
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredictable incorporation of nitrogen into carbon dots: insights from pinacol rearrangement and iminium ion cyclization†
Soohyun
Cho
 a,
Chan-Woo
Jung
b,
Dajin
Lee
a,
Yerim
Byun
a,
Hyemin
Kim
c,
Hyunho
Han
d,
Ji-Hee
Kim
*e and
Woosung
Kwon
*af
a,
Chan-Woo
Jung
b,
Dajin
Lee
a,
Yerim
Byun
a,
Hyemin
Kim
c,
Hyunho
Han
d,
Ji-Hee
Kim
*e and
Woosung
Kwon
*af
aDepartment of Chemical and Biological Engineering, Sookmyung Women's University, Seoul 04310, South Korea. E-mail: wkwon@sookmyung.ac.kr
bDepartment of Energy Science, Sungkyunkwan University, Suwon 16419, South Korea
cDepartment of Cosmetics Engineering, Konkuk University, Seoul 05029, South Korea
dDepartment of Urology, Urological Science Institute, Yonsei University College of Medicine, Seoul 03722, South Korea
eDepartment of Physics, Pusan National University, Busan 46241, South Korea. E-mail: kimjihee@pusan.ac.kr
fInstitute of Advanced Materials and Systems, Sookmyung Women's University, Seoul 04310, South Korea
First published on 21st September 2023
Abstract
Nitrogen-doped carbon dots (CDs) have attracted considerable attention across various research areas and applications due to their enhanced optical properties and photostability. However, the mechanism of nitrogen incorporation in CDs remains elusive, hampering the precise control over nitrogen-incorporated structures and the investigation of the effects of nitrogen on the electronic structure and optical properties of CDs. In this study, we employed a rational design approach, utilizing glucosamine and ethylene glycol as the carbon source and co-reagent, respectively, to synthesize N-doped CDs. Our synthesis strategy involved pinacol rearrangement and iminium ion cyclization reactions, enabling the reliable formation of N-doped CDs. Notably, the resulting CDs exhibited distinctive emissive states attributed to heteroatomic defect structures, including oxygenic and nitrogenic polycyclic aromatic hydrocarbons. To gain further insights into their energy levels and electronic transitions, we conducted comprehensive investigations, employing extended Hückel calculations and pump-probe spectroscopy. The synthesized CDs displayed great promise as bioimaging and photodynamic therapy agents, highlighting their potential for biomedical applications. Moreover, our study significantly contributes valuable insights into the rational design of N-doped CDs with controllable chemical and electronic structures, thereby paving the way for advancements in their diverse range of applications.
Introduction
Carbon dots (CDs) are carbon-based optical nanomaterials that exhibit superior photoluminescence (PL) properties, excellent biological compatibility, and remarkable chemical stability.1–3 These characteristics make CDs highly attractive for diverse applications, including imaging, sensing, and drug delivery.4–9 To expand the application of CDs, researchers explored various methods for enhancing their optical properties.10,11 A significant emphasis has been placed on modifying their chemical structures through the introduction of heteroatomic impurities such as boron, nitrogen, oxygen, and sulfur.12–16 These impurities generate intragap states in CDs, which alter their electronic band structures and shift their PL wavelengths.17–19 Among these impurities, nitrogen is considered the optimal dopant for CDs due to its similar atomic size to carbon20,21 and its ability to form strong covalent bonds with carbon. This bond formation enables the introduction of additional photoexcitable electrons to the ground state.22Nitrogen-containing functional groups such as amino (–NH2) and amide (–CONH2) groups can be found on the surface of CDs and modify their electronic structure by acting as electron donors or acceptors. In addition, nitrogen can be incorporated into the CD core for generating pyridine- or pyrrole-like structures. N-doped CDs exhibit stronger PL intensities and longer PL lifetimes than undoped CDs.23 Various nitrogen-containing organic compounds such as organic amines, amino acids, aminosilanes, and ammonia derivatives24–28 can be used as precursors to produce N-doped CDs; however, the exact mechanism of nitrogen incorporation during the carbonization process remains unclear, and current synthesis strategies for N-doped CDs are often based on intuition or past experience, which lacks a systematic and deliberate design approach that could enable precise control over resulting structures.29 This limits our ability to study the effects of nitrogen on the electronic band structures and optical properties of CDs. Furthermore, the use of nitrogen-containing organic compounds as precursors does not always guarantee the formation of N-doped CDs, suggesting the need for the further exploration of alternative synthetic routes.
We present a rational approach to the synthesis of N-doped CDs derived from glucosamine (GA) and ethylene glycol (EG) (GE-CDs). GA is a readily carbonizable amino monosaccharide,30,31 and EG inhibits the release of the amine group from GA during thermal decomposition by forming an iminium ion, facilitating nitrogen incorporation. We conducted a comprehensive investigation into the characteristics of nitrogen-incorporated structures formed in GE-CDs and compared these structures with those of undoped CDs produced from GA without EG (G-CDs). Transient absorption (TA) spectroscopy and time-resolved PL (TRPL) spectroscopy revealed the effect of nitrogen-incorporated structures on the electronic band structure and optical transitions of GE-CDs. Finally, leveraging the electron transition/transfer properties of GE-CDs, we demonstrated their potential for in vitro biological imaging and photodynamic therapy (PDT) of human prostate cancer (PC3) and lung adenocarcinoma (A549) cells. Our rational design approach provides a promising avenue for the precise control of the structures of N-doped CDs and enables the study of the effects of nitrogen on their electronic band structures and optical properties, opening new possibilities for their use in various applications.
Results and discussion
Synthesis and chemical characterization of GE-CDs
Major products formed via the thermal degradation of GA in water at around pH 4 include furfural-like compounds such as 5-hydroxymethylfurfural, furfural, and furan-2,5-dicarboxaldehyde quinazoline.32 Under these reaction conditions, the amino group of GA is mostly hydrolyzed into ammonia, which is excluded from the reaction by evaporation. The remaining furfural intermediates are carbonized to form undoped CDs or G-CDs. They contain an abundance of oxygen-incorporated structures or oxygen-induced defects (OIDs),33 whereas nitrogen-incorporated structures are not formed.EG was added as a co-reagent to prevent the hydrolysis of the amino group in GA. Scheme 1 presents a proposed mechanism for the production of GE-CDs from GA and EG, involving well-established pinacol rearrangement and iminium ion oligomerization/cyclization reactions. Under acidic conditions, EG undergoes a pinacol rearrangement to generate acetaldehyde,34 which then reacts with GA to form imines via condensation. The protonation of these imines forms iminium ions that are only transiently present in aqueous solutions, but can be stabilized by nucleophiles such as EG.35 The iminium ions then undergo cyclization to create various heterocycles or oligomers with the simultaneous removal of water, which results in the formation of partially graphitized carbon cores.36–38 Ultimately, nitrogenous compounds are incorporated into GE-CDs through carbonization, which results in the formation of nitrogen-incorporated structures or nitrogen-induced defects (NIDs).
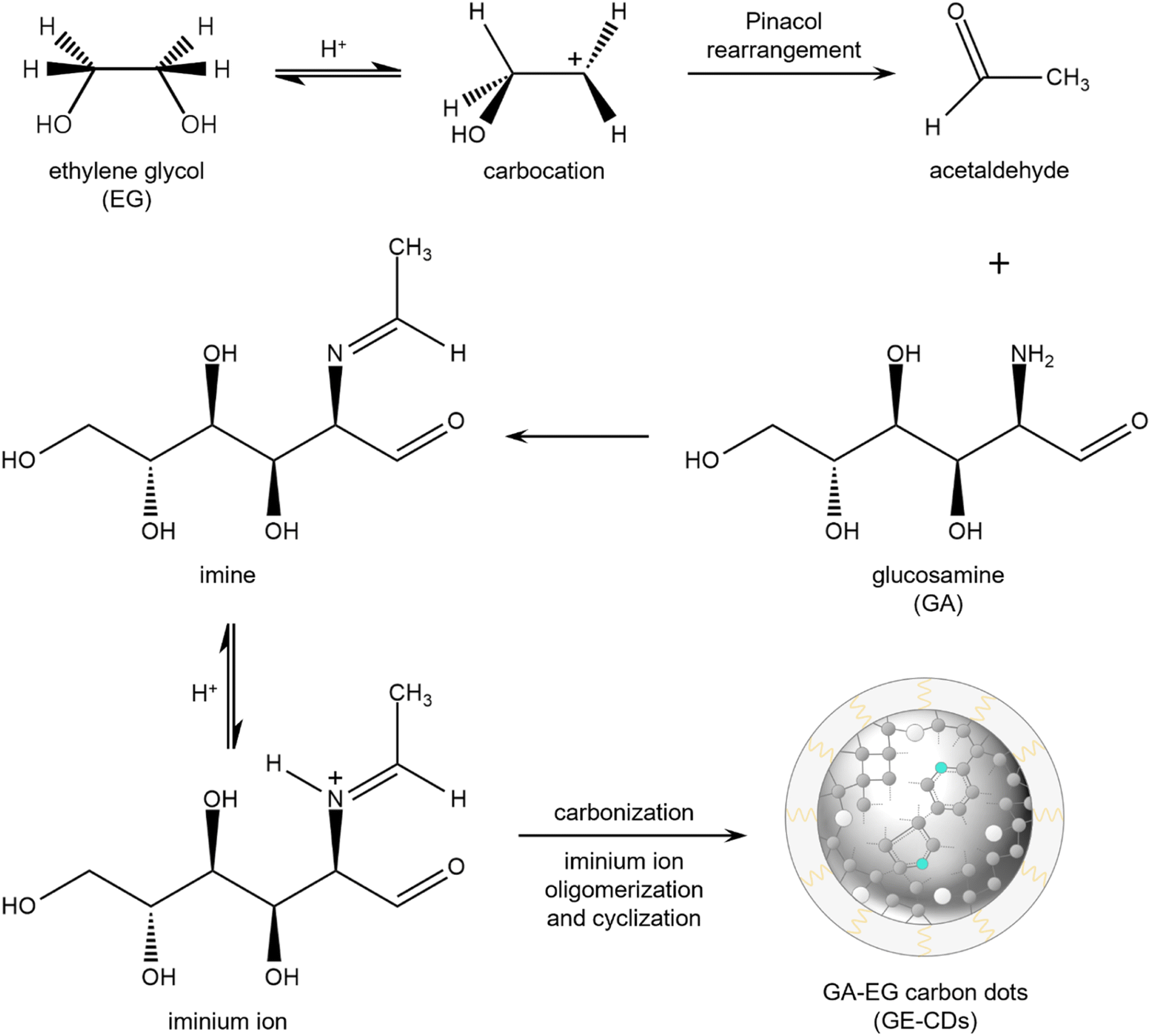 | ||
| Scheme 1 Proposed mechanism for the formation of GE-CDs (grey: carbon, cyan: nitrogen, white: oxygen). | ||
The 1H nuclear magnetic resonance (NMR) structural characterization of the CDs revealed nitrogen incorporation via the formation of an imine intermediate. To better understand the molecular mechanisms underlying GE-CD formation, GE-CDs were synthesized in deuterated EG (EG-d6), and the aliquots of the reaction were extracted at various time intervals (ranging from 1–12 h) and subjected to 1H NMR spectroscopy (Fig. 1a). The formation of an imine was confirmed by a multiplet at δ = 8.0–8.3 ppm (α). Although the imine signal quickly reached a maximum intensity and then diminished over time, the aromatic protons signal at δ = 7.0–7.4 ppm (β) appeared and continued to increase in the reaction duration. The 1H NMR spectra of the GA solution before and after the reaction are presented in Fig. 1b. The peak at 3.58 ppm corresponding to protons attached to the aromatic amines, appeared only in GE-CDs spectrum, indicating that the amine groups were successfully fixed during the formation of GE-CDs. Additional signals at 2.14 ppm observed for both CDs correspond to protons adjoining aromatic or sp2 carbons, indicating the formation of carbon cores with polyaromatic domains. Thus, we propose that the initial stages of the reaction involve iminium formation, followed by the trapping of the iminium electrophile, carbonization, and aromatization, resulting in the synthesis of nitrogen-doped CDs.
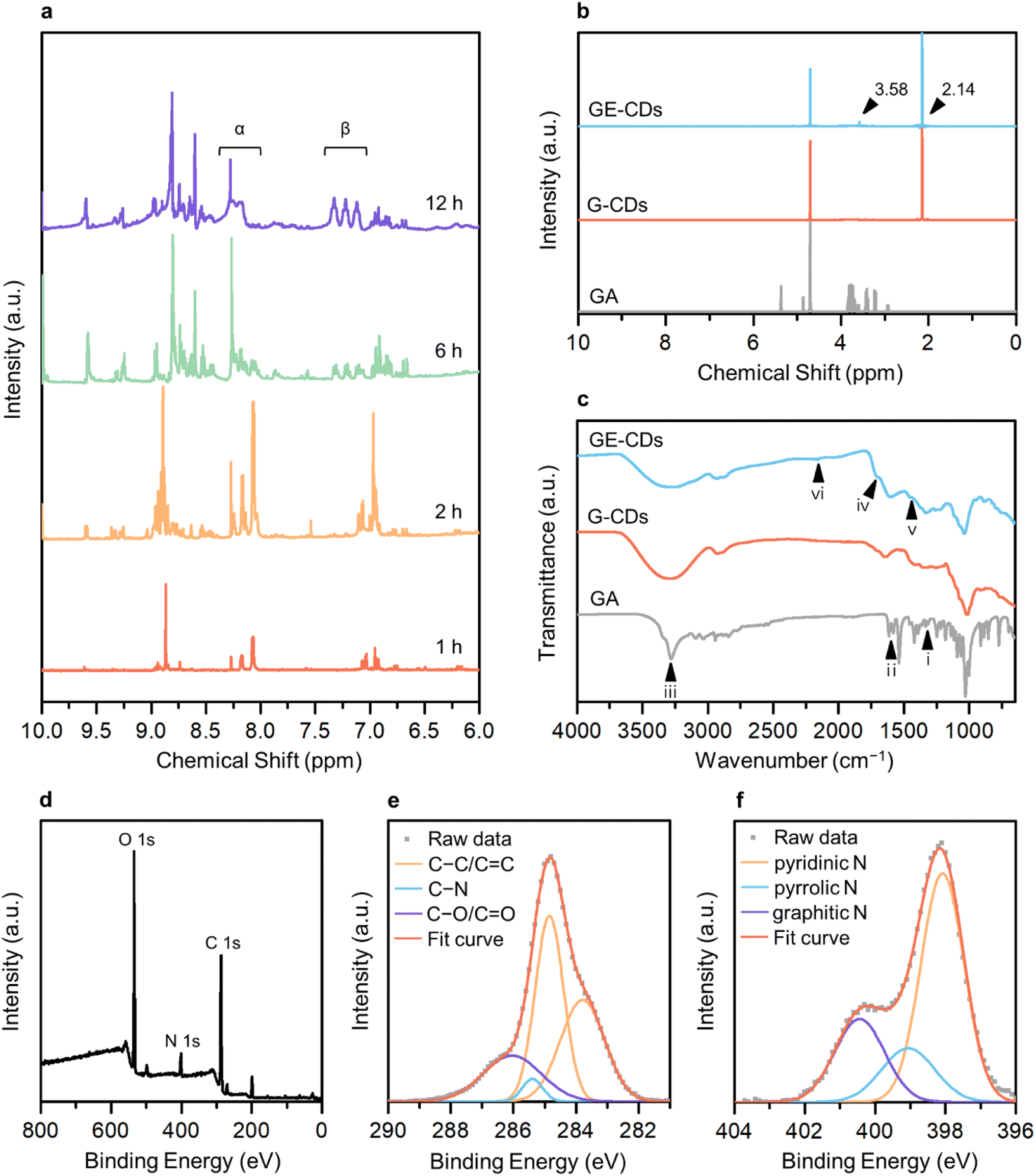 | ||
| Fig. 1 (a) 1H NMR spectra of GE-CDs with different reaction times. (b) 1H NMR spectra of GA, G-CDs, and GE-CDs. (c) FTIR spectra of GA, G-CDs, and GE-CDs. (d) XPS spectrum of GE-CDs. High-resolution XPS spectra of (e) carbon 1s and (f) nitrogen 1s of GE-CDs. | ||
The composition and chemical properties of the CDs were evaluated using Fourier-transform infrared (FTIR) spectroscopy, X-ray photoelectron spectroscopy (XPS) and elemental analysis. The FTIR spectrum of GA exhibits peaks for C–N stretching (1333 cm−1, i), amine N–H bending (1616 cm−1, ii), and amine N–H and hydroxyl O–H stretching (3200–3400 cm−1, iii) (Fig. 1c). In the FTIR spectra of G-CDs and GE-CDs, the hydroxyl O–H stretching (3200–3400 cm−1, iii) peak was broadened as compared to the GA data, indicating that the O–H groups were more developed during the formation of CDs with OID states. Moreover, the peaks around 1709 cm−1 (iv) commonly observed in G-CDs and GE-CDs indicate the existence of aldehyde groups through the formation of furfurals (from the reaction between GA and water) and acetaldehyde (from the reaction between EG and water), respectively. The aromatic cyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1456 cm−1, v) and C–H (2160 cm−1, vi) vibrating peaks were observed in both CDs, indicating the formation of carbon cores with polyaromatic domains. However, the amine C–N (1336 cm−1, i) stretching and N–H (1616 cm−1, ii) bending peaks only appeared on GE-CDs spectra; these results confirmed that the addition of EG successfully prevented the release of ammonia from GA during the reaction.
C (1456 cm−1, v) and C–H (2160 cm−1, vi) vibrating peaks were observed in both CDs, indicating the formation of carbon cores with polyaromatic domains. However, the amine C–N (1336 cm−1, i) stretching and N–H (1616 cm−1, ii) bending peaks only appeared on GE-CDs spectra; these results confirmed that the addition of EG successfully prevented the release of ammonia from GA during the reaction.
XPS measurements were conducted on G-CDs and GE-CDs to evaluate the proposed mechanism for the synthesis reaction (Scheme 1) and verify the effect of EG on nitrogen incorporation. The raw XPS data of G-CDs (Fig. S1a†) demonstrated carbon and oxygen peaks. In the carbon 1s spectra, a signal at 284.8 eV corresponded to the C–C/CC groups, and another peak at 285.5 eV for C–O/CO groups was recorded (Fig. S1b†). As shown in Fig. S1c,† the oxygen 1s spectra displayed peaks at 529.4 and 531.3 eV, which correspond to the C–O and CO groups, respectively. The nitrogen 1s spectrum of G-CDs, shown in Fig. S1d,† displayed a CN signal with a noise-like pattern, which indicates that most of the amine groups in GA were hydrolyzed into ammonia to be excluded from the reaction by evaporation. However, a small portion of them underwent transformation into pyrazine,32 which could potentially be incorporated into G-CDs. The addition of EG resulted in increased intensity of the nitrogen-related signals, suggesting the formation of imine groups through the condensation reaction between GA and the aldehyde from the EG intermediate. The raw XPS data for GE-CDs displayed peaks corresponding to carbon, nitrogen, and oxygen (Fig. 1d). The carbon 1s spectra depicted in Fig. 1e have peaks at 284.8, 285.4, and 286.0 eV, which correspond to the C–C/CC, C–N, and C–O/CO groups, respectively. In Fig. S2,† the oxygen 1s spectra displayed peaks at 529.7 and 531.3 eV, which correspond to the C–O and CO groups, respectively. In Fig. 1f, the nitrogen 1s spectra reveal three distinct peaks at 398.1, 399.1, and 400.4 eV, respectively. These peaks are associated with the presence of pyridinic, pyrrolic, and graphitic nitrogen species,39,40 indicating the successful formation of N-doped CDs through the addition of EG. In addition, Table 1 indicates that G-CDs have negligible nitrogen content, suggesting that the majority of nitrogen was liberated from GA due to the amine hydrolysis. GE-CDs demonstrated a nitrogen content of 7.81%, indicating the effective integration of nitrogen-containing compounds through the inclusion of EG because of iminium ion stabilization.
| Sample | Atomic percentage (%) | ||
|---|---|---|---|
| C | N | O | |
| GA | 50 | 8.33 | 41.67 |
| G-CDs | 58.12 | 0.45 | 41.43 |
| GE-CDs | 60.25 | 7.81 | 31.94 |
Morphological characterization of GE-CDs
Transmission electron microscopy (TEM) imaging was performed to investigate the morphology and size of GE-CDs in the solution. Fig. 2a and S3† display TEM images of GE-CDs exhibiting spherical shapes with sizes ranging from 3 to 6 nm. As shown in the high-resolution TEM images (Fig. 2a), the spacing between adjacent lattice planes for random nanoparticles in the GE-CDs solution was found to be in range of 0.215–0.375 nm. This finding suggests that the as-prepared GE-CDs possess localized graphite-like structures surrounded by amorphous carbon, a characteristic commonly associated with turbostratic carbon.41,42 Dynamic light scattering (DLS) was employed to characterize the hydrodynamic size distribution of GE-CDs. The DLS particle size curve of GE-CDs (Fig. S4†) is monomodal and has a diameter of 9.2 nm at the peak of the distribution. The crystallinity and degree of graphitization of GE-CDs were determined using X-ray diffraction (XRD). The XRD patterns of GE-CDs displayed in Fig. 2b show that there are two Bragg diffraction peaks at approximately 2θ = 24.6° (d = 0.36 nm) and 35.0° (d = 0.26 nm). In the present study, these two peaks were indexed to the (002) and (111) planes, which correspond to the presence of amorphous carbon and graphitic carbon, respectively,43,44 indicating the presence of a large amorphous carbon network in association with the hexagonal graphite lattice. Fig. 2c shows the Raman spectra of GE-CDs. The spectrum exhibited two prominent peaks centered at 1348 and 1561 cm−1, which can be assigned to the D band and G band, respectively. The D band corresponds to the vibrations of sp3-hybridized carbon atoms with dangling bonds or disordered carbon atoms, while the G band associated with the E2g mode of graphite because of the vibration of sp2-bonded carbon atoms in a two-dimensional hexagonal lattice.45 The ratio of the D-band peak intensity (ID) to the G-band peak intensity (IG) or the ID/IG ratio of GE-CDs was 1.006, which indicates the presence of defective carbon structures in GE-CDs. The high values of ID and the broad spectral widths of the D and G peaks provide further confirmation of the turbostratic structure present in the GE-CDs.46,47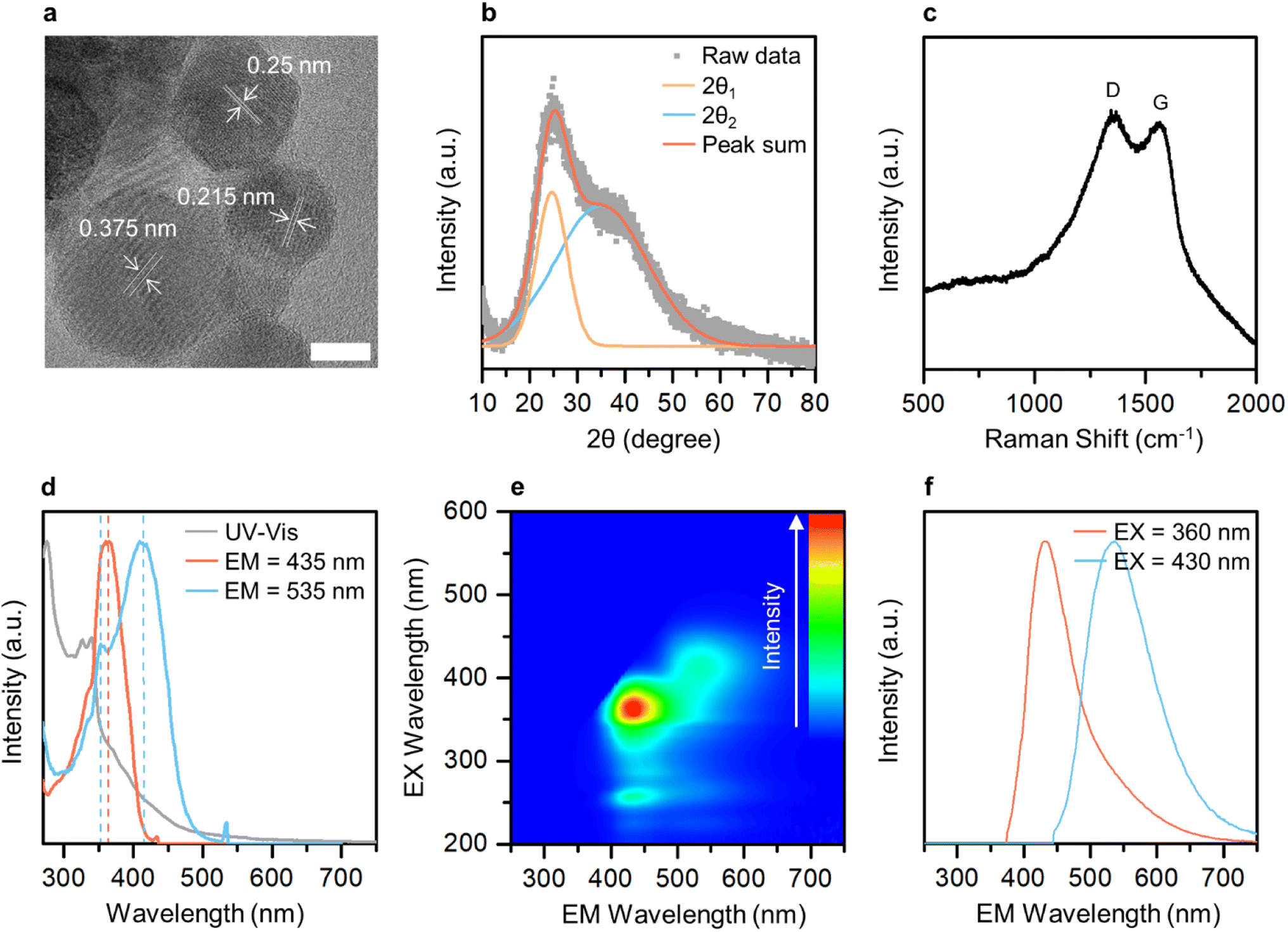 | ||
| Fig. 2 (a) High-resolution TEM image of GE-CDs. (scale bar, 10 nm). (b) XRD pattern of GE-CDs. (c) Raman spectrum of GE-CDs. (d) Normalized UV-visible absorption and PL excitation spectra of GE-CDs. (e) PL emission contour map of GE-CDs. (f) PL emission spectra of GE-CDs at excitation wavelengths of 360 and 430 nm. “EX” and “EM” designate excitation and emission, respectively. | ||
Optical characterization of CDs
The optical properties and electronic band structure of GE-CDs were investigated using UV-visible absorption spectroscopy and PL excitation/emission spectroscopy (Fig. 2d and e, respectively). Fig. 2d shows the UV-visible absorption and PL excitation spectra of GE-CDs. The absorption spectrum displayed a primary peak in the range of 270–300 nm, which can be attributed to the π–π* transition occurring in carbonized cores. Secondary peaks at approximately 330 nm and tertiary bands in the range of 350–550 nm were assigned to interstate (n–π*) electronic transitions involving OID and NID states, respectively. The PL excitation spectrum recorded at an emission wavelength of 435 nm exhibits a single peak at an excitation wavelength of 361 nm, which can be associated with the n orbitals of oxygen atoms in GE-CDs, i.e., lone-paired electrons in OID states were pumped into π* orbitals of the graphitic units of GE-CDs (n[O]–π*) by excitation light. However, the excitation spectrum at an emission wavelength of 535 nm exhibits an additional sharp peak at 412 nm, which can be attributed to the transition of lone-paired electrons in the NID states (n[N]–π*). The PL emission spectra of GE-CDs were examined to investigate the relationship between their PL characteristics and the presence of defect states. The emission contour map (Fig. 2e) and spectra (Fig. 2f) reveal two primary emission peaks observed at excitation wavelengths ranging from approximately 350 nm to 440 nm: one centered at ∼432 nm and the other at ∼537 nm. The 432 nm emission can be attributed to the n[O]–π* transitions in OID states, whereas the 537 nm emission can be attributed to n[N]–π* transitions in NID states.The optical properties of G-CDs were examined and compared to those of GE-CDs in order to gain further insights into the formation and characteristics of NID states (Fig. S5†). G-CDs contain a negligible amount of nitrogen in their carbon matrix, as demonstrated by the XPS and FTIR spectroscopy results. Fig. S5a† shows the absorption and excitation spectra of G-CDs. The broad absorption band with no prominent peak in the range of 330–550 nm can be assigned to n–π* transitions between the n orbitals of various surface groups such as the hydroxyl or carbonyl group and the π* orbitals of the polyatomic domains. This implies that G-CDs contained a significant amount of oxygen atoms that originated from GA as a carbon precursor. Similar to the case of GE-CDs, the excitation spectrum of G-CDs at an emission wavelength of 435 nm showed a peak at an excitation wavelength of 347 nm, which may be associated with the transition of n electrons in OID states. However, the excitation spectrum at an emission wavelength of 535 nm exhibits a broad absorption band in the range of 350–450 nm originating from various OID states, with no distinct NID states developed in G-CDs. Fig. S5b† shows emission bands of G-CDs at various excitations from 350 to 430 nm. Broad emission spectra ranging from 360 to 700 nm are observed, attributed to the cumulative effect of the π–π* transitions of the intrinsic carbon core, as well as the interstate to band (n[O]–π*) transitions between surface groups and the carbon core.48 Moreover, the excitation wavelength-dependent emission is observed in the emission spectra because of the presence of various functional groups on the surface of G-CDs, and this can result in a series of emissive traps between π and π* of C–C that excite at different excitation energies and emit correspondingly. In comparison with G-CDs, an almost excitation wavelength-independent emission behavior was observed in the emission spectra of GE-CDs (Fig. S6†), which was attributed to the formation of uniform OID or NID states and surface groups on the carbon core upon the addition of EG.
To ascertain the distinguishability and controllability of the OID and NID states, we varied the reaction duration (12, 24, and 48 hours) in order to manipulate the formation ratio between these states. The broad light absorption band ranging from 300 to 600 nm increased with an increase in the reaction duration (Fig. S7†). In addition, the relative emission intensity from the NID states increased (Fig. 3a), which indicate the enhanced formation of NID states in GE-CDs. The optical band gap of GE-CDs at different reaction times was assessed using a Tauc plot, which involved plotting the absorbance against the photon energy using the Wood and Tauc method (Fig. 3b). The estimated direct optical bandgap energies of GE-CDs synthesized for reaction durations of 12, 24, and 48 h were 3.52, 3.37, and 3.33 eV, respectively. These values further support the enhanced formation of nonbonding (or intragap) states associated with the NIDs.
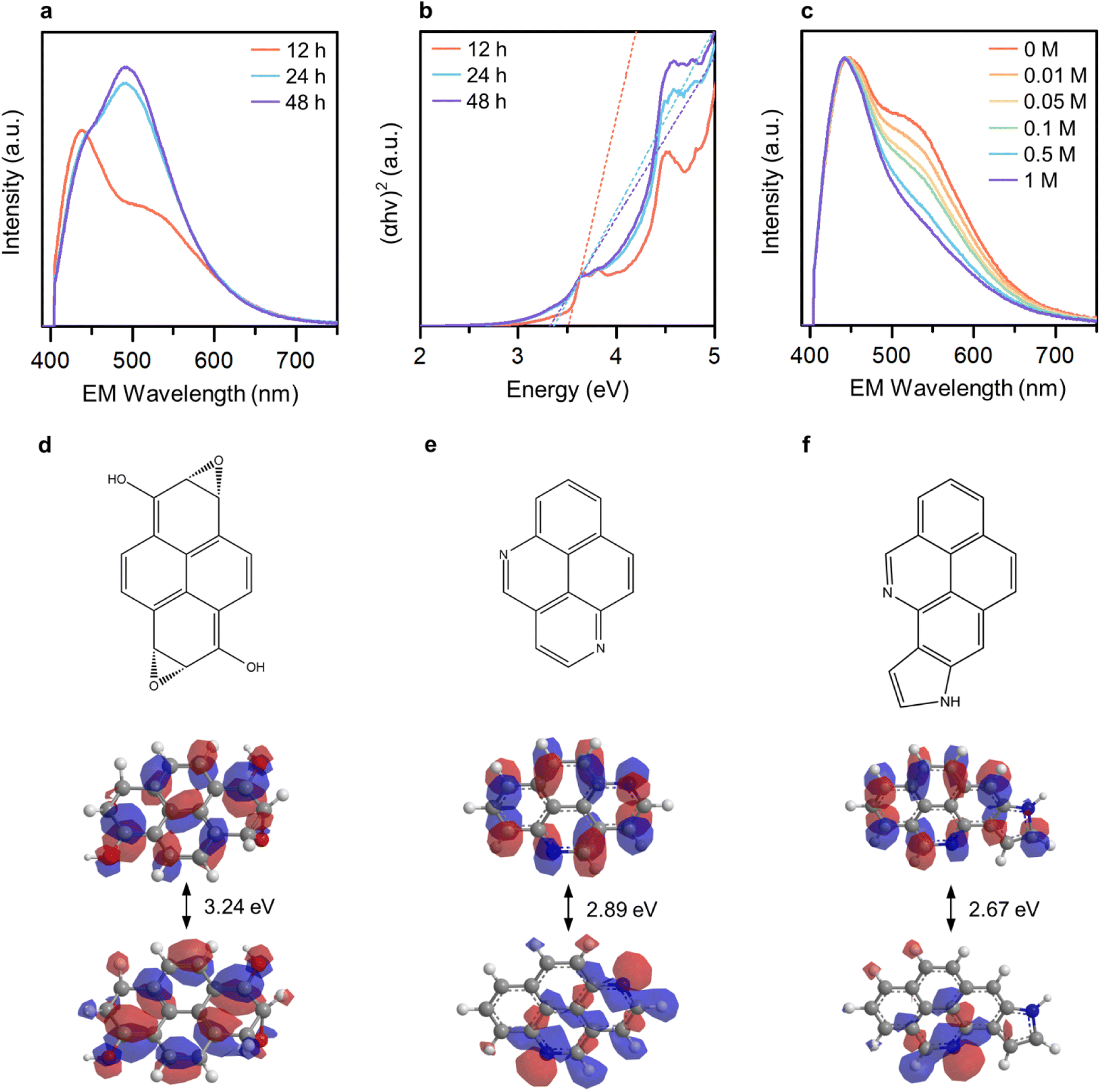 | ||
| Fig. 3 (a) Normalized PL emission spectra at an excitation wavelength of 390 nm and (b) Tauc plots of GE-CDs at different reaction times. (c) Normalized PL emission spectra of GE-CDs at an excitation wavelength of 390 nm in the presence of acetic acid with different concentrations. “EM” designates emission. Molecular orbital energy levels and band structures of (d) oxygenic pyrene, (e) pyridinic pyrene, and (f) pyrrolic pyrene (grey: carbon, blue: nitrogen, red: oxygen, white: hydrogen). | ||
We conducted a series of experiments involving the protonation of GE-CDs to provide additional evidence affirming that the OID and NID states exhibit distinct energy and optical characteristics. Changes in the absorption and emission spectra of GE-CDs caused by protonation were investigated in acidic solutions with various concentrations of acetic acid (Fig. 3c and S8†). The absorption spectra of GE-CDs were acquired by varying the concentration of acetic acid from 0 to 1 M, leading to negligible changes in the absorption band (Fig. S8a†). Fig. S8b† displays the calculated Tauc plot curves for both pristine and protonated GE-CDs, allowing for the determination and comparison of their respective bandgap values before and after the addition of acetic acid. The band gaps of pristine GE-CDs and protonated GE-CDs remained nearly identical at 3.52 eV, suggesting that the core structure of GE-CDs remained unaffected by the protonation process with acid.49
The emission spectra of GE-CDs as a function of acetic acid concentration are presented in Fig. 3c. The relative emission intensity originating from the NID states (upon excitation at 390 nm) of GE-CDs decreases with increasing acetic acid concentration. This observation indicates that the protonation-induced fluorescence quenching is associated with the NID states of GE-CDs. Amine (–NH2) functional groups attached to CDs are widely recognized for their electron-donating properties. However, the electron-donating nature of the amine groups on the CD surface was weakened under acidic conditions.50 In other words, fluorescence quenching arises from diminished electron donation capability from the surface domain to the carbon core. Further, fluorescence quenching was assessed using the Stern–Volmer relationship:
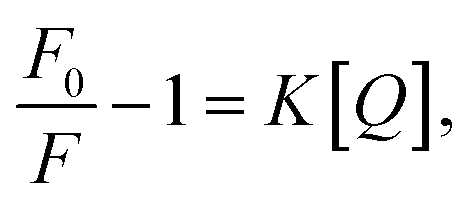
Extended Hückel calculations were conducted to establish the molecular orbital theory descriptions for GE-CDs. Pyrene, a polycyclic aromatic hydrocarbon unit commonly formed under our reaction condition,52 was selected as the base. The molecular orbital energy levels and band structures of the four pyrene variants were computed and compared with the experimental data (Fig. 3d–f and S10†). The energy gap of pyrene was 6.71 eV (Fig. S10a†). Subsequently, we simulated the energy-gap variations resulting from the incorporation of oxygen and nitrogen groups into pyrene. The moderate oxidation of pyrene, which leads to the introduction of alcohol and epoxide groups, decreases the energy gap to 3.24 eV (Fig. 3d). This is in good agreement with the observed electronic transitions involving the OID states in GE-CDs (3.43 eV). Further, we examined several nitrogen heterocyclic compounds arising from the iminium ion cyclization. As demonstrated in Fig. 3e and f, pyrene containing pyridinic nitrogen and pyrrolic nitrogen exhibited energy gaps of 2.89 and 2.67 eV, respectively, corresponding to the electronic transition energies involving the NID states in GE-CDs (3.01 eV). The energy gap of pyrene with graphitic nitrogen was calculated as 1.03 eV, which is unlikely to be observed in GE-CDs (Fig. S10c†).
We conducted pump-probe spectroscopic measurements (e.g., TA and TRPL) to elucidate the PL mechanism of CDs. TA spectroscopy experiments were performed using various excitation wavelengths for investigating the ultrafast electron dynamics in CDs and analyzing differences resulting from the presence of NID states in GE-CDs. Initially, the TA kinetics of CDs with a pump wavelength of 360 nm were monitored using a probe wavelength of 435 nm, which corresponded to OID states (Fig. 4a and b). Interestingly, both samples exhibited a negative change in the absorption signal (ΔA) in the early time-delay region. The negative ΔA coincides with ground-state bleaching (GSB), a photobleaching (PB) mechanism indicating the depletion of ground-state carriers excited by the probe beam, which implies a high probability of occupying OID states.
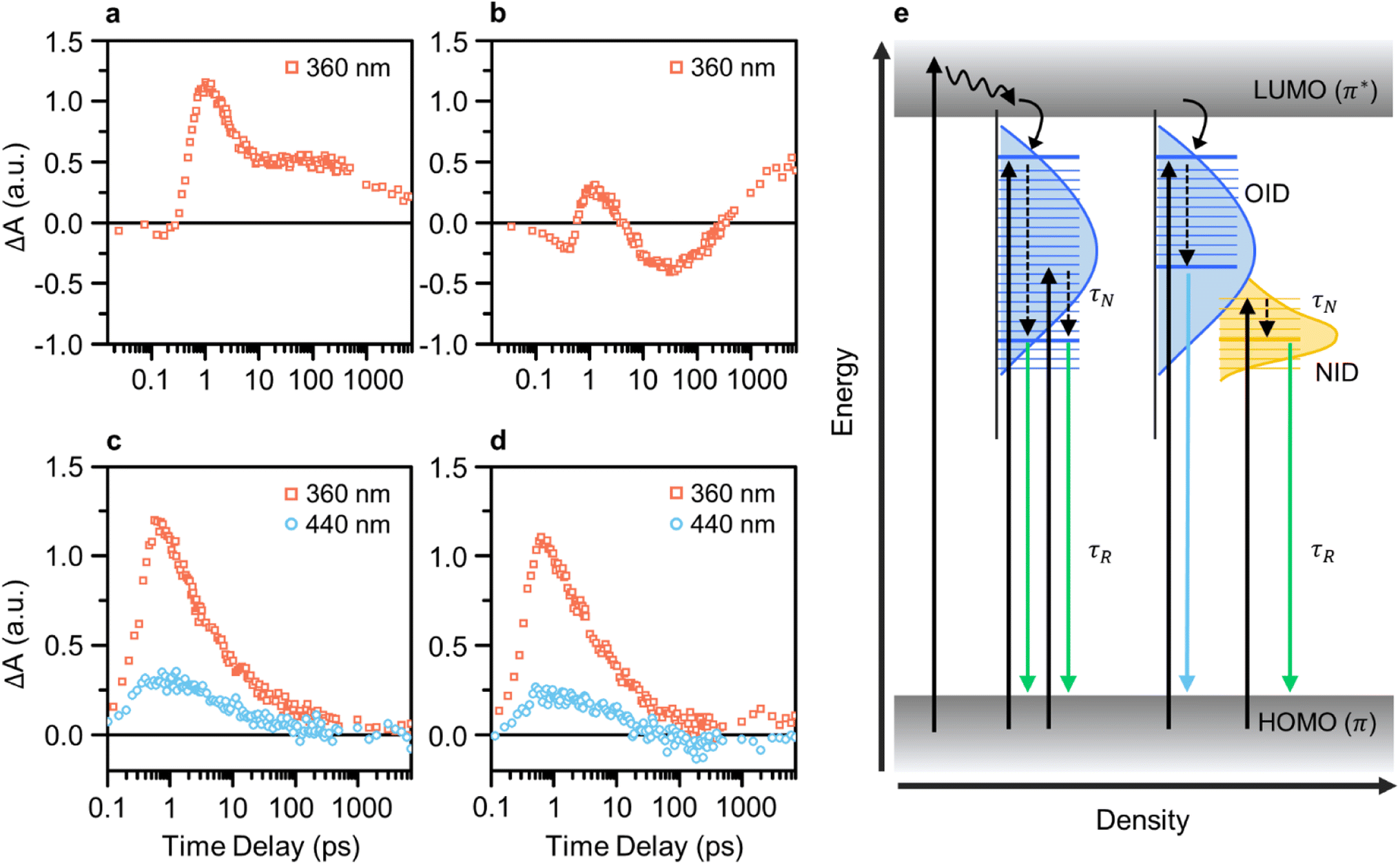 | ||
| Fig. 4 TA kinetics at the 435 nm probe wavelength excited with 360 nm pump wavelength of (a) G-CDs and (b) GE-CDs. TA kinetics at the 535 nm probe wavelength of (c) G-CDs and (d) GE-CDs excited by 360 and 440 nm pump wavelengths. (e) Schematic of the photocarrier relaxation mechanism in G-CDs (left) and GE-CDs (right). | ||
Compared to G-CDs (Fig. 4a), GE-CDs (Fig. 4b) exhibited an additional PB signal that emerged at approximately 4 ps. The observed PB signal can be attributed to stimulated emission (SE), signifying the transfer of energy from OID states to NID states. In the TA contour maps of G-CDs and GE-CDs, excited by a 360 nm pump wavelength (Fig. S11†), we note that the ground-state bleach (GSB) signal in G-CDs persists for up to 100 ps but rapidly diminishes within 10 ps in GE-CDs. In connection with the disappearance of the GSB signal in GE-CDs, a SE signal emerges around 410 nm. Recalling that the excitation spectrum at an emission wavelength of 535 nm shows a sharp peak at 412 nm, corresponding to the transition of lone-paired electrons in the NID states, it is plausible that the SE signal is associated with the energy transfer between OID and NID states.53
Fig. S12† presents a clear and distinctive comparison of G-CDs and GE-CDs in terms of their TA spectra at the selected time delays at a pump wavelength of 360 nm. The energy transfer between the OID and NID states is further corroborated by the PB signal observed within the wavelength range of 400–500 nm in the TA spectra of GE-CDs at prolonged time delays (Fig. S12a–d†). The rate of decrease in the ΔA value around 400–450 nm, which is associated with the OID states, was slower in G-CDs than in GE-CDs, suggesting that OID states were occupied at a slower rate in G-CDs than in GE-CDs (Fig. S12e and f†). This difference can be ascribed to the relatively uniform chemical structures of GE-CDs, which resulted in well-defined emission states, unlike those of G-CDs. Further, this can also cause discrepancies in the decay constants (τ) extracted using single exponential functions, as shown by their calculated values of 2.31 and 5.20 ps for G-CDs and GE-CDs, respectively (Table S1a†). The comparatively longer decay time observed in GE-CDs suggests the formation of well-defined OID and NID states with fewer trap states in comparison to those for G-CDs.
The transition behaviors of the CDs were investigated by monitoring their TA kinetics at pump wavelengths of 360 and 440 nm, while using a fixed probe wavelength of 535 nm corresponding to the low-energy OID states (G-CDs) or NID (partially overlapped with OID) states (GE-CDs) (Fig. 4c and d). The rate at which the ΔA value reaches zero is faster in GE-CDs than in G-CDs, which indicates that GE-CDs may have well-defined and uniformly distributed NID states at the energy level corresponding to the 535 nm probe wavelength. Furthermore, the extracted decay constants associated with G-CDs and GE-CDs varied because of their distinct origins of the 535 nm emission (Table S1b†). Upon fitting the TA spectrum of G-CDs with a pump wavelength of 360 nm to a single exponential decay function, specifically considering the formation of OID states, the τ1 value was determined to be 1.73 ps. In the case of GE-CDs, biexponential decay functions were employed to accommodate potential energy transfer between OID and NID states, yielding the τ1 and τ2 values of 1.6 and 16.1 ps, respectively. This 360 nm pump wavelength is associated with OID states commonly present in both G-CDs and GE-CDs, and therefore, we can anticipate this similarity in the τ1 values. The sole presence of τ2 in GE-CDs, characterized by its prolonged decay time, can be ascribed to the energy transfer between OID and NID states. When using a pump wavelength of 440 nm, the τ1 values were found to be 5.03 and 20.0 ps for G-CDs and GE-CDs, respectively. The prolonged decay time observed in GE-CDs suggests the presence of NID states that can capture photoexcited carriers and extend their lifetime.
The TRPL spectra are recorded at a probe wavelength of 535 nm to compare the recombination dynamics of G-CDs and GE-CDs (Fig. S13†). We used pump wavelengths ranging from 340 to 440 nm, which can excite carriers to different energy states. Fig. S13a and b† show the TRPL decay profiles depending on the excitation wavelengths of G-CDs and GE-CDs, respectively. The profiles were fitted to bi-exponential decay functions, and two decay processes were identified: relatively short and long parameters (Table S2†). A shorter lifetime (τN) indicates a fast PL decay process caused by non-radiative recombination or charge-carrier relaxation of the photocarriers;54,55 a longer lifetime (τR) represents radiative recombination. As shown in Table S2,† for both samples, the PL lifetime increases with an increase in the pump wavelength. However, compared to G-CDs, a slightly prolonged lifetime was observed for GE-CDs. This phenomenon can be explained by the development of stable NID states in GE-CDs and the dominant occupation of photocarriers in NID states at longer excitation wavelengths.
Furthermore, the PL quantum yields (QYs) were theoretically calculated as the ratio of the average rate of radiative recombination to the cumulative average rate of all processes involved in the excited state. The calculated QYs of GE-CDs ranged from 49.9 to 66.7% depending on the excitation wavelength (Fig. S14†). Subsequently, the absolute QYs were measured for GE-CDs to enable a direct comparison with the theoretical QYs (Fig. S15†). These measurements yielded QY values of 58.2 and 52.4% under 360 and 440 nm excitation, respectively, aligning closely with the theoretical values.
In Fig. 4e, we summarize the photocarrier relaxation mechanism in G-CDs and GE-CDs. G-CDs exhibit a diverse range of OID states, spanning broad PL from 400 to 550 nm, which are primarily attributable to the formation of different structures resulting from the uncontrolled carbonization of GA. In contrast, GE-CDs possess clearly distinguishable and precisely defined OID and NID states due to the inclusion of EG, which enables relatively accurate manipulation of their structural properties. Consequently, the OID and NID states of GE-CDs demonstrate distinctive and excitation-independent PL characteristics, and particularly in this instance, the emission in the 500–550 nm range can be attributed solely to the NID states.
In vitro bioimaging and PDT
Cytotoxicity studies play a crucial role in determining material suitability for biological applications. Low cytotoxicity is key for an ideal multifunctional biomaterial. Hence, the inherent cytotoxicity of GE-CDs was evaluated by measuring the relative viability of PC3 and A549 cells treated with different concentrations of GE-CDs using a cell counting kit-8 (CCK-8) assay. Fig. 5a shows the viability of PC3 and A549 cells after incubation with GE-CDs at concentrations of 0.125–1 mg mL−1 for 1 day. The viabilities of both cancer cells remained >90% even at a concentration of 1 mg mL−1, indicating that these GE-CDs are not toxic and highly biocompatible, and thus, they can serve as potential candidates for both in vitro and in vivo applications.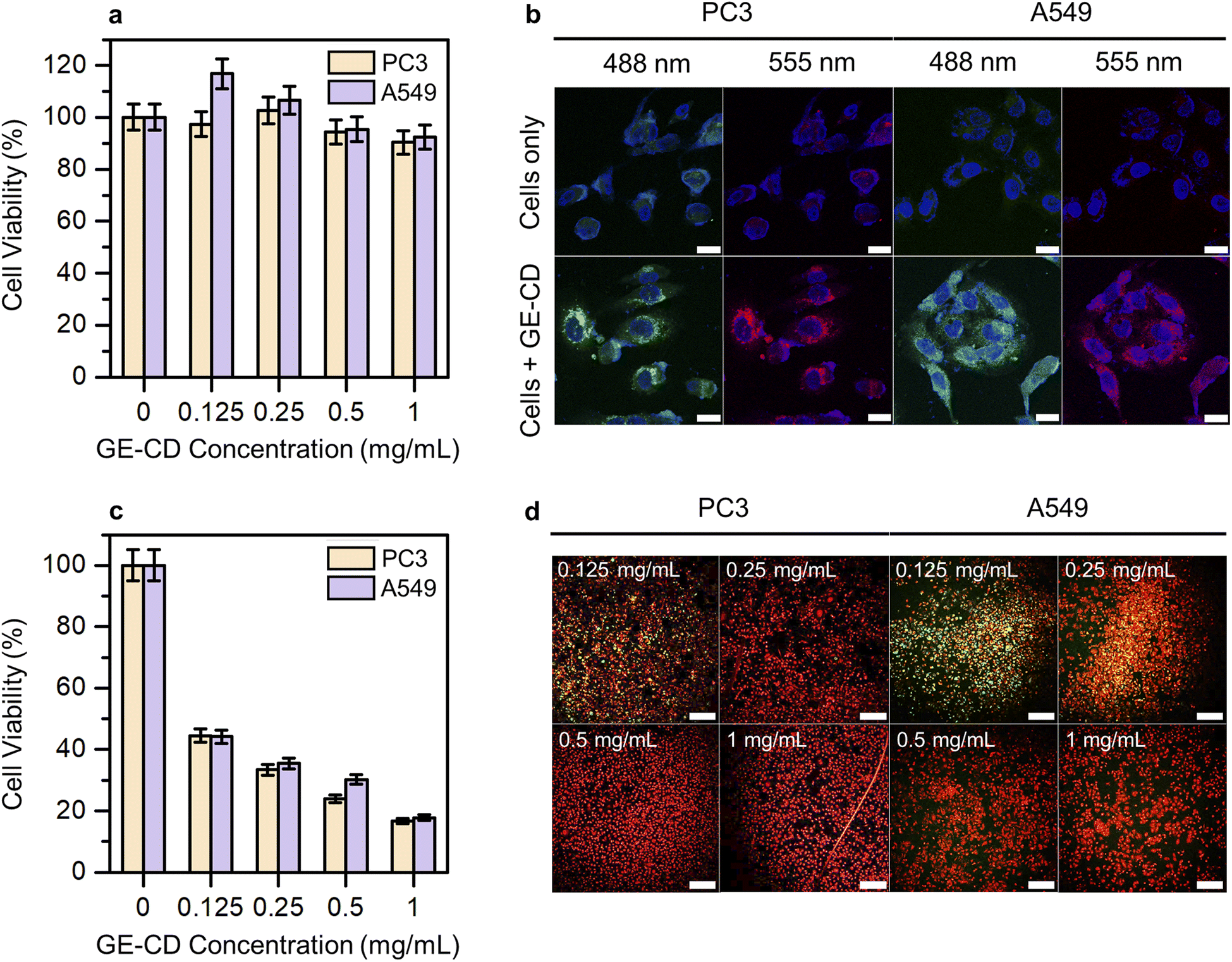 | ||
| Fig. 5 (a) In vitro cell viability of PC3 and A549 cells treated with various concentrations of GE-CDs. The optical density at 0 mg mL−1 was set to 100%. Data are represented as the mean ± standard deviation (n = 6). (b) Confocal microscopy images of PC3 and A549 cells treated with 1 mg mL−1 GE-CDs under 488 nm and 555 nm lasers. The cell nuclei were stained with DAPI. (Scale bar, 20 μm). (c) Cell viability of PC3 and A549 cells incubated with GE-CDs after irradiation with a 473 nm laser for 10 min. The optical density at 0 mg mL−1 was set to 100%. Data are represented as the mean ± standard deviation (n = 4). (d) Fluorescence images of calcein AM (green fluorescence for live cells) and PI (red fluorescence for dead cells) co-stained PC3 and A549 cells treated with various concentrations of GE-CDs. (Scale bar, 200 μm). | ||
In vitro cellular imaging was performed using PC3 and A549 cells to assess the performance of GE-CDs as bioimaging agents. Both cells were incubated in cell culture media containing GE-CDs (1 mg mL−1) for 1 day at 37 °C. Then, the cells were washed and imaged under a confocal laser scanning microscope. The cells exhibited multicolor emissions, as shown by the green and red colors in the images in Fig. 5b, depending on the excitation lasers used (488 and 555 nm, respectively). Control cells without GE-CDs treatment exhibited significantly lower detectable fluorescence emission under the same exposure conditions. Further observation of these images confirmed that the cells retained good morphology, indicating the high biocompatibility and easy integration of GE-CDs. The fluorescence emission was limited to the cytoplasmic region, resulting in weak staining of the nuclear region. These results confirm that GE-CDs can act as potential agents for cell imaging with multicolored emission.
Chemical moieties with incomplete molecular structures containing dangling bonds and radicals can create non-radiative trap states, leading to the production of reactive oxygen species (ROS).56 The absorption and PLE spectra of GE-CDs in Fig. 2d are compared to determine the presence of these non-radiative trap states. The disappearance of a distinct absorption peak at 326 nm in the PLE spectra indicated the decay of excitons generated by the 326 nm excitation through a non-radiative pathway. Further, the energy state associated with this excitation was a non-radiative trap state. We employed a series of assays to assess the ROS production capabilities of GE-CDs and to identify the specific types of ROS they generate (Fig. S16†).57–59 Firstly, we utilized the singlet oxygen sensor green (SOSG) assay to detect the generation of singlet oxygen. In this assay, the SOSG reagent reacts with singlet oxygen, leading to the emission of green fluorescence at approximately 525 nm. Next, we utilized dihydrorhodamine (DHR) 123 to detect the generation of hydrogen peroxide. This compound exhibits intense green fluorescence at 526 nm upon reacting with hydrogen peroxide.60,61 Lastly, we conducted an assay utilizing the Red Fluorometric ROS Kit, which generates a red-fluorescent (emission wavelength of 605 nm) product in proportion to the presence of superoxide and hydroxyl radicals.
We observed a significant increase in PL intensity at ∼525 nm when using SOSG (Fig. S16b†) and the Red Fluorometric ROS Kit (Fig. S16f†), indicating the ability of GE-CDs to generate singlet oxygen, superoxide, and hydroxyl radicals. Remarkably, our control groups exposed to UV radiation alone displayed an inability to generate singlet oxygen (Fig. S16a†) but produced a substantial quantity of superoxide and hydroxyl radicals (Fig. S16e†). This observation led us to conclude that the presence of GE-CDs significantly enhances the generation of singlet oxygen compared to superoxide and hydroxyl radicals. However, the PL intensity remained nearly unchanged when utilizing DHR 123 (Fig. S16c and d†), suggesting that GE-CDs do not generate hydrogen peroxide. Our findings indicate that GE-CDs primarily generate singlet oxygen and partially generate superoxide and hydroxyl radicals when exposed to UV radiation. This makes them a promising candidate for use as a photosensitizer in PDT.
The PDT efficacy through ROS generation by GE-CDs was investigated in PC3 and A549 cells under 473 nm two-photon excitation to further examine the effects of CD-induced ROS on apoptotic cell death. The PC3 and A549 cells were treated with varying concentrations of GE-CDs to assess ROS generation and PDT efficacy. The cell viability of the control group not treated with GE-CDs was set to 100%, and the relative cell viability of the remaining groups treated with 0.125, 0.25, 0.5, and 1 mg mL−1 of GE-CDs was compared (Fig. 5c). The group treated with 0.125 mg mL−1 GE-CDs and irradiated with a 473 nm two-photon laser exhibited a significant PDT effect through ROS generation in each cell. As the concentration increased, the effect of PDT increased, showing a cell viability of approximately 16–18% at 1 mg mL−1 of GE-CDs.
The effect was further demonstrated by using a live/dead assay on both PC3 and A549 cells (Fig. S17† and 5d). Live cells are presented with a green stain, and dead cells are presented in red. As shown in Fig. S17,† in the control group, which was only irradiated by the laser without GE-CDs treatment, the green fluorescence intensity of live cells was confirmed with almost no dead cells. However, with an increase in the concentration of the treated GE-CDs (Fig. 5d), the green fluorescence intensity decreased gradually and the red fluorescence intensity increased, indicating that cell viability decreased with increasing GE-CDs concentration. The low survival rate of GE-CD-treated cancer cells following laser irradiation demonstrates the effective PDT efficiency of GE-CDs.
Conclusion
The rational design approach involving pinacol rearrangement and iminium ion cyclization of GA and EG successfully resulted in the synthesis of N-doped CDs (referred to as GE-CDs). The formation of iminium ions during the dehydration reaction between GA and acetaldehyde confirmed the incorporation of nitrogen in the GE-CDs. Notably, the GE-CDs exhibited chemically distinct and energetically discernible emissive states: OID and NID states associated with 435 and 535 nm PL, respectively. These emissive states originated from oxygenic and nitrogenic polycyclic aromatic hydrocarbons present in the GE-CDs. The electronic band structures and transitions of the GE-CDs were thoroughly investigated through a combination of theoretical and experimental methods, including extended Hückel calculations, dynamic PL quenching experiments, and pump–probe spectroscopy. These investigations provided insights into the energy levels and electronic behavior of the GE-CDs. Furthermore, the high biocompatibility and ability of the GE-CDs to generate ROS demonstrated their potential as bioimaging and PDT agents in PC3 and A549 cells. This highlights their promising application in the biomedical field. In conclusion, our systematic strategy offers a promising approach for achieving precise control over the chemical and electronic structures of nitrogen-doped carbon dots. The study also unveils new possibilities for designing and modulating their optical properties, which hold potential for further mechanism research and broaden the scope of applications, particularly in the biomedical field.Experimental
Materials
GA hydrochloride (≥99%), EG (≥99%), EG-d6 (98 at% D, ≥99%), and acetic acid (≥99%) were purchased from Merck. Cellulose ester dialysis tubes were purchased from Spectra/Par Biotech. All solutions were prepared using triple-distilled water.Synthesis of CDs
0.25 g of GA was dissolved in 15 mL of deionized water to synthesize G-CDs. 0.25 g of GA and 10 mL of EG were dissolved in 5 mL of distilled water to synthesize GE-CDs. Each solution was heated on a hot plate at 200 °C for 12 h until the water evaporated completely. The resulting CD solutions were dialyzed against deionized water for four days using cellulose ester dialysis tubes with a cutoff molecular weight of 100–500 Da. After the dialysis, water was removed from the solution by lyophilization or heating, and G-CD and GE-CDs powders were obtained.Material characterizations
CDs were dispersed in distilled water by sonication and dropped onto a TEM grid for the TEM observations. TEM was performed using a JEOL JEM-2200FS instrument equipped with a Cs corrector. DLS measurements were performed using a Horiba SZ-100 particle size analyzer. XRD was performed using a Bruker D8 Advance diffractometer. Raman spectroscopy was performed on an Alvatek XperRam S spectrometer with a 532 nm laser excitation. XPS measurements were performed using a VG Scientific ESCALAB 250 spectrometer equipped with an A1 X-ray source (1486.6 eV). FTIR spectroscopy was performed using a Thermo Scientific Nicolet iS50 FTIR spectrometer equipped with an attenuated total reflection (ATR) accessory. 1H NMR spectra of GA, G-CDs, and GE-CDs dissolved in D2O were recorded using a Bruker Avance III HD500 spectrometer (500 MHz).Optical characterizations
For all spectroscopic measurements, CD solutions were dispersed in distilled water by sonication. UV-visible absorption and PL spectroscopy of the CDs were performed using a Scinco S-3100 spectrophotometer and Jasco FP-8500 fluorometer with 10 mm × 10 mm QS-grade quartz cuvettes (111-QS, Hellma Analytics), respectively. Extended Hückel calculations were conducted using YAeHMOP software for pyrene, oxygenic pyrene, pyridinic pyrene, and pyrrolic pyrene. For TA spectroscopy, the fundamental pulsed laser from a 1 kHz Ti:Sapphire amplifier (Legend Elite, Coherent) with a 790 nm central wavelength and 30 fs pulse duration were split into two beams. One beam directed toward an optical parametric amplifier (TOPAS Prime, Light Conversion) was tuned to various wavelengths as a pump pulse, with a pulse energy of 80 nJ per pulse. The other beam was focused on a nonlinear crystal for generating a broadband white light continuum as a probe pulse. The probe beam monitored changes in the absorption induced by the pump beam as a function of the time delay between the pump and probe pulses. A TA spectrometer with a 1024-pixel CMOS sensor (HELIOS, Ultrafast Systems) was used to measure the data. The TRPL measurements were performed using a spectrometer coupled to a streak camera (C10910, Hamamatsu). For photoexcitation, a pulsed laser with diverse wavelengths was generated from an optical parametric amplifier (OPA). The absolute QY measurements were acquired using a Jasco FP-8500 fluorometer with a 100 mm integrating sphere setup (ILF-835) and subsequently analyzed utilizing the Jasco Spectra Manager II software.Cytotoxicity assessment of GE-CDs
The cytotoxicity of GE-CDs on PC3 and A549 cells was evaluated using a CCK-8 assay. PC3 and A549 cells were maintained in RPMI-1640 medium supplemented with 1 wt% antibiotic-antimycotic solution and 10 vol% fetal bovine serum (FBS). Subsequently, the cells in the media were seeded in each of the 96 wells of the cell culture plates (1 × 104 cells in 100 μL per well) and incubated for 1 day. GE-CDs were dispersed in RPMI-1640 medium at concentrations of 0.125, 0.25, 0.5, and 1 mg mL−1 and added to each well. The cell culture plates were incubated at 37 °C in a humidified 5% CO2 incubator for 1 day. The cells were washed with fresh phosphate-buffered saline (PBS) and treated with 10% CCK-8 assay solution in FBS-supplemented media. After incubation for 2 h, the optical density of each well was measured at 450 nm using a microplate reader (SpectraMax M2).In vitro bioimaging of GE-CDs
Cell imaging was performed on PC3 and A549 cells using confocal microscopy. PC3 and A549 cells (ATCC) cultured in RPMI-1640 medium and then seeded with 500 μL of cell suspension into each chamber of a 4-chamber confocal slide for 1 day. The medium was replaced with 500 μL RPMI-1640 medium containing 1 mg mL−1 of GE-CDs and incubated at 37 °C in a humidified 5% CO2 incubator for 1 day. Subsequently, cells were washed with PBS twice and fixed with 4% formaldehyde solution. Cells were stained with mounting medium with DAPI (4′,6-diamidine-20-phenylindole dihydrochloride). Confocal laser scanning microscopic analysis of these cells was performed using a Zeiss LSM 700 confocal microscope with diode-pumped solid-state (DPSS) lasers (488 and 555 nm).ROS measurement
SOSG (Thermo Fisher Scientific), DHR 123 (Merck), and the Red Fluorometric Intracellular ROS Kit (Merck) were employed to detect singlet oxygen, hydrogen peroxide, and superoxide/hydroxyl radicals, respectively. The SOSG reagent was prepared by dissolving it in 33 μL of methanol to achieve a concentration of 5 mM. Subsequently, 4 μL of this stock solution was added to a 10 mL solution of standard pH 7 phosphate buffer (Daejung Chemicals & Metals). DHR 123 was dissolved in water to attain a concentration of 20 μM. As for the Red Fluorometric Intracellular ROS Kit, the ROS detection reagent was reconstituted with 40 μL of DMSO, and 20 μL of this resulting stock solution was added to a 10 mL assay buffer solution. To assess the effect of GE-CDs, we dispersed them in a 2 mL solution of one of the ROS detection reagents at a concentration of 0.5 mg mL−1 within a quartz cuvette. After being exposed to a UV lamp (254 nm, 8 W) for a duration of 2 hours, we measured fluorescence at an excitation wavelength of 504 nm and emission wavelengths ranging from 515 to 600 nm for the SOSG and DHR 123 probes. Additionally, fluorescence from the Red Fluorometric Intracellular ROS Kit probe was measured with an excitation wavelength of 520 nm and emission wavelengths ranging from 530 to 700 nm.In vitro PDT test and live/dead assay
The PDT efficacy of GE-CDs was evaluated in the PC3 and A549 cells. Cells were maintained under the same conditions as those used in the cell viability test and seeded onto a 96-well plate. After one day of cell culture time, GE-CDs were treated in each well at 0.125, 0.25, 0.5, and 1 mg mL−1; the cell culture plate was incubated at 37 °C in a humidified 5% CO2 incubator for 1 day. Subsequently, a 473 nm laser of 15 mW cm−1−2 intensity was irradiated for 10 min. The cells were incubated at 37 °C for 1 day. After washing and replacing the cell medium with 100 μL of serum free medium, the CCK-8 assay solution was added to make 10% of the total concentration. After 2 h of incubation, the optical density of each well was measured at 450 nm using a microplate reader. A live/dead cell viability assay was conducted under the same conditions as the PDT test using the CCK-8 assay. After irradiating the cells with the laser, the cell medium was replaced by 100 μL of PBS with calcein AM (0.05%) and ethidium homodimer-1 (0.2%). Then, the cells were observed under a confocal microscope.Author contributions
S. C. and W. K. conceived and planned the experiments. S. C., D. L., and Y. B. performed the synthesis, characterization, and spectroscopic measurements. H. K. and H. H. performed the biological demonstration and in vitro experiments. C.-W. J. and J.-H. K. performed pump-probe measurements and theoretical calculations. S. C., C.-W. J., J.-H. K., and W. K. collaborated in writing the manuscript. W. K. thoroughly supervised the research. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Programs (NRF-2022R1A2C4002403 and RS-2023-00246477) of the National Research Foundation of Korea and the Health Technology R&D Project (HI22C1186000022) of the Korea Health Industry Development Institute funded by the Ministry of Health & Welfare, Korea. W. K. gratefully acknowledges the financial support granted by Hexa FIC.References
- L. Đorđević, F. Arcudi, M. Cacioppo and M. Prato, Nat. Nanotechnol., 2022, 17, 112–130 CrossRef PubMed.
- N. Farshidfar, S. Fooladi, M. H. Nematollahi and S. Iravani, RSC Adv., 2023, 13, 14517–14529 RSC.
- S. Ren, M. Cui, X. Chen, S. Mei and Y. Qiang, J. Colloid Interface Sci., 2022, 628, 384–397 CrossRef CAS PubMed.
- W. Luo, L. Zhang, Z. Yang, X. Guo, Y. Wu, W. Zhang, J. Luo, T. Tang and Y. Wang, J. Nanobiotechnol., 2021, 19, 320 CrossRef CAS PubMed.
- L. Jiang, H. Cai, W. Zhou, Z. Li, L. Zhang and H. Bi, Adv. Mater., 2023, 35, 2210776 CrossRef CAS PubMed.
- J. Belza, A. Opletalová and K. Poláková, Microchim. Acta, 2021, 188, 430 CrossRef CAS PubMed.
- S. K. Tammina, Y. Wan, Y. Li and Y. Yang, J. Photochem. Photobiol., B, 2020, 202, 111734 CrossRef CAS PubMed.
- T. Rasheed, Trends Anal. Chem., 2023, 160, 116957 CrossRef CAS.
- A. B. Talib and M. H. Mohammed, Mater. Today: Proc., 2023, 80, 2327–2333 CrossRef CAS.
- F. Wang, L. Wang, J. Xu, K. Huang and X. Wu, Analyst, 2021, 146, 4418–4435 RSC.
- V. L. John, Y. Nair and T. P. Vinod, Part. Part. Syst. Charact., 2021, 38, 2100170 CrossRef CAS.
- X. Wu, Z. Luo, W. Li, L. Xia and Y. Xiong, Spectrochim. Acta, Part A, 2023, 302, 122995 CrossRef CAS PubMed.
- Z. Liu, Q. Chu, H. Chen, Y. J. Qiang, X. Zhang and Y. W. Ye, Colloids Surf., A, 2023, 669, 131504 CrossRef CAS.
- X. Wang, J. Zeng, S. Xie, L. Tao and X. Sun, New J. Chem., 2023, 47, 9879–9886 RSC.
- G. Kalaiyarasan, J. Joseph and P. Kumar, ACS Omega, 2020, 5, 22278–22288 CrossRef CAS PubMed.
- Anju, A. Rais, K. Rawat, T. Prasad and H. B. Bohidar, Nanotechnology, 2020, 32, 025501 CrossRef PubMed.
- X. Li, Y. Fu, S. Zhao, J. Xiao, M. Lan, B. Wang, K. Zhang, X. Song and L. Zeng, Chem. Eng. J., 2022, 430, 133101 CrossRef CAS.
- Y. Zhang, R. Yuan, M. He, G. Hu, J. Jiang, T. Xu, L. Zhou, W. Chen, W. Xiang and X. Liang, Nanoscale, 2017, 9, 17849–17858 RSC.
- S. Mohandoss, S. Ganesan, S. Palanisamy, S. You, K. Velsankar, S. Sudhahar, H. Lo and Y. R. Lee, Chemosphere, 2023, 313, 137444 CrossRef CAS PubMed.
- X. Liu, J. Liu, B. Zheng, L. Yan, J. Dai, Z. Zhuang, J. Du, Y. Guo and D. Xiao, New J. Chem., 2017, 41, 10607–10612 RSC.
- H. Qi, L. Qiu, X. Zhang, T. Yi, J. Jing, R. Sami, S. F. Alanazi, Z. Alqahtani, M. D. Aljabri and M. M. Rahman, RSC Adv., 2023, 13, 2663–2671 RSC.
- Y. Park, Y. Kim, H. Chang, S. Won, H. Kim and W. Kwon, J. Mater. Chem. B, 2020, 8, 8935–8951 RSC.
- S. Sooksin, V. Promarak, S. Ittisanronnachai and W. Ngeontae, Sens. Actuators, B, 2018, 262, 720–732 CrossRef CAS.
- D. Das and R. K. Dutta, ACS Appl. Nano Mater., 2021, 4, 3444–3454 CrossRef CAS.
- J. Kang and D. Kang, Chem. Eng. J., 2021, 420, 129990 CrossRef CAS.
- B. K. Barman, Ø. S. Handegård, D. Hernández-Pinilla, S. L. Shinde and T. Nagao, ACS Appl. Electron. Mater., 2021, 3, 3761–3773 CrossRef CAS.
- R. Atchudan, T. N. J. I. Edison, K. R. Aseer, S. Perumal, N. Karthik and Y. R. Lee, Biosens. Bioelectron., 2018, 99, 303–311 CrossRef CAS PubMed.
- J. Wu, Y. Feng, Y. Shao and Y. Sun, J. Nanosci. Nanotechnol., 2018, 18, 4196–4203 CrossRef CAS PubMed.
- Y. Liu, L. Jiang, B. Li, X. Fan, W. Wang, P. Liu, S. Xu and X. Luo, J. Mater. Chem. B, 2019, 7, 3053–3058 RSC.
- A. R. Nallayagari, E. Sgreccia, R. Pizzoferrato, M. Cabibbo, S. Kaciulis, E. Bolli, L. Pasquini, P. Knauth and M. L. Di Vona, J. Nanostruct. Chem., 2022, 12, 565–580 CrossRef CAS.
- X. Wang, T. Lin, W. Wu, H. Wu and D. Yan, Environ. Technol., 2022, 43, 4213–4226 CrossRef CAS PubMed.
- C. Shu, J. Agric. Food Chem., 1998, 46, 1129–1131 CrossRef CAS.
- Y. Byun, C. Jung, J. Kim and W. Kwon, Dyes Pigm., 2022, 208, 110895 CrossRef CAS.
- W. B. Smith, Tetrahedron, 2002, 58, 2091–2094 CrossRef CAS.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- J. Royer, M. Bonin and L. Micouin, Chem. Rev., 2004, 104, 2311–2352 CrossRef CAS PubMed.
- G. S. Singh, Green Synthetic Approaches for Biologically Relevant Heterocycles, ed. G. Brahmachari, Elsevier, Second Edition, 2021, pp. 655−687 Search PubMed.
- S. A. Hill, D. Benito-Alifonso, D. J. Morgan, S. A. Davis, M. Berry and M. C. Galan, Nanoscale, 2016, 8, 18630–18634 RSC.
- S. Wang, M. Ma, Q. Liang, X. Wu, K. Abbas, J. Zhu, Q. Xu, A. C. Tedesco and H. Bi, ACS Appl. Nano Mater., 2022, 5, 6679–6690 CrossRef CAS.
- X. Wu, F. Yu, Y. Han, L. Jiang, Z. Li, J. Zhu, Q. Xu, A. C. Tedesco, J. Zhang and H. Bi, Nanoscale, 2023, 15, 376–386 RSC.
- Y. Eom, S. M. Son, Y. E. Kim, J. Lee, S. Hwang and H. G. Chae, Carbon, 2019, 150, 142–152 CrossRef CAS.
- K. Ishimaru, T. Hata, P. Bronsveld, T. Nishizawa and Y. Imamura, J. Wood Sci., 2007, 53, 442–448 CrossRef CAS.
- T. Qiu, J. Yang, X. Bai and Y. Wang, RSC Adv., 2019, 9, 12737–12746 RSC.
- A. N. Emam, S. A. Loutfy, A. A. Mostafa, H. Awad and M. B. Mohamed, RSC Adv., 2017, 7, 23502–23514 RSC.
- T. N. J. I. Edison, R. Atchudan, M. G. Sethuraman, J. Shim and Y. R. Lee, J. Photochem. Photobiol., B, 2016, 161, 154–161 CrossRef CAS PubMed.
- F. J. Chao-Mujica, L. Garcia-Hernández, S. Camacho-López, M. Camacho-López, M. A. Camacho-López, D. Reyes Contreras, A. Pérez-Rodríguez, J. P. Peña-Caravaca, A. Páez-Rodríguez, J. G. Darias-Gonzalez, L. Hernandez-Tabares, O. Arias de Fuentes, E. Prokhorov, N. Torres-Figueredo, E. Reguera and L. F. Desdin-García, J. Appl. Phys., 2021, 129, 163301 CrossRef CAS.
- H. Wu, R. Gakhar, A. Chen, S. Lam, C. P. Marshall and R. O. Scarlat, J. Nucl. Mater., 2020, 528, 151802 CrossRef CAS.
- S. K. Tammina and Y. Yang, J. Photochem. Photobiol., A, 2020, 387, 112134 CrossRef CAS.
- Y. Park, M. D. Tran, Y. Kim, S. Won, Y. Kim, T. Lee, T. Gregorkiewicz, Y. H. Lee, J. Kim and W. Kwon, Carbon, 2022, 188, 229–237 CrossRef CAS.
- N. Dhenadhayalan, K. Lin, R. Suresh and P. Ramamurthy, J. Phys. Chem. C, 2016, 120, 1252–1261 CrossRef CAS.
- Y. Song, S. Zhu, S. Xiang, X. Zhao, J. Zhang, H. Zhang, Y. Fu and B. Yang, Nanoscale, 2014, 6, 4676–4682 RSC.
- A. Oberlin, Carbon, 1984, 22, 521–541 CrossRef CAS.
- R. Berera, R. van Grondelle and J. T. M. Kennis, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS PubMed.
- N. Basu and D. Mandal, Carbon, 2019, 144, 500–508 CrossRef CAS.
- L. Wang, T. Rossi, M. Oppermann, B. Bauer, L. Mewes, D. Zare, T. H. Chow, J. Wang and M. Chergui, J. Phys. Chem. C, 2020, 124, 24322–24330 CrossRef CAS.
- Y. Kim, Y. Park, S. Han, W. Park, M. Kim, K. Kim, J. Joo, S. K. Hahn and W. Kwon, Nanomaterials, 2022, 12, 70 CrossRef CAS PubMed.
- Z. Li, D. Wang, M. Xu, J. Wang, X. Hu, S. Anwar, A. C. Tedesco, P. C. Morais and H. Bi, J. Mater. Chem. B, 2020, 8, 2598–2606 RSC.
- X. Wu, M. Xu, S. Wang, K. Abbas, X. Huang, R. Zhang, A. C. Tedesco and H. Bi, Dalton Trans., 2022, 51, 2296–2303 RSC.
- C. Dong, S. Wang, M. Ma, P. Wei, Y. Chen, A. Wu, Z. Zha and H. Bi, Appl. Mater. Today, 2021, 25, 101178 CrossRef.
- V. Poderys, G. Jarockyte, S. Bagdonas, V. Karabanovas and R. Rotomskis, J. Photochem. Photobiol., B, 2020, 204, 111802 CrossRef CAS PubMed.
- M. C. Ortega-liebana, M. M. Encabo-berzosa, A. Casanova, M. D. Pereboom, J. O. Alda, J. L. Hueso and J. Santamaria, Chem.–Eur. J., 2019, 25, 5539–5546 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00550j |
| This journal is © The Royal Society of Chemistry 2023 |