DOI:
10.1039/D3NA00004D
(Paper)
Nanoscale Adv., 2023,
5, 3336-3347
Co-delivery of a tumor microenvironment-responsive disulfiram prodrug and CuO2 nanoparticles for efficient cancer treatment†
Received
3rd January 2023
, Accepted 8th May 2023
First published on 9th May 2023
Abstract
Disulfiram (DSF) has been used as a hangover drug for more than seven decades and was found to have potential in cancer treatment, especially mediated by copper. However, the uncoordinated delivery of disulfiram with copper and the instability of disulfiram limit its further applications. Herein, we synthesize a DSF prodrug using a simple strategy that could be activated in a specific tumor microenvironment. Poly amino acids are used as a platform to bind the DSF prodrug through the B–N interaction and encapsulate CuO2 nanoparticles (NPs), obtaining a functional nanoplatform Cu@P–B. In the acidic tumor microenvironment, the loaded CuO2 NPs will produce Cu2+ and cause oxidative stress in cells. At the same time, the increased reactive oxygen species (ROS) will accelerate the release and activation of the DSF prodrug and further chelate the released Cu2+ to produce the noxious copper diethyldithiocarbamate complex, which causes cell apoptosis effectively. Cytotoxicity tests show that the DSF prodrug could effectively kill cancer cells with only a small amount of Cu2+ (0.18 μg mL−1), inhibiting the migration and invasion of tumor cells. In vitro and in vivo experiments have demonstrated that this functional nanoplatform could kill tumor cells effectively with limited toxic side effects, showing a new perspective in DSF prodrug design and cancer treatment.
Introduction
Cancer is the leading cause of death,1 with an estimated 19.3 million new cancer cases and 10 million cancer deaths worldwide in 2020.2 At present, various treatments have been developed to treat cancer, such as surgery, radiotherapy, and chemotherapy, the most commonly used method in the clinic. However, chemotherapy has serious adverse reactions and drug resistance, resulting in limited clinical outcomes.3–5 Therefore, there is an urgent need to develop highly effective drugs to treat cancer.6
Disulfiram was approved by the U.S. Food and Drug Administration for the treatment of alcoholism in 1951 and has been used clinically for more than 70 years.7–9 Since the 1970s, clinical trials have also been conducted to evaluate its efficacy against cancer, and the available experimental results revealed its potential for cancer treatment in the clinic.8,10,11 It has been demonstrated that DSF has treatment potential in a variety of tumor models, including breast,12 liver,13 colorectal,14 non-small cell lung,15 and glioblastoma.16 However, DSF has poor stability in vivo, causing unexpected side effects or limited therapeutic effects, which are significant obstacles hindering its implementation for cancer therapy.17,18 DSF is unstable in the acidic gastric environment and the bloodstream. Some in vivo studies have shown that the t1/2 of DSF is only 4 min.19 In the blood, DSF is rapidly degraded to DTC, and then the DTC is metabolized to S-methyl N,N-diethyldithiocarbamate (DTC-Me) by thiol methyltransferase. Furthermore, the DTC-Me is then oxidized to S-methyl N,N-diethyldithiocarbamate sulfoxide (DTC-MeSO) and S-methyl N,N-diethyldithiocarbamate sulfone (DTC-MeSO2),20 which would have toxic side effects in clinical treatment. For example, a clinical trial of DSF in patients with mCRPC showed significant side effects, but the prognosis was not improved.21 Another clinical trial of DSF in treating glioblastoma indicated that patients experienced side effects such as delirium, ataxia, and neuropathy.22 In addition, DTC-MeSO and DTC-MeSO2 are also potent ALDH inhibitors in vivo,19,23 which would maintain anti-alcoholism activity, but lost anticancer activity when DSF is exposed to blood. Therefore, the instability of DSF in the blood is one of the main obstacles to its anticancer activity in clinical applications. Moreover, DSF is a divalent metal ion chelator that is metabolized in the body and degraded to diethyldithiocarbamate (DDC), which chelates with copper to form the copper diethyldithiocarbamate complex (Cu(DDC)2). Cu(DDC)2 induces cancer cell death by targeting the p97 separase linker nucleoprotein localization protein 4 (NPL4) and inhibiting the proteasomal protein degradation pathway.24–26 On the other hand, Cu(DDC)2 could also inhibit superoxide dismutase (SOD), trigger the production of ROS,27,28 and induce apoptosis.29 Therefore, DSF requires copper(II) ions in the treatment of cancer to form Cu(DDC)2, and the addition of copper ions can effectively improve the therapeutic effect of DSF. Nevertheless, copper plays an important role in tumor development, especially for tumor angiogenesis.30 Inappropriate copper ion delivery will promote the growth of tumor cells and reduce the therapeutic effect of disulfiram.31 Importantly, if exogenous supplementation with copper(II) and DSF produces toxic substances in normal tissues, it will cause significant side effects. Thus, the failure of DSF in oncology clinical applications could be attributed to its lack of tumor-targeting specificity and the production of unwanted metabolites.7
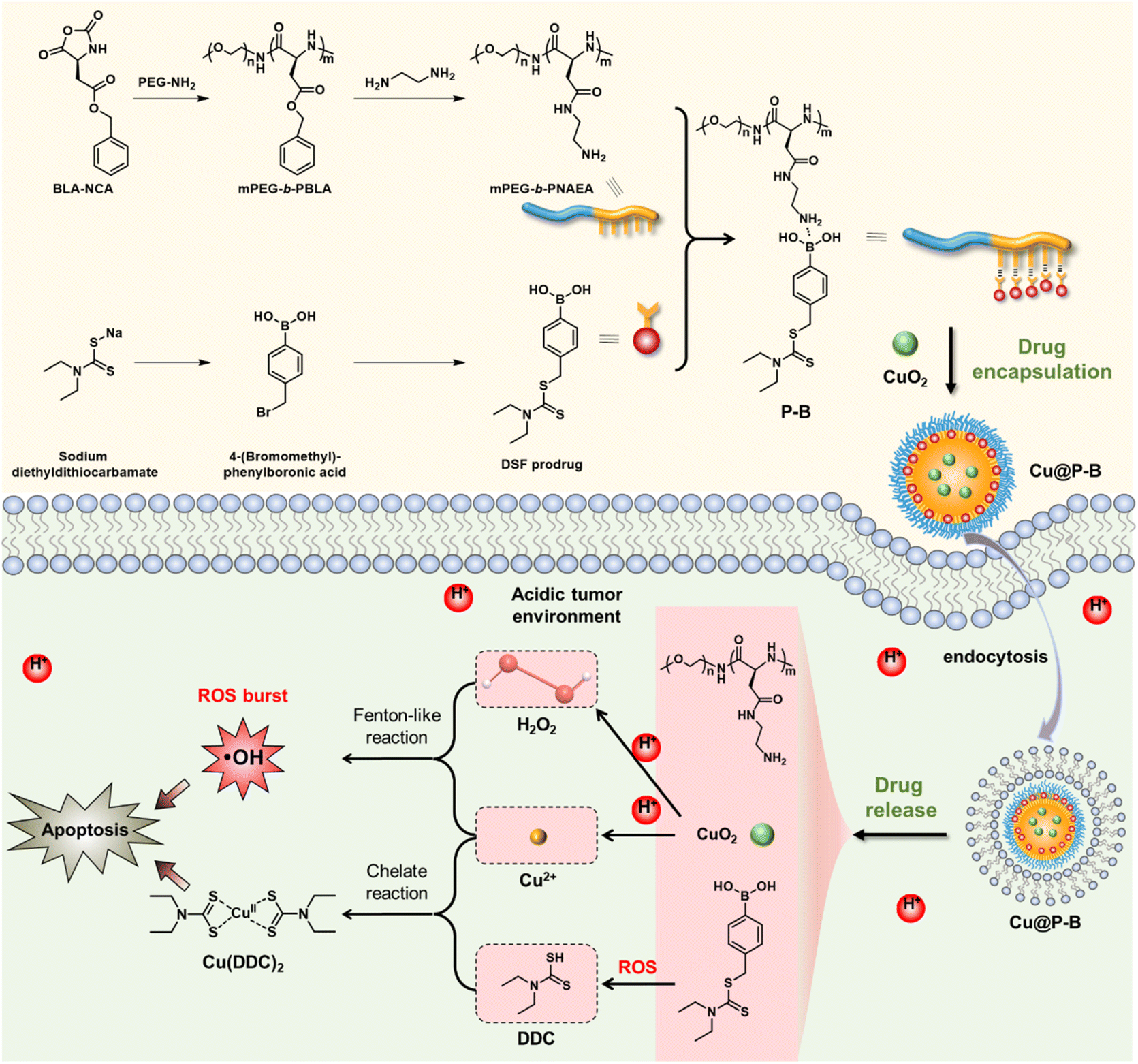
Thus, to enhance the therapeutic effect based on DSF and reduce the side effects, it is necessary to accurately control the delivery of copper ions and DSF to specifically produce toxic substances in tumor tissue. Herein, we constructed a functional nanoplatform to combat cancer based on the DSF prodrug. As shown in Scheme 1, we prepared the polypeptide through ring-opening polymerization based on benzyl(S)-2,5-dioxooxazolidine-4-acetate (BLA-NCA), which was further modified with PEG–NH2 to obtain mPEG-b-PBLA. Thereafter, the amphiphilic polymer was obtained by introducing the DSF prodrug into the nanoplatform through B–N coordination to form Cu@P–B NPs. Because the B–N coordination could be broken in the specific tumor microenvironment, the functional Cu@P–B NPs released the encapsulated DSF prodrug and CuO2 NPs under the tumor microenvironment. Under an acidic environment, CuO2 NPs released Cu2+ and H2O2 and increased tumor ROS levels through a Fenton-like reaction. At the same time, elevated ROS in the tumor tissue promoted cell apoptosis and activated the prodrug to release DDC. Furthermore, DDC produced by the DSF prodrug combined with Cu2+ could generate the poisonous copper complex Cu(DDC)2 (Scheme 1), which further kills the tumor cells efficiently. In vitro and in vivo experiments proved that codelivery of the DSF prodrug and Cu2+ to tumor tissue could effectively kill cells with high biocompatibility, providing a new perspective in cancer therapy and the drug design of DSF.
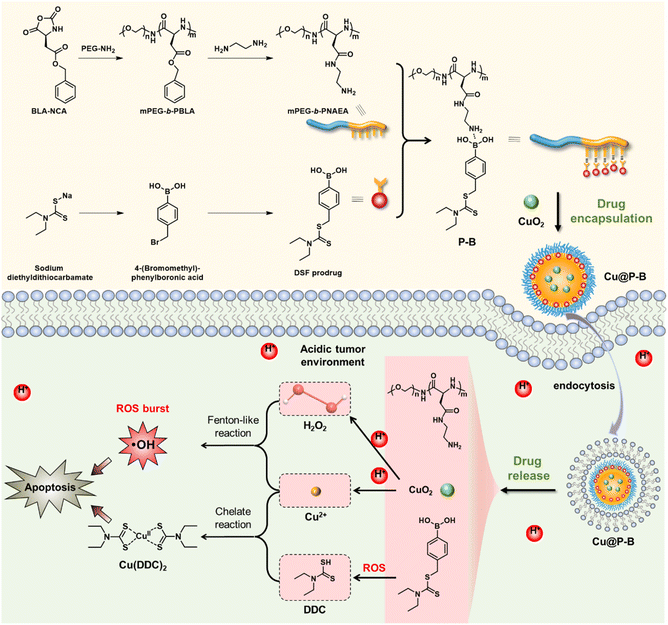 |
| | Scheme 1 Synthetic route of the DSF prodrug and mPEG-b-PNAEA. Illustration of the antitumor activity of the functional Cu@P–B nanoplatform. After internalization into cancer cells, the CuO2 and DSF prodrugs were released to generate Cu(DDC)2 for efficient cancer treatment. | |
Materials and methods
Synthesis of the DSF prodrug
The DSF prodrug was synthesized according to the methods reported in the literature with minor modifications.32 In brief, 4-(bromomethyl)phenylboronic acid (1.202 g, 5.59 mmol) and sodium diethyldithiocarbamate trihydrate (1.259 g, 5.59 mmol) were dissolved in methanol (2.0 mL), and the mixture was stirred in methanol at room temperature for 3 hours. After removal of the solvent by rotary evaporation, the product was dissolved in dichloromethane, and the pH was adjusted to basic by adding sodium hydroxide (NaOH) solution. Thereafter, aqueous hydrochloric acid (HCl, 1.0 M) was then added to adjust the pH to be acidic (pH = 1). The produced white precipitate was dissolved upon the addition of dichloromethane and washed with water. The organic layer was dried over sodium sulfate and evaporated to dryness in vacuo to give the DSF prodrug as a white solid (0.680 g, yield 43%).
Synthesis of CuO2 NPs
CuO2 NPs were prepared by the reaction of copper chloride (CuCl2), H2O2, and NaOH in an aqueous solution containing polyvinyl pyrrolidone (PVP) at room temperature for 30 min. In brief, PVP (2.5 g) was dissolved in an aqueous solution containing CuCl2·2H2O (25 mL, 0.01 M). Then, H2O2 (100 μL) and NaOH (25 mL, 0.01 M) were added sequentially to the above mixture, and precipitated particles were formed at this time.33 After stirring at room temperature for 30 min, a drop of mercaptoethanol was added to the mixture, and the stirring was continued for 3 hours. After stirring, the CuO2 NPs were collected by centrifugation at 5000 rpm for 5 min, washed three times with methanol, and then dispersed by sonication with 20 mL of methanol.
Synthesis of mPEG-b-PNAEA
BLA NCA (398.8 mg, 1.6 mmol) was dissolved in dry THF (16.0 mL) and added to a 1.0 M solution of lithium bis(trimethylsilyl)amide (LiHMDS) in THF (0.2 mL, 0.2 mmol) at room temperature with stirring. The reaction mixture was stirred at room temperature for 3 hours, and then mPEG-NH2 (600 mg, 0.2 mmol) dissolved in dry THF was added, followed by stirring overnight.34 After the reaction, mPEG-b-PBLA was obtained via precipitation in excess methanol. mPEG-b-PBLA was dissolved in DMF (10 mL), and ethylenediamine (1.0 mL) was added. Then the mixture was maintained at room temperature for 10 hours. Upon completion of the reaction, the mixture was purified by dialysis (MWCO, 1000 Da) against methanol for 24 hours.35 After removing most of the solvent by rotary evaporation, mPEG-b-PNAEA was obtained by precipitation in excess diethyl ether and concentrated under vacuum.
Preparation of P–B and Cu@P–B NPs
P–B and Cu@P–B NPs were prepared by the typical nanoprecipitation method. The DSF prodrug (30.0 mg) was dissolved in 1.5 mL methanol and mixed with mPEG-b-PNAEA (30.0 mg in 1.5 mL methanol). The mixture was maintained at room temperature for 6 hours and then added dropwise into DI water under vigorous stirring to obtain P–B micelles. For Cu@P–B NPs, the mixture was blended with CuO2 NPs before the nanoprecipitation method. Methanol and other small molecules were removed by dialysis (MWCO 1000 Da) against DI water for 12 hours.
Characterization of P–B and Cu@P–B NPs
The particle size of the NPs was measured by dynamic light scattering (DLS) using a 90Plus Zeta Nano Particle Size and Zeta Potential Analyzer (NanoBrook 90Plus Zeta). The morphology of the NPs was observed and analyzed using transmission electron microscopy (TEM). To determine the prodrug and CuO2 contents in the micelles, Cu and B contents before and after dialysis were measured by inductively coupled plasma-atomic emission spectrometry (ICP-AES, Optima 7300 DV). The concentrations and drug loadings of the prodrug and CuO2 were determined by calculation. The drug loading content (DLC, wt%) was calculated according to the following formula.
| DLC = (amount of loaded drug/amount of drug-loaded NPs) × 100% |
Cell culture
4T1 and A549 cells were cultured in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum and 1% penicillin and streptomycin. The incubator was maintained at 37 °C in humidified 5% CO2. In all of the studies, the cells were subcultured when the cell density reached 80 to 90%.
Cytotoxicity assays
The cytotoxicity of CuO2, P–B, and Cu@P–B NPs was assessed by MTT assay. A549/4T1/293T cells were seeded in 96-well plates at a density of 4000 cells per well. After overnight incubation, A549/4T1/293T cells were treated with CuO2 (0.12 μg mL−1, 0.18 μg mL−1, 0.24 μg mL−1, and 0.30 μg mL−1), P–B (1.4 μg mL−1, 2.2 μg mL−1, 2.7 μg mL−1, and 3.4 μg mL−1), and Cu@P–B (1.4 μg mL−1, 2.2 μg mL−1, 2.7 μg mL−1, and 3.4 μg mL−1), which contained the corresponding amount of CuO2, for 48 hours. Then, the cells were incubated with MTT (1.0 mg mL−1) for 4 hours. After removing the culture medium, DMSO (150 μL) was added, and the absorbance at 570 nm was measured with a microplate reader (Varioskan Flash).
In vitro ROS generation
DCFH-DA was employed as a fluorescent ROS probe to indicate the cellular ROS induced by Cu@P–B NPs. Briefly, A549 cells were exposed to PBS, 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B and 2.2 μg mL−1 Cu@P–B (containing 0.18 μg mL−1 CuO2) for 4 hours. Then, the cells were washed with PBS once and stained with DCFH-DA (10 μM) for 30 min, and the fluorescence images were recorded by using an inverted fluorescence microscope (Olympus X71). ROS levels were quantitatively determined by flow cytometry using the ROS-sensitive fluorescent dye DCFH-DA. A549 cells were collected, washed, incubated with DCFH-DA (10 μM) for 25 min, and washed with PBS three times, and the intracellular levels of ROS were determined by flow cytometry (CytoFLEX).
The evaluation of GSH
A549 cells were incubated with PBS, 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B, and 2.2 μg mL−1 Cu@P–B for 6 hours, and then collected by centrifugation. Total GSH was determined using a GSH and GSSG Assay Kit (Beyotime, China) according to the manufacturer's protocol. Absorbance was measured at 412 nm with a microplate reader.
Hemolysis test
Fresh whole blood was acquired from mice by using a vacuum blood collection tube containing heparin sodium, which was washed to obtain a red blood cell (RBC) suspension. Then, the RBCs were diluted with PBS (volume ratio, 4%). 200 μL RBC suspension was mixed with different solutions, including 200 μL PBS (blank control), distilled water (positive control), CuO2 (0.12 μg mL−1, 0.18 μg mL−1, 0.24 μg mL−1, 0.30 μg mL−1, 0.36 μg mL−1, and 0.72 μg mL−1), P–B (1.4 μg mL−1, 2.2 μg mL−1, 2.7 μg mL−1, 3.4 μg mL−1, 4.4 μg mL−1, and 8.8 μg mL−1), and Cu@P–B (1.4 μg mL−1, 2.2 μg mL−1, 2.7 μg mL−1, 3.4 μg mL−1, 4.4 μg mL−1, and 8.8 μg mL−1) containing the corresponding amount of CuO2. The mixtures were incubated for 60 min at 37 °C. The supernatant was carefully removed and transferred to 96-well plates. The OD was determined by spectroscopy at 576 nm. Three replicates were set for each sample. The hemolysis ratio was calculated by hemolysis (%) = [OD (sample)−OD (PBS)]/[OD (water)−OD (PBS)] × 100%.
Apoptosis assay
An annexin V-FITC apoptosis analysis kit was used to detect apoptosis by flow cytometry assays performed as previously described.36 A549 cells were incubated with PBS, 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B and 2.2 μg mL−1 Cu@P–B for 6 hours, collected by centrifugation, washed with PBS twice, and then stained with Annexin V-FITC and PI in binding buffer for 15 minutes at room temperature in the dark. All flow cytometry assays were performed on a Beckman CytoFLEX Flow Cytometer, and the results were analyzed using CytExpert software. All assays were conducted in triplicate at least three times.
JC-1 staining
Mitochondrial membrane potential was evaluated by using the fluorescent probe JC-1. A549 cells (4.0 × 105) were incubated after different treatments (PBS, 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B and 2.2 μg mL−1 Cu@P–B) for 6 hours. After washing once with PBS, the cells were incubated with JC-1 dye working solution for 20 min. Then the cells were washed twice with staining buffer (1X) before being observed with an inverted fluorescence microscope.
Live–dead staining assay
Live–dead stains fluorescein diacetate (FDA) and propidium iodide (PI) were employed to evaluate cancer cell death. A549 cells were separately treated with PBS, 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B and 2.2 μg mL−1 Cu@P–B for 6 hours in an incubator. After washing once with PBS, the cells were incubated with FDA (8.0 μg mL−1) and PI (5.0 μg mL−1) for 30 min in the incubator. Then the cells were washed three times with PBS, and fluorescence images were recorded.
Scratch assay
A549 cells were seeded in six-well plates and cultured until they reached 95% confluence. The adherent monolayer cells were scratched using 200 μL pipette tips, and the scratched wells were washed three times with PBS to remove detached debris after scratching. Then, RPMI 1640 containing PBS, 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B and 2.2 μg mL−1 Cu@P–B was added to the scratched cells. The cells were then incubated with 5% CO2 at 37 °C for 24 hours. The initial wounding and the movement of the cells in the scratched area were photographically monitored and imaged using a microscope (Olympus X71) coupled with a digital imaging system at 0, 12, and 24 h.
Cell migration assays
The ability of the cells to migrate was assessed using a cell migration assay. CT26 cells in the logarithmic growth phase were harvested by centrifugation, and 5.0 × 105 cells per mL were resuspended in RPMI-1640 medium supplemented with PBS, 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B and 2.2 μg mL−1 Cu@P–B separately. 200 μL cell suspension was added to the upper chamber of each transwell insert with an 8.0 μm pore size, and 600 μL of RPMI-1640 with 10% FBS was added to the lower chamber of each transwell insert. The transwell inserts were incubated at 37 °C for 24 hours. The invasive cells were fixed with 4% paraformaldehyde in PBS for 20 min, stained with 0.1% crystal violet for 20 min, and photographed under a light microscope.
Biodistribution study and in vivo cancer treatment
Female Balb/c mice (6 to 8 weeks, 18–22 g) were purchased from Slack Laboratory Animal Co. Ltd (Shanghai). All animal experimental procedures were approved by the Institutional Animal Ethics Committee of the University of Science and Technology of China; the ID is 2022-N(A)-021.
Tolerance tests were conducted by injecting normal non-tumor-bearing Balb/c mice with different concentrations of Cu@P–B. The mice were randomly divided into four groups with three mice in each group. 100 μL PBS, 0.044 mg kg−1, 0.088 mg kg−1 and 0.175 mg kg−1 Cu@P–B were injected through the tail vein of mice, once a day for a total of three injections. Mice body weights were measured before injection (day 0) and after injection (days 1, 2, and 3).
For the biodistribution study, 100 μL Cu@P–B NPs containing 0.1 mg kg−1 Cu2+ were injected into 4T1 tumor-bearing mice through the tail vein. At 12 hours post injection, the mice were sacrificed. Tumors and major organs were collected, weighed, and digested in nitric acid. The Cu contents were measured by ICP-AES. Mice that were injected with PBS served as a control.
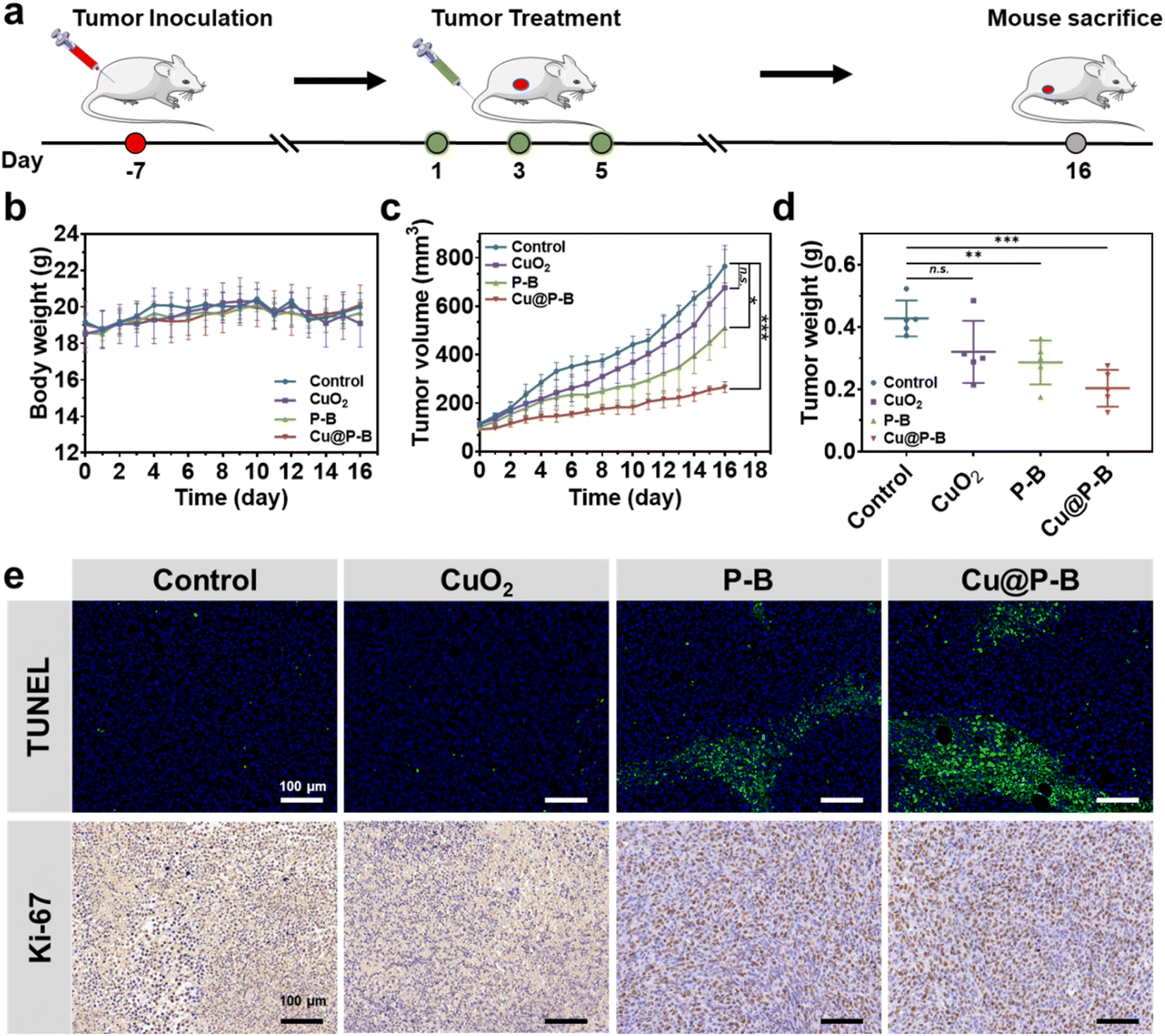
In vivo treatment was performed using 4T1 tumor-bearing mice. To establish the tumor-bearing mouse model, 4T1 cells were harvested by centrifugation and resuspended in sterile PBS. The harvested cells (1.0 × 106) were then implanted inoculated at the right flank of mice. The mice bearing 4T1 tumors (∼100 mm3) were randomly divided into four groups and were intravenously injected with 100 μL PBS, CuO2, P–B, or Cu@P–B at a Cu2+ dosage of 0.1 mg kg−1 every other day for three doses. Each group contained five mice. The size of the tumor and body weight were measured every day. After 16 days of observation, the mice were sacrificed for tumor weight and histological examination. All tumors were then sectioned into slices, and H&E/TUNEL/Ki-67 staining was performed for histological analysis. The heart, liver, spleen, lung and kidney were also harvested for H&E staining.
Statistical analysis
Quantitative data are presented as mean ± SD, and the results were analyzed using GraphPad Prism 8 software. One-way ANOVA was performed to analyze the significance of differences in multiple group comparisons. And comparison between the two groups was performed by a t-test. The statistical significance was set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Results and discussion
Synthesis of the DSF prodrug, CuO2 and mPEG-b-PNAEA
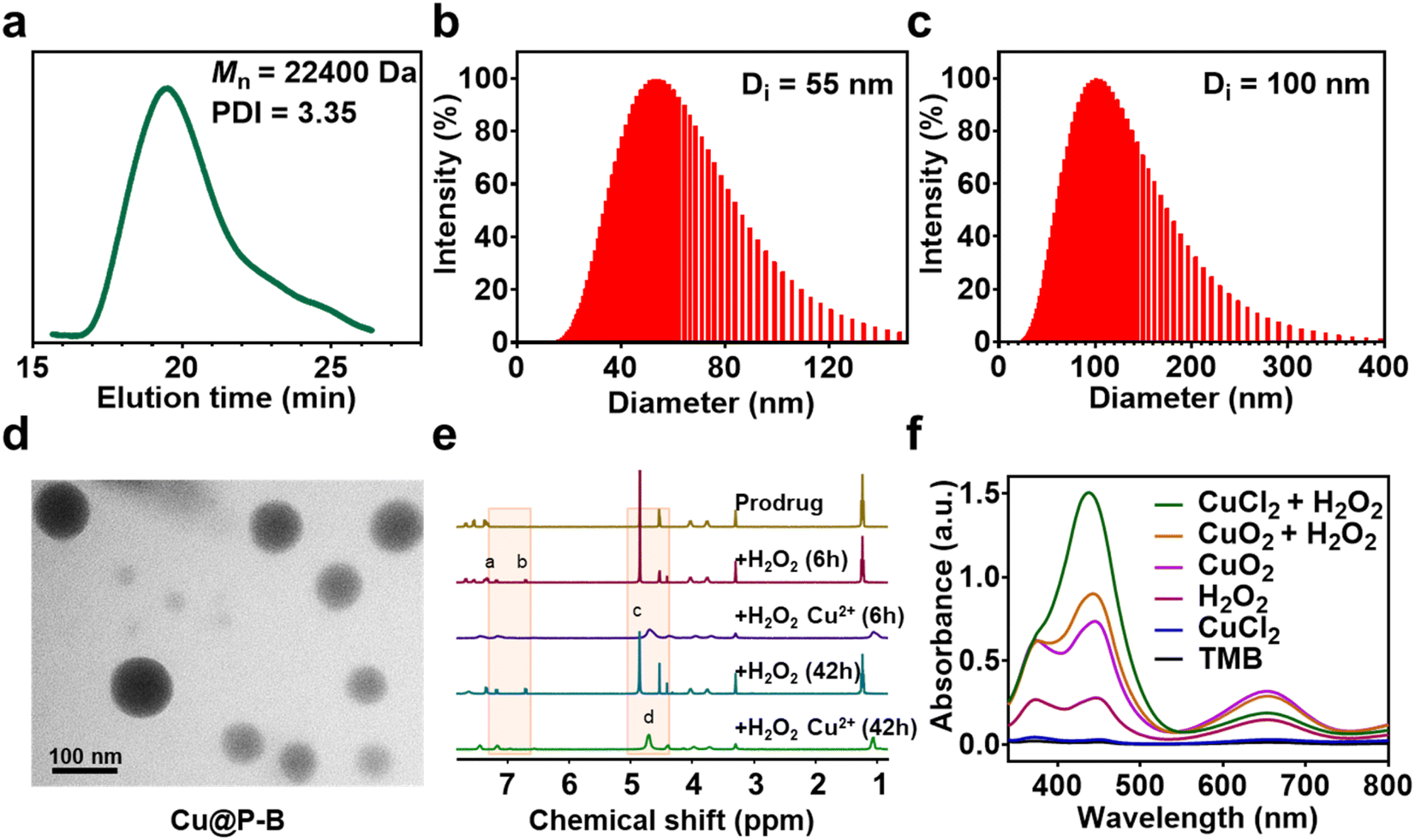
The functional nanoplatform consists of a DSF prodrug, CuO2 NPs, and a drug vector. The DSF prodrug was synthesized by a reaction between 4-(bromomethyl)phenylboronic acid and sodium diethylcarbamodithioate trihydrate. After concentration in vacuo, a white powder was obtained. The synthesis process of the prodrug is shown in Scheme 1. The chemical structure of the DSF prodrug was confirmed by 1H-NMR (solvent: CD3OD) (Fig. S1†). CuO2 NPs were prepared by the reaction of copper(II) chloride (CuCl2), H2O2, and sodium hydroxide (NaOH) in an aqueous solution containing PVP at room temperature for 30 min.33 The drug vector was prepared via ring-opening polymerization of α-amino acid N-carboxyanhydrides (NCAs).34 Specifically, the mPEG-b-PBLA diblock copolymer (designed degree of polymerization = 22) was synthesized through the ROP of the BLA–NCA monomer with mPEG-NH2 as the macroinitiator.37 The Mn and PDI of mPEG-b-PNAEA were 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1 and 3.35, respectively (Fig. 1(a)).
400 g mol−1 and 3.35, respectively (Fig. 1(a)).
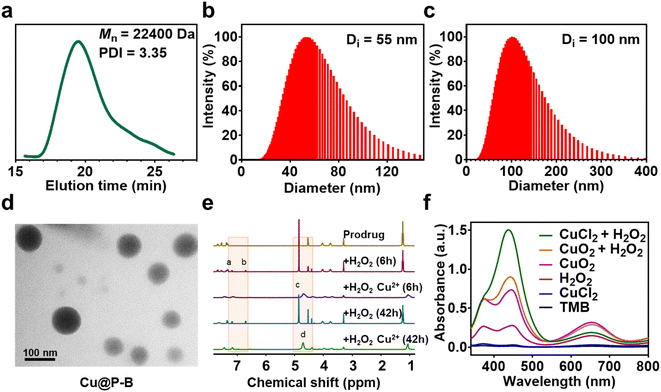 |
| | Fig. 1 (a) GPC curve of mPEG-b-PNAEA, Mn = 22400 g mol−1, and PDI = 3.35. (b) Dynamic light scattering (DLS) measurement of P–B and (c) DLS measurement of Cu@P–B. (d) TEM image of the synthesized Cu@P–B NPs. (e) 1H-NMR spectra of P–B micelles after H2O2 or H2O2/Cu2+ treatment at different times. (f) UV-vis spectra of TMB aqueous solution treated under different conditions (Cu2+, H2O2, CuO2, CuO2/H2O2, and Cu2+/H2O2) for 30 min. Note that the weak absorption between 600 and 800 nm in the Cu2+-treated group is from CuCl2. | |
Drug loading and characterization of P–B and Cu@P–B
A wide range of Lewis acids per bases can be incorporated to manipulate the polymer structure to control drug release. Boronic acid, a common Lewis acid, is of particular interest due to its ability to form reversible interactions with donor atoms such as oxygen and nitrogen. More importantly, boronic acids per esters, such as boronic acid structures in prodrugs, are sensitive to ROS overproduction by cancer cells. The preparation of DSF prodrug-loaded P–B and Cu@P–B NPs and the B–N coordination interaction between the DSF prodrug and mPEG-b-PNAEA enhanced drug loading are shown in Scheme 1. For P–B, the drug loading efficiency of the prodrug was 15.61%. For Cu@P–B, the drug loading efficiency of the prodrug and CuO2 was 15.37% and 42.93%, respectively. After loading CuO2, the size of P–B increased from 55 nm (Fig. 1(b)) to 100 nm (Fig. 1(c)). As shown in Fig. S2,† the particle size of Cu@P–B in PBS showed minimal change within 7 days, indicating that the nanoparticles have good colloidal stability. The transmission electron microscopy (TEM) images showed that Cu@P–B possessed a spherical morphology (Fig. 1(d)). And the TEM image of P–B is shown in Fig. S3.† According to a published paper, aryl boronate esters could be oxidized by ROS (such as H2O2). Therefore, in cancer cells, excess ROS can selectively trigger the release of drugs to kill the cells, reducing systemic toxicity with enhanced antitumor activity. Thus, we further investigated H2O2-responsive activity by 1H-NMR. After H2O2 treatment, peaks of 4-(hydroxymethyl)phenol and mPEG-b-PNAEA (peaks a and b) appeared, demonstrating that the DSF prodrug reacted with H2O2. However, upon incubation with H2O2 in the presence of Cu2+, a peak of Cu(DDC)2 appeared (peak d), while the previously existing peak (c) disappeared (Fig. 1(e)). The reason seems to be that the DSF prodrug reacted with Cu2+ to form Cu(DDC)2. Under the tumor microenvironment, Cu2+ catalyzes H2O2 to produce highly toxic hydroxyl radicals (˙OH) through a Fenton-like reaction. To study the H2O2 responsive activity and ˙OH production, Cu@P–B and H2O2 were investigated using the 3,3′,5,5′-tetramethylbenzidine (TMB) assay, based on the fact that TMB could be oxidized by the highly reactive ˙OH to give a blue–green color with maximum absorbance at approximately 650 nm. As shown in Fig. 1(f), neither Cu2+ nor H2O2 alone had a detectable effect on the absorbance increase of TMB even when the incubation duration was extended to 4 h. In contrast, Cu2+/H2O2 and CuO2/H2O2 led to a rapid color change of TMB aqueous solution, thereby demonstrating the excellent Fenton-like catalytic performance of Cu2+ and efficient ˙OH generation by the Cu2+-driven Fenton reaction. Therefore, the functional nanoplatform was prepared successfully, which could release the DSF prodrug and initiate a Fenton-like reaction to generate highly reactive ˙OH.
Cell viability
The in vitro cytotoxicity of CuO2, P–B and Cu@P–B NPs was evaluated by MTT assay, and the cell viability was expressed as the percentage of absorbance of control (PBS-treated) cells. As shown in Fig. 2(a) and (b), the 4T1 cells and A549 cells were treated with CuO2 (0.12 μg mL−1, 0.18 μg mL−1, 0.24 μg mL−1, and 0.30 μg mL−1), P–B (1.4 μg mL−1, 2.2 μg mL−1, 2.7 μg mL−1, and 3.4 μg mL−1), and Cu@P–B (1.4 μg mL−1, 2.2 μg mL−1, 2.7 μg mL−1, and 3.4 μg mL−1) separately for 48 hours. The results showed that the viability of cancer cells decreased significantly with increasing drug concentrations. In particular, the cancer cell killing activity of Cu@P–B NPs was significantly better than that of CuO2 and P–B alone. Specifically, 1.4 μg mL−1 Cu@P–B (containing 0.12 μg mL−1 CuO2) killed approximately 70% of 4T1 cells, CuO2 and P–B killed 5 and 30%, respectively. Similar results were also obtained in A549 cells. Meanwhile in the normal human cell 293T, neither CuO2 nor P–B showed obvious toxicity, and Cu@P–B was only slightly toxic at the highest concentration of 0.30 μg mL−1 (Fig. S4†). As for cancer cells, 0.12 μg mL−1 Cu@P–B could kill most cells, demonstrating that it was relatively safe for normal cells. Therefore, Cu@P–B has a high anticancer effect. At the same dosage, the Cu@P–B experimental group had a more significant tumor cell inhibitory effect than the CuO2 and P–B groups (p < 0.01) (Fig. 2(a) and (b)), which means that the antitumor activity is associated with the co-delivery of DSF-prodrug and CuO2. To further verify that the cytotoxicity resulting from the coexistence of Cu2+ and P–B, free copper ions were added to the cell medium to investigate the cell killing activity. The MTT results of different concentrations of P–B and CuCl2 acting on A549 cells for 48 hours showed that the presence of Cu2+ could enhance the anticancer activity of P–B NPs (Fig. 2(b) and (c)), which confirmed our hypothesis. In the experiments with the addition of exogenous free copper ions, only when the copper ion concentration reached 0.6 μg mL−1 did it show an obvious antitumor effect. However, when the DSF prodrug and CuO2 were co-delivered with the functional nanoplatform, the copper ion concentration was only 0.12 μg mL−1 (0.18 μg mL−1 CuO2), which killed most tumor cells. Therefore, functional NPs codelivering CuO2 and DSF prodrugs have high copper ion utilization efficiency. The nanoparticles only need to deliver a small number of copper ions to kill all tumor cells, which is conducive to improving the antitumor activity and reducing the toxicity and side effects resulting from copper ions. Cancer cells normally have elevated levels of ROS, increasing their susceptibility to ROS-induced apoptotic signaling.6 Since the literature has reported that CuO2 can kill cancer cells through ROS, we also examined the level of ROS produced in the cells. As revealed in Fig. 2(d), green fluorescence was hardly observed in the control group due to the low concentration of ROS in the cells, whereas the cellular fluorescence intensity of the cells in the experimental group gradually increased after different treatments (including 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B and 2.2 μg mL−1 Cu@P–B) for 4 hours. In addition, we further quantitatively investigated ROS levels in cancer cells. The ROS fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA) was used to evaluate the ROS concentration in A549 cells. As shown in Fig. 2(e) and (f), the results of flow cytometry showed that the fluorescence intensity of Cu@P–B was approximately 1.7 times higher than that of the control, indicating that Cu@P–B contributed to the increase in ROS levels. The CuO2 and P–B groups had higher cell viability, while only the Cu@P–B group had lower cell viability (Fig. 2(a) and (b)), indicating that ROS promoted the activation of the prodrug and accelerated cell death. However, the intracellular GSH levels of CuO2, P–B, and Cu@P–B were slightly lower than that of the control, revealing that the drug could reduce intracellular GSH levels and make cancer cells more susceptible to oxidative stress (Fig. 2(g)). To further evaluate the biocompatibility of Cu@P–B, we performed hemolysis experiments. The results indicated that CuO2, P–B, and Cu@P–B had no hemolytic activity on red blood cells (Fig. S5†), and the hemolysis rates were all lower than 5% (Fig. 2(h)). In conclusion, Cu@P–B induced significant tumor cell death and increased the intracellular ROS level.
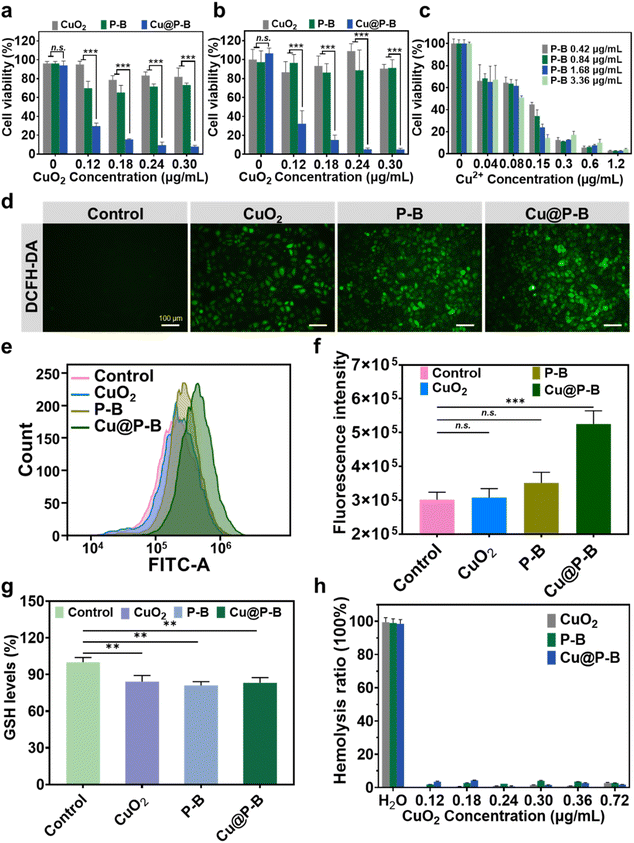 |
| | Fig. 2 (a) The viability of 4T1 cells after different treatments for 48 h (n = 3, *p < 0.05, **p < 0.01, and ***p < 0.001). (b) The viability of A549 cells under various conditions for 48 h (n = 3, *p < 0.05, **p < 0.01, and ***p < 0.001). (c) The viability of A549 cells treated with different concentrations of P–B and Cu2+ for 48 h. (d) Fluorescence images of DCFH-DA-stained A549 cancer cells after exposure to CuO2, P–B, and Cu@P–B for 4 h. The scale bar represents 100 μm. (e) Intracellular ROS levels in 4T1 cells were measured by flow cytometric analysis. (f) Quantification of intracellular ROS intensity calculated from the flow cytometry results (n = 3, *p < 0.05, **p < 0.01, and ***p < 0.001). (g) The intracellular GSH level after the treatment of CuO2, P–B, and Cu@P–B (n = 3, *p < 0.05, **p < 0.01, and ***p < 0.001). (h) The hemolytic activities of CuO2, P–B, and Cu@P–B at different concentrations. | |
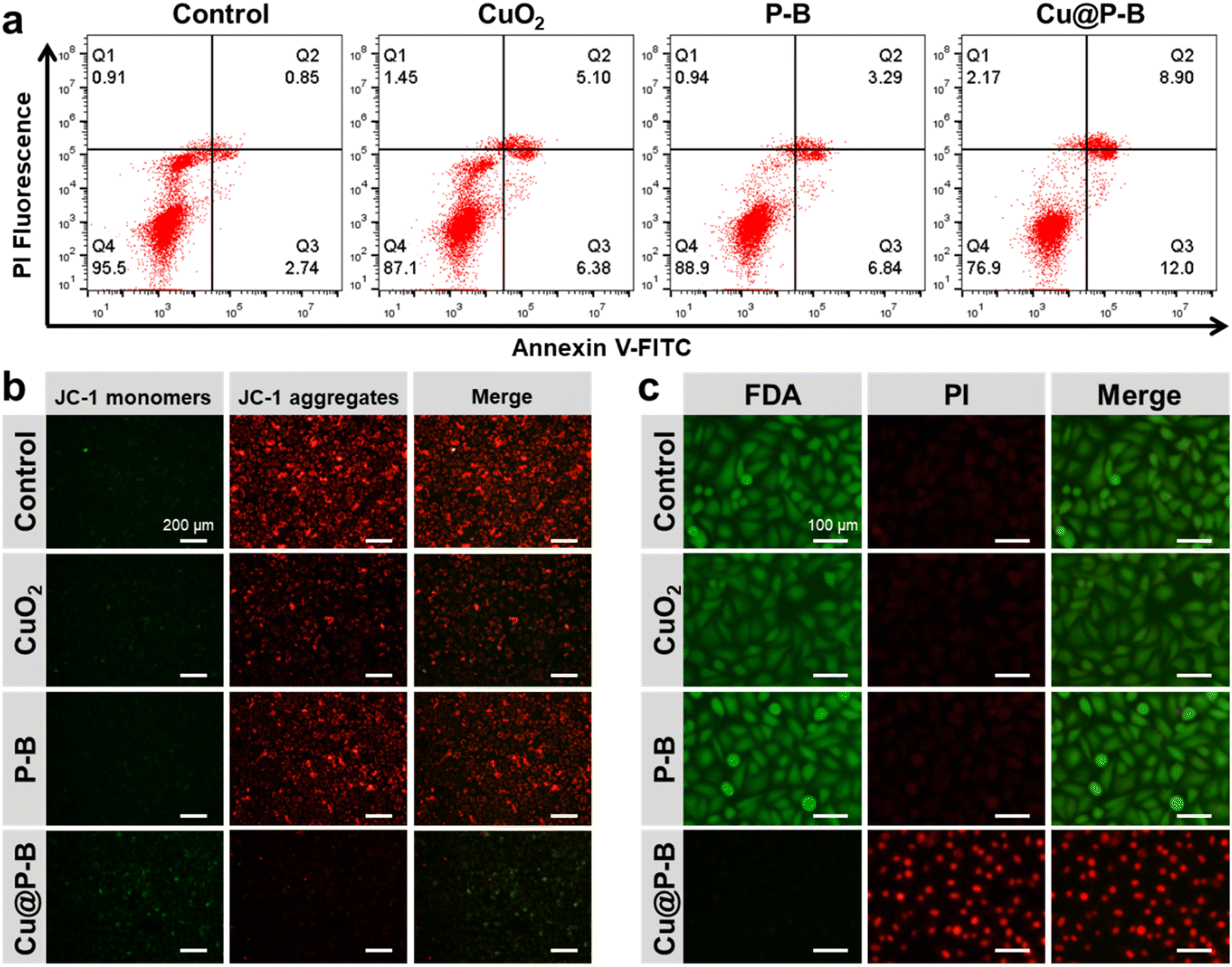
Cu@P–B NP induced cell apoptosis in A549 cells
A flow cytometry assay after annexin V-FITC/PI staining showed that exposure to Cu@P–B NPs increased the percentage of cells in both early (Fig. 3(a), lower right quadrant) and late apoptosis (Fig. 3(a), upper right quadrant). As indicated in Fig. S6,† the quantification results showed that the apoptosis rate of the A549 cancer cells treated with CuO2 or P–B for 6 hours was 10.53 or 9.38%, respectively, which was slightly upregulated compared with the control group (3.48%). However, the apoptosis rate of the A549 cells was significantly increased (19.08%) after Cu@P–B treatment. Another hallmark of apoptotic cell death, the collapse of mitochondrial membrane potential, was also observed in Cu@P–B-treated cells using JC-1 staining. A mitochondrial membrane potential detection kit was used to detect the mitochondrial membrane potential of A549 cells after different treatments (including 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B and 2.2 μg mL−1 Cu@P–B) and incubation for 6 hours. Fig. 3(b) shows that after Cu@P–B NP treatment for 6 hours, the mitochondrial membrane potential decreased significantly compared with that in the CuO2 and P–B experimental groups. The loss of mitochondrial membrane potential was demonstrated by the change in JC-1 fluorescence from red (JC-1 aggregates) to green (JC-1 monomers). This result indicated that A549 cells underwent early apoptosis. Together with the cell viability results, it could be deduced that Cu@P–B NPs would be an effective therapeutic nanoplatform with significant anticancer ability.38 The above results indicate that Cu@P–B NPs triggered A549 cell apoptotic-like cell death and inhibited tumor growth. It has also been demonstrated that Cu(DDC)2 potently inhibits proteasomal activity, induces ROS generation, and suppresses the NF-κB pathway, which is associated with apoptosis in human tumor cells.39 Our results confirmed that Cu@P–B NPs induced more apoptosis, thereby increasing the inhibition of A549 cells. To gain the most intuitive information about the effect of Cu@P–B on cell death, A549 cells were also treated with 0.18 μg mL−1 CuO2, 2.2 μg mL−1 P–B and 2.2 μg mL−1 Cu@P–B for 6 hours and then stained with FDA and PI. Consistent with the outcomes obtained by the MTT assay, Cu@P–B NPs possessed the most effective cancer cell killing ability (Fig. 3(c)), revealing the cooperative work of CuO2 and P–B in promoting cancer cell inhibition.
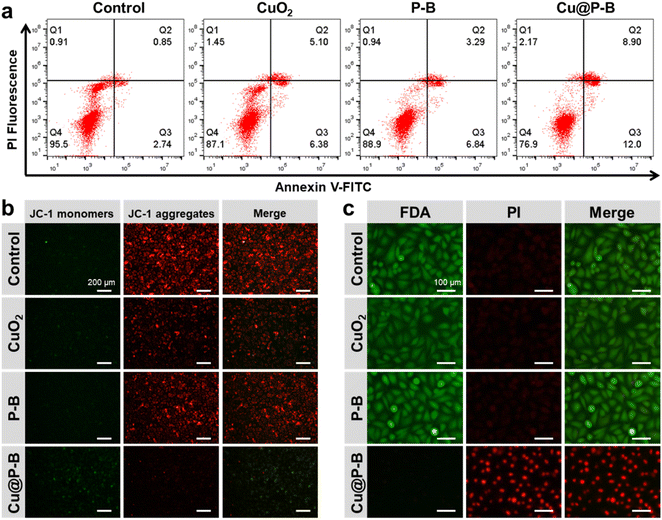 |
| | Fig. 3 (a) The apoptosis of A549 cells was detected using annexin V-FITC/PI staining after treatment with CuO2, P–B, and Cu@P–B for 6 hours. (b) Fluorescence images showed JC-1 staining of A549 cells after treatments with CuO2, P–B, and Cu@P–B (JC-1 monomers, green; JC-1 aggregates, red). The scale bar is 200 μm. (c) Representative live–dead staining images of A549 cells after incubation with PBS, CuO2, P–B, and Cu@P–B for 6 hours (PI, red; FDA, green). The scale bar is 100 μm. | |
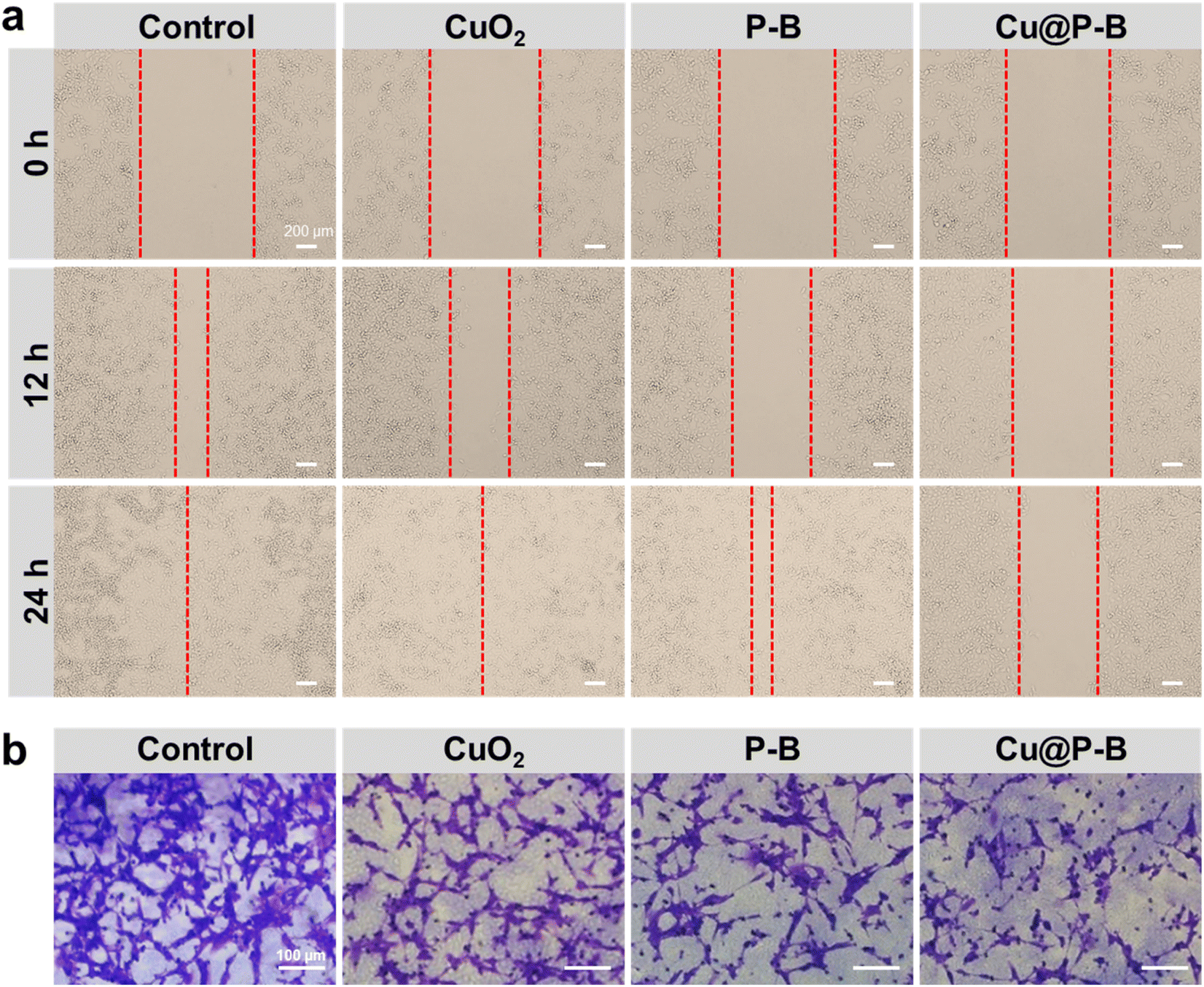
Cell migration and invasion
To investigate the effects of Cu@P–B on epithelial-mesenchymal transformation (EMT) in tumor cells, we performed cell migration and invasion experiments.40 As shown in Fig. 4(a), Cu@P–B (2.2 μg mL−1) greatly repressed the migration of A549 cells. After 24 hours of incubation, the PBS group had been completely fused, while there were still large scratches in the Cu@P–B-treated group. The statistical results of the cell migration rate are shown in Fig. S7,† and treatment with Cu@P–B caused cell migration that was approximately 40% of that of the PBS group. To validate these results, transwell assays were carried out. Significant inhibition of cellular migration was found in cells treated with 2.2 μg mL−1 Cu@P–B for 24 hours, as demonstrated by a dramatic drop in transmigrated cells relative to untreated cells (Fig. 4(b)). The statistical results in Fig. S8† show that treatment with CuO2, P–B, and Cu@P–B separately led to a dramatic decrease in cell invasion compared to PBS, especially Cu@P–B, which resulted in an approximately 9% invasion rate. Cancer metastasis is a rather complex process involving a series of biological programs, of which EMT is generally believed to be the driving force for cancer cell transition from epithelial features into mesenchymal phenotypes.41 In summary, these findings revealed that Cu@P–B greatly enhanced the metastatic behaviors of A549 and CT26 cancer cells.
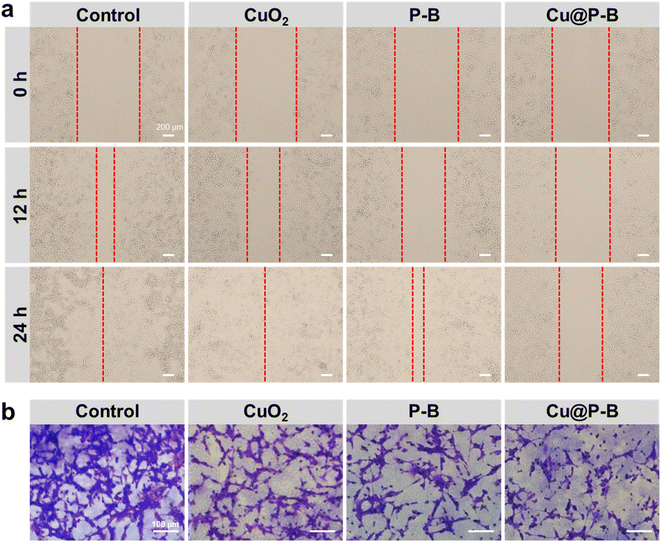 |
| | Fig. 4 (a) Effect of CuO2, P–B, or Cu@P–B treatment on the migration of A549 cancer cells. The scale bar is 200 μm. (b) Effect of CuO2, P–B, or Cu@P–B treatment on the invasion of CT26 cells detected by transwell assay. The scale bar is 100 μm. | |
In vivo antitumor effects of Cu@P–B
The in vivo biological behavior of Cu@P–B, including biocompatibility, was systematically evaluated. To further investigate the antitumor activity of Cu@P–B NPs in vivo, we first performed dose tolerance tests on normal Balb/c mice and found that (Fig. S9†), when an intravenous dose up to 175 μg kg−1 CuO2 (contained in Cu@P–B) was injected, the body weight of normal mice remained stable, compared with the control. The injection of the highest dose of Cu@P–B did not cause obvious toxicity, indicating that the mice could tolerate the highest dose. It is hypothesized that Cu@P–B NPs with a small size can effectively accumulate in tumor tissue through an enhanced permeability and retention (EPR) effect. To confirm this, the accumulation at the tumor site was then evaluated by intravenous injection of Cu@P–B into 4T1 tumor-bearing mice. The tumors and major organs of the mice were harvested 12 hours post injection to evaluate the accumulation of Cu@P–B based on Cu in the tumor and each organ using ICP-AES. As shown in Fig. S10,† Cu@P–B effectively accumulated in tumor tissues during the time course of incubation, with the 12 hour passive-targeting efficiency reaching 4.3% via the typical EPR effect. Based on its effective accumulation in tumors, Cu@P–B, which acted as a carrier of the prodrug and a copper source, could effectively deliver Cu(DDC)2 to the tumor site and increase the number of copper ions at the site, thereby enhancing the chemotherapeutic efficacy of P–B. At the same time, the side effects caused by excess copper were substantially reduced.42 The in vivo anticancer efficacy of Cu@P–B was next evaluated. 4T1 tumor-bearing female Balb/c mice were established and randomly divided into four groups, which were intravenously injected with saline, CuO2, P–B, or Cu@P–B at a Cu2+ dosage of 0.1 mg kg−1 every other day for three doses (Fig. 5(a)). The tumor volume and body weight were monitored every day. During the whole therapeutic process, no significant body-weight changes were observed (Fig. 5(b)), which confirmed the high therapeutic biosafety of Cu@P–B NPs. As shown in Fig. 5(c), (d) and S11,† mice injected with saline (control group) showed rapid tumor growth, whereas the growth of tumors in Cu@P–B-treated mice was significantly suppressed. In addition, the tumor growth inhibition value of Cu@P–B treated mice on day 16 was 65.26%. After the treatment period, tumor and tissue (heart, liver, spleen, lung, and kidney) sections from sacrificed mice were stained with H&E, and the tumors were also stained with TUNEL and Ki-67 antigens. As presented in Fig. S12,† the H&E staining results showed no obvious toxicity in the organs. TUNEL staining results (Fig. 5(e)) showed that the majority of cell necrosis occurred in the tumor sections from the mice treated with Cu@P–B. In contrast, no apparent cell necrosis was observed in the control group. Furthermore, tumor cells in the Cu@P–B group showed greater inhibition of proliferation compared to Ki-67 staining in the other groups (Fig. 5(e)). The remarkable tumor growth inhibitory effect of Cu@P–B can be attributed to efficient tumor accumulation and the production of Cu(DDC)2 in the tumor microenvironment. Therefore, the above results demonstrate the feasibility of using Cu@P–B as a potential agent for cancer therapy in vivo.
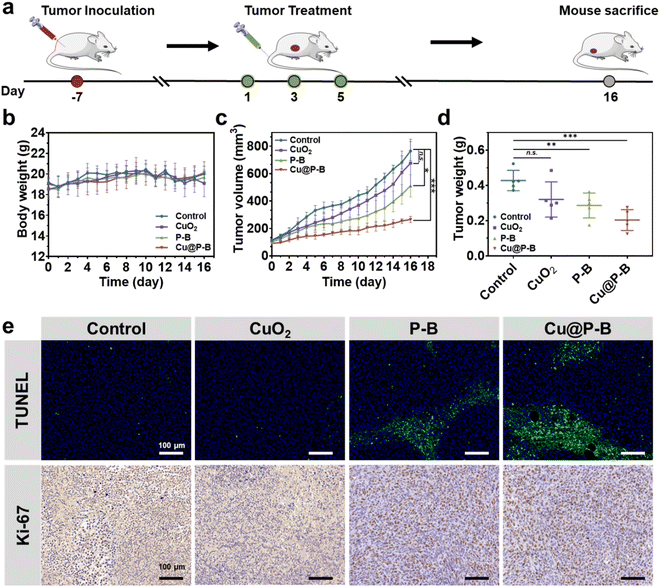 |
| | Fig. 5 (a) Schematic illustration of the 4T1 tumor establishment and treatment process. (b) Body weight and (c) tumor volume change curves of 4T1 tumor-bearing mice after various treatments (n = 5, *p < 0.05, **p < 0.01, and ***p < 0.001). (d) Weight of the collected tumors after different treatments (n = 5, *p < 0.05, **p < 0.01, and ***p < 0.001). (e) Terminal deoxynucleotidyl transferase (TUNEL, top row), and Ki-67 antigen staining (bottom row) of tumor tissues harvested from the corresponding mice after different treatments; the scale bar is 100 μm. Different treatment conditions included saline, CuO2, P–B and Cu@P–B. | |
Conclusions
In this study, we successfully synthesized a functional nanoparticle for synergistic chemotherapy by a simple method that could target the tumor area through the EPR effect, increasing the synergistic effect with copper in cancer therapy. Specifically, the tumor microenvironment-responsive DSF prodrug and CuO2 were co-delivered to tumor tissue to trigger a cascade reaction and kill the cancer cells. ROS overproduction in the tumor microenvironment could disrupt the interaction of the DSF prodrug to release and activate DDC. The loaded CuO2 will release Cu2+ and H2O2 in an acid environment and induce cellular oxidative stress, which will accelerate the release of the DSF prodrug. Importantly, the activated DDC and released Cu2+ will form toxic compounds to kill the cancer cells. The results show that Cu@P–B NPs greatly increase oxidative stress in the tumor site, causing the elevation of ROS and leading to apoptosis, which significantly inhibits the growth of tumor cells. In vitro and in vivo experiments show that Cu@P–B has very potent potential in cancer therapy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by grants from the Anhui Natural Science Fund Project (2108085MH311 and 2208085MH252), Anhui Key Research and Development Program (201904a07020092), Natural Science Self-Financing Project of Anhui Provincial Hospital (2019ZC042), and Provincial Quality Engineering Teaching and Research Project of Anhui Province Colleges and Universities (2019jyxm0972).
Notes and references
- C. Xia, X. Dong, H. Li, M. Cao, D. Sun, S. He, F. Yang, X. Yan, S. Zhang, N. Li and W. Chen, China Med. J., 2022, 135, 584–590 CrossRef PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- R. Baskar, K. A. Lee, R. Yeo and K. W. Yeoh, Int. J. Med. Sci., 2012, 9, 193–199 CrossRef PubMed.
- M. Wolacewicz, R. Hrynkiewicz, E. Grywalska, T. Suchojad, T. Leksowski, J. Rolinski and P. Niedzwiedzka-Rystwej, Cancers, 2020, 12, 1181 CrossRef CAS PubMed.
- M. Arruebo, N. Vilaboa, B. Saez-Gutierrez, J. Lambea, A. Tres, M. Valladares and A. Gonzalez-Fernandez, Cancers, 2011, 3, 3279–3330 CrossRef CAS PubMed.
- L. Sun, Y. Xu, Y. Gao, X. Huang, S. Feng, J. Chen, X. Wang, L. Guo, M. Li, X. Meng, J. Zhang, J. Ge, X. An, D. Ding, Y. Luo, Y. Zhang, Q. Jiang and X. Ning, Small, 2019, 15, 1901156 CrossRef PubMed.
- B. Johansson, Acta Psychiatr. Scand., 1992, 86, 15–26 CrossRef PubMed.
- W. Malin, D. Katarina, R. Linda, G. Joachim, N. Peter, I. Anders, L. Rolf and L. Henrik, Biochem. Pharmacol., 2007, 73, 25–33 CrossRef PubMed.
- C. Conticello, D. Martinetti, L. Adamo, S. Buccheri, R. Giuffrida, N. Parrinello, L. Lombardo, G. Anastasi, G. Amato, M. Cavalli, A. Chiarenza, R. De Maria, R. Giustolisi, M. Gulisano and F. Di Raimondo, Int. J. Cancer, 2012, 131, 2197–2203 CrossRef CAS PubMed.
- P. Dufour, J. M. Lang, C. Giron, B. Duclos, P. Haehnel, D. Jaeck, J. M. Jung and F. Oberling, Biotherapy, 1993, 6, 9–12 CrossRef CAS PubMed.
- H. Nechushtan, Y. Hamamreh, S. Nidal, M. Gotfried, A. Baron, Y. I. Shalev, B. Nisman, T. Peretz and N. Peylan-Ramu, Oncologist, 2015, 20, 366–367 CrossRef PubMed.
- H. Fasehee, R. Dinarvand, A. Ghavamzadeh, M. Esfandyari-Manesh, H. Moradian, S. Faghihi and S. H. Ghaffari, J. Nanobiotechnol., 2016, 14, 1–18 CrossRef PubMed.
- Z. Wang, J. Tan, C. McConville, V. Kannappan, P. E. Tawari, J. Brown, J. Ding, A. L. Armesilla, J. M. Irache, Q. B. Mei, Y. Tan, Y. Liu, W. Jiang, X. W. Bian and W. Wang, Nanomedicine, 2017, 13, 641–657 CrossRef CAS PubMed.
- M. Najlah, A. Said Suliman, I. Tolaymat, S. Kurusamy, V. Kannappan, A. M. A. Elhissi and W. Wang, Pharmaceutics, 2019, 11, 610 CrossRef CAS PubMed.
- X. Wu, X. Xue, L. Wang, W. Wang, J. Han, X. Sun, H. Zhang, Y. Liu, X. Che and J. Yang, Eur. J. Pharmacol., 2018, 827, 1–12 CrossRef CAS PubMed.
- C. C. Liu, C. L. Wu, M. X. Lin, C. I. Sze and P. W. Gean, Int. J. Mol. Sci., 2021, 22, 10496 CrossRef CAS PubMed.
- H. J. Cho, T. S. Lee, J. B. Park, K. K. Park, J. Y. Choe, D. I. Sin, Y. Y. Park, Y. S. Moon, K. G. Lee, J. H. Yeo, S. M. Han, Y. S. Cho, M. R. Choi, N. G. Park, Y. S. Lee and Y. C. Chang, J. Mol. Biol., 2007, 40, 1069–1076 CAS.
- R. Malcolm, M. F. Olive and W. Lechner, Expert Opin. Drug Saf., 2008, 7, 459–472 CrossRef CAS PubMed.
- P. Hu, L. Jin and T. A. Baillie, J. Pharmacol. Exp. Ther., 1997, 281, 611–617 CAS.
- X. Zhuo, T. Lei, L. Miao, W. Chu, X. Li, L. Luo, J. Gou, Y. Zhang, T. Yin, H. He and X. Tang, J. Colloid Interface Sci., 2018, 529, 34–43 CrossRef CAS PubMed.
- M. T. Schweizer, J. Lin, A. Blackford, A. Bardia, S. King, A. J. Armstrong, M. A. Rudek, S. Yegnasubramanian and M. A. Carducci, Prostate Cancer Prostatic Dis., 2013, 16, 357–361 CrossRef CAS PubMed.
- J. Huang, J. L. Campian, A. D. Gujar, D. D. Tran, A. C. Lockhart, T. A. DeWees, C. I. Tsien and A. H. Kim, J. Neurooncol., 2016, 128, 259–266 CrossRef CAS PubMed.
- A. Madan and M. D. Faiman, Alcohol. Clin. Exp. Res., 1994, 18, 1013–1017 CrossRef CAS PubMed.
- A. Bruning and R. E. Kast, Cell Cycle, 2014, 13, 1513–1514 CrossRef PubMed.
- A. McMahon, W. Chen and F. Li, J. Control. Release, 2020, 319, 352–359 CrossRef CAS PubMed.
- Z. Skrott, M. Mistrik, K. K. Andersen, S. Friis, D. Majera, J. Gursky, T. Ozdian, J. Bartkova, Z. Turi, P. Moudry, M. Kraus, M. Michalova, J. Vaclavkova, P. Dzubak, I. Vrobel, P. Pouckova, J. Sedlacek, A. Miklovicova, A. Kutt, J. Li, J. Mattova, C. Driessen, Q. P. Dou, J. Olsen, M. Hajduch, B. Cvek, R. J. Deshaies and J. Bartek, Nature, 2017, 552, 194–199 CrossRef CAS PubMed.
- P. F. John and T. Valerie, Expert Rev. Anticancer Ther., 2008, 8, 1751–1757 CrossRef PubMed.
- Z. Jie, C. Feili, D. Huijuan, S. Pengcheng, Y. Yao, Z. Yanyan, L. Rongwei, W. Shiyun, L. Peng, W. Weiguang and X. Bing, J. Transl. Med., 2014, 12, 163 CrossRef PubMed.
- X. Bing, S. Pengcheng, S. F. Ivo, Z. Yanyan, H. Fen, W. Weiguang and Z. Shuyun, Blood
Cells. Mol. Dis., 2011, 47, 264–269 CrossRef PubMed.
- S. Shao, Q. Zhou, J. Si, J. Tang, X. Liu, M. Wang, J. Gao, K. Wang, R. Xu and Y. Shen, Nat. Biomed. Eng., 2017, 1, 745–757 CrossRef CAS PubMed.
- S. Ishida, P. Andreux, C. Poitry-Yamate, J. Auwerx and D. Hanahan, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 19507–19512 CrossRef CAS PubMed.
- X. Kang, Y. Cai, Q. Wang, C. Wang, W. Chen, W. Yang, A. Suryawanshi, G. Zhou, P. Chen and F. Li, Int. J. Pharm., 2021, 607, 120972 CrossRef CAS PubMed.
- L. Li-Sen, H. Tao, S. Jibin, O. Xiang-Yu, W. Zhangtong, D. Hongzhang, T. Rui, L. Yijing, W. Jun-Feng, L. Yuan, Y. Guocan, Z. Zijian, W. Sheng, N. Gang, Y. Huang-Hao and C. Xiaoyuan, J. Am. Chem. Soc., 2019, 141, 9937–9945 CrossRef PubMed.
- Y. Wu, D. Zhang, P. Ma, R. Zhou, L. Hua and R. Liu, Nat. Commun., 2018, 9, 5297 CrossRef CAS PubMed.
- L. Zhang, B. Tian, Y. Li, T. Lei, J. Meng, L. Yang, Y. Zhang, F. Chen, H. Zhang, H. Xu, Y. Zhang and X. Tang, ACS Appl. Mater. Interfaces, 2015, 7, 25147–25161 CrossRef CAS PubMed.
- J. Zhang, Z. Zou, B. Wang, G. Xu, Q. Wu, Y. Zhang, Z. Yuan, X. Yang and C. Yu, Biomaterials, 2018, 161, 228–239 CrossRef CAS PubMed.
- S. Lv, Y. Wu, K. Cai, H. He, Y. Li, M. Lan, X. Chen, J. Cheng and L. Yin, J. Am. Chem. Soc., 2018, 140, 1235–1238 CrossRef CAS PubMed.
- Z. Yao, B. Zhang, T. Liang, J. Ding, Q. Min and J. J. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 18995–19005 CrossRef CAS PubMed.
- N. C. Yip, I. S. Fombon, P. Liu, S. Brown, V. Kannappan, A. L. Armesilla, B. Xu, J. Cassidy, J. L. Darling and W. Wang, Br. J. Cancer, 2011, 104, 1564–1574 CrossRef CAS PubMed.
- R. Liting, F. Wenya, S. Jie, M. Juan, X. Ming, Z. Ben-Zhan, Z. Nanfeng and L. Sijin, Theranostics, 2020, 10, 6384–6398 CrossRef PubMed.
- M. Luan, J. Chang, W. Pan, Y. Chen, N. Li and B. Tang, Anal. Chem., 2018, 90, 10951–10957 CrossRef CAS PubMed.
- W. Wu, L. Yu, Q. Jiang, M. Huo, H. Lin, L. Wang, Y. Chen and J. Shi, J. Am. Chem. Soc., 2019, 141, 11531–11539 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ya-Di
Geng
a,
Yun-Xiao
Liu
b,
Xuan
Nie
a,
Ya-Di
Geng
a,
Yun-Xiao
Liu
b,
Xuan
Nie