
DLP 3D printed hydrogels with hierarchical structures post-programmed by lyophilization and ionic locking†
Zhuo
Sun
ab,
Qian
Zhao
 b,
Sainan
Ma
a and
Jingjun
Wu
*ab
b,
Sainan
Ma
a and
Jingjun
Wu
*ab
aNingbo Research Institute, Zhejiang University, Ningbo 315807, China. E-mail: jingjunwu@zju.edu.cn
bState Key Laboratory of Chemical Engineering, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China
First published on 21st October 2022
Abstract
Porous hydrogels have been intensively used in energy conversion and storage, catalysis, separation, and biomedical applications. Controlling the porosity of these materials over multiple length scales brings about new functionalities and higher efficiency but is a challenge using the current manufacturing methods. Herein we developed a post-programming method to lock the lyophilized pores of 3D printed hydrogels as an experimental platform towards hierarchically structured pores. 3D printing endows the hydrogels with arbitrary 3D geometries and controllable pores at the millimeter length scale. Lyophilization and ionic crosslinking of the as-printed hydrogel networks are conducted as a post-programming process which results in pores at micrometer length scales beyond the printing resolution. Utilizing this combined manufacturing technology, 3D hydrogel lattices with tunable porosities and mechanical properties can be created, which are further exploited for efficient solar vapor generation.
New conceptsEndowing functional materials, such as hydrogels, with geometries and/or porosities over multiple length scales has been a long-standing endeavor. One of the existing efforts has been devoted to combining traditional pore-forming methods, such as porogen leaching, with the newly developed 3D printing technology. However, the coupling of the fabrication methods of different hierarchies brings about inevitable limitations to fabrication freedom. We report that hydrogels with hierarchical structures can be fabricated via DLP 3D printing and a subsequent two-step post-programming process of lyophilization and ionic locking. While 3D printing endows arbitrary 3D geometries at a coarser length scale, lyophilization and ionic locking result in pores at a length scale below the printing resolution. To the best of our knowledge, this is the first effort to decouple the construction of macroscopic geometry and microstructures. To fully demonstrate the advantage of these hierarchical structures, we present the application of the 3D printed porous hydrogels in solar vapor generation. |
Introduction
Porous hydrogels have shown promise for use in the fields of actuators,1 energy storage,2 energy harvesting,3 water sustainability,4 tissue engineering,5 and supported catalysis.6 The modulation of pore size and morphology is essential for controlling the hydrogel properties. Several strategies have been developed to fabricate porous hydrogels with controlled pore morphology and distribution, such as porogen leaching, phase separation, cryo-templating, gas foaming, etc.7–11 One limitation of all these methods is the difficulty of constructing porous features spanning over multiple length scales. On the other hand, hierarchically structured hydrogels with pores of distinct sizes are required in many of the applications mentioned above. For example, hierarchical porosity is quite desirable for adsorption and separation processes for achieving a balance of the high accessible surface area and ready mass transport.12 So, it is crucial to develop novel routes to improve the level of control over the architecture of synthetic porous hydrogels.3D printing, as an emerging technology of material processing, is attracting great attention due to its ability to precisely create complex 3D geometries at macroscopic length scales.13,14 Among the various 3D printing technologies, vat photopolymerization, with digital light processing (DLP) and stereolithography (SLA) as the best-known examples, is endowed with higher resolution and better surface fidelity,15–19 enabling the digital fabrication of porous materials in the form of lattices or cellular structures. Through a precise control of the light, micro/sub-micron features can also be achieved. Theoretically, it is possible to exploit 3D printing to directly define the multiscale porosity as well as a complex 3D shape. However, it is actually quite challenging to build hierarchical structures from the centimeter-scale down to the micrometer scale because of the trade-off between the UV image resolution and the field of view.20–22 In this case, porogen leaching has been combined with DLP printing to create hierarchical structures in hydrogels.23,24 While the mm-sized pores and overall 3D geometry can be simply controlled by the light pattern during printing, μm-sized micropores are determined by the removal of the porogens incorporated into the printing ink. However, guaranteeing the stability and printability is highly challenging due to the unfavorable rheology characteristics such as high viscosity or thixotropy, especially when high porogen loading is required to get interconnected pores.
Contrarily, the cryo-templating strategy seems to be an ideal alternative because it exploits the water of the hydrogel precursor as porogens and no extra additives are needed.25,26 However, conventional cryo-generation of porous hydrogels requires the polymerization of aqueous solution in the frozen state, which is not compatible with DLP printing. Another process is lyophilization which can convert a nonporous hydrogel into a solvent-free foam. However, the pores in such a foam are temporary and thermodynamically unstable, and disappear upon rehydration due to the swelling-induced recovery of the lyophilized chains.27,28 The retention of the lyophilized pore structure under a hydrated state is nontrivial. Herein, a post-programming method has been developed to lock the lyophilized pores of 3D printed hydrogels as an experimental platform towards hierarchically structured pores. A normally DLP 3D printed poly(N-isopropylacrylamide-co-acrylic acid) hydrogel is lyophilized to get a freeze-dried foam with temporary pores. Subsequently, metal ions, such as Fe3+, are introduced to form ionic coordination with carboxyl groups as secondary crosslinking, preventing the pores from collapsing upon swelling. Using this approach, we produce 3D bulk hierarchically structured hydrogels with tunable pore sizes, pore morphologies, and unprecedented strength for the high porosity achieved. Finally, to fully demonstrate the advantages of the 3D printed hierarchically porous hydrogel, it is utilized as an efficient candidate applied in wastewater contamination and desalination based on interfacial solar vapor generation.29,30
Experimental
Materials
N-Isopropylacrylamide (NIPAM), Rhodamine B (RhB), and dopamine hydrochloride were purchased from Aladdin. N,N′-Methylenebisacrylamide (BIS), acrylic acid (AA), anhydrous ethanol, ethyl acetate (EA), EDTA disodium salt dihydrate and ferric(III) chloride hexahydrate were obtained from Macklin. The light absorber (Basic Violet 8) was purchased from TCI. Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) was purchased from SinoBioPrint Biotech Ltd (Shanghai, China). Real seawater (collected from Qingdao, Shandong) was bought online. All reagents were used as received without purification.Preparation of NIPAM-AA porous hydrogels
NA50 was chosen as a sample to present the detailed synthesis. NIPAM (0.1 g), AA (0.1 g), BIS (0.002 g, 1 wt% of the monomer), and LAP (0.008 g, 4 wt% of the monomer) were dissolved in deionized water (0.8 g) to get a precursor. Different samples were simply prepared by adjusting the mass ratio of AA. The precursor was poured into a glass mold with a 1 mm thick silicon spacer. The photo-curing process was conducted in a UV chamber (IntelliRay 600, 90 mW cm−2, 265–700 nm) for 30 seconds on each side. The as-prepared hydrogel sheets were immersed in deionized water for 24 h to remove residual monomers. Then, they were frozen at −20 °C and dried in a lyophilizer. The control of different freezing temperatures and unidirectional freezing was conducted on the surface of a copper billet. The freeze-dried foams were immersed in FeCl3/EA solution (0.1 M or 1 M) for 24 h. EtOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and DI water were used to replace ethyl acetate for 36 h to get the final porous hydrogels.
:1 v/v) and DI water were used to replace ethyl acetate for 36 h to get the final porous hydrogels.
Preparation of NA50-1 M-PDA porous hydrogels
The as-prepared NA50-1 M hydrogels were immersed in an aqueous solution of dopamine hydrochloride (2 wt%) for 24 hours. The polymerization of dopamine was catalyzed by the intrinsic Fe3+.Erasing of the porosities
Porosity erasing was realized by immersing the hierarchically structured hydrogel in EDTA aqueous solution (0.1 M) for 12 h at 75 °C and then putting in DI water for 3 h to reach equilibrium.3D printing
The printable precursor was prepared by adding Basic Violet 8 (0.01 wt%) to the precursor of NA50 as the light absorber. The precursor solution was bubbled with argon for 30 min to remove dissolved oxygen. The 3D printing process was conducted on a bottom-up 3D printer (MoonRay, SprintRay). The light intensity of the projector is 2 mW cm−2. All the printed models were set at a slice thickness of 100 μm and the exposure time was 30 s for each layer.Solar vapor generation measurement
The size of the flat NA50-1 M-PDA cylinder is 4.5 cm in diameter and 1 cm in thickness. The 3D lattice with micropores is a 2.5 × 2.5 × 2.5 cm lattice (with a surface porosity of ca. 60%) and the one without micropores was prepared by undergoing porosity erasing. 1 sun irradiation was simulated using a xenon (Xe) lamp (CEL-PE300L-3A, CEAULIGHT) with an energy intensity of 1000 W m−2, which was measured by using a solar power meter (SM206-SOLAR). The water after desalination was captured in a customized device (Fig. S11, ESI†). The water evaporation rate was calculated as follows: water evaporation rate = Δm/(T × S), where Δm, T, and S represent the mass change of water, time of illumination, and irradiation area, respectively. The ambient temperature and humidity of the environment were ca. 22.5 °C and 55%, respectively.Characterization
Scanning electron microscopy (SEM) was carried out on SU-3500 and SU-8010 (HITACHI, Japan) instruments. Confocal laser scanning microscopy (CLSM) was conducted using a TCS SP5 II (Leica, Germany). Before the observation, hydrogel samples were dyed by a RhB aqueous solution (5 mg L−1) for 12 h and then washed with DI water for 48 h to eliminate the residual RhB. ATR-FTIR spectroscopy was conducted on a Fourier transform infrared spectrometer (Nicolet 5700). The UV-vis-NIR spectra were measured using a UV-vis spectrometer (U4800, HITACHI). The content of Fe3+ in NA50-0.1 M and NA50-1 M was tested on a thermal gravimetric analyzer (TA Q500, USA). The water evaporation enthalpy was tested on a differential scanning calorimeter (TA Q200, USA). The contents of four salt ions (Na+, Mg2+, Ca2+, and K+) were tested on an inductive coupled plasma emission spectrometer (ICP-OES/MS, Thermo ICAP PRO).Mechanical tests
Compression tests were conducted on a UTM2503 electronic universal testing machine (Shenzhen SUNS Technology Stock Co. Ltd) at a compression rate of 50 mm min−1. The compressive modulus was calculated by the linear fitting of the strain–stress curves ranging from 5% to 15%. Tests were repeated until each composition was successfully measured at least three times.Results and discussion
The hydrogel ink consists of N-isopropylacrylamide (NIPAM) and acrylic acid (AA) as monomers, N,N′-methylenebisacrylamide (BIS) as the chemical crosslinker, and lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) as the photoinitiator (water content: 80 wt%). Fig. 1a schematically demonstrates how to endow a 3D printed hydrogel of the above common formulation31–33 with a porous structure in the fully swollen state. The hydrogel is firstly lyophilized to get a freeze-dried foam. Then, the foam is treated with Fe3+/ethyl acetate solution. The porous structure of the foam is locked through the Fe3+–carboxyl interaction as secondary crosslinking. After the ethyl acetate is replaced by water, a porous dual-crosslinked hydrogel can be obtained. Directly swelling the dried foam in water will lead to the disappearance of pores because of the swelling-induced recovery of the lyophilized chains.27 Notably, a conventional Fe3+ aqueous solution is not used here, because the freeze-dried NIPAM-AA foam absorbs water immediately (Fig. S1, ESI†) and there will be an inevitable and intractable competition between the swelling of the polymer network and Fe3+–carboxyl interaction-induced network locking. Ethyl acetate is exploited because of its good solubility in FeCl3 and contrarily, poor affinity for the NIPAM-AA network. A bottom-up DLP 3D printer with a light source of 405 nm is utilized to print with the NIPAM-AA hydrogel ink (Fig. 1b). The smallest printable feature is several hundreds of microns (Fig. S2, ESI†), which is decent for a quite soft hydrogel. Of course, the resolution can be improved in future investigations by employing a higher resolution light source. A series of 3D hydrogels with complex geometries are printed and endowed with porous structures after being post-programmed by the two-step lyophilization and ionic locking (Fig. 1c).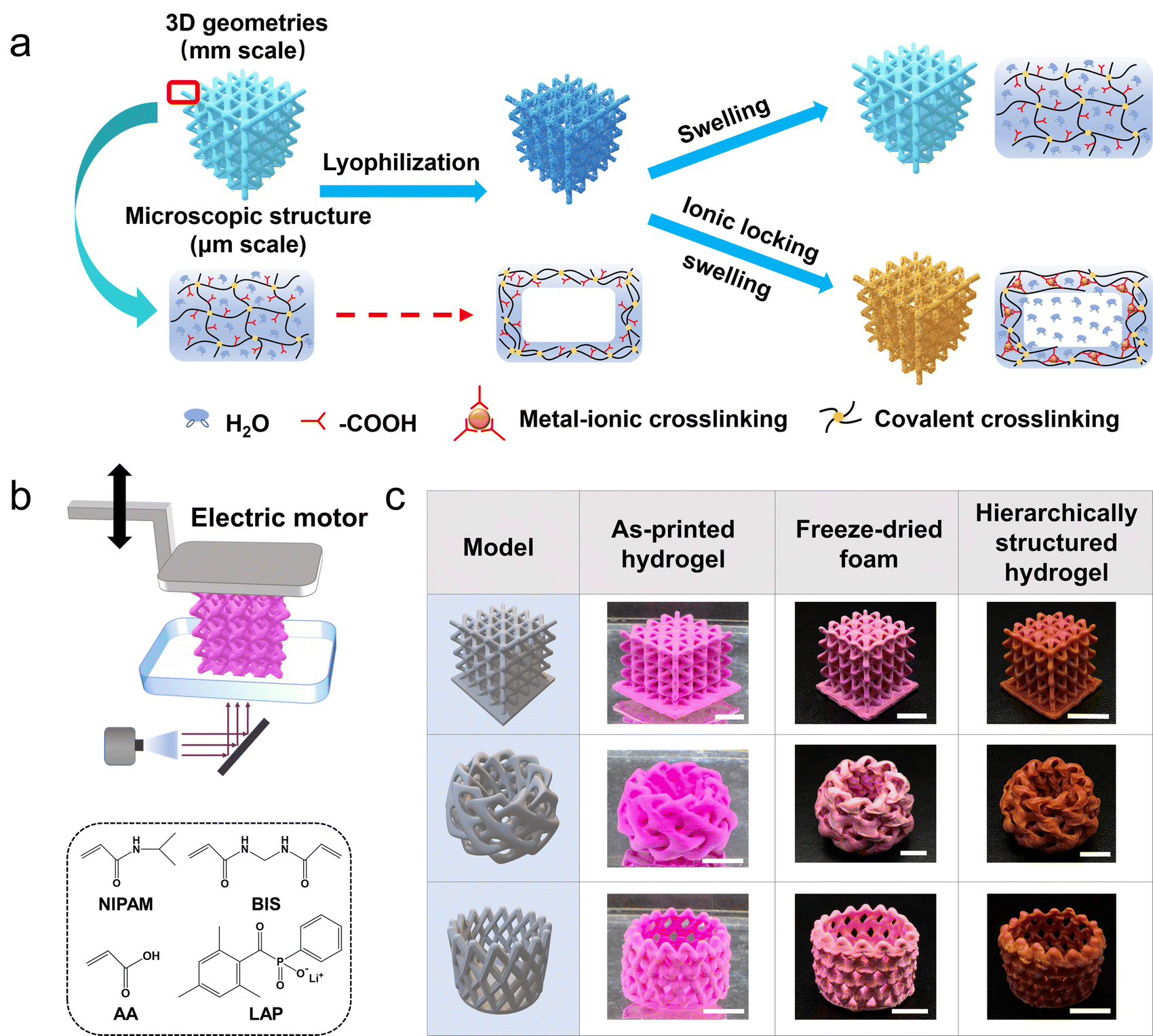 | ||
| Fig. 1 Schematic of lyophilization-assisted DLP 3D printing of hierarchically structured hydrogels. (a) Schematic demonstration of pore-formation. (b) DLP 3D printing process and formulation of the hydrogel ink. (c) 3D printed porous hydrogels. Scale bar: 1 cm. | ||
To prevent the stressed primary network from elastic recovery upon swelling, i.e. transforming from a porous foam to a nonporous hydrogel, the strength of the secondary metal–carboxyl crosslink should be as large as possible. On the one hand, Fe3+ is employed because the Fe3+–carboxyl interaction is much larger than that of others such as Al3+–carboxyl and Ca2+–carboxyl.17,34,35 On the other hand, the density of the secondary crosslink can be adjusted by the content of the AA in the hydrogel ink (hydrogel samples are denoted as NAm, where m represents the mass ratio of AA in the total mass of NIPAM and AA). The coordination interaction of Fe3+–carboxyl is confirmed via ATR-FTIR spectroscopy. The peak at 1592 cm−1 is assigned to Fe3+–COO− coordination,36 and the peaks at 1750–1600 and 1500–1400 cm−1 are assigned to the asymmetric and symmetric stretching vibrations of carbonyl groups.37 As the content of AA increases, more Fe3+–carboxyl crosslinks are formed (Fig. 2a). For the sample NA10 with the lowest content of AA, the Fe3+–carboxyl interaction is too weak to be detected. Due to the presence of the Fe3+–carboxyl crosslinks, the lyophilized and thus stretched primary chemical crosslinked network is locked, and the lyophilization-induced temporary porous structures are fixed and preserved after being fully swollen in water. As shown in Fig. 2b, CLSM images clearly show that the NA40, NA50, and NA60 hydrogel samples exhibit an interconnected porous structure in the swollen state, which coincides well with the morphology of the freeze-dried sample (Fig. S3, ESI†), verifying the effectiveness of the proposed post-programming strategy. In contrast, the porous structure of NA50 foams disappears when it is immersed in pure water, demonstrating the critical role of Fe3+–carboxyl interaction in locking porous structures (Fig. S4, ESI†). Because of the interconnected open porous structure, water can be squeezed out from the hydrogels easily and absorbed back quickly (Movie S1, ESI†). For NA10, pores with clear outlines can hardly be observed. Considering the undetectable Fe3+–carboxyl interaction in FTIR spectroscopy, it can be concluded that the crosslinking density of Fe3+–carboxyl coordination for NA10 is too small to lock the primary chemical network during swelling. For hydrogels with a larger content of AA than NA60, samples are found difficult to be ideally lyophilized into a uniform freeze-dried foam, probably because the hydrogels have too much affinity for water. So, NA50 with 50 wt% AA is used for further investigation. Obviously, as the secondary crosslink shows an effective locking of the primary chemically crosslinking network, the lyophilization process directly determines the porous structures of the hydrogels because the size and morphology of the pores are the replicas of the ice crystals. It is found that NA50 hydrogels lyophilized in a lower freezing temperature exhibit smaller pores (Fig. 2c), which can be attributed to the more amount of crystal nucleus and smaller crystal sizes. In addition, aligned pores can be obtained when unidirectional freezing is applied. As shown in Fig. 2d, a 3D printed nonporous hydrogel lattice is unidirectionally frozen, lyophilized, and Fe3+ treated as mentioned above, resulting in an anisotropic porous structure. It can be clearly observed that aligned pores are available along the freezing direction. Because of the small, but nonnegligible difference in the temperature gradient in the samples along the freezing direction, the pores away from the cooling source are apparently larger than those near the cooling source. These unique 3D aligned pores with a gradual size distribution may be exploited for directional fluid transportation.38,39
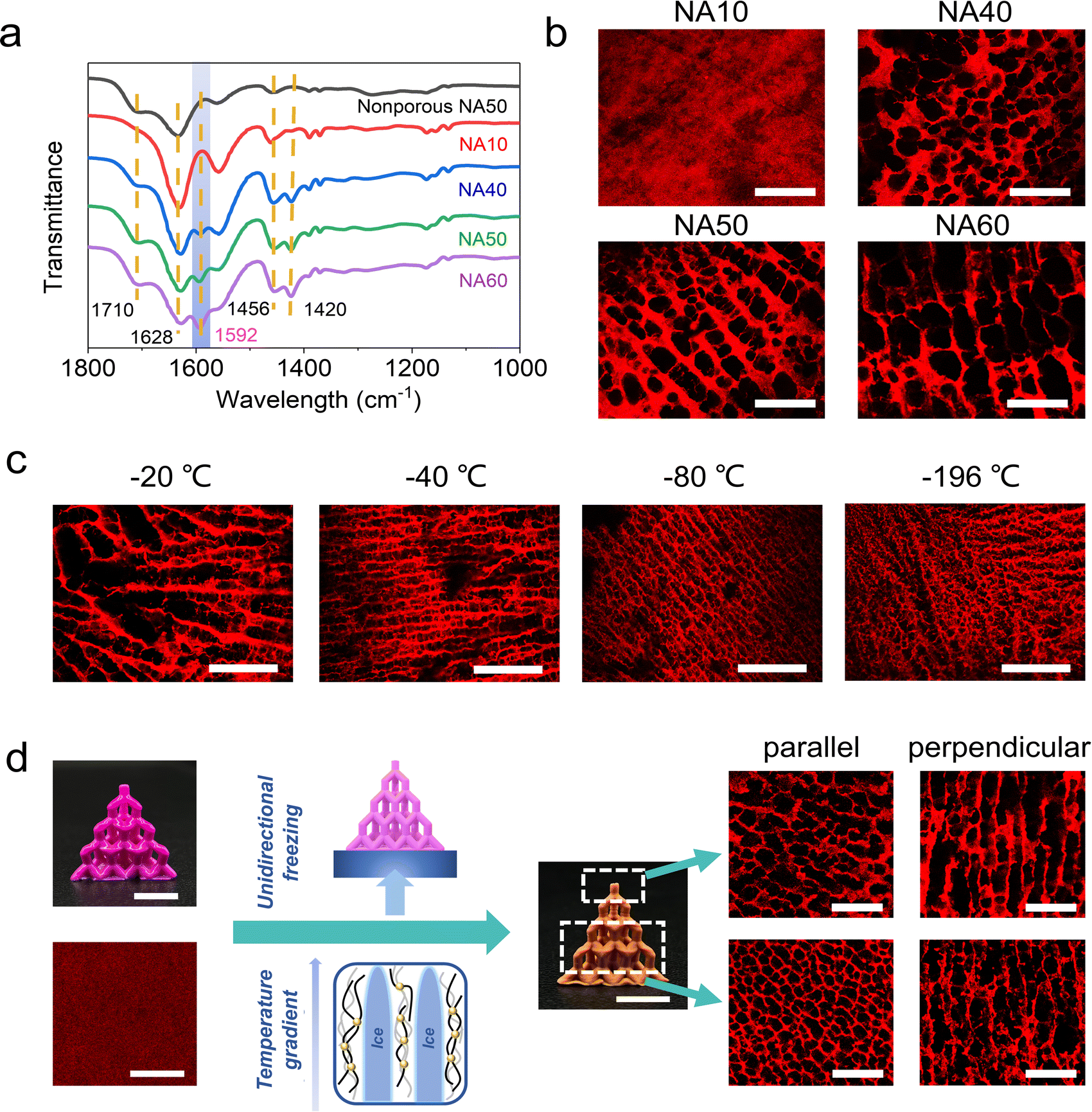 | ||
| Fig. 2 Morphology of microscopic porous hydrogels. (a) ATR-FTIR spectra of the nonporous hydrogel without Fe3+ and Fe3+-loaded porous hydrogels with different AA contents. (b) Confocal laser scanning microscopy (CLSM) images of NAm porous hydrogels (scale bar: 100 μm). (c) Morphologies of NA50 sample lyophilized in different freezing temperatures (scale bar: 100 μm). (d) Anisotropic porous structures enabled by unidirectional freezing. Scale bar: 1 cm for the optical images and 50 μm for the CLSM images. | ||
The Fe3+ loaded porous NA50 hydrogels exhibit an unusually high stiffness compared with common porous hydrogels. In addition, the modulus of the hydrogels is closely related to the Fe3+ concentration of the Fe3+/ethyl acetate solution. Fig. 3a shows the compressive stress–strain curves of the obtained porous hydrogels treated with different Fe3+/ethyl acetate solutions. When the Fe3+ concentration is larger than 0.5 M (denoted as NA50-x, where x is 0.05, 0.1, 0.5, and 1 M), the compressive modulus can reach 5.32 ± 0.47 MPa, which is higher than vast majority of the reported hydrogels (Fig. S5, ESI†). A 1 × 1 × 1 cm cubic porous hydrogel can support as large as a one-kilogram weight (Fig. 3b). Such a high stiffness may be attributed to the high Fe3+ loading (18.2 wt% and 7.3 wt% for freeze-dried NA50-1 M and NA50-0.1 M, TGA, Fig. S6, ESI†) and possible hydrolysis of Fe3+ when ethyl acetate is replaced by water. When the Fe3+ concentration decreases to 0.05 M, the compressive modulus decreases to 0.21 ± 0.02 MPa, which is still a relatively stiff porous hydrogel. Another interesting finding lies in the fact that the secondary Fe3+–carboxyl crosslink, which is directly related to the porous structure, is a dynamic, reversible bond. So, it is possible for a 3D printed hydrogel to reversibly switch between a nonporous structure and a porous one. When an as-prepared 3D porous NA50-0.1 M hydrogel lattice is treated with an EDTA aqueous solution (pH = 4.5), EDTA will react with Fe3+ to form a more stable Fe3+-EDTA chelate.40 The primary NIPAM-AA network swells and turns into a nonporous hydrogel in the absence of a secondary crosslink (Fig. 3c). In contrast, the porous structure is reserved when the same porous hydrogel is treated with an aqueous HCl solution, indicating the role of Fe3+-EDTA chelation in erasing of the pores (Fig. S7, ESI†). In addition, the obtained nonporous hydrogel can be further changed into a porous one (Fig. 3c).
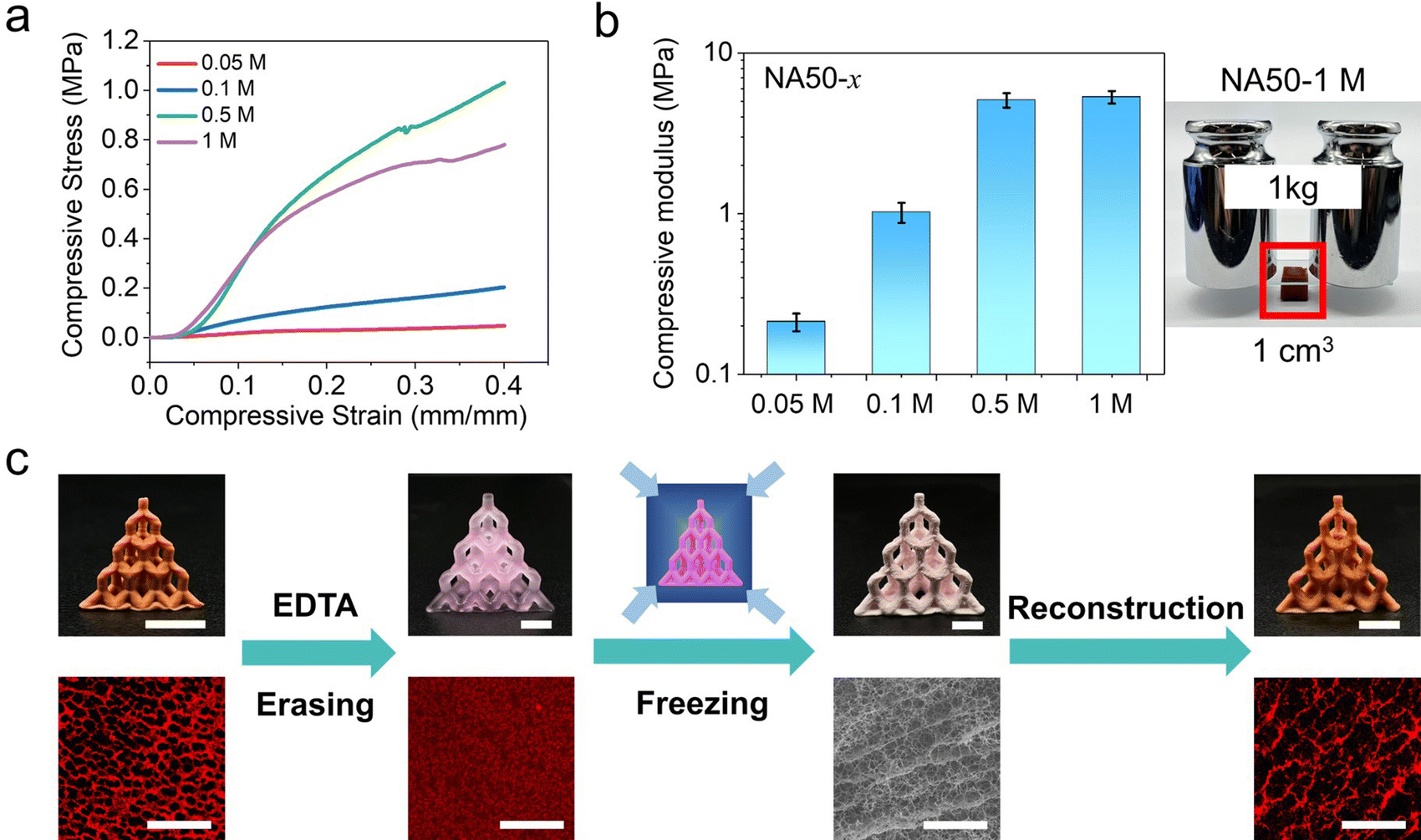 | ||
| Fig. 3 Mechanical performance and reversible reconfiguration of the porous morphology. (a) The compressive stress–strain curves and (b) the compressive modulus of NA50-x. (c) Reversible switching between porous and nonporous morphologies. Scale bar: 1 cm for the optical images and 100 μm for the CLSM and SEM images. | ||
To fully utilize the 3D printed hierarchical structures, a 2.5 × 2.5 × 2.5 cm NA50-1 M hydrogel lattice is used as an efficient water evaporation matrix for interfacial solar vapor generation (SVG). The lyophilization-induced microscopic pores can profit an efficient water transport due to the capillary force and easier water evaporation due to a decreased evaporation enthalpy, both of which are critical for an efficient SVG (Fig. S8, ESI†).41–43 On the other hand, the significantly increased surface area of the 3D printed lattice geometry benefits both energy harvesting and water evaporation (Fig. 4a). To achieve a high photothermal conversion efficiency, polydopamine (PDA) is in situ deposited on the pore walls through Fe-mediated redox polymerization (Fig. 4b and Fig. S9, ESI†).44–46 The UV-vis-IR spectra show that NA50-1 M-PDA exhibits a high light absorption of up to 95% among all the solar irradiance spectra (Fig. 4c). A customized device is designed to measure the water evaporation rate (Fig. S10, ESI†). Under 1 sun irradiation (1 kW m−2), the top layer of NA50-1 M-PDA hydrogel is quickly heated to an equilibrium temperature of 32 °C within 8 minutes and the remaining part maintains a temperature of around 25 °C (Fig. 4d). The rapid temperature rise is owing to the good photothermal effect of PDA and ensures that NA50-1 M-PDA hydrogels reach a stable water evaporation rate in a shorter time. The water evaporation rate of the 3D hydrogel lattice is around 2.85 kg m−2 h−1, which is among the best in this field.47–51 In contrast, the water evaporation rate of pure water and a flat hydrogel is 0.37 kg m−2 h−1 and 0.62 kg m−2 h−1, respectively (Fig. 4e and f). Considering the same energy input and similar microscopic porous structures, the huge difference of the water evaporation rate between hydrogels of different geometries mainly results from the different surface areas. 3D hydrogel lattices possess a surface area as large as 86.26 cm2, which is 5.4 times larger than that of the 2D flat hydrogel (15.90 cm2). In addition, a 3D hydrogel lattice without micropores is also fabricated as a control to show the effect of micropores on the water evaporation rate. There is an obvious decrease of the water evaporation rate from 2.85 kg m−2 h−1 to 1.60 kg m−2 h−1 when there are no micropores. Furthermore, pure water is replaced by seawater to evaluate the desalination efficiency using the above-mentioned hydrogel lattice. Fig. 4g shows that the water evaporation rate is 2.55 kg m−2 h−1 for seawater and remains quite stable for at least 8 hours. The concentrations of four salt ions (Na+, Mg2+, Ca2+, and K+) are reduced by over three orders of magnitude after one cycle of desalination is performed (Fig. 4h). After 8 hours of irradiation, NA50-1 M-PDA samples maintained the microscopic structures, indicating the good stability of the materials (Fig. S12, ESI†). All of them meet the standards of the World Health Organization (WHO) and the U.S. Environmental Protection Agency (EPA) for drinkable water. Simultaneously, in comparison to the current membrane and heat-based seawater desalination methods, SVG has a more obvious advantage in desalination.7,52
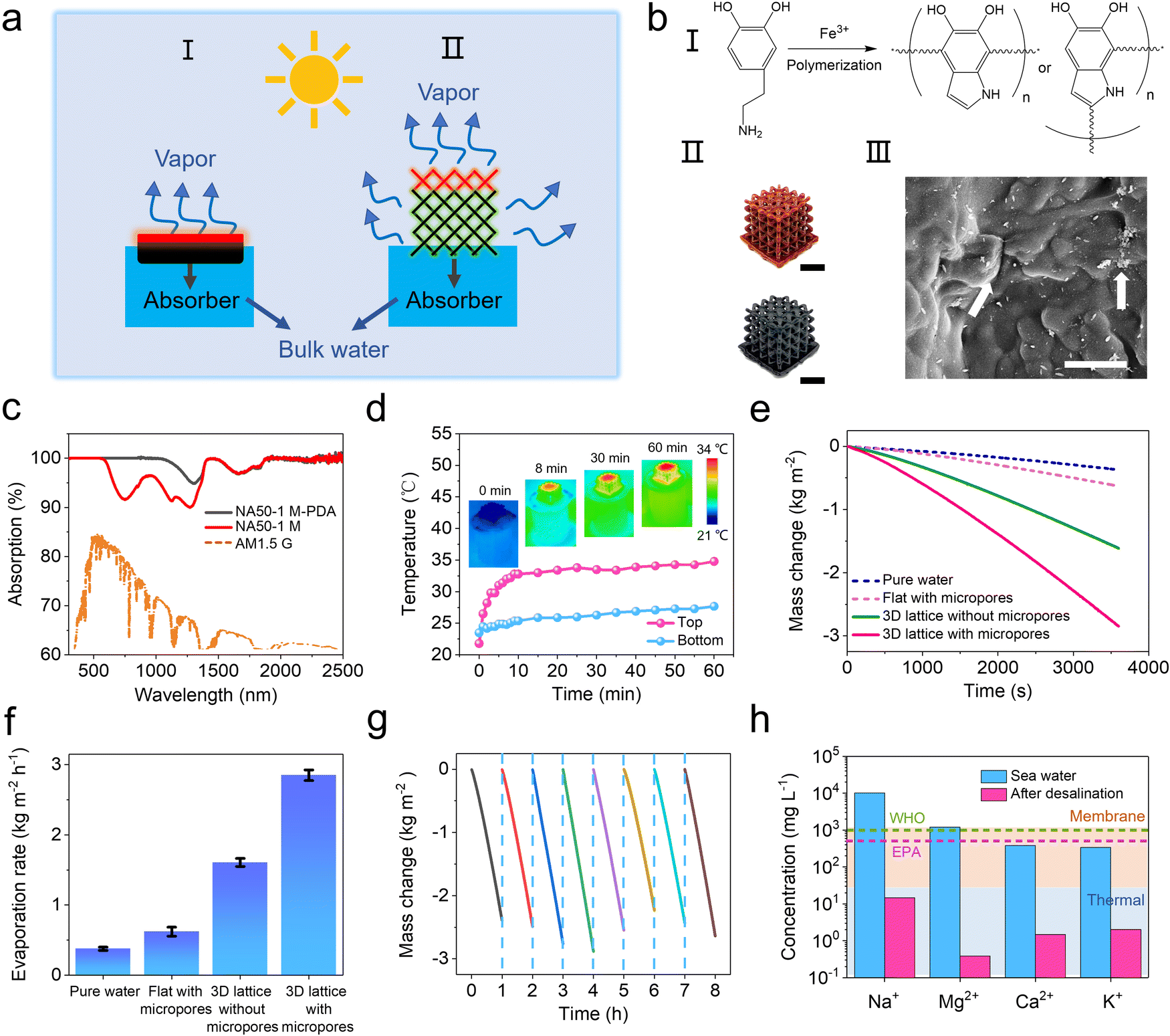 | ||
| Fig. 4 Interfacial solar vapor generation performance of the hierarchically structured hydrogel. (a) Scheme of solar vapor generation utilizing porous hydrogels of different geometries. (b) In situ deposition of PDA on the NA50-1 M hydrogel surface. Scale bar: 1 cm for the optical images and 1 μm for the SEM image. (c) UV-vis-IR spectra of NA50-1 M and NA50-1 M-PDA and the normalized spectral solar irradiance density of the air mass 1.5 global (AM 1.5G) tilt solar spectrum. (d) Heating rate curve of the top of 3D NA50-1 M-PDA hydrogel lattice and bulk water at the bottom. Insets: Infrared images showing the temperature distribution. (e and f) Water evaporation rates of pure water and hydrogels with different geometries and porous structures. (g) Water evaporation rate for real seawater using 3D NA50-1 M-PDA hydrogel lattice. (h) Concentrations of four primary ions (Na+, Mg2+, Ca2+, and K+) in seawater before and after desalination. | ||
Conclusions
In summary, we propose a method for fabricating hierarchically structured hydrogels via DLP 3D printing and a post two-step programming process of lyophilization and ionic locking. The overall 3D geometry and millimeter-sized pores are defined by the light pattern during 3D printing, while micrometer-sized pores are endowed by lyophilization. Fe3+–carboxyl coordination is exploited as a secondary crosslinking to lock the lyophilization-induced pores. The pore sizes, pore morphologies, and mechanical properties can be modulated. As a proof of concept, a hierarchically porous hydrogel lattice is applied as a water evaporation matrix for efficient solar vapor generation. Future work may extend this technology to biocompatible hydrogels, such as the alginate–Ca2+ hydrogel, to investigate the effectiveness of hierarchically porous structures in the field of tissue engineering.Author contributions
Zhuo Sun: investigation, data curation, writing – original draft. Qian Zhao: funding acquisition, supervision. Sainan Ma: data curation. Jingju Wu: conceptualization, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. U20A6001). We thank Yingying Zhang (Testing and Analysis Center of the Department of Polymer Science and Engineering, Zhejiang University) for assistance with performing CLSM measurement and Lu Zong (Qingdao University of Science & Technology) for the guidance on solar vapor generation.Notes and references
- R. Luo, J. Wu, N. Dinh and C. Chen, Adv. Funct. Mater., 2015, 25, 7272 CrossRef CAS.
- J. Lim, G. Y. Lee, H. J. Lee, S. K. Cha, S. H. Koo, W. J. Lee and S. O. Kim, Energy Storage Mater., 2019, 16, 251 CrossRef.
- X. Yin, Y. Zhang, Q. Guo, X. Cai, J. Xiao, Z. Ding and J. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 10998 CrossRef CAS PubMed.
- Y. Duo, J. Bae, Z. Fang, P. Li, F. Zhao and G. Yu, Chem. Rev., 2020, 120, 7642 CrossRef PubMed.
- S. J. Hollister, Nat. Mater., 2005, 4, 518 CrossRef CAS PubMed.
- L. Zhang, H. Lu, J. Chu, J. Ma, Y. Fan, Z. Wang and Y. Ni, ACS Sustainable Chem. Eng., 2020, 8, 12655 CrossRef CAS.
- Y. Guo, L. S. Vasconcelos, N. Manohar, J. Geng, K. P. Johnston and G. Yu, Angew. Chem., Int. Ed., 2022, 61, e202114074 CAS.
- Y. Alsaid, S. Wu, D. Wu, Y. Du, L. Shi, R. Khodambashi, R. Rico, M. Hua, Y. Yan, Y. Zhao, D. Aukes and X. He, Adv. Mater., 2021, 33, 2008235 CrossRef CAS PubMed.
- J. Wu, Q. Zhao, J. Sun and Q. Zhou, Soft Matter, 2012, 8, 3620 RSC.
- C. W. Visser, D. N. Amato, J. Mueller and J. A. Lewis, Adv. Mater., 2019, 31, 1904668 CrossRef CAS.
- X. Yang, L. Chen, Y. Li, J. C. Rooke, C. Sanchez and B. Su, Chem. Soc. Rev., 2017, 46, 481 RSC.
- M. A. Lsaacs, N. Robinson, B. Barbero, L. J. Durndell, J. C. Manayil, C. M. A. Parlett, C. D’Agostino, K. Wilson and A. F. Lee, J. Mater. Chem. A, 2019, 7, 11814 RSC.
- E. MacDonald and R. Wicker, Science, 2016, 353, 1512 CrossRef.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mulhaupt, Chem. Rev., 2017, 117, 10212 CrossRef CAS.
- B. Zhang, S. Li, H. Hingorani, A. Serjouei, L. Larush, A. A. Pawar, W. H. Goh, A. H. Sakhaei, M. Hashimoto, K. Kowsari, S. Magdassi and Q. Ge, J. Mater. Chem. B, 2018, 6, 3246 RSC.
- S. Deng, J. Wu, M. D. Dickey, Q. Zhao and T. Xie, Adv. Mater., 2019, 31, 1903970 CrossRef.
- Z. Sun, Y. Lu, Q. Zhao and J. Wu, Addit. Manuf., 2022, 50, 102563 CAS.
- J. Wu, J. Guo, C. Linghu, Y. Lu, J. Song, T. Xie and Q. Zhao, Nat. Commun., 2021, 12, 6070 CrossRef CAS.
- C. Yu, J. Schimelman, P. Wang, K. L. Miller, X. Ma, S. You, J. Guan, B. Sun, W. Zhu and S. Chen, Chem. Rev., 2020, 120, 10695 CrossRef CAS.
- M. Dong, Y. Han, X. P. Hao, H. C. Yu, J. Yin, M. Du, Q. Zheng and Z. L. Wu, Adv. Mater., 2022, 34, 2204333 CrossRef CAS.
- J. Odent, T. J. Wallin, W. Pan, K. Kruemplestaedter, R. F. Shepherd and E. P. Giannelis, Adv. Funct. Mater., 2017, 27, 1701807 CrossRef.
- M. Caprioli, I. Roppolo, A. Chiappone, L. Larush, C. F. Pirri and S. Magdassi, Nat. Commun., 2021, 12, 2462 CrossRef CAS PubMed.
- N. T. H. Men, T. H. Jeong, S. Y. Kim, K. B. Kim, T. H. Ha, S. J. Ahn and Y. H. Kim, J. Manuf. Process., 2021, 67, 46 CrossRef.
- X. Mu, T. Bertron, C. Dunn, H. Qiao, J. Wu, Z. Zhao, C. Saldana and H. J. Qi, Mater. Horiz., 2017, 4, 442 RSC.
- Q. Zhao, J. Sun, Q. Ling and Q. Zhou, Langmuir, 2009, 25, 3249 CrossRef CAS.
- Q. Zhao, J. Sun and Q. Zhou, J. Appl. Polym. Sci., 2007, 104, 4080 CrossRef CAS.
- D. Chen, X. Xia, T. W. Wong, H. Bai, M. Behl, Q. Zhao, A. Lendlein and T. Xie, Macromol. Rapid Commun., 2017, 38, 1600746 CrossRef.
- D. Chen, Y. Zhang, C. Ni, C. Ma, J. Yin, H. Bai, Y. Luo, F. Huang, T. Xie and Q. Zhao, Mater. Horiz., 2019, 6, 1013 RSC.
- C. Ma, Q. Liu, Q. Peng, G. Yang, M. Jiang, L. Zong and J. Zhang, ACS Nano, 2021, 15, 19877 CrossRef CAS.
- X. Li, R. Lin, G. Ni, N. Xu, X. Hu, B. Zhu, G. Lv, J. Li, S. Zhu and J. Zhu, Natl. Sci. Rev., 2018, 5, 70 CrossRef CAS.
- F. F. M. Cabrini, M. Champeau and M. G. Oliveira, J. Appl. Polym. Sci., 2020, 137, e49056 CrossRef.
- M. A. Haq, Y. Su and D. Wang, Mater. Sci. Eng., C, 2017, 70, 842 CrossRef PubMed.
- R. Fei, J. T. George, J. Park and M. A. Grunlan, Soft Mater., 2012, 8, 481 RSC.
- P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054 CrossRef CAS PubMed.
- J. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133 CrossRef CAS PubMed.
- C. Li, C. Wang, C. Keplinger, J. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. You and Z. Bao, Nat. Mater., 2016, 8, 618 CAS.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637 CrossRef CAS.
- Y. Ni, X. Zhou, J. Gong, L. Xue and Q. Zhao, Adv. Funct. Mater., 2021, 31, 2103818 CrossRef CAS.
- W. Wang, Y. Wang, J. Zheng, X. Yu, W. Chen, J. Li and Y. Liu, J. Mater. Chem. A, 2022, 10, 12608 RSC.
- X. Le, W. Lu, H. Xiao, L. Wang, C. Ma, J. Zhang, Y. Huang and T. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 9038 CrossRef CAS PubMed.
- Y. Xu, J. Xu, J. Zhang, X. Li, B. Fu, C. Song, W. Shang, P. Tao and T. Deng, Nano Energy, 2022, 93, 106882 CrossRef CAS.
- M. Gao, L. Zhu, C. K. Peh and G. W. Ho, Energy Environ. Sci., 2019, 12, 841 RSC.
- A. S. Alketbi, A. Raza, M. Sajjad, H. Li, F. AlMarzooqi and T. Zhang, EcoMat, 2022, 4, e12157 CrossRef CAS.
- L. Han, L. Yan, M. Wang, K. Wang, L. Fang, J. Zhou, J. Fang, F. Ren and X. Lu, Chem. Mater., 2018, 30, 5561 CrossRef CAS.
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353 CrossRef CAS.
- J. H. Ryu, P. B. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523 CrossRef CAS.
- D. Ding, H. Wu, X. He, F. Yang, C. Gao, Y. Yin and S. Ding, J. Mater. Chem. A, 2021, 9, 11241 RSC.
- P. Mu, Z. Zhang, W. Bai, J. He, H. Sun, Z. Zhu, W. Liang and A. Li, Adv. Mater., 2019, 9, 1802158 Search PubMed.
- M. Zhu, Y. Li, F. Chen, X. Zhu, J. Dai, Y. Li, Z. Yang, X. Yan, J. Song, Y. Wang, E. Hitz, W. Luo, M. Lu, B. Yang and L. Hu, Adv. Energy Mater., 2018, 8, 1701028 CrossRef.
- N. Xu, X. Hu, W. Xu, X. Li, L. Zhou, S. Zhu and J. Zhu, Adv. Mater., 2017, 29, 1606762 CrossRef PubMed.
- Y. Geng, W. Sun, P. Ying, Y. Zheng, J. Ding, K. Sun, L. Li and M. Li, Adv. Funct. Mater., 2021, 31, 2007648 CrossRef CAS.
- F. Zhao, X. Zhou, Y. Shi, X. Qian, M. Alexander, X. Zhao, S. Mendez, R. Yang, L. Qu and G. Yu, Nat. Nanotechnol., 2018, 13, 489 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mh00962e |
| This journal is © The Royal Society of Chemistry 2023 |