
High-contrast reversible multiple color-tunable solid luminescent ionic polymers for dynamic multilevel anti-counterfeiting†
Xiao
Ma
 *,
Mingyue
Zhou
,
Ling
Jia
,
Guangkun
Ling
,
Jiashu
Li
,
Wei
Huang
and
Dayu
Wu
*
*,
Mingyue
Zhou
,
Ling
Jia
,
Guangkun
Ling
,
Jiashu
Li
,
Wei
Huang
and
Dayu
Wu
*
Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology, Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, School of Petrochemical Engineering, Changzhou University, Changzhou, Jiangsu 213164, China. E-mail: maxiao@cczu.edu.cn; wudy@cczu.edu.cn
First published on 18th October 2022
Abstract
Dynamic color-tunable luminescent materials, which possess huge potential applications in advanced multilevel luminescence anti-counterfeiting, are of considerable interest. However, it remains challenging to develop simple high-contrast reversible multiple (triple or more than triple) color-tunable high-efficiency solid luminescent materials with low cost, facile synthesis, and good processability. Herein, by simply grafting charged multi-color AIEgen-based chromophores into polymers, a series of high-efficiency multiple color-tunable luminescent single ionic polymers are constructed through tuning feed ratios, counter anions and reaction solvents. Remarkably, some ionic polymers can not only achieve rare high-contrast reversible multiple color-tunable emission in solid states in response to different solvent stimuli, but also could realize excitation-dependent color-tunable emission. To the best of our knowledge, such charming multiple (triple or more than triple) color-tunable solid polymers responding to multiple external stimuli are still rare. Based on comparative studies of emission spectra, excitation spectra and fluorescence lifetimes before and after swelling, it could be inferred that solvent stimuli could induce microstructure changes of these ionic polymers and then change the aggregated-states of their corresponding AIE-active emission centers. Moreover, the different solvent stimuli could induce to produce different degrees of microstructure changes, resulting in their unique multiple color-tunable emission. More significantly, these smart color-tunable ionic polymers show great promise for applications in dynamic multilevel (three-level or even more than three-level) anti-counterfeiting.
New conceptsAlthough some reported luminescent polymers could realize color-tunable emission in solid states, they still have less switchable emission color, bad reversibility, low-contrast, and low emission efficiency, which are disadvantages in terms of practical applications in advanced luminescence anti-counterfeiting. Here, an innovative approach to integrate electrostatic interaction and multi-color aggregation-induced emission (AIE) chromophores into polymer chains endows AIE-based luminescent ionic polymers synchronously with high-efficiency emission, high-contrast triple (even more than triple) color-tunable emission, and excellent optical reversibility in solid states. More significantly, a fast and non-destructive approach to modulate multi-color emission of solid ionic polymers, which utilizes the fact that different solvent stimuli could induce ionic polymers to produce different degrees of microstructure changes, was proposed to achieve reversible multiple color-tunable emission of ionic polymers. In comparison with existing approaches, the following innovations have been presented: first, achieving reversible emission color regulation of ionic polymers via simple solvent swelling/de-swelling due to electrostatic interactions; second, achieving multiple color-tunable emission by only tuning different common solvents; and finally, facilely achieving dynamic three-level (even more than three-level) anti-counterfeiting in response to different solvent stimuli. These results pave the way to developing more novel multifunctional color-tunable emission materials based on the charged AIE-active chromophores for anti-counterfeiting, smart displays, and optical sensing. |
Introduction
Anti-counterfeiting is highly in demand in modern society due to the fact that counterfeit goods are seriously influencing human daily life.1 To prevent counterfeiting, luminescence anti-counterfeiting technology has attracted extensive attention, since plenty of luminescent materials displaying unique optical characteristics can be used.2 However, at present, the commercial static luminescent anti-counterfeiting materials, which exhibit a single emission color and can be easily counterfeited, already cannot meet advanced anti-counterfeiting technology demands. Alternatively, dynamic luminescent materials, which exhibit color-tunable emission under external stimuli (such as excitation light, chemical reagents, mechanical stimuli, heat stimuli, and other factors), possess huge potential applications in advanced luminescence anti-counterfeiting.3 Thus, much effort has been devoted to constructing fascinating color-tunable luminescent materials for advanced luminescence anti-counterfeiting technology in recent years. Fortunately, many different kinds of interesting color-tunable luminescent materials, including inorganic luminescent materials,4 carbon dots,5 organic small molecule dyes,6 supramolecular complexes,7 metal–organic frameworks,8 polymers,9 and so on, have been successfully developed so far. Although great progress has been achieved, to the best of our knowledge, many of the reported color-tunable luminescent materials require sophisticated and tedious synthesis, some color-tunable crystalline-state luminescent materials are not suitable for processability and some color-tunable multi-component luminescent materials show poor reversibility and optical instability. For practical applications in luminescence anti-counterfeiting technology, the development of simple color-tunable solid luminescent materials with low cost, facile synthesis, easy processability, good reversibility and excellent stability is urgently needed, but is still challenging.Among the various color-tunable luminescent materials, single luminescent polymers have received intense interest because they are not only be easily synthesized to offer diverse molecular structures, realizing multicolor emissions, but they also have good processability and excellent stability for applications.10 However, most single luminescent polymers can only achieve a kind of emission color in the aggregated solid states upon external stimuli, which could only serve as a single-level luminescence anti-counterfeiting material and it is difficult to meet the requirements of advanced multilevel luminescence anti-counterfeiting technology. Hence, the current important subject in this field is to exploit an effective synthetic strategy to construct multiple color-tunable solid single luminescent polymers in response to external stimuli.11
As far as we know, the emission behaviours of some small molecule organic luminescent materials are greatly dependent on their molecular structures/conformations or packing modes.12 Upon exposure to external stimuli, these small molecule organic luminescent materials could achieve color-tunable emission due to the fact that external stimuli in some degree could induce changes in their molecular structures/conformations or packing modes. In view of this, some believe that incorporating such color-tunable organic chromophores into polymers would be an effective strategy to create color-tunable luminescent polymers. Excitingly, on the basis of this strategy, some novel intriguing color-tunable luminescent polymers have been successfully designed.13 Specifically, some of them which could realize color-tunable emission are mainly ascribed to the structural transformations upon protonation or deprotonation and the presence of light-switchable chromophores in the polymer backbones (such as light switchable azobenzene, spiropyran, and diarylethene, and so on).14 Unlike those polymers, Li and coworkers have developed a kind of novel multicolor emission polymer by introducing coordination-switchable boron chromophores, which display temperature and solvent-dependent emission color changes in solution.15 More recently, they have further realized that the interesting color-tunable solid state polymeric materials responded to solvent stimuli and high pressure by controlling the packing density of the coordination-switchable boron chromophores.16 Relatively speaking, most of those structure switchable luminescent polymers in solid states still have a low emission efficiency, which is a disadvantage to their practical application in luminescence anti-counterfeiting.
To address this problem, it would be feasible to develop unconventional high-efficiency chromophores and then incorporate them into polymers. Significantly, a kind of organic luminogen with aggregation-induced emission (AIE) (called AIEgens), which displays enhanced emission in the aggregated solid states, has received intense attention in the last two decades.2h,17 In particular, the AIE properties of those AIEgens are highly sensitive to microenvironmental variations. Accordingly, by integrating AIEgens into a polymer system, a great number of color-tunable high-efficient luminescent polymers or polymeric gels have been fabricated using three synthetic methods, including covalently bonding AIEgens into polymer chains, physically doping AIEgens into polymer matrixes, and supramolecular polymerization of AIEgens.18 Therein, covalently bonding AIEgens into polymer chains should be the more effective strategy to develop color-tunable single luminescent polymers. It should be noted that many color-tunable single luminescent polymers obtained by this strategy could merely exhibit multi-color emission in solution states.19 By contrast, solid color-tunable AIEgen-based single luminescent polymers have been very limited up until now.20 More recently, Gu and coworkers constructed high-efficiency color-tunable AIEgen-based solid core–shell fluorescent polymeric particles based on aggregation microenvironment manipulation.21 In fact, the related color-tunable AIEgen-based polymeric particles in solid states display irreversible emission from white to blue then to orange before and after wetting. From the practical application point of view, it would be desirable to exploit more reversible color-tunable solid AIEgen-based single luminescent polymers.
In addition, from the point of view of security capability, most of the reported color-tunable AIEgen-based single luminescent polymers could usually display two different emission colors in solid states in response to external stimuli,20 which may be applied to the double-level luminescence anti-counterfeiting mode. Relatively speaking, they should have a higher anti-counterfeiting capability than the static single-level luminescent anti-counterfeiting materials. In particular, if color-tunable AIEgen-based single luminescent polymers could achieve three or more different emission colors in solid states in response to single stimuli or multiple different stimuli, they would be used for multilevel luminescence anti-counterfeiting technology. To some extent, a more multilevel luminescence anti-counterfeiting mode should be harder to forge compared with a single-level or double-level luminescence anti-counterfeiting mode, which would be the most promising next-generation advanced luminescence anti-counterfeiting technology.2a,9b,9h,22 Nevertheless, such multiple color-tunable solid single luminescent polymers are still very rare to date. In particular, we could envisage that such multiple color-tunable AIEgen-based solid single luminescent polymers responding to multiple external stimuli should be accessed, given that some AIEgen-based chromophores with three or more emission colors in solid states could be covalently bound to polymer chains.
Herein, by covalently grafting novel charged multi-color AIEgen-based chromophores into polymer chains, we have successfully obtained a series of simple multiple color-tunable solid luminescent ionic polymers through tuning the feed ratios, counter anions and reaction solvents. Significantly, some high-efficiency ionic polymers could not only achieve rare high-contrast reversible multiple color-tunable emission in solid states in response to different solvent stimuli, but also could realize excitation-dependent color-tunable emission. To the best of our knowledge, such charming multiple color-tunable solid polymers responding to multiple external stimuli are still not frequently reported to date. Due to various emission characteristics, these AIEgen-based color-tunable solid single luminescent ionic polymers would possess great potential in dynamic multilevel anti-counterfeiting.
Results and discussion
Emission properties of small-molecule ionic compounds
In principle, covalently bonding multicolor AIEgens into polymer chains could construct multiple color-tunable single luminescent polymers, whereas one of the key issues faced is to exploit such novel functional AIEgens which could display multicolor emission in the different conditions. Herein, three AIE-active small-molecule ionic compounds with different anions (L-Cl, L-PF6 and L-BF4) have been synthesized (Fig. 1a, see Scheme S1 and the ESI†). Firstly, L-Cl could be facilely obtained by the reaction of ligand L (L = 2,6-di(naphthalen-2-yl)-4,4′-bipyridine) and benzyl chloride in acetonitrile (CH3CN). Subsequently, by exchanging Cl− with [PF6]− or [BF4]− in methanol, L-PF6 and L-BF4 could be gained, respectively. They have been characterized using NMR data, mass spectra and IR analysis (see the ESI†). For L-Cl, L-PF6 and L-BF4, their solutions exhibit similar weak orange emission peaks at about 628 nm because their multiple rotatable aromatic (pyridine/naphthalene) rings could serve as non-radiative pathways to deactivate the excited states in solution (Fig. 1b and Table S1, ESI†). Interestingly, after adding water into their solution, the resulting aggregation sates show enhanced different emission compared to their corresponding solution (Fig. 1b and Table S1, ESI†), which should be mainly due to the fact that their multiple rotational motions could be restricted in solution aggregation states. This demonstrates that these ionic compounds with the same emission center L+ (L-Cl, L-PF6 and L-BF4) display similar AIE characteristics.23 In particular, in crystalline states, the three ionic compounds exhibit different bright emission color under a 365 nm UV lamp (Fig. 1c and Table S1, ESI†). Specifically, L-Cl emits cyan light with a maximum emission peak at 477 nm, L-PF6 emits yellow green light with an emission peak at 529 nm and L-BF4 emits yellow light with an emission peak at 548 nm (Fig. 1c). Moreover, L-BF4 has the highest quantum yield of up to 84.6% under 365 nm excitation. Such distinct emission differences suggest that the counter anion effects have a great influence on the emission properties of the AIE-active emission center L+ of the ionic compounds in this system, which are mainly due to the fact that the different counter anions could lead to their different molecule packing modes in the solid states and then the different intro-molecular/intermolecular interactions (Fig. S1, ESI†). Similar phenomena have also been observed in some ionic compounds.24 In other words, the emission properties of the AIE-active emission center L+ are sensitive to the local microenvironment. From this, we assume that incorporating this charged AIE-active multi-color emission center L+ into polymers may construct the desired color-tunable single luminescent ionic polymers responding to external stimuli. | ||
| Fig. 1 (a) Molecular structures of L-Cl, L-PF6 and L-BF4; (b) emission spectra of L-Cl, L-PF6 and L-BF4 in solution states (10−4 mol L−1) and the corresponding solution aggregation states after increasing the water volume fraction to 90%. Inset images: their corresponding emission photographs under 365 nm UV lamp. (c) Emission spectra of L-Cl, L-PF6 and L-BF4 in the crystalline solid sates and their corresponding emission photographs under a 365 nm UV lamp (inset images). | ||
Emission properties of ionic polymers prepared in solvent CH3CN
To obtain color-tunable solid single luminescent ionic polymers, a simple polymer post-modification strategy has been employed.25 At first, the emissive ionic polymers Pn-Cl (mass ratios of L/P = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, n = 1; 1:2, n = 2) were facilely synthesized by grafting ligand L on the commercially available chloromethyl polystyrene resin P through tuning the mass ratios of L and P in CH3CN (Fig. 2a, see Scheme S2 and the ESI†). At room temperature, those ionic polymers are nearly insoluble in some common solvents, such as DCM, EtOH (ethanol), MeOH (methanol), EA (ethyl acetate), DMF, and DMSO. Details for the syntheses and characterizations of the ionic polymers are shown in the ESI† (see Fig. S2 and S3). X-Ray powder diffraction measurements reveal that the two ionic polymers are in amorphous states (Fig. S3, ESI†). It is observed that the ionic polymers tend to aggregate into microsphere particles. The SEM graph of P1-Cl further indicates that the sizes of the microsphere particles are in the range of about 10–100 μm in diameter (Fig. 2b). Additionally, the emission of both P1-Cl and P2-Cl has been measured, as shown in Fig. 2c. Upon excitation at 365 nm, P1-Cl exhibits a maximum emission peak at 542 nm with a quantum yield of 11.2%, and P2-Cl displays a slight red-shifted emission peak at 568 nm with a quantum yield of 7.0%. Clearly, both emissions are different from the emission of the precursors P and L (Fig. 2c), which demonstrates the formation of the new ionic polymers. By comparison, it also could be concluded that the emission of P1-Cl and P2-Cl should originate from the emission of the newly produced AIE-active emission center L+. Moreover, we note that both P1-Cl and P2-Cl with different feed-ratios exhibit different emission, suggesting that the ratios of the suspended AIE-active emission center (L+) could affect the emission of the related ionic polymers. Furthermore, the emission properties of both P1-Cl and P2-Cl in response to solvent DCM stimuli have been explored, respectively (Fig. S4, ESI†). It is found that both emission peaks show small red-shifts (20 nm red-shift for P1-Cl, 10 nm red-shift for P2-Cl) after wetting them with DCM. Thus, no high-contrast emission color changes could be observed in those ionic polymers containing Cl− anion.
:10, n = 1; 1:2, n = 2) were facilely synthesized by grafting ligand L on the commercially available chloromethyl polystyrene resin P through tuning the mass ratios of L and P in CH3CN (Fig. 2a, see Scheme S2 and the ESI†). At room temperature, those ionic polymers are nearly insoluble in some common solvents, such as DCM, EtOH (ethanol), MeOH (methanol), EA (ethyl acetate), DMF, and DMSO. Details for the syntheses and characterizations of the ionic polymers are shown in the ESI† (see Fig. S2 and S3). X-Ray powder diffraction measurements reveal that the two ionic polymers are in amorphous states (Fig. S3, ESI†). It is observed that the ionic polymers tend to aggregate into microsphere particles. The SEM graph of P1-Cl further indicates that the sizes of the microsphere particles are in the range of about 10–100 μm in diameter (Fig. 2b). Additionally, the emission of both P1-Cl and P2-Cl has been measured, as shown in Fig. 2c. Upon excitation at 365 nm, P1-Cl exhibits a maximum emission peak at 542 nm with a quantum yield of 11.2%, and P2-Cl displays a slight red-shifted emission peak at 568 nm with a quantum yield of 7.0%. Clearly, both emissions are different from the emission of the precursors P and L (Fig. 2c), which demonstrates the formation of the new ionic polymers. By comparison, it also could be concluded that the emission of P1-Cl and P2-Cl should originate from the emission of the newly produced AIE-active emission center L+. Moreover, we note that both P1-Cl and P2-Cl with different feed-ratios exhibit different emission, suggesting that the ratios of the suspended AIE-active emission center (L+) could affect the emission of the related ionic polymers. Furthermore, the emission properties of both P1-Cl and P2-Cl in response to solvent DCM stimuli have been explored, respectively (Fig. S4, ESI†). It is found that both emission peaks show small red-shifts (20 nm red-shift for P1-Cl, 10 nm red-shift for P2-Cl) after wetting them with DCM. Thus, no high-contrast emission color changes could be observed in those ionic polymers containing Cl− anion.
 | ||
| Fig. 2 (a) Molecular structures of ionic polymers Pn-Cl (n = 1, 2), Pn-BF4 (n = 1–7) and Pn-PF6 (n = 1–7); (b) SEM images of P1-Cl, P1-PF6 and P1-BF4; (c) emission spectra of P, L, P1-Cl, P1-Cl, P1-PF6 and P1-BF4 under 365 nm excitation; (d and e) emission photographs of P1-PF6 and P1-BF4 under different conditions; (f and g) emission spectra of P1-PF6 and P1-BF4 in the dry states and the wetted states by the different solvents; (h and i) excitation spectra of P1-PF6 and P1-BF4 in the dry states and the wetted states by solvent DCM and DMF; (j and k) emission lifetime of P1-PF6 and P1-BF4 in the dry states and the wetted states by solvent DCM and DMF. | ||
In order to further develop high-contrast color-tunable emission materials, ionic polymers (P1-PF6 and P1-BF4) have been synthesized by grafting ligand L on polymer P through exchanging Cl− with [PF6]− or [BF4]− in CH3CN (Fig. 2a, and Scheme S2, ESI†), respectively. They have been characterized using solid state 13C NMR data, TGA, SEM and SEM-EDS (Fig. 2b and Fig. S5–S8, ESI†). We find that the two new amorphous polymers also tend to aggregate into microsphere particles of about 10–100 μm in diameter and have excellent thermal stability (Fig. 2b and Fig. S6, S7, ESI†). Typically, SEM-EDS analysis indicates that P1-PF6 contains element F, confirming the formation of the new ionic polymer (Fig. S8, ESI†). Similar to P1-Cl, both P1-PF6 and P1-BF4 are also nearly insoluble in some common solvents at room temperature, so they would not fade away in solid states in response to various solvent stimuli. Furthermore, emission spectra measurements reveal that P1-PF6 exhibits cyan light with a maximum emission peak at 501 nm and P1-BF4 displays a green peak at 527 nm under 365 nm excitation (Fig. 2c). Moreover, both ionic polymers exhibit high quantum yields (31% for P1-PF6 and 20% for P1-BF4) (Fig. S9, ESI†). Notably, three ionic polymers (P1-Cl, P1-PF6 and P1-BF4) display different emissions even though they have the same emission center (L+) and the same feed ratios, implying that the emission properties of the ionic polymers in this system could be regulated by their counter anions. This is similar to some related small-molecule ionic compounds containing different counter anions.24a,b,d From this, we may deduce that the emission differences of those ionic polymers should be closely correlated with their different aggregation ground states (see below).
To our excitement, when adding a drop of DCM into P1-PF6 and P1-BF4, respectively, we find that both P1-PF6 and P1-BF4 begin to swell, and at the same time, their emission colors change greatly and quickly under 365 nm excitation (Fig. 2d and e). That is, the emission of P1-PF6 changes from cyan to yellow green and the emission of P1-BF4 changes from yellowish green to yellow. After further adding DCM, the swelling P1-PF6 and P1-BF4 emit off-white light and yellow light, respectively (Fig. 2d and e). Their emission spectra reveal that the swelling P1-PF6 and P1-BF4 exhibit the very broad emission bands (Fig. 2f and g). Compared to the corresponding dry ionic polymers, the swelling P1-PF6 displays an about 41 nm red-shift emission peak at 542 nm, and the swelling P1-BF4 exhibits an about 49 nm red-shift emission peak at 576 nm. Significantly, after 3 minutes, their emission colors almost recover to the initial states because volatilization of DCM leads to deswelling of the swelling P1-PF6 and P1-BF4. In short, introduction/volatilization of solvent DCM in this system could induce swelling/deswelling of the ionic polymers in solid states, thereby causing the high-contrast reversible color-tunable emission. What's more, only by simply adjusting the counter anions, two different reversible color-tunable emission materials have been obtained easily. On the other hand, it could be inferred that their reversible emission color changes before and after adding DCM should be mainly attributed to the fact that the swelling/deswelling of ionic polymers could modify the microenvironment of the AIE-active emission center L+. Or rather, their corresponding aggregation ground states could be regulated in some degree before and after wetting, resulting in the different intramolecular/intermolecular interactions. This may be supported by their changed excitation spectra from the dry states to the wetted states (Fig. 2h and i). Additionally, the fluorescence lifetimes of both P1-PF6 and P1-BF4 before and after wetting have been detected, respectively (Fig. 2j and k). It is found that the fluorescence lifetime of P1-PF6 reduces from 8.9 ns in the dry state to 5.4 ns in the wetted state with DCM. Likewise, the fluorescence lifetime of P1-BF4 reduces from 9.8 ns in the dry state to 3.1 ns in the wetted state with DCM. Their varied fluorescence lifetimes could further support changes of their corresponding aggregation microenvironment. Moreover, it is worth mentioning that after 10 cycles of DCM uptake and release, it is found that both emission spectra almost completely recover to the initial dry states, suggesting their excellent optical reversibility (Fig. S10, ESI†). On the basis of many previous studies which indicate that some charged ionic polymers could exhibit good self-recovery because of electrostatic interactions,26 we presume that the excellent optical reversibility in this system may also be mainly due to the presence of electrostatic interactions in these charged ionic polymers. That is, the abundant electrostatic interactions are beneficial to enable these swelling ionic polymers to recover to the initial aggregated states after deswelling.
For advanced multilevel anti-counterfeiting, achieving multiple stimuli-responsive emission of luminescent materials is highly needed.2a In view of the fact that both P1-PF6 and P1-BF4 in solid states have realized interesting color-tunable emission in response to solvent DCM stimuli, we have further studied the stimuli-responsive emission properties of both P1-PF6 and P1-BF4 in response to many other solvents (Fig. 2d and e). After wetting P1-PF6 and P1-BF4 with EtOH, respectively, the wet P1-PF6 does not exhibit distinct emission color changes and the wet P1-BF4 shows little emission color change under 365 nm excitation. As expected, the emission spectra of P1-PF6 wetted by EtOH is nearly identical to that of the dry P1-PF6, and P1-BF4 wetted by EtOH shows a little blue-shifted emission peak at 518 nm (Fig. 2f and g). After adding EA into the dry P1-PF6 and P1-BF4, respectively, it can be seen that the emission color of P1-PF6 changes from cyan to yellow green and P1-BF4 exhibits the little emission color change. The corresponding emission spectra indicate that P1-PF6 wetted by EA shows a red-shifted emission band with a maximum emission peak at 530 nm and P1-BF4 wetted by EA show a broadened emission band with a maximum emission peak at 536 nm (Fig. 2f and g). By contrast, after wetting P1-PF6 and P1-BF4 with DMF, respectively, both their emission intensities clearly decrease (Fig. 2d and e). Specifically, upon excitation at 365 nm, P1-PF6 wetted with DMF exhibits weak white light emission and P1-BF4 wetted with DMF displays weak yellow green light. Furthermore, their corresponding emission spectra have been recorded, respectively (Fig. 2f and g). Fig. 2f indicates that P1-PF6 wetted with DMF shows a very broad emission band with a maximum emission peak at 460 nm and a shoulder peak at about 550 nm. The corresponding Commission Internationale de l’Eclairage (CIE) coordinates of (0.26, 0.28) ascertain its white light emission. It is noted that the maximum emission peak at 460 nm is close to the maximum emission peak of P (λem = 450 nm) in the dry state and the wetted state under 365 nm excitation (Fig. S11, ESI†), which demonstrates that the emission peak at 460 nm should be a result of the emission of the polymer chain P. The shoulder peak at about 550 nm should be attributed to the emission of the suspended AIE-active emission center L+. Fig. 2g indicates that P1-BF4 wetted with DMF shows a slightly red-shifted and broadened emission band with a maximum peak at 536 nm compared with the dry P1-BF4. Furthermore, the fluorescence lifetimes of both P1-PF6 wetted with DMF and P1-BF4 wetted with DMF have been measured, respectively. It is found that the fluorescence lifetime of the former for the emission peak at 550 nm reduces to 2.0 ns and the fluorescence lifetime of the latter for the emission peak at 536 nm reduces to 2.2 ns (Fig. 2j and k). In addition, we find that their related excitation spectra show many changes before and after wetting with DMF (Fig. 2h and i). All these changes confirm that solvent DMF stimuli could also induce microstructure changes of P1-PF6 and P1-BF4. It should be noted that both P1-PF6 and P1-BF4 wetted with DMF would take a relatively long time to restore to their initial emission color because solvent DMF evaporates slowly. Interestingly, after adding EtOH into both P1-PF6 and P1-BF4 wetted with DMF, respectively, we find that P1-PF6 wetted with DMF firstly switches to green emission and then recovers to the initial emission color after one day, and P1-BF4 wetted with DMF immediately recovers to the enhanced yellow green emission. These studies also verified their excellent optical reversibility. All in all, both P1-PF6 and P1-BF4 could respond to the different solvents, achieving the intriguing high-contrast reversible multiple color-tunable emission in solid states. To the best of our knowledge, such high-contrast reversible multiple color-tunable solid emission materials responding to different solvent stimuli are still not frequently reported so far.
Besides solvent stimuli-responsive emission, exploiting excitation-wavelength dependent color-tunable emission materials for anti-counterfeiting is also fascinating from theoretical research to practical application.2a,9j,27 Therefore, we further explore excitation-wavelength dependent emission of both P1-PF6 and P1-BF4 in the dry states and the wetted states, respectively (Fig. 3 and Fig. S12, ESI†). In the dry states, both P1-PF6 and P1-BF4 do not exhibit obvious excitation-dependent color-tunable emission (Fig. S12, ESI†). After wetting them with DCM or DMF, some interesting excitation-dependent emission phenomenon have been observed (Fig. 3). Therein, P1-BF4 wetted with DCM or DMF has no obvious excitation-dependent color-tunable emission (Fig. 3e and f). Significantly, P1-PF6 wetted with DCM and P1-PF6 wetted with DMF show excitation-dependent color-tunable emission (Fig. 3a–c). As shown in Fig. 3a, under 330 nm excitation, P1-PF6 wetted with DCM exhibits broad dual emission peaks at 386 nm and 542 nm, so white-light emission could be achieved, which is also supported by the calculated CIE coordinates of (0.32, 0.39) (Fig. 3b). Notably, the emission peak at 386 nm is close to the maximum emission peak of P (λem = 390 nm) in the dry state and the wetted state under 330 nm excitation (Fig. S11, ESI†), implying that the emission peak at 386 nm should arise from the emission of the polymer chain P. Moreover, it could be inferred that the emission peak at 542 nm should be ascribed to the emission of the AIE-active emission center L+. With increasing the excitation wavelength to 350 nm, the emission peak at 386 nm gradually decreases, and then a broad emission band with a maximum emission peak at 542 nm could still be seen. Thus, the emission is still white-light. With further increasing the excitation wavelength to 400 nm, the emission peak at 386 nm gradually fades, and the broad emission band becomes narrow. Consequently, P1-PF6 wetted with DCM emits yellow green light under 400 nm excitation. After increasing to 420 nm, P1-PF6 wetted with DCM still emits yellow green light. The relevant emission changes could be further verified by the corresponding CIE coordinates (Fig. 3b). On the whole, by changing the excitation wavelength from 330 nm to 420 nm, P1-PF6 wetted with DCM could achieve continuous color-tunable emission from white to yellow green. For P1-PF6 wetted with DMF, it displays a broad emission band with a strong emission peak at 389 nm and a weak shoulder peak at about 540 nm under 330 nm excitation. Thus, blue light emission could be observed, which is supported by the corresponding CIE coordinates of (0.22, 0.18) (Fig. 3d). After gradually increasing the excitation wavelength to 365 nm, the emission peak at 389 nm constantly decreases, and a new emission peak at about 460 nm with the shoulder peak at 540 nm could be seen. Hence, the weak white light emission could be realized. After increasing the excitation wavelength to 390 nm, P1-PF6 wetting with DMF displays a very broad emission band and also emits white light. After gradually increasing excitation wavelength to 430 nm, P1-PF6 wetted with DMF emits a yellow emission peak at 545 nm with CIE coordinates of (0.35, 0.45). In short, P1-PF6 wetted with DMF could achieve the tunable multi-color changes from blue to white and then to yellow green by changing the excitation wavelength from 330 nm to 430 nm (Fig. 3d). It is worth mentioning that it is very rare that such a simple single ionic polymer P1-PF6 in solid states could simultaneously achieve multiple color-tunable emission in response to several different solvent stimuli and excitation-dependent color-tunable emission. Moreover, these encouraging results demonstrate that integrating the AIE-active chromophores into the emissive polymer chains would be a good strategy to construct novel multiple color-tunable smart emission materials.
 | ||
| Fig. 3 (a and b) Emission spectra of P1-PF6 wetted with DCM under the different excitation wavelengths and the corresponding CIE coordinates; (c and d) emission spectra of P1-PF6 wetted with DMF under different excitation wavelengths and the corresponding CIE coordinates; (e and f) emission spectra of P1-BF4 wetted with DCM and P1-BF4 wetted with DMF under the different excitation wavelengths. | ||
Inspired by the foregoing encouraging results, we have tried to carry out different mass-ratio reactions between L and P to acquire more color-tunable emission materials with different emission color, which would meet various application requirements. Here, six ionic polymers with different feed ratios, Pn-BF4, Pn-PF6 (mass ratios of L to P = 1/2, n = 2; 2/3, n = 3; 1/1, n = 4) have been synthesized in CH3CN, and their emission properties have been studied in detail (Fig. 4 and see the ESI†). Typically, X-ray powder diffraction, SEM images and TGA measurements of ionic polymers (P2-PF6, P2-BF4) further reveal that the series of amorphous ionic polymers also tend to aggregate into microsphere particles and show excellent thermal stability (Fig. S8 and S13–S15, ESI†). As expected, those amorphous ionic polymers are also nearly insoluble in some common solvents at room temperature, which would be advantageous in response to some solvent stimuli in solid states. Fig. 4a discloses that, in comparison with P1-PF6 and P1-BF4, six ionic polymers (P2-PF6, P2-BF4, P3-PF6, P3-BF4, P4-PF6, P4-BF4) display the red-shifted maximum emission peaks under 365 nm excitation. Overall, their maximum emission peaks display large red-shifts of about 105 nm with increasing feed-ratios from 1/10 to 1/1. Notably, although the first four ionic polymers (P2-PF6, P2-BF4, P3-PF6, P3-BF4) have been obtained from the different feed-ratios, they emit similar yellow light with the approximate maximum emission peaks at 543–550 nm. Those imply that, with increasing feed ratios of L+ to a certain range, the produced ionic polymers in the dry states may tend to form similar aggregation ground states by self-regulation. This could be supported by their similar excitation spectra in the dry states (Fig. 4b). Moreover, we find that four ionic polymers (P2-PF6, P2-BF4, P3-PF6, P3-BF4) in the dry states similarly show no obvious color-tunable emission changes upon excitation at 330–455 nm (Fig. S16, ESI†). For both P4-PF6 and P4-BF4 with the higher feed-ratios, they also emit yellow light, but they exhibit a broader emission band (Fig. 4a). Their corresponding excitation energy spectra for the emission peak at 590 nm are even extended to a longer wavelength compared with those ionic polymers (P1-PF6, P1-BF4, P2-PF6, P2-BF4, P3-PF6, P3-BF4) (Fig. 4b). In view of their broad excitation energy spectra, the excitation-dependent emission of P4-PF6 and P4-BF4 has also been studied, respectively. It is found that both P4-PF6 and P4-BF4 show no obvious emission changes upon excitation at 365–460 nm. With further increasing the excitation wavelength to 520 nm, their emission bands become narrow and the corresponding maximum emission peaks display some red-shifts. Consequently, the emission color has changed from yellow to orange, which could be supported by the corresponding CIE coordinates (Fig. S17, ESI†). Importantly, we find that those ionic polymers also display good luminescence efficiency and their corresponding luminescence efficiencies slightly decrease with increasing the feed-ratios (35% for P2-PF6, 21% for P2-BF4, 18% for P3-PF6, 16% for P3-BF4, 15% for P4-PF6 and 11% for P4-BF4) (Fig. S18, ESI†). In brief, those results prove that tuning the feed-ratios of the AIE-active emission center (L+) could modify the emission properties of the related ionic polymers, finally developing a wide-range of high-efficiency multi-color emission materials. Furthermore, the solvent stimuli responsive emission of those ionic polymers (P2-PF6, P2-BF4, P3-PF6, P3-BF4, P4-PF6, P4-BF4) have been explored, respectively, as shown in Fig. 4c–f and Fig. S19 (ESI†). After wetting those ionic polymers with DCM, the maximum emission peaks of the first four swelling ionic polymers display large red-shifts (about 40 nm red-shift from 543 nm to 583 nm for P2-PF6, about 34 nm red-shift from 546 nm to 580 nm for P2-BF4, about 45 nm red-shift from 545 nm to 590 nm for P3-PF6 and about 43 nm red-shift from 546 nm to 589 nm for P3-BF4). Thus, high-contrast emission color changes could be observed in the first four ionic polymers before and after wetting with DCM (Fig. 4c). For both P4-BF4 and P4-PF6 with the broader emission band, after wetting them with DCM, both emission spectra do not exhibit large shifts, but it could be seen that both P4-BF4 and P4-PF6 wetted with DCM have relatively narrow emission bands with maximum emission peaks at about 590–610 nm, so the emission color changes are still evident. Notably, due to rapid evaporation of the solvent DCM, all those deswelling ionic polymers recover to their initial emission color, confirming their reversible color-tunable emission (Fig. 4c). After wetting those ionic polymers with EA or EtOH, no high-contrast emission changes could be found mainly because of no obvious swelling (Fig. 4c–f and Fig. S19, ESI†). In particular, after wetting all those ionic polymers with DMF, it is found that the emission intensity of those swelling ionic polymers have been largely weakened. Accordingly, they display red-shifted broad weak emission bands with maximum emission peaks at about 600–620 nm. Then, after adding EtOH into those ionic polymers wetted with DMF, they quickly return to the near initial emission color. All in all, these different ionic polymers could achieve high-contrast reversible multiple color-tunable emission in response to different solvent stimuli, implying that they would also be potentially applied to advanced multi-level anti-counterfeiting.
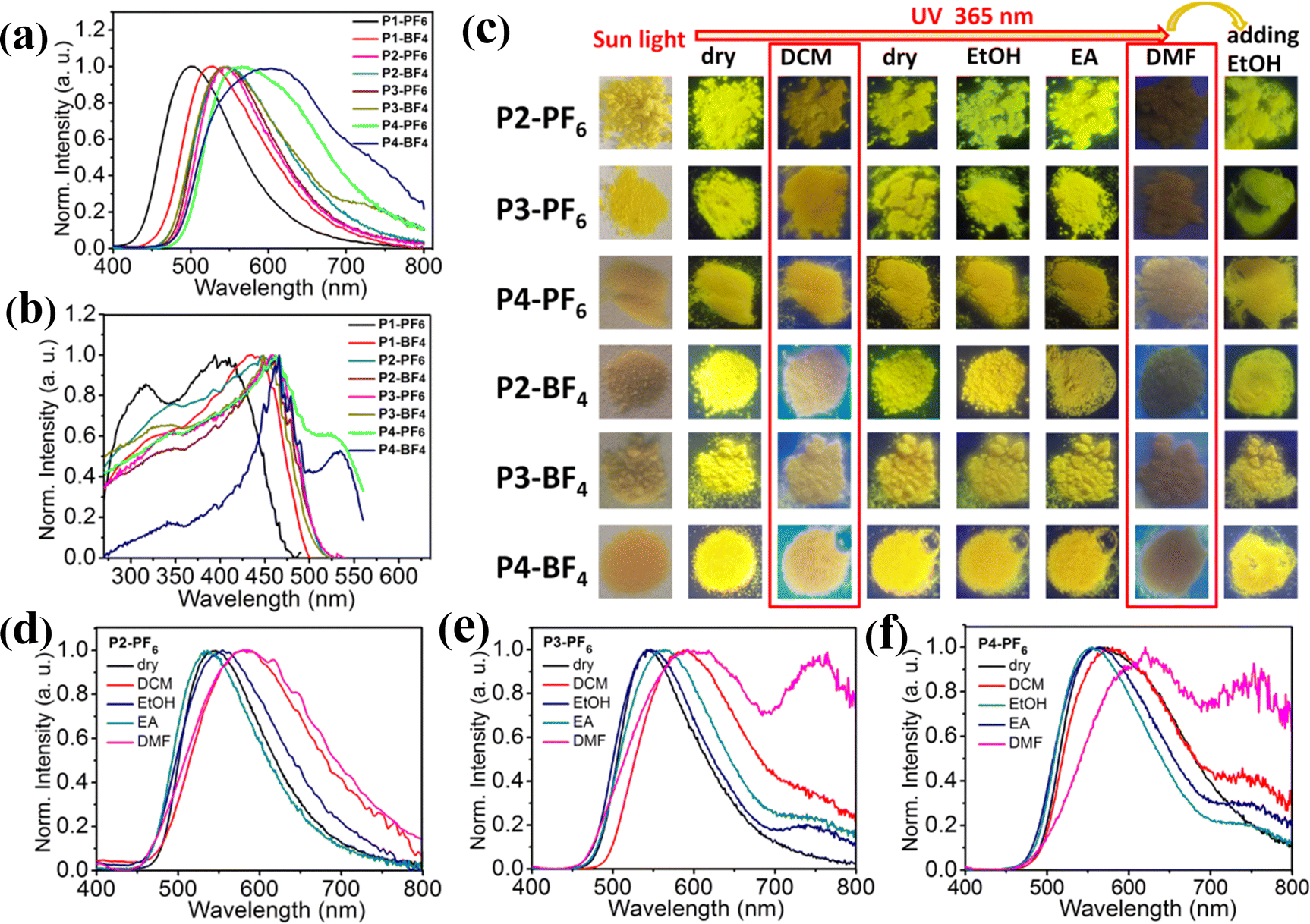 | ||
| Fig. 4 (a) Emission spectra of Pn-BF4 and Pn-PF6 (n = 1–4) under 365 nm excitation; (b) excitation spectra for P1-PF6 (λem = 501 nm), P1-BF4 (λem = 527 nm), P2-PF6 (λem = 545 nm), P2-BF4 (λem = 545 nm), P3-PF6 (λem = 545 nm), P3-BF4 (λem = 545 nm), P4-PF6 (λem = 590 nm), and P4-BF4 (λem = 590 nm); (c) emission photographs of P2-PF6, P2-BF4, P3-PF6, P3-BF4, P4-PF6 and P4-BF4 in the dry states and the various wetted states responding to the different solvents; (d–f) emission spectra of P2-PF6, P3-PF6 and P4-PF6 in the dry states and the various wetted states responding to the different solvents. | ||
Notably, after wetting those ionic polymers (P2-PF6, P2-BF4, P3-PF6, P3-BF4, P4-PF6, P4-BF4) with DCM or DMF, their emission becomes relatively weak and even their red-shifted emission peaks at about 580–620 nm are close to the weak emission peaks at about 628 nm of those AIE-active small-molecule ionic compounds (L-Cl, L-PF6 and L-BF4) in solution (Fig. 1b, 4d–f and Fig. S19, ESI†). This phenomenon should be understandable, since the introduction of DCM or DMF could enable those ionic polymers to swell, concurrently, and the aggregation microstructure (such as packing models or conformations) of the suspended AIE-active emission center L+ could be changed in some degree and their related aromatic (pyridine/naphthalene) rings could moderately rotate similar to solution states. In addition, we also note that the emission intensity of those ionic polymers wetted with DMF is weaker than that of the ionic polymers wetted with DCM. This may be due to the fact that these ionic polymers have a greater swelling degree in DMF, resulting in the fact that their related aromatic rings could more easily rotate and then the excited states could decay rapidly by non-emissive paths (see below).
In order to further reveal their microstructure changes before and after swelling, we have done a comparative study on the excitation energy spectra of these ionic polymers (P2-PF6, P2-BF4, P3-PF6, P3-BF4, P4-PF6, P4-BF4) in the dry states and the wetted states with DCM and the wetted states with DMF, as shown in Fig. 5. For P2-PF6 with a relatively low feed-ratio, after wetting with either DCM or DMF, the related maximum excitation energy bands are slightly blue-shifted (Fig. 5a). Similar changes have been observed in P1-PF6 with a lower feed-ratio. Whereas, for P3-PF6 with the relatively high feed-ratio, the maximum excitation energy band has an obvious red-shift after wetting with DCM. After wetting with DMF, the excitation band is also red-shifted, but the high energy excitation band from 270 nm to 450 nm relatively diminishes (Fig. 5b). Moreover, P4-PF6 shows a similar change with P3-PF6 before and after wetting. On the whole, for these ionic polymers containing [PF6]−, their excitation spectra exhibit some visible changes before and after wetting, suggesting their microstructure changes in response to solvent DCM or DMF stimuli. For the ionic polymers containing [BF4]−, their excitation spectra also exhibit some similar interesting change trends from the dry states to the wetted states with DCM then to the wetted states with DMF (Fig. S20, ESI†). As shown in Fig. S20 (ESI†), compared with the dry P2-BF4, P2-BF4 wetted by DCM exhibits a little red-shifted excitation band and P2-BF4 wetted by DMF display a clearly red-shifted broader excitation band. In particular, P3-BF4 wetted by DCM shows broader excitation spectra with a low-energy shoulder peak in comparison with the dry P3-BF4. Moreover, the low-energy shoulder peak largely increases in P3-BF4 wetted by DMF. In addition, we find that the low-energy shoulder peak of the excitation band of the dry P4-BF4 also greatly increases after wetting by DCM, and then further elevates after wetting by DMF. Taken together, changes of these excitation energy spectra not only reveal the microstructure changes of those ionic polymers before and after wetting, but also suggest that their microstructures may undergo different degrees of transformation from the dry states to the wetted states with DCM then to the wetted states with DMF, which is in line with changes of the foregoing corresponding emission color. In addition, the fluorescent lifetimes of those ionic polymers (P2-PF6, P2-BF4, P3-PF6, P3-BF4, P4-PF6, P4-BF4) in the dry states and the wetted states with DCM and the wetted states with DMF have been detected, respectively, and it is found that their fluorescent lifetimes gradually decreased from the dry states to the wetted states with DCM then to the wetted states with DMF (Fig. 5d–f and Fig. S20, ESI†), which further support their microstructure changes from the dry states to the wetted states with DCM or then to the wetted states with DMF.
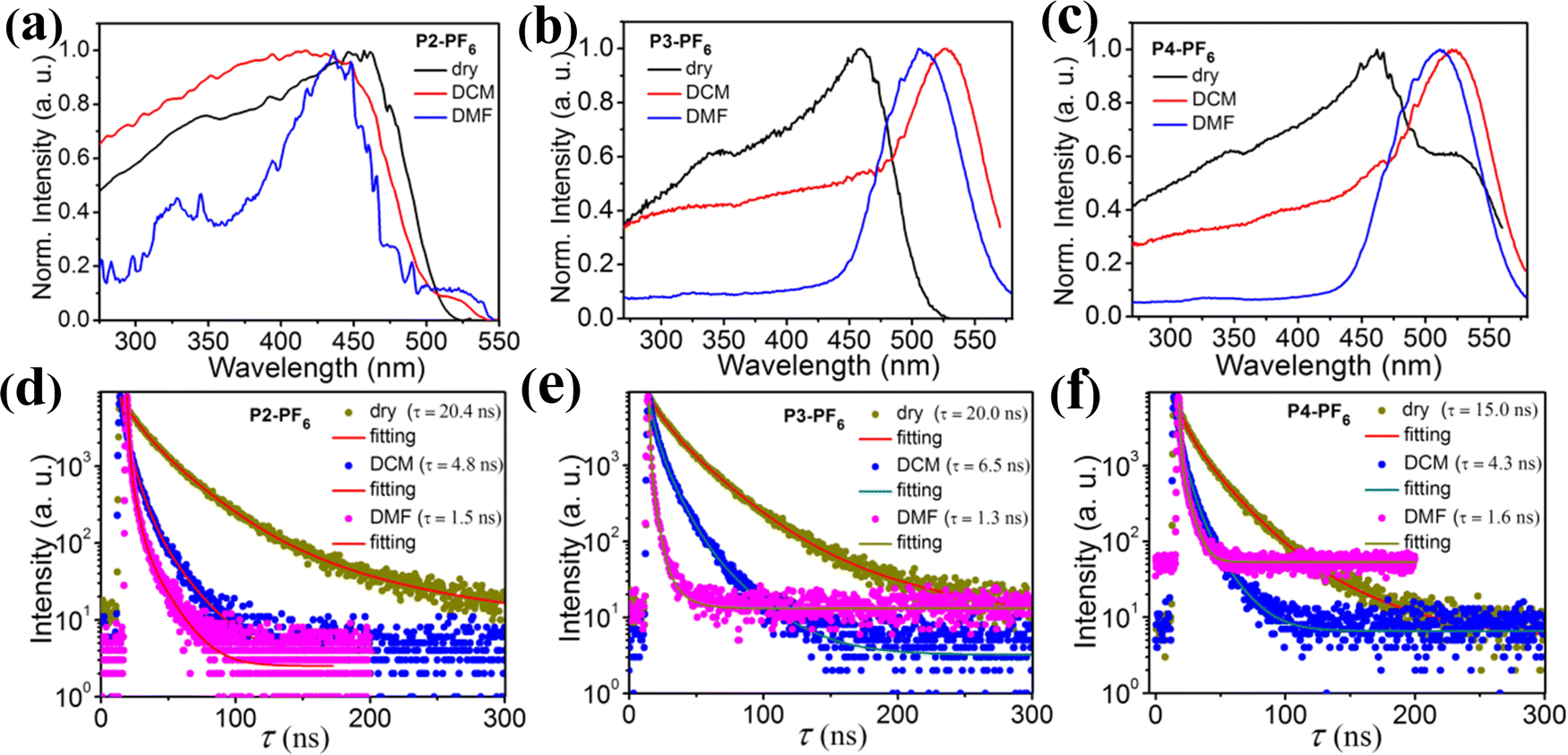 | ||
| Fig. 5 (a–c) Excitation spectra of those ionic polymers (P2-PF6, P3-PF6, P4-PF6) in the dry states, the wetted states with DCM and the wetted states with DMF. (d and f) Emission lifetimes of those ionic polymers (P2-PF6, P3-PF6, P4-PF6) in the dry states, the wetted states with DCM and the wetted states with DMF. | ||
In light of the fact that some ionic polymers have exhibited broadened excitation energy bands in the swelling states, we have considered probing the excitation wavelength dependent emission properties of those ionic polymers in the swelling states. It is observed that both P2-PF6 and P2-BF4 wetted with DCM or DMF do not exhibit evident excitation-dependent color-tunable emission upon excitation at 330–450 nm (Fig. S21, ESI†). For both P3-PF6 and P3-BF4 with higher feed-ratios, after wetting them with DCM or DMF, they show no obvious excitation-dependent color-tunable emission upon excitation at 365–460 nm. After increasing their excitation wavelength to 520 nm, their emission bands become narrow. As a result, we could still observe small emission color changes (Fig. S22, ESI†). Similarly, after wetting P4-PF6 and P4-BF4 with either DCM or DMF, little excitation-dependent color-tunable emission could also be observed with increasing the excitation wavelength from 365 nm to 540 nm (Fig. S23, ESI†). On the whole, some ionic polymers in the wetted states have exhibited excitation-dependent color-tunable emission, but the corresponding contrast is not high. It would still be necessary to develop high-contrast excitation-dependent color-tunable emission materials.
Emission properties of ionic polymers prepared in solvent DMF
As we know, some polymers could exhibit different swelling aggregation states in different solvents, and accordingly, the grafting polymers with different microstructures may be prepared by grafting other functional groups in different reaction solvents.28 From this, different functional polymers may be gained for the various application requirements. To our excitement, when we have attempted to execute the above similar reactions in solvent DMF, a series of different color-tunable ionic polymers (P5-PF6, P5-BF4, P6-PF6, P6-BF4, P7-PF6, P7-BF4) (L/P = 1:10, n = 5; 1:1, n = 6; 2:1, n = 7) have been obtained by tuning the mass-ratios of L to P (Scheme S2, ESI†). Typically, X-ray powder diffractions, SEM images and TGA measurements of ionic polymers (P5-PF6, P5-BF4) reveal that those ionic polymers also tend to aggregate into amorphous microsphere particles and display good thermal stability (Fig. S24–S26, ESI†). As shown in Fig. 6a, among those ionic polymers, P5-PF6 displays the shortest maximum emission peak at 495 nm under 365 nm excitation, which is close to that of P1-PF6 (λem = 501 nm), and P7-BF4 has the longest maximum emission peak at 530 nm, which is close to that of P1-BF4 (λem = 527 nm). Overall, with increasing feed-ratios from 1/10 to 2/1, their maximum emission peaks display about 34 nm red-shifts. Clearly, compared to the foregoing ionic polymers prepared in CH3CN, those ionic polymers prepared in DMF show a smaller degree of red-shifts despite the larger feed-ratio. These results imply that modifying the reaction solvent could indeed effectively tune the emission properties of those ionic polymers in this system. We think that this may be due to the fact that different solvents with different polarities, which have different swelling abilities for polymer P, could induce the formation of different microstructure ionic polymers during the reaction process.28 More specifically, due to the fact that CH3CN is a relatively poor swelling agent for polymer P, the grafting of L onto P mainly takes place on the surface of the polymer microsphere. In contrast, DMF is a good swelling agent for polymer P, so the grafting may take place deep within P and then L has been grafted dispersedly onto the polymer chain.28b Consequently, the suspended AIE-active emission center L+ in those ionic polymers prepared in DMF is relatively dispersive. In addition, we also note that, in comparison with P5-BF4, P5-PF6, P6-BF4 and P6-PF6, both P7-PF6 and P7-BF4 with the higher feed-ratios display relatively broad emission bands, and P7-BF4 has the broadest emission band (Fig. 6a). Consequently, the emission color of these ionic polymers has been tuned from cyan to green then to yellow green by modifying the feed-ratios (Fig. 6a). Furthermore, the excitation spectra of these ionic polymers have been obtained, as shown in Fig. 6b. It is found that the first five ionic polymers (P5-PF6, P5-BF4, P6-PF6, P6-BF4, P7-PF6) exhibit similar broad excitation bands with a maximum peak at 380–420 nm. From this, upon excitation at 315–420 nm, they have displayed no obvious excitation-dependent color-tunable emission (Fig. S27, ESI†). However, the dry P7-BF4 has a red-shifted broader excitation band with a maximum peak at 450 nm for the emission peak at 530 nm. Moreover, we find that the shoulder emission peak at 590 nm for the dry P7-BF4 has a broader excitation band with multiple peaks (Fig. S28, ESI†), which may suggest the presence of multiple aggregation ground states in the dry P7-BF4 with a high feed-ratio. Thereby, the dry P7-BF4 has realized excitation-dependent emission color changes from yellow green to orange upon excitation at 365–530 nm, which could be supported by the corresponding CIE coordinates (Fig. S29, ESI†). Additionally, the quantum yield measurements of these ionic polymers indicate that they show excellent luminescence efficiency (28% for P5-PF6, 34% for P5-BF4, 28% for P6-PF6, 40% for P6-BF4, 25% for P7-PF6 and 20% for P7-BF4) (Fig. S30, ESI†). In a word, these results further verify that adjusting the feed-ratio of AIE-active group L+ in reaction solvent DMF could achieve a wide-range of high efficiency multi-color emission and interesting excitation-dependent color-tunable emission.
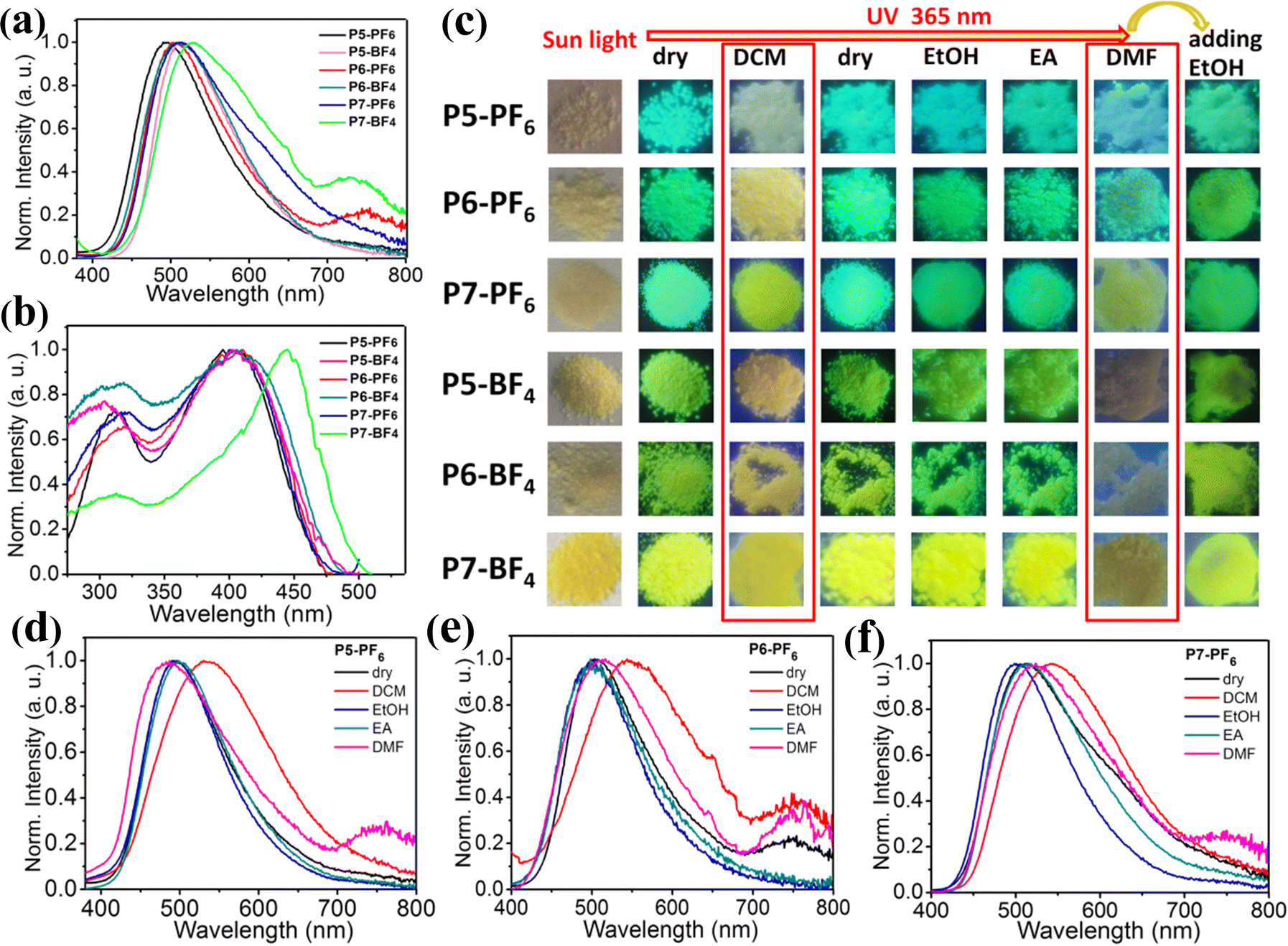 | ||
| Fig. 6 (a) Emission spectra of Pn-BF4 and Pn-PF6 (n = 5–7) under 365 nm excitation; (b) excitation spectra for P5-PF6 (λem = 495 nm), P5-BF4 (λem = 510 nm), P6-PF6 (λem = 507 nm), P6-BF4 (λem = 510 nm), P7-PF6 (λem = 514 nm), and P7-BF4 (λem = 530 nm); (c) emission photographs of ionic polymers (P5-PF6, P5-BF4, P6-PF6, P6-BF4, P7-PF6, P7-BF4) in the dry states and the various wetted states responding to the different solvents; (d–f) emission spectra of the ionic polymers (P5-PF6, P6-PF6, P7-PF6) in the dry states and the various wetted states responding to the different solvents. | ||
As expected, these ionic polymers (P5-PF6, P5-BF4, P6-PF6, P6-BF4, P7-PF6, P7-BF4) also exhibit fascinating solvent stimuli responsive emission, and their related emission spectra and emission photographs have been given in Fig. 6c–f and Fig. S31 (ESI†). When these ionic polymers have been wetted with EtOH or EA, their emission peaks only show little shifts, so no high-contrast emission color changes could be observed before and after wetting. In particular, these ionic polymers responding to DCM exhibit a clearly red-shifted broad emission band, and therefore the bright high-contrast emission color changes could be discernible. When the solvent DCM quickly evaporates, these re-dried ionic polymers recover to their initial emission color. By contrast, when these polymers have been wetted with DMF, their emission intensity is evidently reduced and the related emission bands also show some changes. Thus, another kind of different emission states could be presented. After further adding EtOH into those ionic polymers wetted with DMF, their weak emission rapidly transforms into the enhanced initial emission. In short, those ionic polymers prepared in DMF also could respond to different solvents, achieving great high-contrast reversible multiple color-tunable emission in solid states.
Furthermore, we have compared the excitation spectra of these ionic polymers (P5-PF6, P5-BF4, P6-PF6, P6-BF4, P7-PF6, P7-BF4) in the dry states, the wetted states with DCM and the wetted states with DMF, respectively, as shown in Fig. S32 (ESI†). Broadly speaking, the excitation spectra of these ionic polymers in three different states more or less display some differences, which also suggests that their microstructures (such as packing models or conformations) should have been altered by wetting them with the different solvents of DCM or DMF. Notably, for a series of ionic polymers containing [PF6]− (P5-PF6, P6-PF6, P7-PF6), their excitation spectra exhibit relatively little differences before and after wetting. Whereas, for those ionic polymers containing [BF4]− (P5-BF4, P6-BF4, P7-BF4), their excitation spectra have relatively large changes from the dry states to the wetted states with DCM then to the wetted states with DMF. Moreover, for the ionic polymers wetted with either DCM or DMF, the maximum peaks of their excitation spectra display some red-shifts with increasing feed ratios. All those changes imply that the counter ions and feed-ratio could effectively modify the microstructure of these ionic polymers.
Accordingly, the different ionic polymers in different wetted states exhibit different excitation-dependent emission characteristics (Fig. 7 and Fig. S33, S34, ESI†). Among the ionic polymers containing [PF6]− (P5-PF6, P6-PF6, P7-PF6), P5-PF6 with a relatively low feed-ratio shows high contrast excitation-dependent color-tunable emission after wetting with either DCM or DMF. That is, P5-PF6 wetted with DCM displays the excitation-dependent emission color changes from white to yellow green (Fig. 7a) and P5-PF6 wetted with DMF shows the excitation-dependent emission color changes from cold white to green (Fig. 7c), which have been supported by their corresponding CIE coordinates (Fig. 7b and d), respectively. In contrast, both P6-PF6 and P7-PF6 with the higher feed-ratios have not presented distinct excitation-dependent color-tunable emission after wetting with either DCM or DMF (Fig. S33, ESI†). Significantly, for those ionic polymers containing [BF4]− (P5-BF4, P6-BF4, P7-BF4), after wetting them with either DCM or DMF, it is found that P5-BF4 wetted with DMF and both P6-BF4 and P7-BF4 wetted with either DCM or DMF exhibit high contrast excitation-dependent color-tunable emission except that P5-BF4 wetted with DCM shows no obvious excitation-dependent color-tunable emission (Fig. 7e and f). Specifically speaking, P5-BF4 wetted with DMF shows the excitation-dependent emission color changes from white to yellow then to orange. P6-BF4 wetted with DCM shows excitation-dependent emission color changes from white to yellow then to orange. P6-BF4 wetted with DMF shows excitation-dependent emission color changes from white to yellow. P7-BF4 wetted with DCM shows excitation-dependent emission color changes from yellow to red. P7-BF4 wetted with DMF also shows excitation-dependent emission color changes from yellow to red. All these emission changes could be further supported by their corresponding CIE coordinates, respectively (Fig. 7 and Fig. S34, ESI†). Moreover, these results further demonstrate that modifying counter ions and feed ratios of ionic polymers could develop the excitation-dependent color-tunable emission materials for the desirable anti-counterfeiting applications. Remarkably, some of the ionic polymers bearing AIE-active emission center L+ have simultaneously achieved the attractive high contrast solvent stimuli responsive emission and high contrast excitation-dependent color-tunable emission.
 | ||
| Fig. 7 (a, c, e and g) Emission spectra of ionic polymers (P5-PF6, P5-BF4) in the wetted states with DCM and the wetted states with DMF under the different excitation wavelengths, respectively; (b, d, f and h) their corresponding CIE coordinates. | ||
Dynamic luminescence anti-counterfeiting
In view of the fact that these ionic polymers could exhibit reversible multiple color-tunable emission in solid states, their applications for dynamic multi-level anti-counterfeiting have been detected. As a proof-of-concept of the dynamic multi-level anti-counterfeiting mode, at first, one kind of color-tunable ionic polymer P2-PF6 has been applied to writing a thin letter ‘C’ on the filter paper without background fluorescence (Fig. 8a). Due to the fact that the filter paper has enough coarse surface, the amorphous polymer microsphere particles can be entrapped into the gaps between the paper fibers. Under sunlight, only a pale yellow letter ‘C’ could be seen. When letter ‘C’ has been irradiated by 365 nm UV light, bright yellow green light could be observed, which could be regarded as level 1 anti-counterfeiting. After wetting letter ‘C’ with DCM, the yellow green emission promptly turns into orange emission. After about 3 minutes, the orange emission recovers to the initial bright yellow green light due to evaporation of DCM. Accordingly, one high-contrast dynamic emission change could be detected, which could be regarded as level 2 anti-counterfeiting. When the letter ‘C’ has been wetted with DMF, the largely weakened orange emission could be found. As a result, another high-contrast dynamic emission change could be detected, which could be regarded as level 3 anti-counterfeiting. After 5 min, due to a small portion of DMF evaporates, we could still only observe the weak yellow emission. Subsequently, when EtOH has been added into the undried letter ‘C’, the bright yellow green emission quickly reappears. After drying, letter ‘C’ maintains the initial bright yellow green emission. In a word, it is verified that high contrast reversible dynamic three-level luminescence anti-counterfeiting could be achieved by using one kind of color-tunable ionic polymer.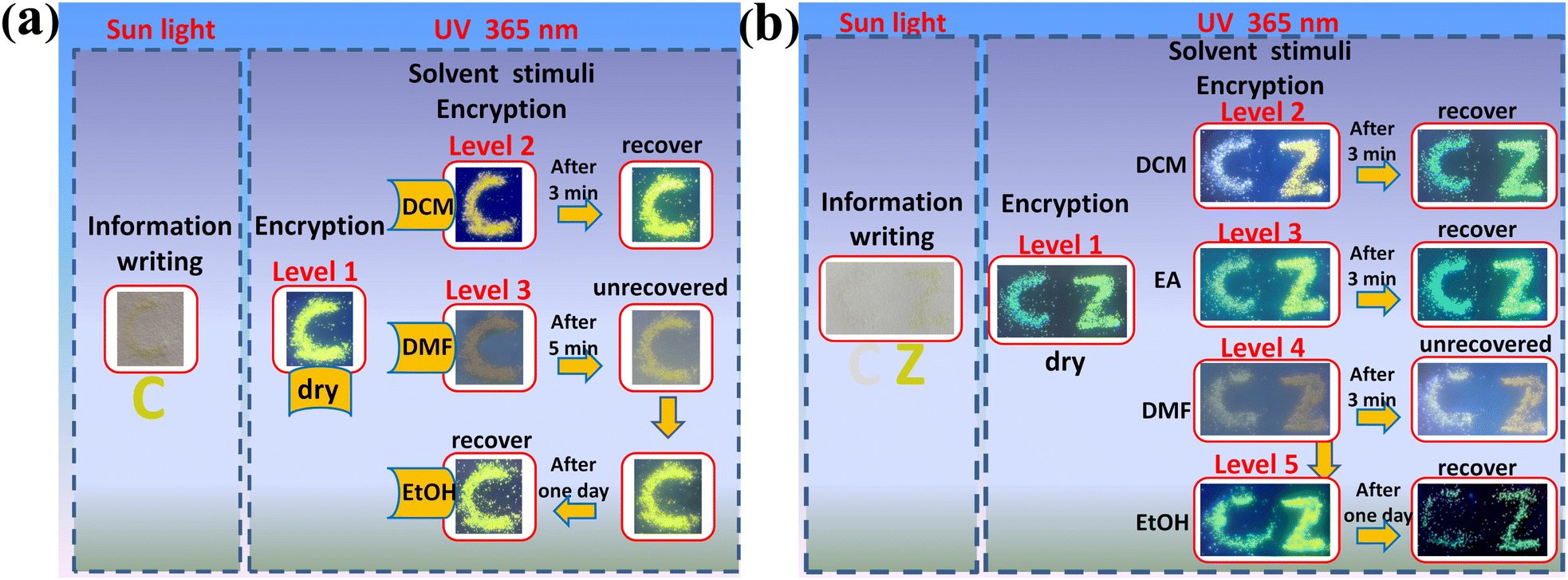 | ||
| Fig. 8 Design and concept of dynamic multi-level luminescence anti-counterfeiting using the color-tunable ionic polymers. (a) Dynamic three-level anti-counterfeiting from one kind of color-tunable ionic polymer. (b) The complicated dynamic multi-level (more than three-level) anti-counterfeiting from two kinds of color-tunable ionic polymers. | ||
In addition, the more complicated dynamic anti-counterfeiting mode could be made by using several kinds of different color-tunable emission materials, which could make forgers more difficult to counterfeit. Likewise, as a proof-of-concept mode, we have selected two kinds of different color-tunable emission ionic polymers (P1-PF6 and P1-BF4) to write two thin letters ‘CZ’ onto filter paper (Fig. 8b). Therein, P1-PF6 is for letter ‘C’, and P1-BF4 is for letter ‘Z’. Under sunlight, the off-white letter ‘C’ could almost not be seen, and the pale yellow letter ‘Z’ could be seen. When irradiated by 365 nm UV light, the two letters emit two different emission colors, which could be regarded as level 1 anti-counterfeiting. After wetting them with DCM, the two different emission colors promptly change into the other two different emission colors, which could be regarded as level 2 anti-counterfeiting. When DCM evaporates dry after about 3 minutes, the emission color recovers to the initial two different emission colors again. After wetting ‘CZ’ with EA, although both only show small emission color changes, in fact, the other two different emission colors could still be distinguished, which could be regarded as level 3 anti-counterfeiting. After evaporating EA, the emission color also recovers to the initial emission color. After wetting two letters ‘CZ’ with DMF, the other two weak emission colors could be found, which could be regarded as level 4 anti-counterfeiting. After 3 min, their emission color shows no obvious changes. Then, when we dropped EtOH into the undried letters ‘CZ’, their emission rapidly enhanced and their emission colors are different from the initial emission color, which could be regarded as level 5 anti-counterfeiting. After one day, we could observe the initial emission color due to evaporation of the solvent. All in all, these multiple changes confirm that a more complicated dynamic multilevel (more than triple) anti-counterfeiting could be realized by combing the various different color-tunable emission materials.
Conclusions
In summary, a series of high-contrast reversible multiple color-tunable emission single ionic polymers with high emission efficiency have been obtained by simply grafting the charged multi-color AIE-active chromophores into polymers through tuning counter anions, feed-ratios and reaction solvents. It is still rare that some of those ionic polymers not only exhibit high contrast reversible multiple color-tunable emission in solid states in response to multiple solvent stimuli, but also realize high-contrast excitation-dependent color-tunable emission. On the basis of the comparative studies of excitation spectra and fluorescent lifetimes in the different states, it could be inferred that solvent stimuli could induce microstructure changes in these ionic polymers and modify the aggregated-states of their corresponding AIE-active emission centers. Moreover, the different solvent stimuli could induce these ionic polymers to produce a different degree of microstructure changes, resulting in their attractive multiple color-tunable emission. Significantly, as a proof-of-concept of the dynamic multilevel anti-counterfeiting mode, it is indicative that these smart color-tunable emission ionic polymers could achieve dynamic multilevel (three-level or even more than three-level) luminescence anti-counterfeiting. In addition, these results pave the way to developing more novel multifunctional color-tunable emission materials based on charged AIE-active chromophores for anti-counterfeiting, smart displays, and optical sensing.Author contributions
X. M. conceived the idea. M. Z., G. L., synthesized the materials and completed the characterization. X. M., M. Z., and J. L. performed the photophysical experiments. X. M., W. H. and D. W. initiated and supervised the work. X. M. wrote the manuscript. X. M., W. H. and D. W. revised the manuscript with input from all authors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors are thankful for financial support from the Priority Academic Program Development of Jiangsu Higher Education Institutions. This work was supported by the National Natural Science Foundation of China (21671027), the Natural Science Foundation of Jiangsu Province (BK20170290), and the Natural Science Foundation of the Higher Education Institutions of Jiangsu Province (17KJB150002). The authors are thankful for the measurement service from the Engineering Center of School of Petrochemical Engineering in Changzhou University.Notes and references
- R. Arppe and T. J. Sørensen, Nat. Rev. Chem., 2017, 1, 0031 CrossRef CAS.
- (a) X. Yu, H. Zhang and J. Yu, Aggregate, 2021, 2, 20–34 CrossRef; (b) H. Zhang, D. Hua, C. Huang, S. K. Samal, R. Xiong, F. Sauvage, K. Braeckmans, K. Remaut and S. C. De Smedt, Adv. Mater., 2020, 32, 1905486 CrossRef CAS; (c) N. R. Visaveliya and J. M. Köhler, Adv. Opt. Mater., 2021, 9, 2002219 CrossRef CAS; (d) J. Andréasson and U. Pischel, Chem. Soc. Rev., 2018, 47, 2266–2279 RSC; (e) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS; (f) S. Mukherjee and P. Thilagar, Angew. Chem., Int. Ed., 2019, 58, 7922–7932 CrossRef CAS; (g) Z. Wu, J. Nitsch and T. B. Marder, Adv. Opt. Mater., 2021, 9, 2100411 CrossRef CAS; (h) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (i) G. Guan, M. Wu and M. Y. Han, Adv. Funct. Mater., 2019, 30, 1903439 CrossRef; (j) S. X. Wei, Z. Li, W. Lu, H. Liu, J. W. Zhang, T. Chen and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 8608–8624 CrossRef CAS; (k) V. W.-W. Yam and A. S.-Y. Law, Coord. Chem. Rev., 2020, 414, 213298 CrossRef CAS.
- (a) T. Ma, T. Li, L. Zhou, X. Ma, J. Yin and X. Jiang, Nat. Commun., 2020, 11, 1811 CrossRef CAS; (b) J. Wang, J. Ma, J. Zhang, Y. Fan, W. Wang, J. Sang, Z. Ma and H. Li, ACS Appl. Mater. Interfaces, 2019, 11, 35871–35878 CrossRef CAS PubMed; (c) O. Guillou, C. Daiguebonne, G. Calvez and K. Bernot, Acc. Chem. Res., 2016, 49, 844–856 CrossRef CAS; (d) Y. Sun, X. Le, S. Zhou and T. Chen, Adv. Mater., 2022, e2201262 CrossRef PubMed; (e) X. X. Le, H. Shang, S. R. Gu, G. Q. Yin, F. Q. Shan, D. Y. Li and T. Chen, Chin. J. Chem., 2022, 40, 337–342 CrossRef CAS.
- (a) Z. Zeng, B. Huang, X. Wang, L. Lu, Q. Lu, M. Sun, T. Wu, T. Ma, J. Xu, Y. Xu, S. Wang, Y. Du and C. H. Yan, Adv. Mater., 2020, 32, 2004506 CrossRef CAS; (b) Y. Zhuang, L. Wang, Y. Lv, T.-L. Zhou and R.-J. Xie, Adv. Funct. Mater., 2018, 28, 1705769 CrossRef.
- (a) C. L. Shen, Q. Lou, C. F. Lv, J. H. Zang, S. N. Qu, L. Dong and C. X. Shan, Adv. Sci., 2019, 6, 1802331 CrossRef; (b) K. Jiang, L. Zhang, J. Lu, C. Xu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2016, 55, 7231–7235 CrossRef CAS.
- (a) G. Huang, Q. Xia, W. Huang, J. Tian, Z. He, B. S. Li and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 17814–17819 CrossRef CAS PubMed; (b) Q. Qi, C. Li, X. Liu, S. Jiang, Z. Xu, R. Lee, M. Zhu, B. Xu and W. Tian, J. Am. Chem. Soc., 2017, 139, 16036–16039 CrossRef CAS PubMed; (c) J. Ji, D. Hu, J. Yuan and Y. Wei, Adv. Mater., 2020, 32, 2004616 CrossRef CAS PubMed; (d) L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS; (e) J. Yuan, P. R. Christensen and M. O. Wolf, Chem. Sci., 2019, 10, 10113–10121 RSC; (f) Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054–16060 CrossRef CAS; (g) M. Jian, Z. Song, X. Chen, J. Zhao, B. Xu and Z. Chi, Chem. Eng. J., 2022, 429, 132346 CrossRef CAS; (h) S. Yang, C.-X. Zhao, S. Crespi, X. Li, Q. Zhang, Z.-Y. Zhang, J. Mei, H. Tian and D.-H. Qu, Chemistry, 2021, 7, 1544–1556 CrossRef CAS; (i) E. Li, K. Jie, M. Liu, X. Sheng, W. Zhu and F. Huang, Chem. Soc. Rev., 2020, 49, 1517–1544 RSC; (j) Y. Qi, N. Ding, Z. Wang, L. Xu and Y. Fang, ACS Appl. Mater. Interfaces, 2019, 11, 8676–8684 CrossRef CAS PubMed.
- (a) X.-K. Ma and Y. Liu, Acc. Chem. Res., 2021, 54, 3403–3414 CrossRef CAS; (b) X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Nat. Commun., 2015, 6, 6884 CrossRef; (c) Z. Li, X. Liu, G. Wang, B. Li, H. Chen, H. Li and Y. Zhao, Nat. Commun., 2021, 12, 1363 CrossRef CAS PubMed; (d) L. Qin, X. Liu, K. He, G. Yu, H. Yuan, M. Xu, F. Li and Y. Yu, Nat. Commun., 2021, 12, 699 CrossRef CAS; (e) M. B. Wang, Q. Li, E. R. Li, J. Y. Liu, J. Zhou and F. H. Huang, Angew. Chem., Int. Ed., 2021, 60, 8115–8120 CrossRef CAS PubMed; (f) M. Zuo, W. Qian, T. Li, X.-Y. Hu, J. Jiang and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 39214–39221 CrossRef CAS; (g) S. J. D. Lugger, S. J. A. Houben, Y. Foelen, M. G. Debije, A. P. H. J. Schenning and D. J. Mulder, Chem. Rev., 2022, 122, 4946–4975 CrossRef CAS; (h) Z. Li, P. Liu, X. Ji, J. Gong, Y. Hu, W. Wu, X. Wang, H. Q. Peng, R. T. K. Kwok, J. W. Y. Lam, J. Lu and B. Z. Tang, Adv. Mater., 2020, 32, 1906493 CrossRef CAS; (i) H. Jin, H. Li, Z. Zhu, J. Huang, Y. Xiao and Y. Yan, Angew. Chem., Int. Ed., 2020, 59, 10081–10086 CrossRef CAS PubMed.
- (a) R. Gao, M. S. Kodaimati and D. Yan, Chem. Soc. Rev., 2021, 50, 5564–5589 RSC; (b) Z. H. Gao, B. Y. Xu, T. J. Zhang, Z. Liu, W. G. Zhang, X. Sun, Y. Liu, X. Wang, Z. F. Wang, Y. L. Yan, F. Q. Hu, X. G. Meng and Y. S. Zhao, Angew. Chem., Int. Ed., 2020, 59, 19060–19064 CrossRef CAS; (c) J. Liu, Y. Zhuang, L. Wang, T. Zhou, N. Hirosaki and R.-J. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 1802–1809 CrossRef CAS; (d) J. I. Deneff, K. S. Butler, L. E. S. Rohwer, C. J. Pearce, N. R. Valdez, M. A. Rodriguez, T. S. Luk and D. S. F. Gallis, Angew. Chem., Int. Ed., 2021, 60, 1203–1211 CrossRef CAS; (e) Y. Y. Liu, X. Zhang, K. Li, Q. C. Peng, Y. J. Qin, H. W. Hou, S. Q. Zang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 22417–22423 CrossRef CAS.
- (a) T. Ogoshi, H. Tsuchida, T. Kakuta, T. A. Yamagishi, A. Taema, T. Ono, M. Sugimoto and M. Mizuno, Adv. Funct. Mater., 2018, 28, 1707369 CrossRef; (b) W. Tian, J. Zhang, J. Yu, J. Wu, J. Zhang, J. He and F. Wang, Adv. Funct. Mater., 2018, 28, 1703548 CrossRef; (c) X. Ma, C. Xu, J. Wang and H. Tian, Angew. Chem., Int. Ed., 2018, 57, 10854–10858 CrossRef CAS PubMed; (d) X. Yao, J. Wang, D. Jiao, Z. Huang, O. Mhirsi, F. Lossada, L. Chen, B. Haehnle, A. J. C. Kuehne, X. Ma, H. Tian and A. Walther, Adv. Mater., 2020, 33, 2005973 CrossRef; (e) X. Le, H. Shang, S. Wu, J. Zhang, M. Liu, Y. Zheng and T. Chen, Adv. Funct. Mater., 2021, 31, 2108365 CrossRef CAS; (f) J. Wang, X. Y. Lou, Y. Wang, J. Tang and Y. W. Yang, Macromol. Rapid Commun., 2021, 42, 2100021 CrossRef CAS; (g) P. Verma, A. Singh and T. K. Maji, Chem. Sci., 2021, 12, 2674–2682 RSC; (h) Y. Zhang, X. Le, Y. Jian, W. Lu, J. Zhang and T. Chen, Adv. Funct. Mater., 2019, 29, 1905514 CrossRef CAS; (i) C. Calvino, A. Guha, C. Weder and S. Schrettl, Adv. Mater., 2018, 30, 1704603 CrossRef PubMed; (j) X. Dou, T. Zhu, Z. Wang, W. Sun, Y. Lai, K. Sui, Y. Tan, Y. Zhang and W. Z. Yuan, Adv. Mater., 2020, 32, 2004768 CrossRef CAS PubMed; (k) L. Gu, W. Ye, X. Liang, A. Lv, H. Ma, M. Singh, W. Jia, Z. Shen, Y. Guo, Y. Gao, H. Chen, D. Wang, Y. Wu, J. Liu, H. Wang, Y.-X. Zheng, Z. An, W. Huang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 18527–18535 CrossRef CAS; (l) W. Lu, M. Si, X. Le and T. Chen, Acc. Chem. Res., 2022, 55, 2291–2303 CrossRef CAS.
- (a) S. Y. Ye, T. Tian, A. J. Christofferson, S. Erikson, J. Jagielski, Z. Luo, S. Kumar, C. J. Shih, J. C. Leroux and Y. Y. Bao, Sci. Adv., 2021, 7, eabd1794 CrossRef CAS; (b) J. Hu, Q. Li, X. Wang, S. Shao, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2019, 58, 8405–8409 CrossRef CAS; (c) J. Lawrence, E. Goto, J. M. Ren, B. McDearmon, D. S. Kim, Y. Ochiai, P. G. Clark, D. Laitar, T. Higashihara and C. J. Hawker, J. Am. Chem. Soc., 2017, 139, 13735–13739 CrossRef CAS; (d) B. Liu, H. Zhang, S. Liu, J. Sun, X. Zhang and B. Z. Tang, Mater. Horiz., 2020, 7, 987–998 RSC; (e) W. Wu and B. Liu, Mater. Horiz., 2021, 9, 99–111 RSC; (f) S. Y. Tao, S. J. Zhu, T. L. Feng, C. Y. Zheng and B. Yang, Angew. Chem., Int. Ed., 2020, 59, 9826–9840 CrossRef CAS.
- (a) T. Han, X. Wang, D. Wang and B. Z. Tang, Top. Curr. Chem., 2021, 379, 7 CrossRef CAS; (b) K. M. Herbert, S. Schrettl, S. J. Rowan and C. Weder, Macromolecules, 2017, 50, 8845–8870 CrossRef CAS; (c) C. I. Aguirre, E. Reguera and A. Stein, Adv. Funct. Mater., 2010, 20, 2565–2578 CrossRef CAS.
- (a) J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Park, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS; (b) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CrossRef CAS PubMed; (c) Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef; (d) S. K. Mellerupab and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC; (e) R. Ramakrishnan, M. A. Niyas, M. P. Lijina and M. Hariharan, Acc. Chem. Res., 2019, 52, 3075–3086 CrossRef CAS PubMed.
- (a) W. Luo and G. Wang, Adv. Opt. Mater., 2020, 8, 2001362 CrossRef CAS; (b) D. Kim and S. Y. Park, Adv. Opt. Mater., 2018, 6, 1800678 CrossRef; (c) Z. Li, X. Ji, H. Xie and B. Z. Tang, Adv. Mater., 2021, 33, 2100021 CrossRef CAS; (d) H. Traeger, D. J. Kiebala, C. Weder and S. Schrettl, Macromol. Rapid Commun., 2020, 42, 2000573 CrossRef; (e) A. Abdollahi, H. Roghani-Mamaqani and B. Razavi, Prog. Polym. Sci., 2019, 98, 101149 CrossRef CAS.
- (a) J. Wang, F. Tang, Y. Wang, S. Liu and L. Li, Adv. Opt. Mater., 2020, 8, 1901571 CrossRef CAS; (b) S. Uchiyama, N. Kawai, A. P. D. Silva and K. Iwai, J. Am. Chem. Soc., 2004, 126, 3032–3033 CrossRef CAS PubMed; (c) C. Chen, X. Zhao, Y. Chen, X. Wang, Z. Chen, H. Li, K. Wang, X. Zheng and H. Liu, Adv. Funct. Mater., 2021, 31, 2104784 CrossRef CAS; (d) P. Q. Nhien, W.-L. Chou, T. T. K. Cuc, T. M. Khang, C.-H. Wu, N. Thirumalaivasan, B. T. B. Hue, J. I. Wu, S.-P. Wu and H.-C. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 10959–10972 CrossRef CAS PubMed; (e) Y. Yuan, H. Zhang and J. Qu, ACS Appl. Polym. Mater., 2021, 3, 4512–4522 CrossRef CAS.
- J. Wang, N. Wang, G. Wu, S. Wang and X. Li, Angew. Chem., Int. Ed., 2019, 58, 3082–3086 CrossRef CAS PubMed.
- J. Wang, R. Yan, Y. Hu, G. Du, G. Liao, H. Yang, Y. Luo, X. Zheng, Y. Chen, S. Wang and X. Li, Angew. Chem., Int. Ed., 2022, 61, e202112290 CAS.
- D. Yan, Q. Wu, D. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 15724–15742 CrossRef CAS PubMed.
- (a) J. Tavakoli, A. J. Ghahfarokhi and Y. Tang, Top. Curr. Chem., 2021, 379, 9 CrossRef CAS; (b) W. Lu, S. Wei, H. Shi, X. Le, G. Yin and T. Chen, Aggregate, 2021, 2, e37 Search PubMed; (c) T. Han, L. Liu, D. Wang, J. Yang and B. Z. Tang, Macromol. Rapid Commun., 2020, 42, 2000311 CrossRef PubMed.
- (a) J. Yang, K. Gu, C. Shi, M. Li, P. Zhao and W.-H. Zhu, Mater. Chem. Front., 2019, 3, 1503–1509 RSC; (b) Y. B. Hu, J. W. Y. Lam and B. Z. Tang, Chin. J. Polym. Sci., 2019, 37, 289–301 CrossRef CAS; (c) T. Li, S. He, J. Qu, H. Wu, S. Wu, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 2964–2970 RSC.
- (a) R. Singh, H.-Y. Wu, A. Kumar Dwivedi, A. Singh, C.-M. Lin, P. Raghunath, M.-C. Lin, T.-K. Wu, K.-H. Wei and H.-C. Lin, J. Mater. Chem. C, 2017, 5, 9952–9962 RSC; (b) J. R. Chen, J. Zhao, B. J. Xu, Z. Y. Yang, S. W. Liu, J. R. Xu, Y. Zhang, Y. C. Wu, P. Y. Lv and Z. G. Chi, Chin. J. Polym. Sci., 2017, 35, 282–292 CrossRef CAS.
- G. Wang, H. Yu, L. Yang, Z. He, L. Zhou, J. Sun, X. Gu, W. Yang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 25246–25251 CrossRef CAS.
- M. Ding, B. Dong, Y. Lu, X. Yang, Y. Yuan, W. Bai, S. Wu, Z. Ji, C. Lu, K. Zhang and H. Zeng, Adv. Mater., 2020, 32, 2002121 CrossRef CAS PubMed.
- (a) C. Zhou, M. Jiang, J. Du, H. Bai, G. Shan, R. T. K. Kwok, J. H. C. Chau, J. Zhang, J. W. Y. Lam, P. Huang and B. Z. Tang, Chem. Sci., 2020, 11, 4730–4740 RSC; (b) Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS PubMed.
- (a) J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed; (b) G. Zhang, X. Zhang, L. Kong, S. Wang, Y. Tian, X. Tao and J. Yang, Sci. Rep., 2016, 6, 37609 CrossRef CAS PubMed; (c) T. Hinoue, Y. Shigenoi, M. Sugino, Y. Mizobe, I. Hisaki, M. Miyata and N. Tohnai, Chem. – Eur. J., 2012, 18, 4634–4643 CrossRef CAS; (d) F. Hu, G. Zhang, C. Zhan, W. Zhang, Y. Yan, Y. Zhao, H. Fu and D. Zhang, Small, 2015, 11, 1335–1344 CrossRef CAS PubMed.
- (a) Q.-y Cao, R. Jiang, M. Liu, Q. Wan, D. Xu, J. Tian, H. Huang, Y. Wen, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 80, 578–583 CrossRef CAS PubMed; (b) C. Du, C. S. Cheung, H. Zheng, D. Li, W. Du, H. Gao, G. Liang and H. Gao, ACS Appl. Mater. Interfaces, 2021, 13, 59288–59297 CrossRef CAS PubMed.
- (a) T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS; (b) W. Zhang, B. Wu, S. Sun and P. Wu, Nat. Commun., 2021, 12, 4082 CrossRef CAS.
- Z. Wang, Y. Zhang, C. Wang, X. Zheng, Y. Zheng, L. Gao, C. Yang, Y. Li, L. Qu and Y. Zhao, Adv. Mater., 2020, 32, 1907355 CrossRef CAS.
- (a) H. M. Ma, R. H. Davis and C. N. Bowman, Polymer, 2001, 42, 8333–8338 CrossRef CAS; (b) H. L. Wang and H. R. Brown, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 263–270 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mh00986b |
| This journal is © The Royal Society of Chemistry 2023 |