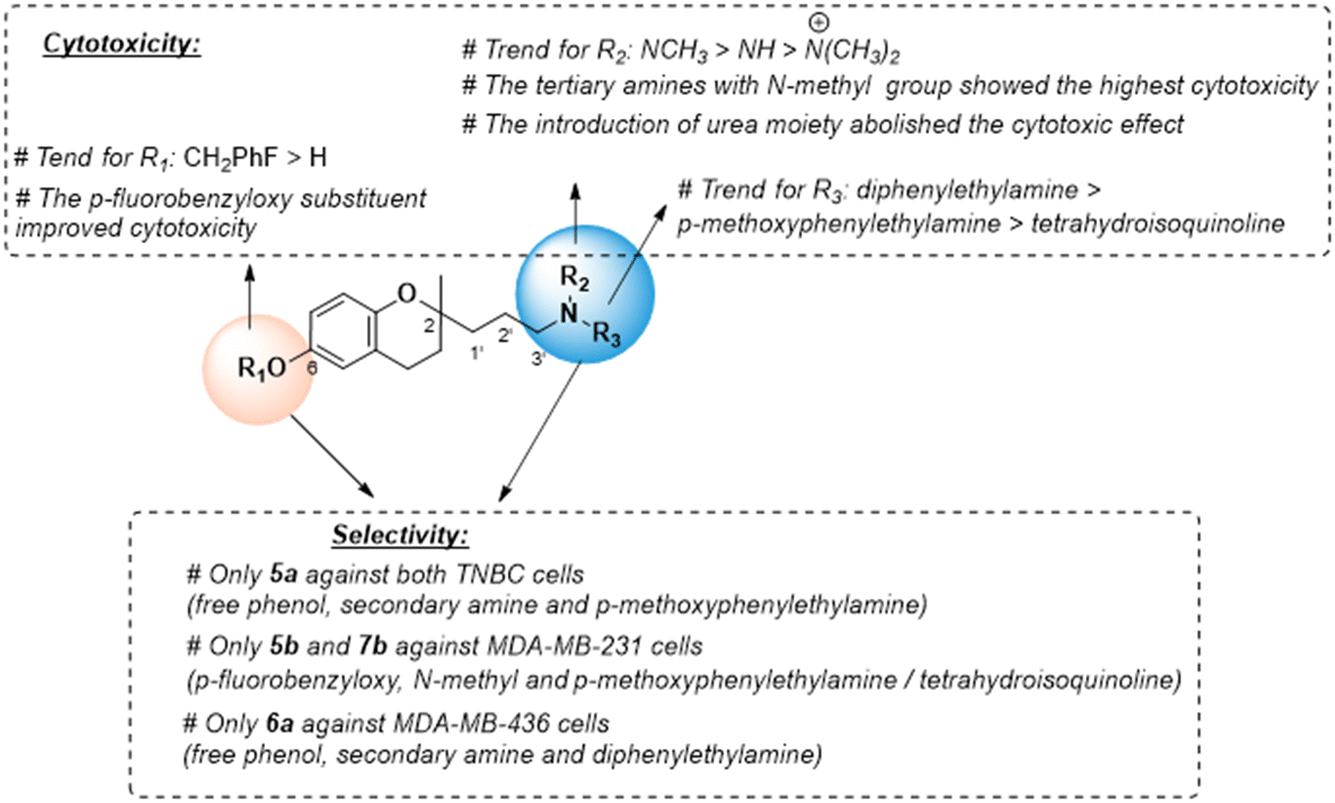
For the three series, the structure–activity relationships (SAR) indicated that the nitrogen atom in the 2-propyl chain and the phenolic group in the benzopyran nucleus play a key role in cytotoxicity (Fig. 2). The tertiary or secondary amine function led to the most potent cytotoxic compounds, especially those bearing the N-(2,2-diphenylethyl) moiety (series 2), while its quaternization giving a ‘permanent cation’ decreased the cytotoxicity, which was most likely due to the difficulty in diffusing across cell membranes.
 | ||
| Fig. 2 SAR study of 2-aminopropyl benzopyran derivatives as potential anti-TNBC agents. | ||
Urea-based derivatives are of growing interest for drug design and development, including anticancer drugs.31 However, the introduction in the 2-aminopropyl benzopyran of the p-chlorophenyl urea moiety increased the steric hindrance, which was detrimental for cytotoxic activity. The presence of the lipophilic p-fluorobenzyloxy substituent in the chromanol ring favored cytotoxic activity, and its removal to a free phenolic group diminished the antitumor effect.
We also examined the selectivity of each compound against non-tumorigenic MCF10A cells. The selectivity index (SI) was calculated as the IC50 ratio of normal cells to cancer cells, SI = IC50 (MCF-10A)/IC50 (MDA-MB-231 or MDA-MBD-436) (Table 1). Four compounds showed slight selectivity against malignant cells. Of those, compound 5a (secondary amine and free phenolic group) bearing the p-methoxyethylamine moiety (series 1) suppressed the growth of both cancer cell lines with higher SI, and 5b (tertiary amine and p-fluorobenzyloxy) displayed selectivity against MDA-MB-231 cells. Similarly, compound 6a (secondary amine and free phenolic group) bearing the diphenylethylamine moiety (series 2) and compound 7b (tertiary amine and p-fluorobenzyloxy) with the isoquinoline moiety (series 3) were selective against MDA-MB-436 or MDA-MB-231 cells, respectively. However, the most cytotoxic compound, 6b, did not exhibit selectivity against cancer cells (Fig. 2). Compounds 5a, 5b, 6a, and 7b with higher SI values were selected to carry out further mechanistic studies.
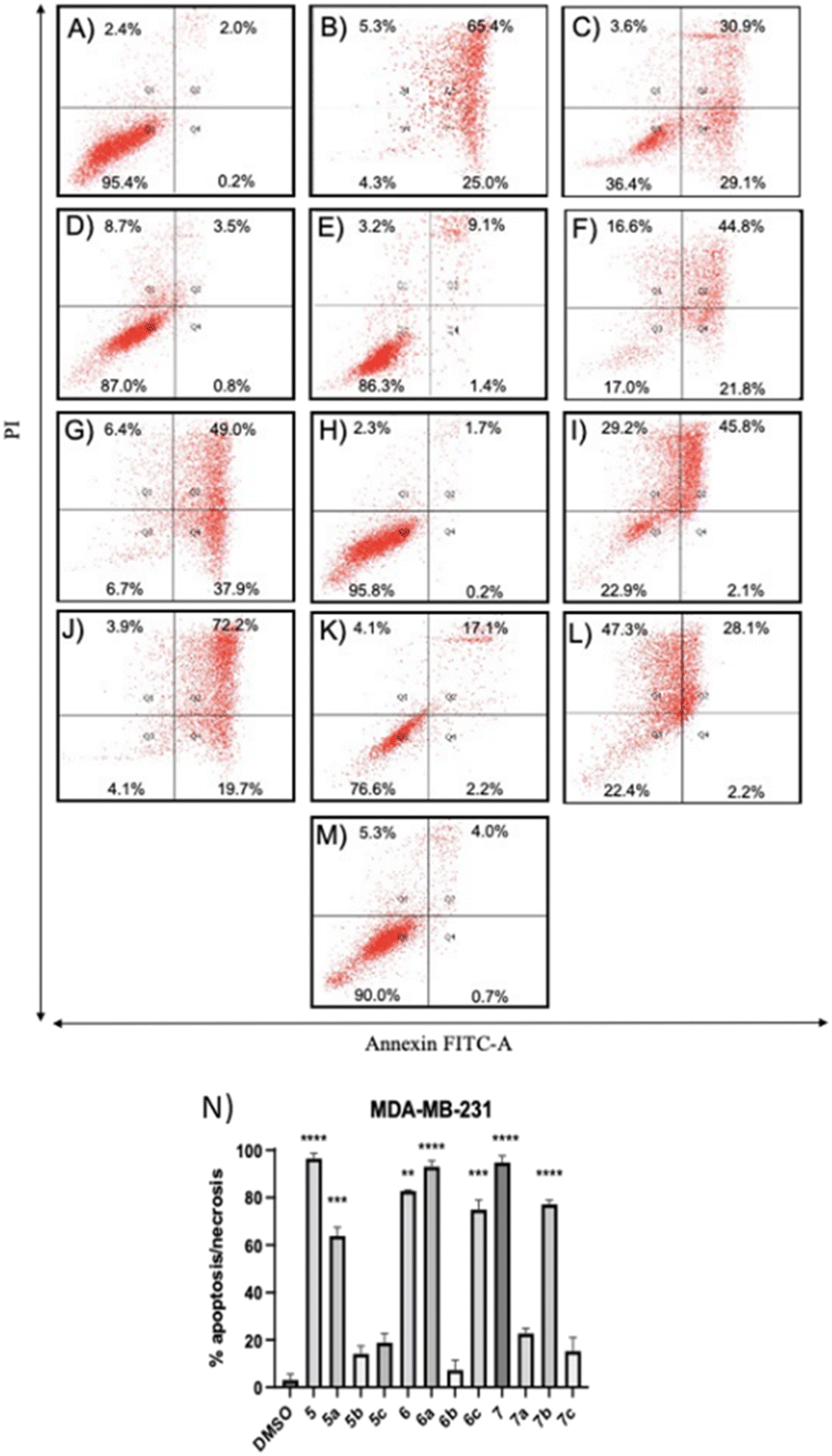 | ||
| Fig. 3 Apoptosis/necrosis analyses of TNBC cells treated with compounds at their respective IC50 values of cytotoxicity after Annexin V-FITC/PI staining. (A–M) Representative flow cytometry dot plots showing the effects of vehicle or compounds on MDA-MB-231 cell apoptosis/necrosis/survival have been included: (A) 0.1% DMSO, (B) 5 at 13.2 μM, (C) 5a at 51.0 μM, (D) 5b at 7.3 μM, (E) 5c at 25.0 μM, (F) 6 at 11.0 μM, (G) 6a at 48.3 μM, (H) 6b at 1.5 μM, (I) 6c at 19.2 μM, (J) 7 at 22.2 μM, (K) 7a at 58.4 μM, (L) 7b at 14.0 μM, (M) 7c at 42.2 μM. (N) Histograms show the percentage of apoptotic and necrotic cell distribution. The data are expressed as the mean ± SD of three independent experiments performed in triplicate. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. DMSO group. | ||
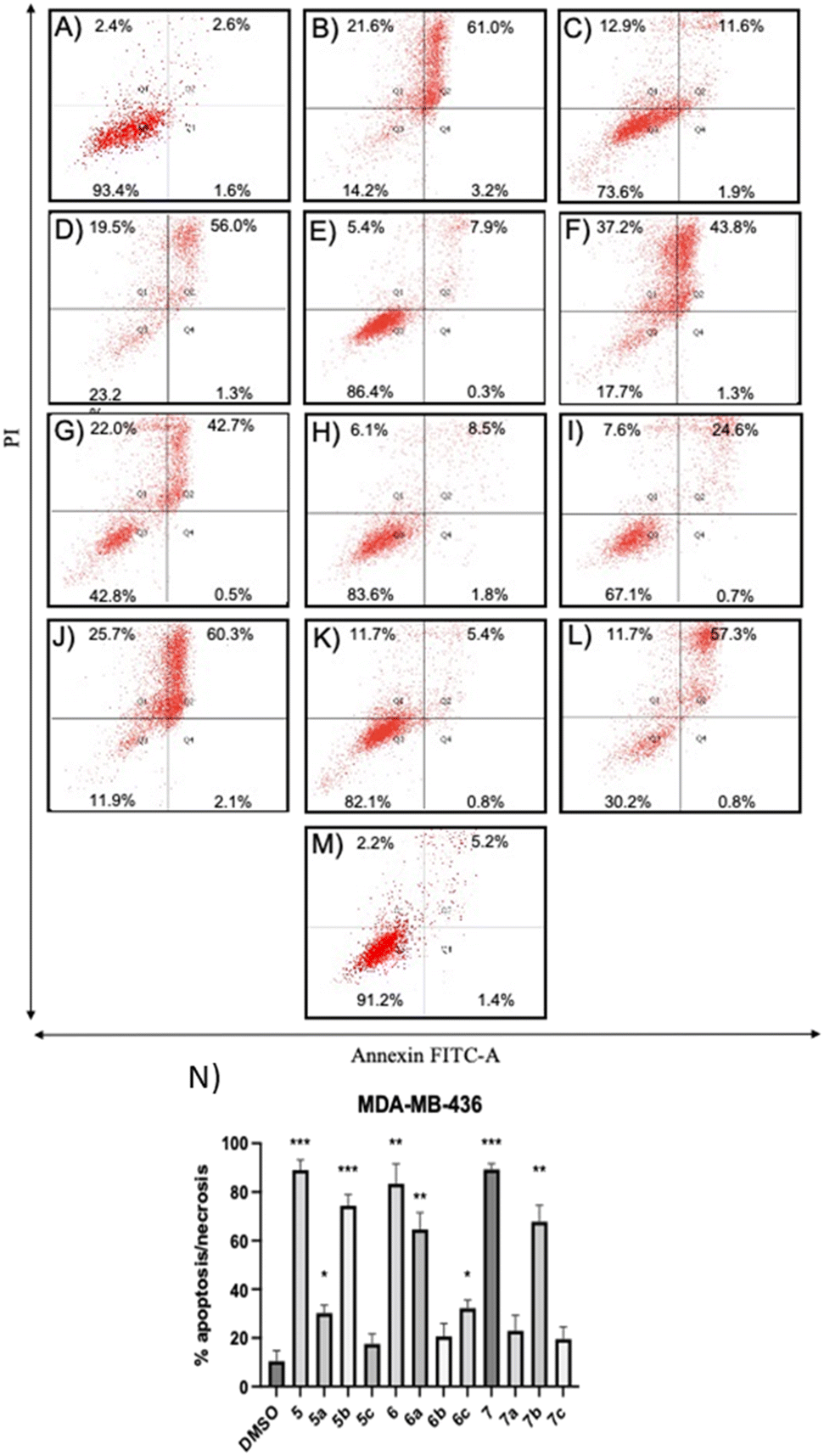 | ||
| Fig. 4 Apoptosis/necrosis analyses of TNBC cells treated with compounds at their respective IC50 values of cytotoxicity after Annexin V-FITC/PI staining. (A–M) Representative flow cytometry dot plots showing the effects of vehicle or compounds on MDA-MB-436 cell apoptosis/necrosis/survival have been included: (A) 0.1% DMSO, (B) 5 at 13.0 μM, (C) 5a at 30.9 μM, (D) 5b at 12.6 μM, (E) 5c at 24.0 μM, (F) 6 at 19.3 μM, (G) 6a at 31.1 μM, (H) 6b at 2.0 μM, (I) 6c at 24.6 μM, (J) 7 at 22.0 μM, (K) 7a at 55.2 μM, (L) 7b at 21.8 μM, and (M) 7c at 53.5 μM. (N) Histograms show the percentage of apoptotic/necrotic cell distribution. The data are expressed as the mean ± SD of three independent experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001 vs. DMSO group. | ||
Compounds 5a and 6c induced a significant percentage of apoptotic and necrotic cells in the MDA-MB-231 line (64% and 75%, respectively), and to a lesser extent in the MDA-MB-436 line (30% and 32%, respectively). Significant apoptosis and necrosis were elicited by compound 5b, but only in the MDA-MB-436 cell line (74%). In addition, ROS are crucial in inducing apoptosis through several pathways. Therefore, the capacity of the apoptotic and necrotic compounds (5, 6, 6a, 7, and 7b) to trigger oxidative stress was further studied in both cancer cell lines, whereas non-apoptotic but cytotoxic compounds (5c, 6b, 7a, and 7c for both cancer cell lines, and 5b for MDA-MB-231 cells) were studied to evaluate their influence on cell cycle distribution.
The positive control, tert-butyl hydroperoxide (TBHP), significantly increased the mean fluorescence level, thus confirming its action mechanism. The results demonstrated that the apoptotic compounds 5, 5a, 6, 6a, 6c, 7, and 7b significantly increased the level of ROS production in both TNBC cell lines (Fig. 5), as compared to control cells. The results suggested that these compounds can induce apoptosis by the generation of ROS in TNBC cells.
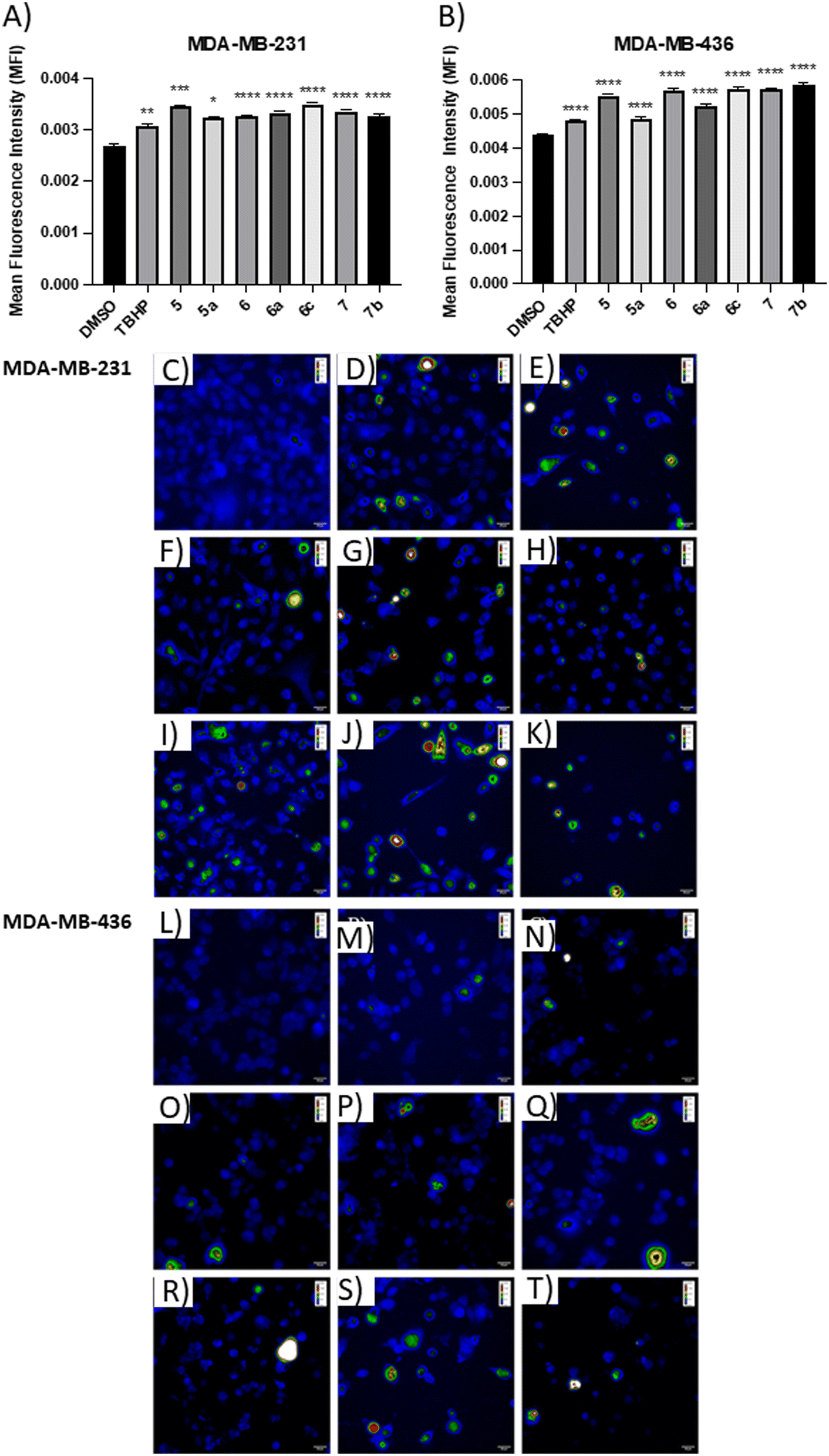 | ||
| Fig. 5 Effect of compounds on intracellular ROS levels. Histograms display the quantification of mean fluorescence intensity (MFI) for vehicle or compounds in (A) MDA-MB-231 cells and (B) MDA-MB-436 cells. Representative confocal fluorescence microscopy images of ROS production in (C–K) MDA-MB-231 cells and (L–T) MDA-MB-436 cells. (C and L) 0.05% DMSO, (D and M) TBHP at 100 μM, (E) 5 at 6.6 μM, (F) 5a at 25.5 μM, (G) 6 at 5.5 μM, (H) 6a at 24.2 μM, (I) 6c at 9.6 μM, (J) 7 at 11.1 μM, (K) 7b at 7.0 μM, (N) 5 at 6.5 μM, (O) 5a at 15.5 μM, (P) 6 at 9.7 μM, (Q) 6a at 15.6 μM, (R) 6c at 12.3 μM, (S) 7 at 11.0 μM, and (T) 7b at 10.9 μM. The data are expressed as the mean ± SEM of three independent experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. DMSO group. | ||
The results showed that all non-apoptotic compounds significantly increased the percentage of the cells in the G1 phase, and lowered the percentage of the cells in the S phase. Regarding the MDA-MB-231 cell line, the percentage of the cells in the G1 phase increased from 49.81% for the control group to 63.02%, 63.73%, 56.12%, 62.18%, and 60.36% for compounds 5b, 5c, 6b, 7a, and 7c, respectively (Fig. 6A and C–H). In the MDA-MB-436 cell line, the percentage of the cells in the G1 phase rose from 41.01% for the control group to 57.87%, 45.81%, 43.89%, and 55.78% for compounds 5c, 6b, 7a, and 7c, respectively (Fig. 6B and I–M). The percentage of the cells in the S phase in the MDA-MB-231 cell line decreased from 42.68% for the control group to 24.84%, 18.38%, 29.33%, 22.81%, and 23.05% for compounds 5b, 5c, 6b, 7a, and 7c, respectively (Fig. 6A and C–H), and in MDA-MB-436 cells, decreased from 40.08% for the control group to 24.25%, 34.56%, 36.58%, and 30.13% for compounds 5c, 6b, 7a, and 7c, respectively (Fig. 6B and I–M). These results suggest that non-apoptotic 2-aminopropyl benzopyrans 5b, 5c, 6b, 7a, and 7c caused cell cycle arrest at the G1 phase.
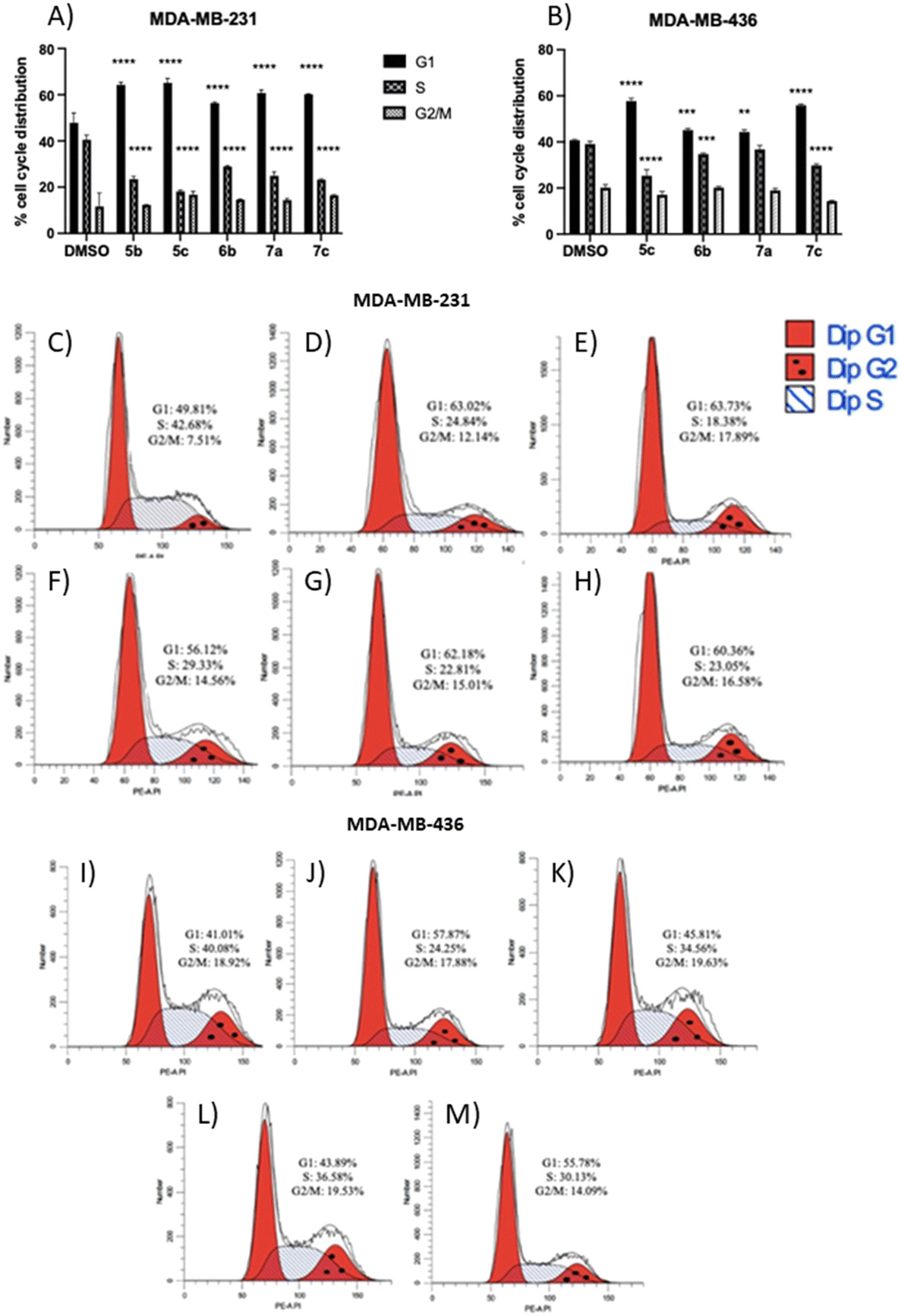 | ||
| Fig. 6 Effect of non-apoptotic/necrotic compounds on cell cycle progression. The analysis of cell cycle distribution was performed by flow cytometry using PI staining. Histograms display the percentage of cell cycle distribution for (A) MDA-MB-231 cells and (B) MDA-MB-436 cells. Representative flow cytometry plots of cell cycle progression experiments with vehicle or non-apoptotic compounds for the (C–H) MDA-MB-231 cell line and (I–M) MDA-MB-436 cell line. (C and I) 0.1% DMSO, (D) 5b at 7.3 μM, (E) 5c at 25.0 μM, (F) 6b at 1.5 μM, (G) 7a at 58.4 μM, (H) 7c at 42.2 μM, (J) 5c at 24.0 μM, (K) 6b at 2.0 μM, (L) 7a at 55.2 μM, and (M) 7c at 53.5 μM. The data are expressed as the mean ± SD of three independent experiments performed in triplicate. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. DMSO group. | ||
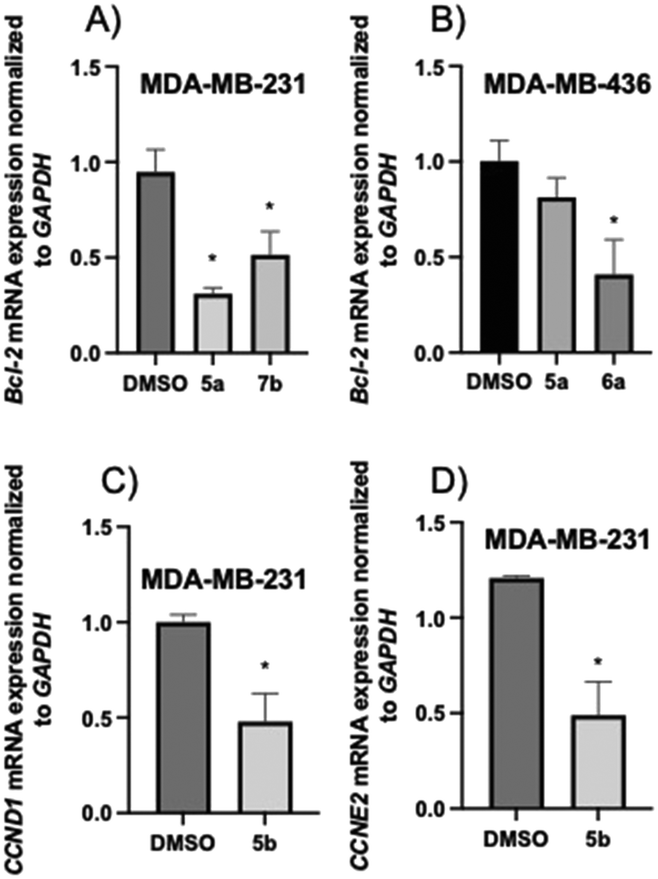 | ||
| Fig. 7 Gene expression in TNBC cells treated with 2-aminopropyl benzopyrans 5a, 5b, 6a, and 7b at their respective IC25 values: (A) 5a at 25.5 μM and 7b at 7.0 μM, (B) 5a at 15.5 μM and 6a at 15.6 μM, and (C and D) 5b at 3.7 μM. The data are expressed as the mean ± SD of three independent experiments performed in triplicate. *P < 0.05 vs. DMSO group. | ||
3. Conclusions
Three series of 2-aminopropyl benzopyran derivatives were synthesised (Scheme 1) and evaluated for their antitumor activity against the MDA-MB-231 and MDA-MB-436 TNBC cell lines. The SAR studies showed that the presence of an amino group (secondary, tertiary, or quaternary) in the structure seems to be essential for cytotoxicity in both TNBC cell lines, and its modification to a urea function renders the derivatives devoid of an anti-tumor effect. For the three series, the cytotoxic activity was as follows: N-methylated derivatives (tertiary amines) 5b, 6b, and 7b > secondary amines 5, 6, and 7 > quaternary amine salts 5c, 6c, and 7c > free phenolic derivatives 5a, 6a, and 7a. In addition, 5a, 5b, 6a, and 7b displayed slight selectivity for tumor cells.An apoptotic and necrotic effect was observed for benzopyrans 5, 5a, 5b, 6, 6a, 6c, 7, and 7b due to, at least in part, an increase in ROS generation, while 5b, 5c, 6b, 7a, and 7c caused cell cycle arrest in the G1 phase. Further cell-based mechanistic studies revealed that the most promising compounds, 5a, 6a, and 7b, downregulated the expression of Bcl-2, and 5b downregulated the expression of cyclins CCND1 and CCND2. Therefore, 2-aminopropyl benzopyrans emerge as new hits and can be used for designing new potential lead compounds in the development of useful agents against TNBC.
4. Experimental section
4.1. Chemistry
Electrospray ionization high-resolution mass spectrometry (ESI-HRMS) was performed using a TripleTOF™ 5600 liquid chromatography-tandem mass spectrometry (LC/MS/MS) System (AB SCIEX) (Toronto, Canada). LC-MS detection was performed using an ultra-high performance liquid chromatography (UHPLC) apparatus (Shimadzu, LCMS-8040) coupled to a tandem mass spectrometry (MS/MS) triple quadrupole equipped with an ESI ion source (Shimadzu, Kyoto, Japan).1H NMR and 13C NMR spectra were recorded on a Bruker AC-300 (Bruker Instruments, Kennewick, WA). The assignments in 1H and 13C NMR were made by correlation spectroscopy (COSY) 45, heteronuclear single quantum correlation (HSQC), and heteronuclear multiple bond correlation (HMBC) recorded at 300 MHz. Chemical shifts (δ) are reported in ppm relative to an internal deuterated solvent reference, with multiplicities indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), or dd (double doublet). All reactions were monitored by analytical thin-layer chromatography (TLC) with silica gel 60 F254 (Merck 5554). Residues were purified by silica gel column chromatography (40–63 μm, Merck Group).
Solvents and reagents were purchased from the commercial sources Scharlab S.L. (Barcelona, Spain) and Sigma-Aldrich (St. Louis, MO, USA), respectively, and used without further purification unless otherwise noted. Dry and freshly distilled solvents were used in those reactions performed under N2. Quoted yields are of purified material. Final compounds were purified to ≥95% as assessed by 1H NMR and LC-MS/MS analysis.
:10) to yield the O-protected benzopyran ester (2) (1.7 g, 79%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.41–7.36 (m, 2H, CH-2′′, CH-6′′), 7.09–7.03 (m, 2H, CH-3′′, CH-5′′), 6.72–6.67 (m, 3H, CH-5, CH-7, CH-8), 4.94 (s, 2H, OCH2Ph-p-F), 4.13 (q, J = 7.1 Hz, 2H, CO2CH2CH3), 2.76 (t, J = 6.7 Hz, 2H, CH2-4), 2.48 (t, J = 7.7 Hz, 2H, CH2-2′), 2.0–1.75 (m, 4H, CH2-3, CH2-1′), 1.24 (t, J = 7.2 Hz, 3H, CO2CH2CH3), 1.23 (s, 3H, CH3-2); 13C NMR (75 MHz, CDCl3) δ 173.6 (CO2CH2CH3), 162.3 (d, JCF = 244 Hz, C-4′′), 152.0 (C-6), 147.9 (C-8a), 133.1 (d, JCF = 3 Hz, C-1′′), 129.1 (d, JCF = 8 Hz, CH-2′′, CH-6′′), 121.4 (C-4a), 117.7 (CH-5), 115.2 (d, JCF = 25 Hz, CH-3′′, CH-5′′), 115.1 (CH-7), 114.4 (CH-8), 74.6 (C-2), 69.9 (OCH2Ph-p-F), 60.3 (CO2CH2CH3), 34.3 (CH2-1′), 31.0 (CH2-3), 28.7 (CH2-2′), 23.6 (CH3-2), 22.2 (CH2-4), 14.1 (CO2CH2CH3); HREIMS m/z calculated for C22H25O4F [M]+ 372.1737, found: 372.1730.
:10) to afford 492 mg of the corresponding aldehyde 3 (406 mg, 92%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 9.78 (t, J = 1.6 Hz, 1H, CHO), 7.39–7.37 (m, 2H, H-2′′, H-6′′), 7.08–7.04 (m, 2H, H-3′′, H-5′′), 6.74–6.67 (m, 3H, H-5, H-7, H-8), 4.94 (s, 2H, OCH2Ph-p-F), 2.77–2.74 (m, 2H, CH2-4), 2.62–2.59 (m, 2H, CH2-2′), 2.03–1.75 (m, 4H, CH2-3, CH2-1′), 1.26 (s, 3H, CH3-2); 13C NMR (75 MHz, CDCl3) δ 202.2 (CHO), 162.4 (d, JCF = 244 Hz, C-4′′), 152.1 (C-6), 147.7 (C-8a), 133.1 (d, JCF = 3 Hz, C-1′′), 129.3 (d, JCF = 8 Hz, C-2′′, C-6′′), 121.4 (C-4a), 117.7 (CH-5), 115.2 (d, JCF = 25 Hz, CH-3′′, CH-5′′), 115.2 (CH-7), 114.5 (CH-8), 74.6 (C-2), 69.9 (OCH2Ph-p-F), 38.4 (CH2-2′), 31.8 (CH2-1′), 31.2 (CH2-3), 23.7 (CH3-2), 22.3 (CH2-4); HRMS (ESI) m/z calculated for C20H21O3F [M + H]+ 329.1547, found: 329.1552.
:40) to afford compound 4 (976 mg, 63% yield) as a white oil.1H NMR (300 MHz, CDCl3) δ 7.41–7.33 (m, 2H, CH-2′′, CH-6′′), 7.08–7.02 (m, 2H, CH-3′′, CH-5′′), 6.80–6.62 (m, 6H, CH-5, CH-7, CH-8, CH-2′′′, CH-5′′′, CH-6′′′), 5.69 (s, NH), 4.92 (s, 2H, OCH2Ph-p-F), 3.85 (s, 6H, 2xOCH3), 3.48–3.45 (m, 2H, CH2-α), 2.81–2.67 (m, 4H, CH2-4, CH2-β), 2.32–2.27 (CH2-2′), 2.03–1.83 (m, 2H, CH2-1′), 1.80–1.67 (m, 2H, CH2-3), 1.25 (s, 3H, CH3-2). 13C NMR (75 MHz, CDCl3) δ 172.9 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 162.0 (d, JCF = 244 Hz, C-4′′), 158.3 (C-3′′′, C-4′′′), 152.1 (C-6), 147.9 (C-8a), 133.2 (d, JCF = 3 Hz, C-1′′), 130.9 (C-1′′′), 129.7 (CH-6′′′), 129.3 (d, JCF = 8 Hz, CH-2′′, CH-6′′), 121.6 (C-4a), 117.5 (CH-5), 115.4 (d, JCF = 21 Hz, CH-3′′, CH-5′′), 115.2 (CH-7), 114.5 (CH-8), 114.1 (CH-2′′′, CH-5′′′), 75.0 (C-2), 70.0 (OCH2Ph-p-F), 55.3 (2xOCH3), 40.8 (CH2-α), 35.2 (CH2-1′), 34.8 (CH2-β), 31.2 (CH2-3), 30.8 (CH2-2′), 23.7 (CH3-2), 22.3 (CH2-4). HRMS (ESI) m/z calculated for C30H35FNO5 [M + H]+ 508.2494, found: 508.2481.
:2) to afford compound 5 (244 mg, 72% yield) or 6 (201 mg, 54% yield) as colorless oils.
:2) to afford compound 7 (443.8 mg, 53% yield) as a colorless oil.
:EtOH in a 1:1 ratio, and refluxed under N2 for 3 hours. Reaction mixtures were stirred for an additional 30 minutes at room temperature, and ammonia 15% was added for alkalinization. Mixtures were concentrated in vacuo, and then, water was added, and extraction proceeded with EtOAc (3 × 15 mL). The combined organic layers were dried over anhydrous Na2SO4 and evaporated to dryness. The residue was purified by C18 column chromatography to afford 36.1 mg, 42.5 mg, and 40.6 mg of the corresponding compound 5a, 6a, and 7a (92–96%) as colorless oils.
:2) to afford 40 mg, 46 mg, and 47.9 mg of 5b, 6b, and 7b, respectively, (76–86% yield) as colorless oils.
:5) to afford 42.8 mg, 47.4 mg, and 55 mg of the respective compounds 5c, 6c, and 7c (79–96% yield) as brown oils.
:2, 9:1, and 6:4 respectively) to yield 46 mg, 67 mg, and 65 mg of compound 5d, 6d, and 7d (68–92% yield), respectively, as colorless oils.
O), 152.0 (C-6), 147.6 (C-8a), 137.5 (C-7′′′), 132.9 (d, J = 3 Hz, C-1′′), 130.9 (C-10′′′), 129.7 (C-1′′′), 129.2 (d, JCF = 8 Hz, CH-2′′, CH-6′′), 128.6 (CH-2′′′, CH-6′′′), 121.7 (C-4a), 121.3 (CH-9′′′, CH-11′′′), 120.0 (CH-8′′′, CH-12′′′), 117.4 (CH-5), 115.2 (CH-7), 115.2 (d, JCF = 21 Hz, CH-3′′, CH-5′′), 114.4 (CH-8), 114.1 (CH-3′′′, CH-5′′′), 75.8 (C-2), 69.9 (OCH2Ph-p-F), 55.1 (OCH3), 49.7 (CH2-α), 47.7 (CH2-3′), 36.0 (CH2-1′), 33.8 (CH2-β), 31.0 (CH2-3), 23.6 (CH3-2), 22.2 (CH2-2′), 22.1 (CH2-4). HRMS (ESI) m/z calculated for C36H38ClFN2O4 [M]+ 617.2575, found: 617.2576.
O), 152.2 (C-6), 147.8 (C-8a), 142.0 (d, J = 2 Hz, C–Ph), 137.8 (C-1′′′), 133.1 (d, JCF = 3 Hz, C-1′′), 129.8 (C-4′′′), 129.4 (d, JCF = 8 Hz, CH-2′′, CH-6′′), 128.6 and 127.5 (CH–Ph), 127.1 (CH-9′′′, CH-11′′′), 121.7 (C-4a), 121.0 (CH-8′′′, CH-12′′′), 117.7 (CH-5), 115.6 (d, JCF = 19 Hz, CH-3′′, CH-5′′), 115.3 (CH-7), 114.5 (CH-8), 75.9 (C-2), 70.0 (OCH2Ph-p-F), 53.8 (CH2-α), 49.9 (CH-β), 48.2 (CH2-3′), 36.3 (CH2-1′), 31.2 (CH2-3), 24.0 (CH3-2), 22.4 (CH2-2′), 22.1 (CH2-4). HRMS (ESI) m/z calculated for C41H40ClFN2O3 [M]+ 663.2784, found: 663.2764.
O), 152.3 (C-6), 148.1 (C-8a), 147.5 (C-3′′′, C-4′′′), 138.0 (C-7′′′), 133.2 (C-1′′), 131.2 (C-10′′′), 131.0 (CH-2′′, CH-6′′), 127.1 (C-1′′′, C-6′′′), 121.6 (CH-9′′′, CH-11′′′), 120.3 (CH-8′′′, CH-12′′′), 117.7 (CH-5), 115.5 (CH-3′′, CH-5′′), 115.2 (CH-7), 114.4 (CH-8), 111.9 and 109.2 (CH-2′′′, CH-5′′′), 75.6 (C-2), 70.0 (OCH2Ph-p-F), 57.0 (CH-3′), 55.8 and 55.7 (OCH3), 32.1 (CH2-1′), 31.4 (CH2-3), 30.4 (CH2-2′), 28.1 (CH2-α), 24.0 (CH3-2), 23.1 (CH2-4, CH2-β). HRMS (ESI) m/z calculated for C37H38ClFN2O2 [M]+ 645.2526, found: 645.2512.
O), 162.0 (d, JCF = 244 Hz, C-4′′), 158.3 (C-3′′′, C-4′′′), 152.1 (C-6), 147.9 (C-8a), 133.2 (d, JCF = 3 Hz, C-1′′), 130.9 (C-1′′′), 129.7 (CH-6′′′), 129.3 (d, JCF = 8 Hz, CH-2′′, CH-6′′), 121.6 (C-4a), 117.5 (CH-5), 115.4 (d, JCF = 21 Hz, CH-3′′, CH-5′′), 115.2 (CH-7), 114.5 (CH-8), 114.1 (CH-2′′′, CH-5′′′), 75.0 (C-2), 70.0 (OCH2Ph-p-F), 55.3 (2xOCH3), 40.8 (CH2-α), 35.2 (CH2-1′), 34.8 (CH2-β), 31.2 (CH2-3), 30.8 (CH2-2′), 23.7 (CH3-2), 22.3 (CH2-4). HRMS (ESI) m/z calculated for C30H35FNO5 [M + H]+ 508.2494, found: 508.2481.
:2) to afford compound 5 (244 mg, 72% yield) or 6 (201 mg, 54% yield) as colorless oils.
:2) to afford compound 7 (443.8 mg, 53% yield) as a colorless oil.
:EtOH in a 1:1 ratio, and refluxed under N2 for 3 hours. Reaction mixtures were stirred for an additional 30 minutes at room temperature, and ammonia 15% was added for alkalinization. Mixtures were concentrated in vacuo, and then, water was added, and extraction proceeded with EtOAc (3 × 15 mL). The combined organic layers were dried over anhydrous Na2SO4 and evaporated to dryness. The residue was purified by C18 column chromatography to afford 36.1 mg, 42.5 mg, and 40.6 mg of the corresponding compound 5a, 6a, and 7a (92–96%) as colorless oils.
:2) to afford 40 mg, 46 mg, and 47.9 mg of 5b, 6b, and 7b, respectively, (76–86% yield) as colorless oils.
:5) to afford 42.8 mg, 47.4 mg, and 55 mg of the respective compounds 5c, 6c, and 7c (79–96% yield) as brown oils.
:2, 9:1, and 6:4 respectively) to yield 46 mg, 67 mg, and 65 mg of compound 5d, 6d, and 7d (68–92% yield), respectively, as colorless oils.
O), 152.0 (C-6), 147.6 (C-8a), 137.5 (C-7′′′), 132.9 (d, J = 3 Hz, C-1′′), 130.9 (C-10′′′), 129.7 (C-1′′′), 129.2 (d, JCF = 8 Hz, CH-2′′, CH-6′′), 128.6 (CH-2′′′, CH-6′′′), 121.7 (C-4a), 121.3 (CH-9′′′, CH-11′′′), 120.0 (CH-8′′′, CH-12′′′), 117.4 (CH-5), 115.2 (CH-7), 115.2 (d, JCF = 21 Hz, CH-3′′, CH-5′′), 114.4 (CH-8), 114.1 (CH-3′′′, CH-5′′′), 75.8 (C-2), 69.9 (OCH2Ph-p-F), 55.1 (OCH3), 49.7 (CH2-α), 47.7 (CH2-3′), 36.0 (CH2-1′), 33.8 (CH2-β), 31.0 (CH2-3), 23.6 (CH3-2), 22.2 (CH2-2′), 22.1 (CH2-4). HRMS (ESI) m/z calculated for C36H38ClFN2O4 [M]+ 617.2575, found: 617.2576.
O), 152.2 (C-6), 147.8 (C-8a), 142.0 (d, J = 2 Hz, C–Ph), 137.8 (C-1′′′), 133.1 (d, JCF = 3 Hz, C-1′′), 129.8 (C-4′′′), 129.4 (d, JCF = 8 Hz, CH-2′′, CH-6′′), 128.6 and 127.5 (CH–Ph), 127.1 (CH-9′′′, CH-11′′′), 121.7 (C-4a), 121.0 (CH-8′′′, CH-12′′′), 117.7 (CH-5), 115.6 (d, JCF = 19 Hz, CH-3′′, CH-5′′), 115.3 (CH-7), 114.5 (CH-8), 75.9 (C-2), 70.0 (OCH2Ph-p-F), 53.8 (CH2-α), 49.9 (CH-β), 48.2 (CH2-3′), 36.3 (CH2-1′), 31.2 (CH2-3), 24.0 (CH3-2), 22.4 (CH2-2′), 22.1 (CH2-4). HRMS (ESI) m/z calculated for C41H40ClFN2O3 [M]+ 663.2784, found: 663.2764.
O), 152.3 (C-6), 148.1 (C-8a), 147.5 (C-3′′′, C-4′′′), 138.0 (C-7′′′), 133.2 (C-1′′), 131.2 (C-10′′′), 131.0 (CH-2′′, CH-6′′), 127.1 (C-1′′′, C-6′′′), 121.6 (CH-9′′′, CH-11′′′), 120.3 (CH-8′′′, CH-12′′′), 117.7 (CH-5), 115.5 (CH-3′′, CH-5′′), 115.2 (CH-7), 114.4 (CH-8), 111.9 and 109.2 (CH-2′′′, CH-5′′′), 75.6 (C-2), 70.0 (OCH2Ph-p-F), 57.0 (CH-3′), 55.8 and 55.7 (OCH3), 32.1 (CH2-1′), 31.4 (CH2-3), 30.4 (CH2-2′), 28.1 (CH2-α), 24.0 (CH3-2), 23.1 (CH2-4, CH2-β). HRMS (ESI) m/z calculated for C37H38ClFN2O2 [M]+ 645.2526, found: 645.2512.
4.2. Pharmacology
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Carlos III Health Institute (ISCIII) and the European Regional Development Fund (FEDER) by grant numbers PI21/02045 and PI21/01351, and the Generalitat Valenciana (GVA) by grant numbers AICO/2021/081 and APOTIP/2020/011. Nuria Cabedo was funded by the ISCIII Miguel Servet programme (CPII20/00010), co-funded by the European Social Fund. Carlos Villarroel-Vicente was funded by the ISCIII (PFIS – FI19/00153), Ainhoa García by the GVA (AICO/2021/081), and Sandra Torres-Ruiz by a pre-doctoral grant (ACIF/2019/119).References
- H. Şenol, P. Tulay, M. Ç. Ergören, A. Hanoğlu, İ. Çalış and G. Mocan, Cytotoxic effects of verbascoside on MCF-7 and MDA-MB-231, Turk. J. Pharm. Sci., 2020, 18, 637–644, DOI:10.4274/tjps.galenos.2021.36599.
- K. R. Senwar, T. S. Reddy, D. Thummuri, P. Sharma, V. G. Naidu, G. Srinivasulu and N. Shankaraiah, Design, synthesis and apoptosis inducing effect of novel (Z)-3-(3′-methoxy-4′-(2-amino-2-oxoethoxy)-benzylidene)indolin-2-ones as potential antitumor agents, Eur. J. Med. Chem., 2026, 118, 34–36, DOI:10.1016/j.ejmech.2016.04.025.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2015, Ca-Cancer J. Clin., 2015, 65, 5–29, DOI:10.3322/caac.21254.
- I. Ahmad and W. Shagufta, Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer, Eur. J. Med. Chem., 2015, 102, 375–386, DOI:10.1016/j.ejmech.2015.08.010.
- A. G. Waks and E. P. Winer, Breast cancer treatment: a review, JAMA, J. Am. Med. Assoc., 2019, 321, 288–300, DOI:10.1001/jama.2018.19323.
- O. A. Bamodu, W. C. Huang, D. T. Tzeng, A. Wu, L. S. Wang, C. T. Yeh and T. Y. Chao, Ovatodiolide sensitizes aggressive breast cancer cells to doxorubicin, eliminates their cancer stem cell-like phenotype, and reduces doxorubicin-associated toxicity, Cancer Lett., 2015, 364, 125–134, DOI:10.1016/j.canlet.2015.05.006.
- M. Gallorini, A. Cataldi and A. di Giacomo, Cyclin-dependent kinase modulators and cancer therapy, BioDrugs, 2012, 26, 377–391, DOI:10.1007/BF03261895.
- D. Nandini, A. Jennifer and D. Pradip, Therapeutic strategies for metastatic triple-negative breast cancers: from negative to positive, Pharmaceuticals, 2021, 14, 455–472, DOI:10.3390/ph14050455.
- M. Brown, A. Tsodikov, K. R. Bauer, C. A. Parise and V. Caggiano, The role of human epidermal growth factor receptor 2 in the survival of women with estrogen and progesterone receptor-negative, invasive breast cancer, The California Cancer Registry, 1999-2004, Cancer, 2008, 112, 737–747, DOI:10.1002/cncr.23243.
- M. Liao, J. Zhang, G. Wang, L. Wang, J. Liu, L. Ouyang and B. Liu, Small-molecule drug discovery in triple negative breast cancer: current situation and future directions, J. Med. Chem., 2021, 64, 2382–2418, DOI:10.1021/acs.jmedchem.0c01180.
- O. Gluz, C. Liedtke, N. Gottschalk, L. Pusztail, U. Nitz and N. Harbeck, Triple-negative breast cancer – current status and future directions, Ann. Oncol., 2009, 20, 1913–1927, DOI:10.1093/annonc/mdp492.
- Y. Li, S. Li, X. Meng, R. Y. Gan, J. J. Zhang and H. B. Li, Dietary natural products for prevention and treatment of breast cancer, Nutrients, 2017, 9, 728, DOI:10.3390/nu9070728.
- Q. Zhao, M. Zhao, A. B. Parris, Y. Xing and X. Yang, Genistein targets the cancerous inhibitor of PP2A to induce growth inhibition and apoptosis in breast cancer cells, Int. J. Oncol., 2016, 49, 1203–1210, DOI:10.3892/ijo.2016.3588.
- J. Chen, Y. Duan, X. Zhang, Y. Ye, B. Ge and J. Chen, Genistein induces apoptosis by the inactivation of the IGF-1R/p-Akt signaling pathway in MCF-7 human breast cancer cells, Food Funct., 2015, 6, 995–1000, 10.1039/c4fo01141d.
- S. S. Bhat, S. K. Prasad, C. Shivamallu, K. S. Prasad, A. Syed, P. Reddy, C. A. Cull and R. G. Amachawadi, Genistein: a potent anti-breast cancer agent, Curr. Issues Mol. Biol., 2021, 43, 1502–1517, DOI:10.3390/cimb43030106.
- Y. Safdari, M. Khalili, M. A. Ebrahimzadeh, Y. Yazdani and S. Farajnia, Natural inhibitors of PI3K/AKT signaling in breast cancer: emphasis on newly-discovered molecular mechanisms of action, Pharmacol. Res., 2015, 93, 1–10, DOI:10.1016/j.phrs.2014.12.004.
- A. A. Abd El-Hafeez, H. O. Khalifa, E. A. M. Mahdy, V. Sharma, T. Hosoi, P. Ghosh, K. Ozawa, M. M. Montano, T. Fujimura, A. R. N. Ibrahim, M. A. A. Abdelhamid, S. P. Pack, S. A. Shouman and S. Kawamoto, Anticancer effect of nor-wogonin (5, 7, 8-trihydroxyflavone) on human triple-negative breast cancer cells via downregulation of TAK1, NF-κB, and STAT3, Pharmacol. Rep., 2019, 71, 289–298, DOI:10.1016/j.pharep.2019.01.001.
- S. Sordon, J. Popłoński, M. Milczarek, M. Stachowicz, T. Tronina, A. Z. Kucharska, J. Wietrzyk and E. Huszcza, Structure-antioxidant-antiproliferative activity relationships of natural C7 and C7-C8 hydroxylated flavones and flavanones, Antioxidants, 2019, 8, 210, DOI:10.3390/antiox8070210.
- K. M. Yap, M. Sekar, Y. S. Wu, S. H. Gan, N. N. I. M. Rani, L. J. Seow, V. Subramaniyan, N. K. Fuloria, S. Fuloria and P. T. Lum, Hesperidin and its aglycone hesperetin in breast cancer therapy: A review of recent developments and future prospects, Saudi J. Biol. Sci., 2021, 28, 6730–6747, DOI:10.1016/j.sjbs.2021.07.046.
- S. Rani, K. Raheja, V. Luxami and K. Paul, A review on diverse heterocyclic compounds as the privileged scaffolds in non-steroidal aromatase inhibitors, Bioorg. Chem., 2021, 113, 105017, DOI:10.1016/j.bioorg.2021.105017.
- C. Zhang, Z. Wang, Y. Shi, B. Yu and Y. Song, Recent advances of LSD1/KDM1A inhibitors for disease therapy, Bioorg. Chem., 2023, 134, 106443, DOI:10.1016/j.bioorg.2023.106443.
- M. C. Gonzalez, A. Serrano, M. C. Zafra-Polo, D. Cortes and K. S. Rao, Polycerasoidin and polycerasoidol, two new prenylated benzopyran derivatives from Polyalthia cerasoides, J. Nat. Prod., 1995, 58, 1278–1284, DOI:10.1021/np50122a022.
- M. C. Zafra-Polo, M. C. González, J. R. Tormo, E. Estornell and D. Cortes, Polyalthidin: new prenylated benzopyran inhibitor of the mammalian mitochondrial respiratory chain, J. Nat. Prod., 1996, 59, 913–916, DOI:10.1021/np960492m.
- A. Bermejo, A. Collado, I. Barrachina, P. Marqués, N. El Aouad, X. Franck, F. Garibotto, C. Dacquet, D. H. Caignard, F. D. Suvire, R. D. Enriz, L. Piqueras, B. Figadère, M. J. Sanz, N. Cabedo and D. Cortes, Polycerasoidol, a natural prenylated benzopyran with a dual PPARα/PPARγ agonist activity and anti-inflammatory effect, J. Nat. Prod., 2019, 82, 1802–1812, DOI:10.1021/acs.jnatprod.9b00003.
- P. Marques, C. Villarroel-Vicente, A. Collado, A. García, L. Vila, I. Duplan, N. Hennuyer, F. Garibotto, R. D. Enriz, C. Dacquet, B. Staels, L. Piqueras, D. Cortes, M. J. Sanz and N. Cabedo, Anti-inflammatory effects and improved metabolic derangements in ob/ob mice by a newly synthesized prenylated benzopyran with pan-PPAR activity, Pharmacol. Res., 2023, 187, 106638, DOI:10.1016/j.phrs.2022.106638.
- H. Taha, C. Y. Looi, A. Arya, W. F. Wong, L. F. Yap, M. Hasanpourghadi, M. A. Mohd, I. C. Paterson and H. Mohd Ali, (6E,10E) Isopolycerasoidol and (6E,10E) isopolycerasoidol methyl ester, prenylated benzopyran derivatives from pseuduvaria monticola induce mitochondrial-mediated apoptosis in human breast adenocarcinoma cells, PLoS One, 2015, 10, e0126126, DOI:10.1371/journal.pone.0126126.
- B. C. Pearce, R. A. Parker, M. E. Deason, D. D. Dischino, E. Gillespie, A. A. Qureshi, K. Volk and J. J. Wright, Inhibitors of cholesterol biosynthesis. 2. Hypocholesterolemic and antioxidant activities of benzopyran and tetrahydronaphthalene analogues of the tocotrienols, J. Med. Chem., 1994, 37, 526–541, DOI:10.1021/jm00030a012.
- A. Bermejo, I. Barrachina, N. El Aouad, X. Franck, N. Chahboune, I. Andreu, B. Figadère, L. Vila, N. Hennuyer, B. Staels, C. Dacquet, D. H. Caignard, M. J. Sanz, D. Cortes and N. Cabedo, Synthesis of benzopyran derivatives as PPARα and/or PPARγ activators, Bioorg. Med. Chem., 2019, 27, 115162, DOI:10.1016/j.bmc.2019.115162.
- A. García, L. Vila, P. Marín, Á Bernabeu, C. Villarroel-Vicente, N. Hennuyer, B. Staels, X. Franck, B. Figadère, N. Cabedo and D. Cortes, Synthesis of 2-prenylated alkoxylated benzopyrans by horner-wadsworth-emmons olefination with PPARα/γ agonist activity, ACS Med. Chem. Lett., 2021, 12, 1783–1786, DOI:10.1021/acsmedchemlett.1c00400.
- L. Vila, N. Cabedo, C. Villarroel-Vicente, A. García, Á Bernabeu, N. Hennuyer, B. Staels, X. Franck, B. Figadère, M. J. Sanz and D. Cortes, Synthesis and biological studies of “Polycerasoidol” and “trans-δ-Tocotrienolic acid” derivatives as PPARα and/or PPARγ agonists, Bioorg. Med. Chem., 2022, 53, 116532, DOI:10.1016/j.bmc.2021.116532.
- A. K. Ghosh and M. Brindisi, Urea derivatives in modern drug discovery and medicinal chemistry, J. Med. Chem., 2020, 63, 2751–2788, DOI:10.1021/acs.jmedchem.9b01541.
- M. Redza-Dutordoir and D. A. Averill-Bates, Activation of apoptosis signalling pathways by reactive oxygen species, Biochim. Biophys. Acta, 2016, 1863, 2977–2992, DOI:10.1016/j.bbamcr.2016.09.012.
- J. M. Adams and S. Cory, The BCL-2 arbiters of apoptosis and their growing role as cancer targets, Cell Death Differ., 2018, 25, 27–36, DOI:10.1038/cdd.2017.161.
- J. C. Martinou and R. J. Youle, Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics, Dev. Cell, 2011, 21, 92–101, DOI:10.1016/j.devcel.2011.06.017.
- S. Cory and J. M. Adams, The Bcl2 family: Regulators of the cellular life-or-death switch, Nat. Rev. Cancer, 2002, 2, 647–656, DOI:10.1038/nrc883.
- H. Guo, H. Cui, X. Peng, J. Fang, Z. Zuo, J. Deng, X. Wang, B. Wu, K. Chen and J. Deng, Modulation of the PI3K/Akt Pathway and Bcl-2 Family Proteins Involved in Chicken's Tubular Apoptosis Induced by Nickel Chloride (NiCl2), Int. J. Mol. Sci., 2015, 16, 22989–23011, DOI:10.3390/ijms160922989.
- I. Neganova and M. Lako, G1 to S phase cell cycle transition in somatic and embryonic stem cells, J. Anat., 2008, 213, 30–44, DOI:10.1111/j.1469-7580.2008.00931.x.
- B. Stecca and E. Rovida, Impact of ERK5 on the Hallmarks of Cancer, Int. J. Mol. Sci., 2019, 20, 1426, DOI:10.3390/ijms20061426.
- R. Paudel, L. Fusi and M. Schmidt, The MEK5/ERK5 Pathway in Health and Disease, Int. J. Mol. Sci., 2021, 22, 7594, DOI:10.3390/ijms22147594.
- W. Song, L. Tang, Y. Xu, J. Xu, W. Zhang, H. Xie, S. Wang and X. Guan, PARP inhibitor increases chemosensitivity by upregulating miR-664b-5p in BRCA1-mutated triple-negative breast cancer, Sci. Rep., 2017, 7, 42319, DOI:10.1038/srep42319.
- W. Song, L. Tang, Y. Xu, J. Xu, W. Zhang, H. Xie, S. Wang and X. Guan, PARP inhibitor increases chemosensitivity by upregulating miR-664b-5p in BRCA1-mutated triple-negative breast cancer, Sci. Rep., 2017, 7, 42319, DOI:10.1038/srep42319.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00385j |
| This journal is © The Royal Society of Chemistry 2023 |