
Critical roles of linker length in determining the chemical and self-assembly stability of SN38 homodimeric nanoprodrugs†
Xin
Wang‡
ab,
Tian
Liu‡
a,
Yuetong
Huang
a,
Fudan
Dong
c,
Lingxiao
Li
a,
Jiaxuan
Song
a,
Shiyi
Zuo
a,
Zhengyang
Zhu
b,
Ken-ichiro
Kamei
 a,
Zhonggui
He
a,
Bingjun
Sun
*a and
Jin
Sun
*a
a,
Zhonggui
He
a,
Bingjun
Sun
*a and
Jin
Sun
*a
aWuya College of Innovation, Shenyang Pharmaceutical University, Shenyang, 110016, P. R. China
bDepartment of Radiology, Affiliated Nanjing Drum Tower Hospital of Nanjing University Medical School, Nanjing, 210008, P. R. China
cHenan Provincial People's Hospital, Zhengzhou, 450003, P. R. China
First published on 20th December 2022
Abstract
Homodimeric prodrug nanoassemblies (HDPNs) have been widely studied for efficient cancer therapy by virtue of their ultra-high drug loading and distinct nanostructure. However, the development of SN38 HDPNs is still a great challenge due to the rigid planar aromatic ring structure. Improving the structural flexibility of homodimeric prodrugs by increasing the linker length may be a potential strategy for constructing SN38 HDPNs. Herein, three SN38 homodimeric prodrugs with different linker lengths were synthesized. The number of carbon atoms from the disulfide bond to the adjacent ester bond is 1 (denoted as α-SN38-SS-SN38), 2 (β-SN38-SS-SN38), and 3 (γ-SN38-SS-SN38), respectively. Interestingly, we found that α-SN38-SS-SN38 exhibited extremely low yield and poor chemical stability. Additionally, β-SN38-SS-SN38 demonstrated suitable chemical stability but poor self-assembly stability. In comparison, γ-SN38-SS-SN38 possessed good chemical and self-assembly stability, thereby improving the tumor accumulation and antitumor efficacy of SN38. We developed the SN38 HDPNs for the first time and illustrated the underlying molecular mechanism of increasing the linker length to enhance the chemical and self-assembly stability of homodimeric prodrugs. These findings would provide new insights for the rational design of HDPNs with superior performance.
New conceptsInspired by the fact that increasing the number of freely rotatable bonds can improve the flexibility of a rigid structure, we proposed the concept that the structural rigidity of a homodimeric prodrug such as SN38 can be compromised by increasing the linker length and then stable homodimeric prodrug nanoassemblies can be self-assembled. In this article, SN38 HDPNs were developed simply by increasing the linker length. Compared with common nanoparticles, these SN38 homodimeric prodrug nanoassemblies retain the advantages of prodrug nanoassemblies, such as high drug loading, facile preparation, long circulation time, tumor-targeted accumulation and activation. In addition, the concept would be beneficial for constructing stable HDPNs which would promote the translation of prodrug nanomedicines from bench to bedside. For the first time, the critical roles of linker length in determining the chemical and self-assembly stability of SN38 homodimeric nanoprodrugs were found and elucidated from the molecular mechanism. These findings provide new insights into the rational design of stable HDPNs, especially for those prodrugs that are difficult to self-assemble into nanoassemblies. |
1. Introduction
Chemotherapy remains one of the main treatment options for most cancer patients.1,2 However, despite great progress in the study of chemotherapy, the outcomes are still far from satisfactory for most chemo-drugs due to the inefficient drug delivery and non-selective toxicity.3,4 For example, SN38, a compound with ultra-high activity in the camptothecin family, which causes DNA single- or double-strand breaks during DNA replication, has been considered as a potential candidate for a variety of cancers.5 However, so far, there is no SN38-based formulation for clinical use because SN38 exhibits extremely low solubility in water, even in any pharmaceutically accepted excipient. It is insoluble in Cremophor, only 11–38 μg mL−1 in water, and 36 μg mL−1 in phosphate buffered saline (pH 7.4), which strictly limits its formulation development. In addition, the non-specific biodistribution and non-selective cytotoxicity of SN38 leads to serious systemic toxicity such as myelosuppression and delayed-onset diarrhea.6 The above shortcomings prevent SN38 from being directly used in the clinic.To address the current dilemma, prodrug-based nanoassemblies (PNs) integrating the superiorities of prodrugs and nanomedicines have attracted much attention.7–11 They demonstrated obvious advantages over traditional nanomedicines:12,13 (1) high drug loading, mostly more than 50% for parent drug; (2) fewer side effects; and (3) a simple preparation process, which is easy for industrial production. In particular, homodimeric prodrug nanoassemblies (HDPNs), as a new branch of PNs, have been extensively investigated for drug delivery due to their ultra-high drug loading and distinct nanostructure in recent years.14–16 Homodimeric prodrugs were obtained by linking two of the same drug molecules via a suitable linker, requiring only one reaction step, which greatly simplified the synthesis process. Moreover, due to the large proportion of the parent drug, the drug loading of HDPNs has been as high as nearly 70% or even higher.17 These excellent properties contributed to HDPNs’ great potentiality towards clinical translation.
However, during formulation development, self-assembly stability is a critical factor limiting the translation of prodrug nanomedicines from bench to bedside, especially for SN38 HDPNs. Previous studies have reported taxanes and doxorubicin HDPNs but so far SN38 HDPNs have not been reported because SN38 has a huge planar aromatic ring structure, which makes its structure extremely rigid and difficult to self-assemble into stable nanostructures.14,18,19 Therefore, improving the structural flexibility of homodimeric prodrugs may be a potential strategy for constructing SN38 HDPNs. Compared with their parent drugs, homodimeric prodrugs have only one more linker, which can rotate freely to improve their structural flexibility, thereby preventing the fast self-aggregation of rigid molecules during the self-assembly process. We wondered whether the self-assembly stability, in vivo fate, and antitumor efficacy of SN38 homodimeric prodrugs could be improved by increasing the linker length. More importantly, what is the underlying molecular mechanism?
Based on the above considerations, we aimed to develop SN38 HDPNs and elucidate the molecular mechanism of linker length on the self-assembly stability of SN38 homodimeric prodrugs for efficient tumor therapy. For specific application, the disulfide bond,9 sensitive to the redox microenvironment of tumor cells, was applied to construct a redox-responsive delivery nano-system. As shown in Fig. 1, three SN38 homodimeric prodrugs bridged by different lengths of disulfide-containing linkers were designed and denoted as α-SN38-SS-SN38 (with the shortest linker), β-SN38-SS-SN38, and γ-SN38-SS-SN38 (with the longest linker), respectively. Interestingly, we found that the linker length could affect the chemical stability of the homodimeric prodrug. α-SN38-SS-SN38 exhibited an extremely low yield and the worst chemical stability. In addition, γ-SN38-SS-SN38 demonstrated better a self-assembly ability due to the long linker, thereby improving the tumor accumulation and antitumor efficacy of SN38, compared with β-SN38-SS-SN38.
 | ||
| Fig. 1 A schematic illustration of the different linker lengths of disulfide bond-bridged SN38 homodimeric prodrug nanoassemblies for cancer therapy. γ-SN38-SS-SN38 demonstrated better chemical and self-assembly ability, thereby improving the tumor accumulation and antitumor efficacy of SN38, compared with α-SN38-SS-SN38 and β-SN38-SS-SN38. | ||
2. Results and discussion
2.1 Synthesis of SN38 homodimeric prodrugs
The synthetic routes of SN38 homodimeric prodrugs are displayed in Fig. S1 (ESI†). The corresponding prodrugs were denoted as α-SN38-SS-SN38, β-SN38-SS-SN38, and γ-SN38-SS-SN38, respectively. As displayed in Fig. S2–S4 (ESI†), the results of MS and 1H NMR showed that the prodrugs were successfully synthesized. However, we unexpectedly found that the yield of α-SN38-SS-SN38 was extremely low, less than 1%, much lower than that of β-SN38-SS-SN38 (61.8%) and γ-SN38-SS-SN38 (61.0%). This may be due to the fact that the linker length of α-SN38-SS-SN38 is too short, leading to a large steric hindrance which strictly blocks the reaction process. Further, the chemical stability of the three prodrugs was evaluated. As shown in Fig. S5 (ESI†), the purity of α-SN38-SS-SN38 dropped rapidly to about 60% after 9 h at room temperature. In comparison, the purity of β-SN38-SS-SN38 and γ-SN38-SS-SN38 remained basically unchanged, demonstrating that the linker length in the homodimeric prodrug could significantly affect the chemical stability of the SN38 homodimeric prodrug. The shorter the linker, the more unstable the SN38 homodimeric prodrug. The poor stability of α-SN38-SS-SN38 was not suitable for drug development. Therefore, only β-SN38-SS-SN38 and γ-SN38-SS-SN38 were used for subsequent experiments. Next, the purities of β-SN38-SS-SN38 and γ-SN38-SS-SN38 were determined by high-performance liquid chromatography (HPLC). As shown in Fig. S6 (ESI†), the purities of both prodrugs were more than 99%, meeting the requirements of subsequent experiments.2.2 Preparation, characterization, and self-assembly stability of SN38 HDPNs
Firstly, to elucidate the critical role of linker length on the self-assembly performance of the SN38 HDPNs, pure prodrug nanoassemblies (non-PEGylated) were prepared through one-step nanoprecipitation (Fig. 2A). β-SN38-SS-SN38 and γ-SN38-SS-SN38 could spontaneously self-assemble into uniform nanoassemblies in aqueous solution. It could be observed from Fig. 2B that the two HDPNs were spherical particles with uniform particle size distribution. Due to the absence of any other excipients, the drug loading of non-PEGylated β-SN38-SS-SN38 NPs and non-PEGylated γ-SN38-SS-SN38 NPs was as high as about 80% for the parent drug SN38 (Table S1, ESI†). The particle size of non-PEGylated β-SN38-SS-SN38 NPs and γ-SN38-SS-SN38 NPs was about 115 nm and 90 nm, respectively, which demonstrated that a longer linker length could improve the self-assembly performance of SN38 homodimeric prodrugs. Then, the stability of non-PEGylated β-SN38-SS-SN38 NPs and non-PEGylated γ-SN38-SS-SN38 NPs stored at room temperature was investigated. After 48 h, the particle size of non-PEGylated γ-SN38-SS-SN38 NPs remained basically unchanged, and the properties of the preparation did not change significantly. However, non-PEGylated β-SN38-SS-SN38 NPs precipitated out at 3 h, as displayed in Fig. 2D and Fig. S7A (ESI†). These results further suggested that the extended linker length improved the self-assembly ability of SN38 homodimeric prodrugs.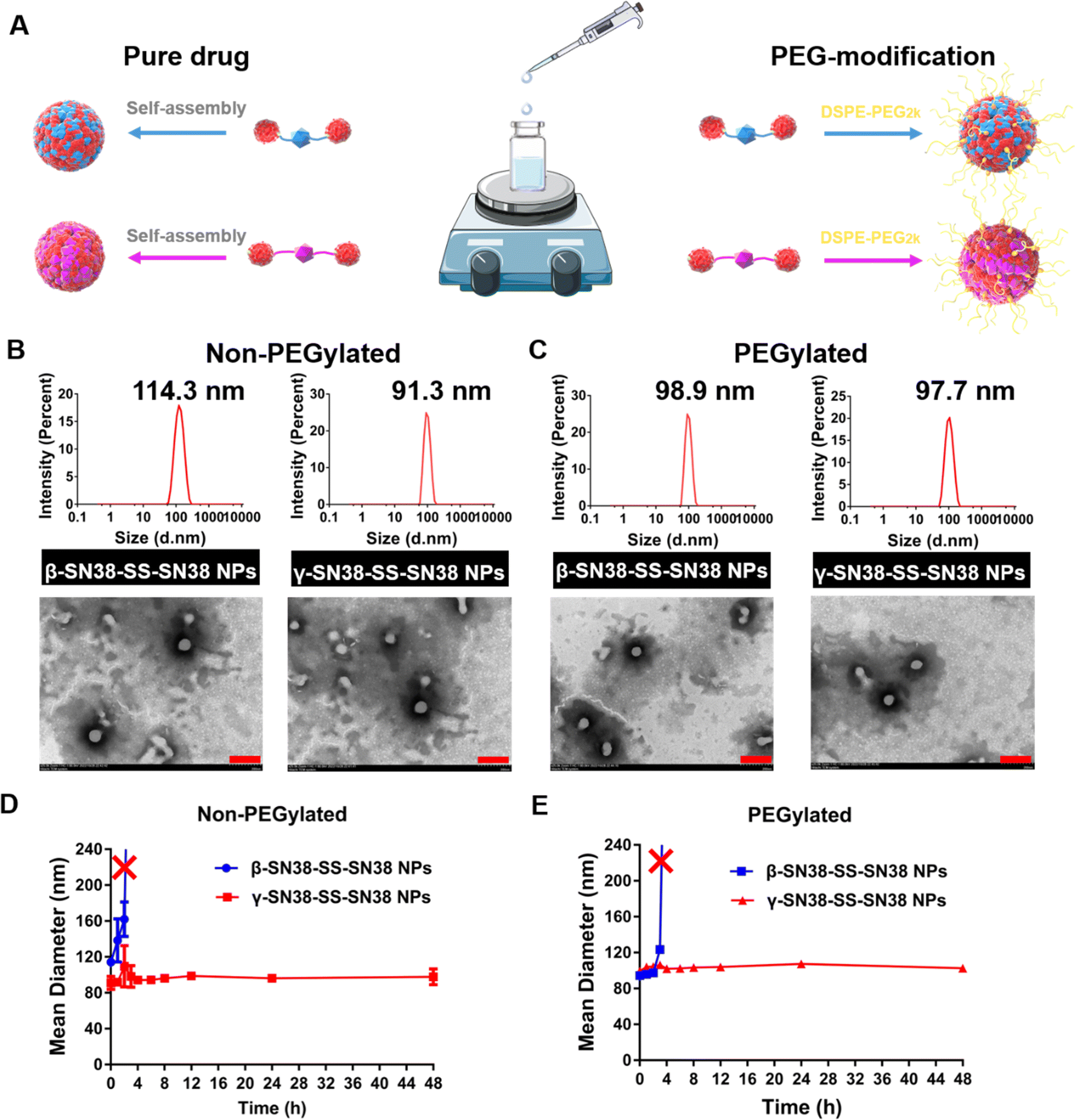 | ||
| Fig. 2 (A) Preparation of HDPNs by a one-step nanoprecipitation method. (B) Particle size distribution and TEM images of the non-PEGylated prodrug nanoassemblies. (C) Particle size distribution and TEM images of the PEGylated prodrug nanoassemblies. Scale bar represents 200 nm. Stability of (D) non-PEGylated and (E) PEGylated prodrug nanoassemblies at room temperature for 48 h. | ||
The difference between the two SN38 HDPNs was only the linker length. It has been demonstrated that the hydrophobic forces drive prodrug self-assembly.20–22 Therefore, the oil-water partition coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) reflecting the hydrophobicity were calculated by MarvinSketch. As shown in Fig. S8A (ESI†), the logP of β-SN38-SS-SN38 and γ-SN38-SS-SN38 was 4.75 and 5.32, respectively. The highly hydrophobic γ-SN38-SS-SN38 facilitated the self-assembly process. Furthermore, the bond angle/dihedral angle of the disulfide bond of γ-SN38-SS-SN38 was closer to 90°, as shown in Fig. S8B (ESI†), which would weaken the structural rigidity of the prodrug and enhance the colloidal stability of the prodrug nanoassemblies. Moreover, compared with β-SN38-SS-SN38, γ-SN38-SS-SN38 had more σ bonds (Fig. S8C, ESI†), which is beneficial for the prodrug to rotate to its lowest energy state during self-assembly.23
P) reflecting the hydrophobicity were calculated by MarvinSketch. As shown in Fig. S8A (ESI†), the logP of β-SN38-SS-SN38 and γ-SN38-SS-SN38 was 4.75 and 5.32, respectively. The highly hydrophobic γ-SN38-SS-SN38 facilitated the self-assembly process. Furthermore, the bond angle/dihedral angle of the disulfide bond of γ-SN38-SS-SN38 was closer to 90°, as shown in Fig. S8B (ESI†), which would weaken the structural rigidity of the prodrug and enhance the colloidal stability of the prodrug nanoassemblies. Moreover, compared with β-SN38-SS-SN38, γ-SN38-SS-SN38 had more σ bonds (Fig. S8C, ESI†), which is beneficial for the prodrug to rotate to its lowest energy state during self-assembly.23
In order to further explain the difference in the self-assembly stability of the SN38 HDPNs, molecular docking and molecular dynamics simulations were used to investigate the self-assembly mechanism of β-SN38-SS-SN38 and γ-SN38-SS-SN38. As shown in Fig. 3A, multiple intermolecular forces facilitated the self-assembly, including hydrogen bonds between the hydroxyl structures, π–π stacking between the SN38 molecules, and alkyl hydrophobic, π-alkyl and sulfur bonds between the linkers. γ-SN38-SS-SN38 formed into stable nanostructure within 10 ns, while β-SN38-SS-SN38 self-assembled a similar nanostructure until 50 ns (Fig. 3B and C), further demonstrating that γ-SN38-SS-SN38 had an obviously better self-assembly performance. Moreover, γ-SN38-SS-SN38 NPs exhibited a much lower free binding energy (−22.855 kcal mol−1) than that of β-SN38-SS-SN38 NPs (−20.981 kcal mol−1, Fig. 3D). According to thermodynamic laws, the more negative the potential energy values, the more stable the formed structure would be.24,25 These findings illustrated that increasing the linker length could effectively improve the self-assembly capability of SN38 HDPNs.
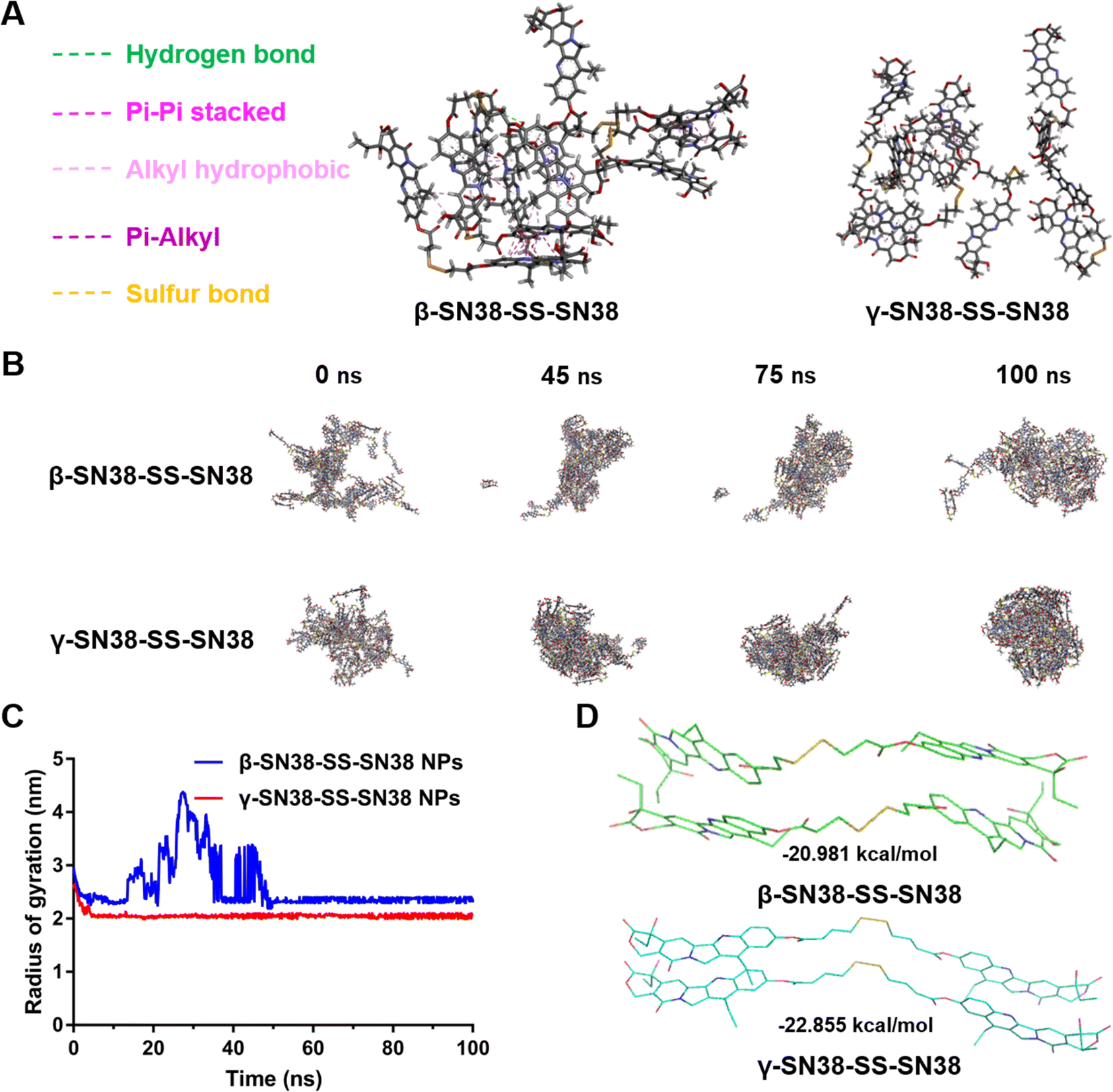 | ||
| Fig. 3 Molecular dynamics simulations of HDPNs. (A) Intermolecular forces of HDPNs. (B) A schematic diagram of the aggregation process of HDPNs from 0 ns to 100 ns. (C) Changes of the radius of gyration during aggregation. (D) The free binding energy of HDPNs. | ||
To improve the blood circulation time of HDPNs, DSPE-PEG2K was used to form a hydration layer on the surface of prodrug nanoassemblies, which could reduce the phagocytosis of the reticuloendothelial system. The PEGylated HDPNs were used for the following studies. As shown in Fig. 2C, both the prodrug nanoassemblies were spherical in structure (100 nm) with a uniform particle size distribution. The change of surface charge and the similar zeta potential to that of DSPE-PEG2K proved that PEG was successfully modified on the surface of nanoparticles (Table S2, ESI†). In spite of the addition of a small amount of DSPE-PEG2K (20%, w/w) during the preparation, the drug loading of the two HDPNs was still more than 60%. Interestingly, after PEG modification, the particle size of β-SN38-SS-SN38 NPs was significantly reduced, reaching the same level as γ-SN38-SS-SN38 NPs, indicating that the addition of DSPE-PEG2K could improve the self-assembly performance of β-SN38-SS-SN38 with poor self-assembly properties. In comparison, the particle size of γ-SN38-SS-SN38 NPs remained basically unchanged.
Then, the stability of PEGylated β-SN38-SS-SN38 NPs and γ-SN38-SS-SN38 NPs stored at room temperature was also investigated. As shown in Fig. 2E and Fig. S7B (ESI†), β-SN38-SS-SN38 NPs precipitated out at 4 h, slightly stronger than non-PEGylated (3 h) in colloidal stability. Similar to the results of non-PEGylated HDPNs, PEGylated γ-SN38-SS-SN38 NPs (hereafter, γ-SN38-SS-SN38 NPs refers to PEGylated γ-SN38-SS-SN38 NPs) still demonstrated better colloidal stability, which may be beneficial for the in vivo fate. This result also indicated that the colloidal stability of HDPNs depends on the molecular structure of the prodrug. The addition of surfactants, such as DSPE-PEG2K, could only facilitate the self-assembly of the prodrug to a certain extent, not fundamentally.
2.3 Redox-responsive release
As shown in Fig. 4A, the release rate of β-SN38-SS-SN38 NPs was much higher than that of γ-SN38-SS-SN38 NPs in blank medium. This was probably because β-SN38-SS-SN38 NPs exhibited poor colloidal stability and were prone to disintegrate. Also, due to the strong polarity of SN38, it was easy for water molecules to attack the ester bond, resulting in the release of SN38.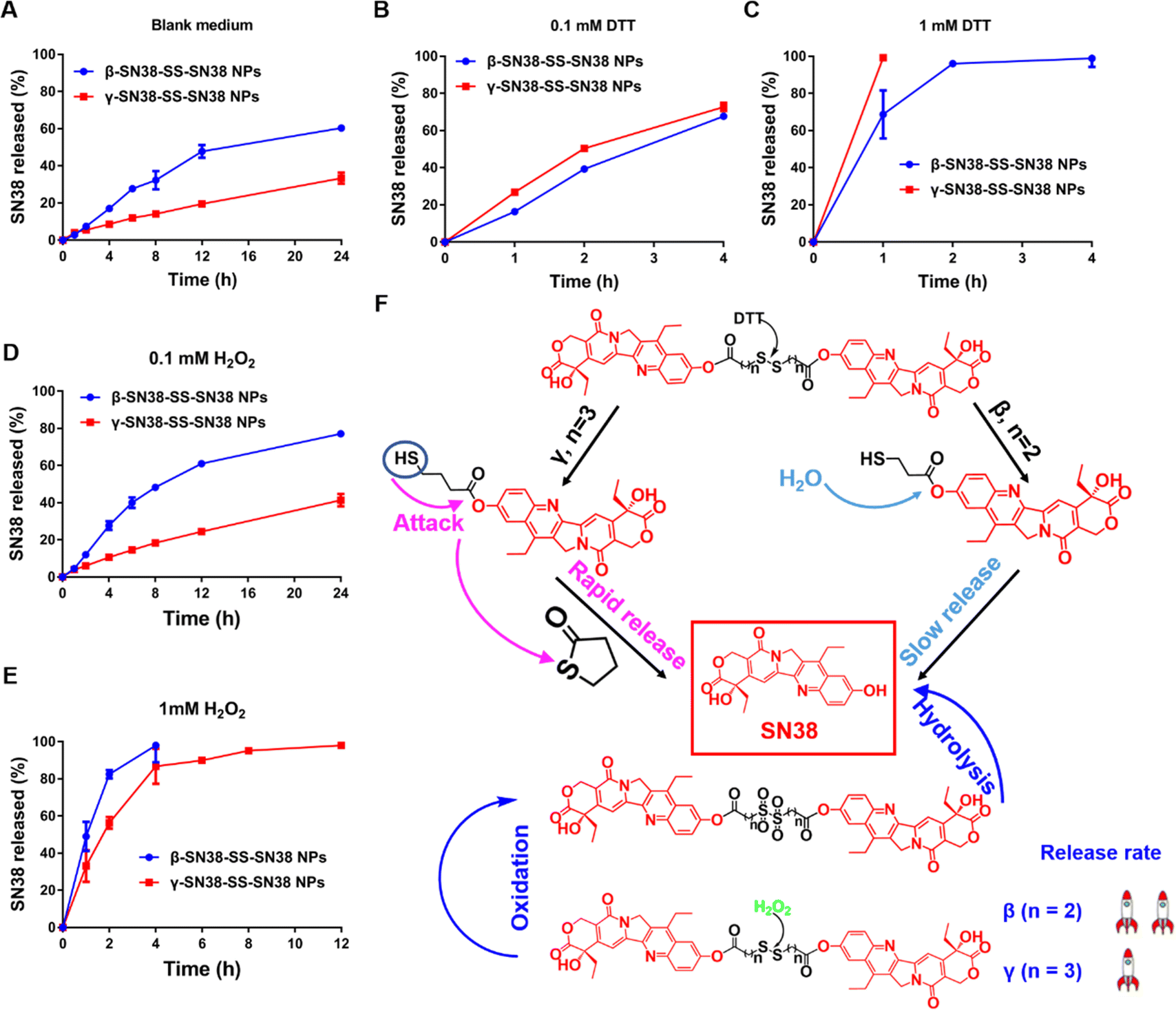 | ||
| Fig. 4 In vitro redox-responsive drug release behavior in the presence of (A) blank medium, (B) 0.1 mM DTT, (C) 1 mM DTT, (D) 0.1 mM H2O2, and (E) 1 mM H2O2. (F) The mechanism of redox-triggered drug release. | ||
Under the stimulus of DTT, the drug release rate of γ-SN38-SS-SN38 NPs was higher than that of β-SN38-SS-SN38 NPs (Fig. 4B and C), which was related to the reduction-responsive mechanism of the disulfide bond. As shown in Fig. 4F, under the attack of DTT, the disulfide bond was degraded into a hydrophilic thiol intermediate (SN38-SH), which promoted the release of SN38. For γ-SN38-SS-SN38, the SN38-SH would quickly generate a five-membered thiolactone ring through intramolecular nucleophilic acyl substitution, facilitating the fast release of SN38, as illustrated in our previous study.9 The results indicated that β-SN38-SS-SN38 NPs would release drugs into blood and normal tissue. In comparison, γ-SN38-SS-SN38 NPs remained stable under normal conditions but were more sensitive to high concentrations of GSH in the tumor microenvironment. The superior selectivity would not only ensure the antitumor effect, but also reduce the non-selective toxicity in normal tissue. As displayed in Fig. S9 (ESI†), the corresponding intermediates were captured using mass spectrometry.
The oxidation-responsive release results of the HDPNs are shown in Fig. 4D and E. No matter whether the conditions were a low concentration or high concentration of H2O2, the drug release rate of the β-SN38-SS-SN38 NPs was faster than that of the γ-SN38-SS-SN38 NPs, consistent with the oxidization-responsive mechanism of the disulfide bond.9 Under the presence of H2O2, the disulfide bond was oxidized to hydrophilic sulfoxide or sulphone, which further promoted the hydrolysis of the adjacent ester bond. The ester bond of the β-SN38-SS-SN38 was closer to the sulfone or sulfoxide, so its hydrolysis rate was faster, as shown in Fig. 4F. The corresponding intermediates were confirmed using mass spectrometry, as shown in Fig. S10 (ESI†).
2.4 Cell uptake, cytotoxicity assays, and DNA damage
Efficient uptake of HDPNs by tumor cells is a prerequisite for their antitumor effects. Therefore, 4T1 cells were used as a tumor cell model to investigate the uptake efficiency of HDPNs via confocal microscopy and flow cytometry. As shown in Fig. 5A, B and Fig. S11 (ESI†), γ-SN38-SS-SN38 NPs showed higher intracellular fluorescence intensity than β-SN38-SS-SN38 NPs and free coumarin-6, demonstrating that γ-SN38-SS-SN38 NPs could be taken up efficiently by tumor cells. The intracellular fluorescence intensity of the β-SN38-SS-SN38 NPs group was not statistically different from that of the free coumarin-6 group, probably because β-SN38-SS-SN38 NPs were easily disintegrated and degraded during incubation.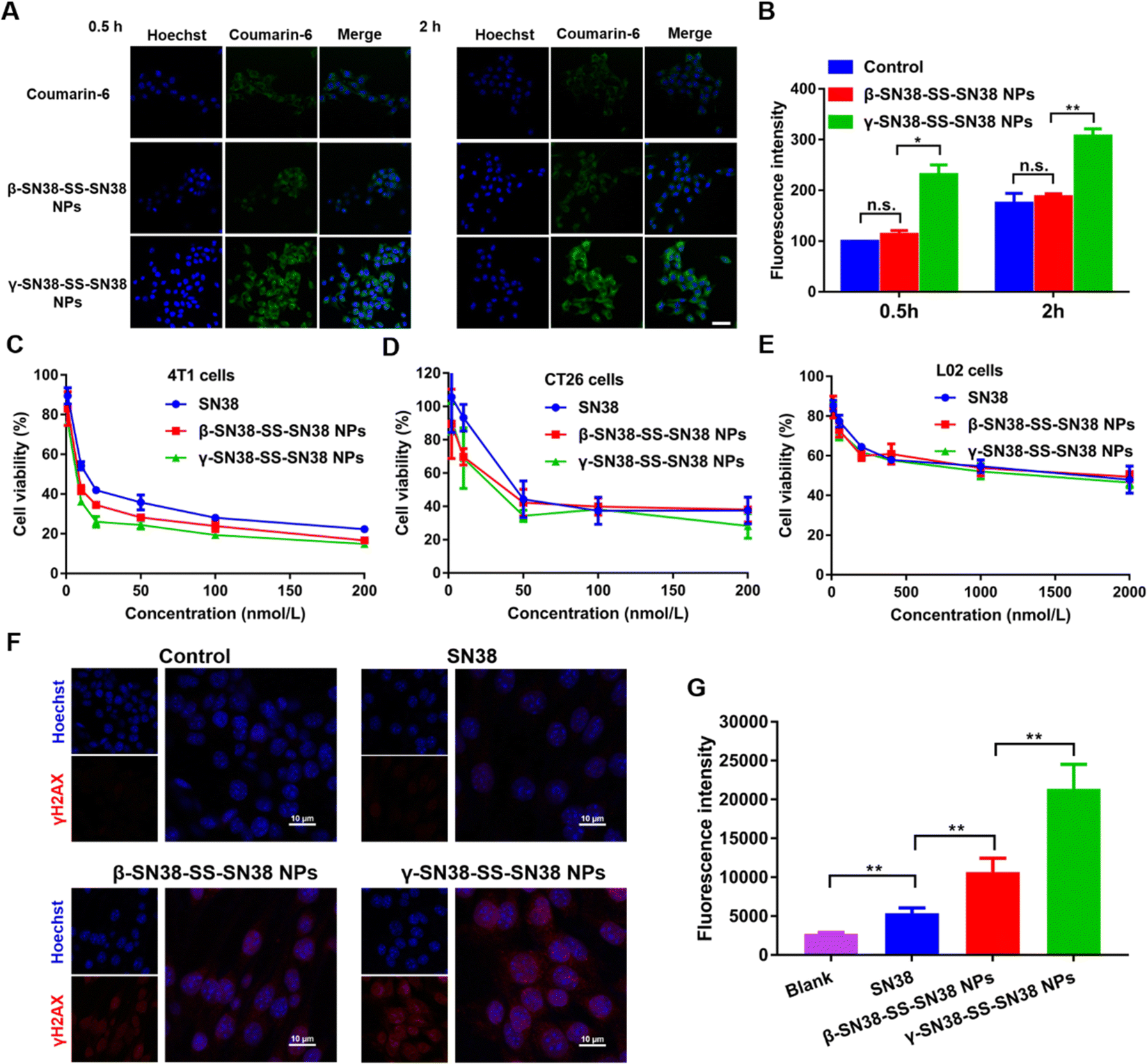 | ||
| Fig. 5 Cellular uptake in 4T1 cells after incubation with free coumarin-6 or coumarin-6-labled prodrug nanoassemblies for 0.5 h and 2 h by (A) confocal laser scanning microscopy (CLSM) images and (B) flow cytometry. Scale bar represents 20 μm. Cell viability treated with various concentrations of SN38 and HDPNs. (C) 4T1 cells, (D) CT26 cells, and (E) L02 cells. (F) CLSM images and (G) quantitative results of immunofluorescence of γH2AX on 4T1 cells after treatment with SN38 and HDPNs at 24 h. Scale bar = 10 μm. **p < 0.005. | ||
Then, the cytotoxicity of the HDPNs to two different tumor cells and one normal cell was investigated to explore whether the HDPNs could effectively inhibit tumor cells, and distinguish tumor cells from normal cells. The results are shown in Table S3 and Fig. 5C–E. Both β-SN38-SS-SN38 NPs and γ-SN38-SS-SN38 NPs had stronger inhibitory effects on tumor cells than the parent drug SN38. In addition, the viability of the L02 cell line was higher than 4T1 and CT26 after various treatments due to the slow hydrolysis rate of active SN38 (Fig. 4) caused by the much higher redox state in tumor cells than normal cells. Furthermore, γ-SN38-SS-SN38 NPs were more cytotoxic than β-SN38-SS-SN38 NPs due to the higher cellular uptake efficiency of γ-SN38-SS-SN38 NPs. The DNA damage results were consistent with the order of cytotoxicity (Fig. 5F and G). As shown in Table S4, the ESI† of the two prodrug nanoassemblies was larger than that of the parent drug SN38, suggesting that prodrug nanoassemblies would discriminate tumor cells from normal cells.
2.5 Hemolysis assay, in vivo pharmacokinetics, and biodistribution
A hemolysis assay was conducted to preliminarily investigate the safety of the prodrug nanoassemblies prior to vein injection. As shown in Fig. S12 (ESI†), the β-SN38-SS-SN38 NPs and γ-SN38-SS-SN38 NPs did not cause hemolysis, and there was no significant difference from the saline group, preliminarily indicating that the prepared prodrug nanoassemblies were safe. Moreover, the hemolysis percentage (HP%) of β-SN38-SS-SN38 NPs and γ-SN38-SS-SN38 NPs was 2.02 and 2.73, respectively, far less than 5%, meeting the requirements of intravenous administration. The method of the hemolysis assay was added in the revised manuscript.Due to the large molecular weight of the SN38 homodimeric prodrug, with the existing mass spectrometry conditions it was difficult to detect stable ion peaks and it was not possible to quantify the prodrug in plasma, so only the plasma concentration of free SN38 was determined, as shown in Fig. 6A and Table S5 (ESI†). Both the area under curve (AUC) and peak plasma concentration (Cmax) of SN38 released from the HDPNs were higher than those of the SN38 solution. Moreover, the AUC and Cmax of SN38 released by γ-SN38-SS-SN38 NPs were significantly higher than those of the β-SN38-SS-SN38 NPs, which was attributed to the better colloidal stability of γ-SN38-SS-SN38 NPs. These results demonstrated that the linker length had a significant effect on the pharmacokinetic behavior of the SN38 homodimeric prodrug. Suitable extension of the linker length would significantly improve the better pharmacokinetic behavior of HDPNs due to the improved self-assembly performance.
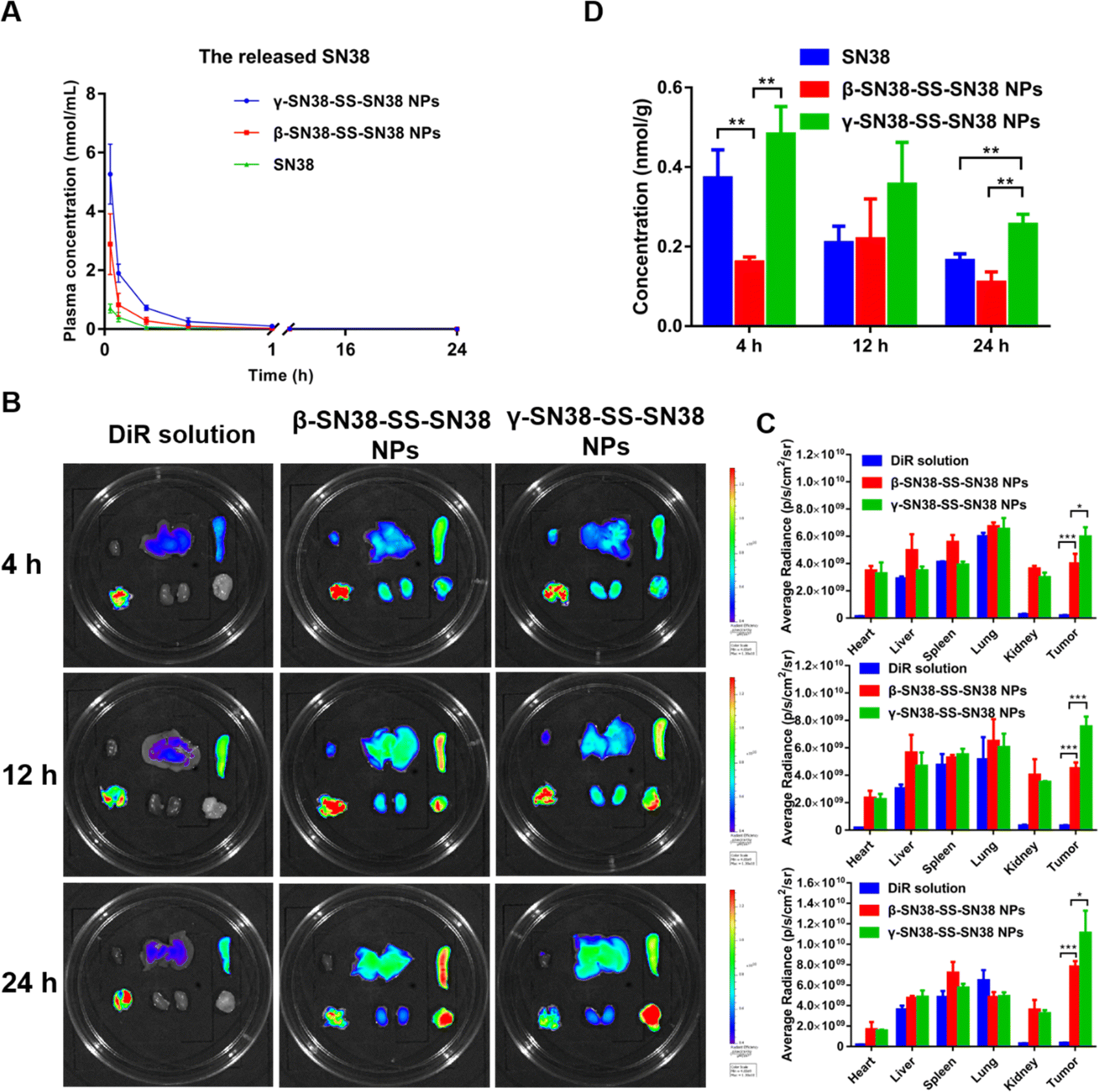 | ||
| Fig. 6 In vivo fate. (A) Molar concentration–time curves of the released SN38 in rat plasma (n = 5). (B) Biodistribution of DiR-labeled prodrug nanoassemblies. (C) Quantitative results of relative organ and tumor accumulation. (D) Tumor accumulation of SN38 (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001 by two-tailed Student's t test, n.s., no significance. | ||
Tissue distribution and tumor accumulation were investigated using DiR-labeled HDPNs. The results are shown in Fig. 6B and C, the DiR solution was mainly distributed in the lungs and there was very little in the tumor tissue. In contrast, the fluorescence intensity of DiR-labeled HDPNs in the tumor tissue was significantly increased due to the EPR effect, which is beneficial for improving the antitumor efficacy of the HDPNs and reducing the side effects. In addition, the fluorescence intensity of γ-SN38-SS-SN38 NPs at the tumor site was higher than that of β-SN38-SS-SN38 NPs, agreeing well with the pharmacokinetic behavior as demonstrated above.
The released parent drug at the tumor site determined the antitumor effect. Therefore, the content of free SN38 in the tumor was quantified by UPLC-MS-MS, and the results are shown in Fig. 6D. γ-SN38-SS-SN38 NPs displayed more SN38 at the tumor site than β-SN38-SS-SN38 NPs and SN38 solution. This may be related to the good stability of γ-SN38-SS-SN38 NPs, which effectively improve the pharmacokinetic behavior and tumor accumulation of HDPNs.
2.6 In vivo antitumor efficacy
As shown in Fig. 7A–C, γ-SN38-SS-SN38 NPs exhibited the best anti-tumor effect, significantly inhibiting tumor growth, and the tumor-bearing rate was significantly lower than that of β-SN38-SS-SN38 NPs and free SN38. The results of tumor cell apoptosis and proliferation are shown in Fig. S13 and S14 (ESI†), respectively. γ-SN38-SS-SN38 NPs caused a large area of tumor cell apoptosis, and the apoptosis rate reached 28.9%, a lot higher than that of the β-SN38-SS-SN38 NPs (13.5%), SN38 solution (10.1%), and saline group (2.7%). The tumor tissue of the γ-SN38-SS-SN38 NPs group showed only a small amount of tumor cell proliferation (28.1%), much lower than that of the β-SN38-SS-SN38 NPs (53.3%), SN38 solution (55.8%), and saline group (85.9%). γ-SN38-SS-SN38 NPs showed a stronger antitumor effect than free SN38 due to the following advantages: (1) longer circulation time; (2) stronger tumor accumulation capacity; and (3) rapid drug release in tumor cells, as illustrated in Fig. 1. In comparison, β-SN38-SS-SN38 NPs with poor colloidal stability would disintegrate rapidly when entering the systemic circulation, losing the advantages of prodrug nanoassemblies and exhibiting a limited antitumor effect.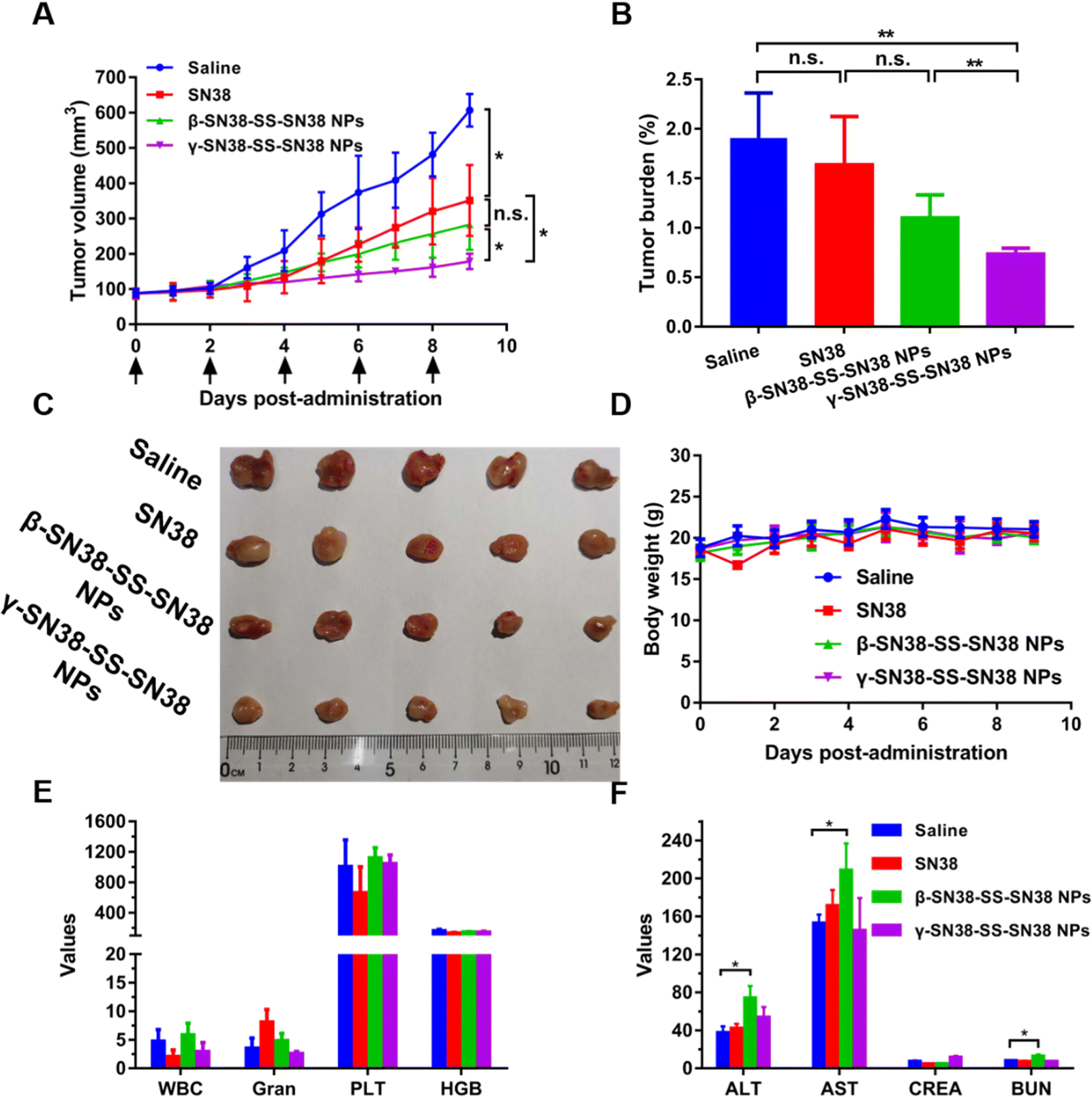 | ||
| Fig. 7 Antitumor efficacy and safety evaluation of HDPNs. (A) Tumor volume. (B) Tumor burden. (C) Images of tumors. (D) Body weight changes. (E) Routine blood examination for 4T1 xenograft tumor bearing mice. WBC: white blood cell count (109 L−1); Grant: neutrophil count (109 L−1); PLT: platelet (109 L−1); HGB: hematocrit (g L−1). (F) Hepatorenal function parameters for 4T1 xenograft tumor bearing mice. AST: aspartate aminotransferase (U L−1); ALT: alanine aminotransferase (U L−1); BUN: blood urea nitrogen (mmol L−1); and CREA: creatinine (μmol L−1). Data were presented as mean ± SD (three independent experiments). *P < 0.05, **P < 0.01. | ||
The safety of the HDPNs was comprehensively evaluated using the body weight change, blood routine, liver and kidney function index, and tissue H&E staining of 4T1 tumor-bearing mice as indicators. As shown in Fig. 7D and E, the body weight and blood routine index of the mice treated with γ-SN38-SS-SN38 NPs were not significantly different from those treated with saline. In contrast, the levels of ALT and AST in mice treated with β-SN38-SS-SN38 NPs were significantly higher than those of normal saline (Fig. 7F), indicating that β-SN38-SS-SN38 NPs caused certain damage to liver function. It might be related to the large accumulation of HDPNs in the liver and the poor stability of β-SN38-SS-SN38 NPs, resulting in more parent drug being released in the liver. H&E staining (Fig. S15, ESI†) exhibited no obvious tissue damage in each group. These results demonstrated that γ-SN38-SS-SN38 NPs exerted superior therapeutic effects but negligible toxicity.
3. Conclusions
To develop SN38 HDPNs and elucidate the molecular mechanism of linker length on chemical and self-assembly stability of the SN38 homodimeric prodrug for efficient tumor therapy, three SN38 homodimeric prodrugs (α-SN38-SS-SN38, β-SN38-SS-SN38, and γ-SN38-SS-SN38) bridged by different lengths of disulfide-containing linkers were synthesized. Interestingly, we found that α-SN38-SS-SN38 exhibited poor chemical stability due to the large steric hindrance caused by a short linker for the first time. In addition, the extension of the linker could improve the self-assembly stability of the SN38 homodimeric prodrug. The γ-SN38-SS-SN38 NPs exhibited better self-assembly stability, thereby performing better in pharmacokinetic behavior and tumor accumulation, resulting in efficient tumor therapy and negligible toxicity. This work comprehensively elucidated the critical roles of the linker length in the chemical and self-assembly stability of homodimeric prodrugs, which would be beneficial for the rational design of HDPNs with superior performance.Author contributions
Xin Wang: conceptualization, visualization, investigation, software, validation, data curation, writing – original draft. Tian Liu: conceptualization, writing, review – original draft. Yuetong Huang: software, investigation. Fudan Dong: software, investigation, data curation. Lingxiao Li: software, investigation, validation. Jiaxuan Song: investigation, data curation. Shiyi Zuo: investigation, data curation. Zhengyang Zhu: review. Ken-ichiro Kamei: funding acquisition. Zhonggui He: funding acquisition. Bingjun Sun: supervision, writing – review & editing. Jin Sun: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Key R&D Program of China (No. 2022YFE0111600), National Natural Science Foundation of China (No. 82272151 and 82204318), Doctoral Scientific Research Staring Foundation of Liaoning Province (No. 2021-BS-130), General Program of Department of Education of Liaoning Province (No. LJKZ0953), Liaoning Revitalization Talents Program (No. XLYC1902061), Jiangsu Funding Program for Excellent Postdoctoral Talent, and National Science Foundation of Jiangsu Province (No. BK20220181).References
- M. D. Hellmann, B. T. Li, J. E. Chaft and M. G. Kris, Ann. Oncol., 2016, 27, 1829–1835 CrossRef PubMed.
- G. Bocci and R. S. Kerbel, Nat. Rev. Clin. Oncol., 2016, 13, 659–673 CrossRef PubMed.
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef PubMed.
- G. Wang, L. Xie, B. Li, W. Sang, J. Yan, J. Li, H. Tian, W. Li, Z. Zhan, Y. Tian and Y. Dai, Nat. Commun., 2021, 12, 5733 CrossRef PubMed.
- Q. Huang, X. Liu, H. Wang, X. Liu, Q. Zhang, K. Li, Y. Chen, Q. Zhu, Y. Shen and M. Sui, Acta Biomater., 2022, 137, 262–275 CrossRef PubMed.
- Y. Matsumura, Adv. Drug Delivery Rev., 2011, 63, 184–192 CrossRef PubMed.
- T. Ramasamy, H. B. Ruttala, B. Gupta, B. K. Poudel, H. G. Choi, C. S. Yong and J. O. Kim, J. Controlled Release, 2017, 258, 226–253 CrossRef CAS.
- B. Sun, C. Luo, X. Zhang, M. Guo, M. Sun, H. Yu, Q. Chen, W. Yang, M. Wang, S. Zuo, P. Chen, Q. Kan, H. Zhang, Y. Wang, Z. He and J. Sun, Nat. Commun., 2019, 10, 3211 CrossRef.
- B. Sun, C. Luo, H. Yu, X. Zhang, Q. Chen, W. Yang, M. Wang, Q. Kan, H. Zhang, Y. Wang, Z. He and J. Sun, Nano Lett., 2018, 18, 3643–3650 CrossRef CAS PubMed.
- X. Wang, L. Li, D. Wang, S. Zuo, T. Liu, F. Dong, X. Zhang, Z. He, B. Sun and J. Sun, Nano Res., 2022, 15, 3367–3375 CrossRef CAS.
- T. Liu, H. Zou, J. Mu, N. Yu, Y. Xu, G. Liu, X. Liang and S. Guo, Chin. Chem. Lett, 2021, 32, 1751–1754 CrossRef.
- B. Sun, Y. Chen, H. Yu, C. Wang, X. Zhang, H. Zhao, Q. Chen, Z. He, C. Luo and J. Sun, Acta Biomater., 2019, 92, 219–228 CrossRef PubMed.
- X. Wang, B. Yang, L. Li, T. Liu, S. Zuo, D. Chi, Z. He, B. Sun and J. Sun, Theranostics, 2021, 11, 7896–7910 CrossRef PubMed.
- Q. Pei, X. Hu, S. Liu, Y. Li, Z. Xie and X. Jing, J. Controlled Release, 2017, 254, 23–33 CrossRef.
- Y. Yang, B. Sun, S. Zuo, X. Li, S. Zhou, L. Li, C. Luo, H. Liu, M. Cheng, Y. Wang, S. Wang, Z. He and J. Sun, Sci. Adv., 2020, 6, eabc1725 CrossRef PubMed.
- L. Li, S. Zuo, F. Dong, T. Liu, Y. Gao, Y. Yang, X. Wang, J. Sun, B. Sun and Z. He, Asian J. Pharm. Sci., 2021, 16, 337–349 CrossRef PubMed.
- Y. Yang, S. Zuo, J. Zhang, T. Liu, X. Li, H. Zhang, M. Cheng, S. Wang, Z. He, B. Sun and J. Sun, Nano Today, 2022, 44, 101480 CrossRef CAS.
- S. Zuo, B. Sun, Y. Yang, S. Zhou, Y. Zhang, M. Guo, M. Sun, C. Luo, Z. He and J. Sun, Small, 2020, 16, e2005039 CrossRef PubMed.
- Y. Wang, X. Wang, F. Deng, N. Zheng, Y. Liang, H. Zhang, B. He, W. Dai, X. Wang and Q. Zhang, J. Controlled Release, 2018, 279, 136–146 CrossRef CAS.
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Pérez-Juste, S. Bals, G. Van Tendeloo, S. H. Donaldson, Jr., B. F. Chmelka, J. N. Israelachvili and L. M. Liz-Marzán, ACS Nano, 2012, 6, 11059–11065 CrossRef.
- Y. Gao, S. Zuo, L. Li, T. Liu, F. Dong, X. Wang, X. Zhang, Z. He, Y. Zhai, B. Sun and J. Sun, Nanoscale, 2021, 13, 10536–10543 RSC.
- W. Han, S. Zhang, J. Qian, J. Zhang, X. Wang, Z. Xie, B. Xu, Y. Han and W. Tian, Chem. – Asian J., 2019, 14, 1745–1753 CrossRef PubMed.
- C. Demangeat, Y. Dou, B. Hu, Y. Bretonnière, C. Andraud, A. D’Aléo, J. W. Wu, E. Kim, T. Le Bahers and A. J. Attias, Angew. Chem., Int. Ed., 2021, 60, 2446–2454 CrossRef PubMed.
- M. Feng, Y. Song, S. H. Chen, Y. Zhang and R. Zhou, Chem. Sci., 2021, 12, 6107–6116 RSC.
- F. Biedermann and H. J. Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00425a |
| ‡ These authors contributed equally and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2023 |