
Hydroxide ion-conducting viologen–bakelite organic frameworks for flexible solid-state zinc–air battery applications†
Deepak
Rase‡
 ab,
Rajith
Illathvalappil‡
ab,
Himan Dev
Singh
ab,
Pragalbh
Shekhar
ab,
Liya S
Leo
ab,
Debanjan
Chakraborty
a,
Sattwick
Haldar
ab,
Ankita
Shelke
c,
Thalasseril G.
Ajithkumar
c and
Ramanathan
Vaidhyanathan
*ab
ab,
Rajith
Illathvalappil‡
ab,
Himan Dev
Singh
ab,
Pragalbh
Shekhar
ab,
Liya S
Leo
ab,
Debanjan
Chakraborty
a,
Sattwick
Haldar
ab,
Ankita
Shelke
c,
Thalasseril G.
Ajithkumar
c and
Ramanathan
Vaidhyanathan
*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research, Dr Homi Bhabha Road, Pashan, Pune 411008, India. E-mail: vaidhya@iiserpune.ac.in; vaidhya@acads.iiserpune.ac.in
bCentre for Energy Science, Indian Institute of Science Education and Research, Dr Homi Bhabha Road, Pashan, Pune 411008, India
cCentral NMR Facility and Physical/Materials Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune 411008, India
First published on 25th November 2022
Abstract
Adaptable polymer-based solid-state electrolytes can be a game-changer toward safe, lightweight flexible batteries. We present a robust Bakelite-type organic polymer covalently decked with viologen, triazine, and phenolic moieties. Its flexible structure with cationic viologen centers incorporates counter-balancing free hydroxide ions into the polymeric framework. By design, the aromatic groups and heteroatoms in the framework can be activated under an applied potential to prompt a push–pull drive, setting off the towing of hydroxide ions via weak electrostatic, van der Waals, and hydrogen-bond interactions. The frontier orbitals from a DFT-modeled structure certify this. The hydroxyl-polymer requires minimal KOH wetting to maintain a humid environment for Grotthuss-type transport. The hydroxide ion conductivity reaches a value of 1.4 × 10−2 S cm−1 at 80 °C and 95% RH, which is retained for over 15 h. We enhanced its practical utility by coating it as a thin solid-state separator-cum-electrolyte on readily available filter paper. The composite exhibits a conductivity of 4.5 × 10−3 S cm−1 at 80 °C and 95% RH. A zinc–air battery (ZAB) constructed using this polymer-coated paper as electrolyte yields a maximum power density of 115 mW cm−2 and high specific capacitance of 435 mA h g−1. The power density recorded for our ZAB is among the best reported for polymer electrolyte–based batteries. Subsequently, the flexible battery fabricated with IISERP-POF11_OH@FilterPaper exhibits an OCV of 1.44 V, and three batteries in series power a demo traffic signal. To underscore the efficiency of hydroxide ion transport through the complex multifunctional backbone of the polymer, we calculated the diffusion coefficient for OH− (Exp: 2.9 × 10−5 cm2 s−1; Comp. 5.2 × 10−6 cm2 s−1) using electrochemical methods and MD simulations. Climbing-edge NEB calculations reveal a large energy barrier of 2.11 eV for Zn2+ to penetrate the polymer and identify hydroxide ions within the polymer, suggesting no undesirable Zn2+ crossover. Our findings assert the readily accessible C–C-linked cationic polymer's capacity as a solid-state electrolyte for ZABs and any anion-conducting membrane.
New conceptsIn metal–air batteries, hydroxide ions produced at the cathode via the reduction of oxygen from the air need to be efficiently transported to combine with metal ions generated by anodic oxidation. A liquid electrolyte such as KOH(aq) typically transports ions. This transport of ions suffers from evaporation of the solvent over time; KOH is gellified to minimize this. However, in the gel, the electrostatic interaction between K+ and OH− restricts facile movement of the latter ion. A passivating layer of K2CO3 also forms on the anode surface, creating internal resistance. To overcome this, we developed a cationic polymer with counter-balancing OH− ions in their nanopores. The advantages are many. For example, the polymer is a steady source of OH− ions since they are in a stoichiometric ratio with the cationic sites. There is no major solvent dependence, so there are fewer problems with electrolyte evaporation. Minimal use of KOH avoids K2CO3 formation. Our insoluble polymer is a safe and humidity-withstanding solid-state electrolyte. This cationic polymer transports OH− ions effortlessly and prevents undesirable crossover of Zn2+ ions, yielding high power density. Furthermore, this bulk cationic framework does not compete with hydroxide-ion diffusion during operation, which lowers internal resistance. |
Introduction
Hydroxide ion-conducting materials are receiving substantial attention owing to their significance in green technologies such as anion exchange membrane fuel cells (AEMFCs), metal–air batteries and water electrolyzers.1–3 Typically, hydroxide ions exhibit low conductivity compared to protons owing to their sluggish diffusion and higher mass.4 Proton-conducting materials are also well established and commercialized for various energy applications.5–7 In comparison, hydroxide ion-conducting materials are still in their infancy. Hence, the rational design of hydroxide ion-conducting materials is beneficial and would pave the way for application-specific charge-storage devices.The rapid increase in energy demand has drawn ample attention to charge storage devices.8,9 To manage the ever-increasing energy demand, it is necessary to develop efficient and cost-effective charge storage devices.10,11 In this regard, zinc–air batteries (ZABs) are receiving wide attention owing to their high energy density, low cost and safety. Furthermore, they have been used for many applications such as hearing-aid devices, cameras, and grid-scale energy storage.12–15 The development of high-performing ZABs requires the advancement of electrodes and separators.16,17 Besides the electrode kinetics, hydroxide ion transport through the electrolyte largely determines the efficiency of ZABs.18 Liquid-based electrolytes create complications in fabricating or handling the devices, and achieving a long cycle life.19,20 In comparison, the solid-state electrolytes are less problematic in this regard. Furthermore, their leakage-proof framework offers superior safety features.21 Therefore, developing ZABs with solid-state separators that concomitantly possess improved performance and flexibility would benefit applications.22 However, the key bottleneck associated with the hydroxide ion-conducting materials is their low ionic conductivity and poor stability under harsh chemical environments.23 Several ion-conducting (H+ or OH−) materials with better conductivity have been reported in the literature in the pelletized state, but their practical potential as a thin and free-standing solid-state separator has not been explored.4,24–27 Hence, we developed a thin hydroxide ion-conducting solid-state separator-cum-electrolyte for ZAB from the cationic polymer by coating it on filter paper (FP), adopting a simple dip-coating strategy.
Various kinds of separators have been employed for ZABs to transport OH− ions, and some porous separators provide satisfactory battery performance.14,28 A fundamental issue associated with the porous separators is the passage of zincate ions (Zn(OH)42−) through the pores.29 For example, the Celgard separators have good chemical stability and low cost. However, the significant transport of zincate ions from the anode to the cathode through the porous separator does occur, resulting in performance loss during the ZAB operation. One can overcome this by integrating the porous separators with the hydroxide ion-conducting insoluble materials. This would block the pores, allowing the passage of OH− ions, and restrict the movement of zincate ions.30 Polymers have been intensely probed for their stability advantage.31 Recently, amide polymers constructed from simple monomers (acyl chloride + melamine) were shown to possess strengths comparable to steel.32 This inspired us to explore the potential of simple systems such as Bakelite and viologen for this task-specific application. Towards this approach, we have synthesized triazine-resorcinol and viologen-based cationic polymeric organic frameworks (POFs) with enriched hydroxyl (–OH) groups.33 These POFs can be obtained in a large scale via a one-pot synthesis using Bakelite chemistry: forming robust C–C bonds without any catalyst.34 Such types of POFs are chemically robust, thermally stable and have found use in a wide range of applications, including energy storage, gas storage, catalysis, and anion exchange.35–38 The viologen-based quaternary center is counterbalanced by the larger anion like Br−, which finds easy exchange with OH− ions. Recently, our group reported a family of cationic POFs, among which IISERP-POF11 (1) demonstrated excellent performance for the anion exchange.38 Therefore, in the present work, the hydroxide ion-exchanged IISERP-POF11, IISERP-POF11_OH (1_OH), was integrated with cellulose paper (IISERP-POF11_OH@Filter Paper, 1_OH@FP) and employed as the solid-state separator-cum-electrolyte for zinc–air battery applications. The integrated system displayed an improved performance for the ZAB, and validated the suitability of employing 1_OH for energy applications.
Results and discussion
Synthesis and bulk characterizations of as-made IISERP-POF11 (1) and hydroxide exchanged IISERP-POF11_OH (1_OH)
The synthesis of 1 from the monomers and its further conversion into 1_OH after the exchange of the Br− with OH− ions is illustrated in Scheme 1 (the detailed synthesis procedure is provided in the ESI†). The formation of the as-synthesized polymer was confirmed by various characterization techniques, such as NMR, FTIR, FESEM, HRTEM, TGA, and N2 adsorption isotherm measurements. 13C cross-polarization (CP) magic angle spinning (MAS) solid-state nuclear magnetic resonance (NMR) studies indicated the presence of Bakelite type coupling between triazine-resorcinol and viologen-based bipyridinium aldehyde (Fig. 1a). Furthermore, some unsubstituted sites on the phloroglucinol rings led to the formation of a characteristic aliphatic C–C bond, which was observed around 26 ppm (Fig. 1a).39–41 It was followed by the other characteristic peaks from aromatic groups like triazine around 172 ppm (i) and aromatic peak around 151 ppm (h). The aliphatic peaks for the building blocks around 65 ppm (b) were intact. This demonstrates that the polymerization is taking place. However, to confirm the stability of the polymer powder, we recorded the IR and NMR spectra of the material subjected to boiling in a THF + DMF mixture, followed by a 24 h soak in 6 M KOH solution. All of the characteristic peaks remained intact, and were positioned at similar chemical shifts with matching intensity. This exceptional chemical stability and the functional group integrity of the polymer, especially towards harsh treatments, is highly desirable for electrolyte applications (Fig. S1, ESI†). Subsequent to the anion exchange (Br− with OH− ions), a significant color change from brown to yellow was observed, and is represented with the photographic images in Scheme 1.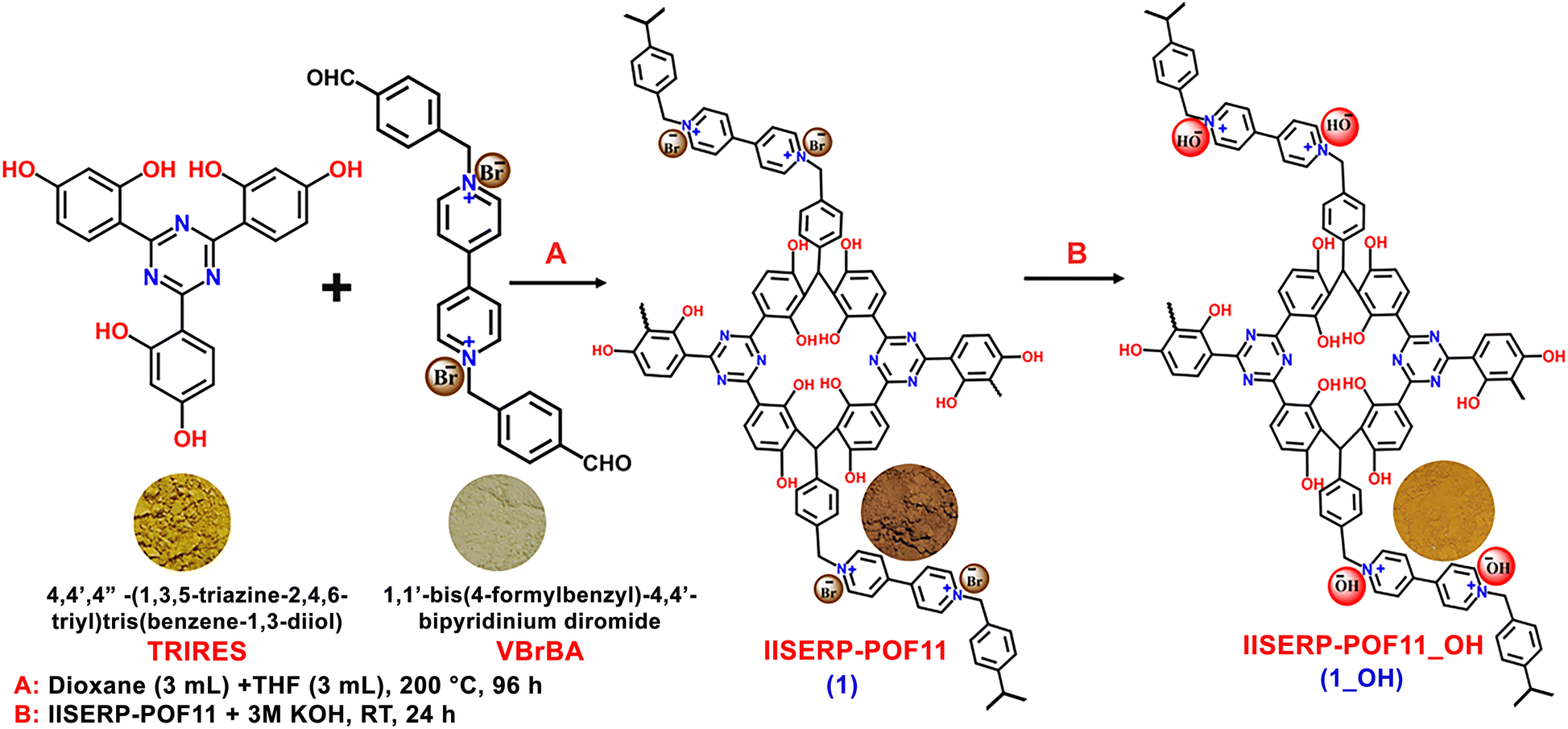 | ||
| Scheme 1 Synthetic procedure of 1_OH. Insets present photographs of the corresponding compounds. | ||
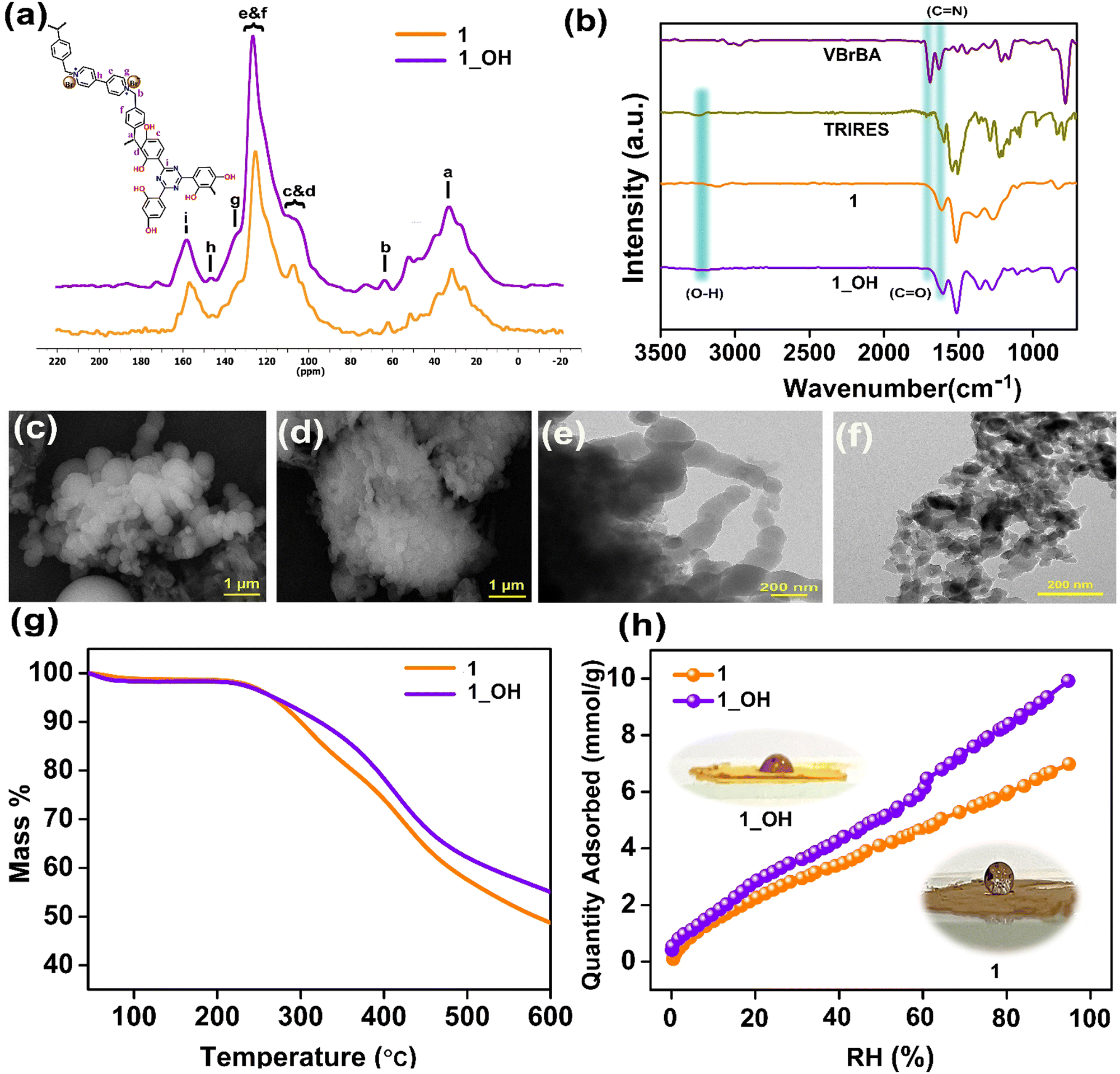 | ||
| Fig. 1 (a) 13C-CPMAS spectra of 1 and 1_OH; (b) IR spectra of TRIRES, VBrBA, 1, and 1_OH; (c and d) FESEM images of 1 and 1_OH; (e and f) HRTEM images of 1 and 1_OH; (g) thermogravimetric analysis of 1 and 1_OH under N2 atmosphere at a ramp rate of 5 °C min−1; and (h) water adsorption isotherms of 1 and 1_OH with insets presenting photographs from the contact angle measurements. | ||
Fig. 1b shows the comparative IR spectra of the monomers, i.e., 1,1′-bis(4-formylbenzyl)-4,4′-bipyridiniumdibromide (VBrBA), 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tris(benzene-1,3-diol) (TRIRES), the as-synthesized cationic polymer (1), and its hydroxide exchanged form, 1_OH. The disappearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1690 cm−1) feature from the monomer VBrBA with the polymerization confirms the C–C bond formation between the aldehyde and the phenolic moieties (Scheme 1). Furthermore, the presence of CN (1620 cm−1) assures that the building blocks are intact in 1 and 1_OH (Fig. 1b). The field emission scanning electron microscopy (FESEM) images of 1 indicate that the particles are either connected or intergrown microspheres of different diameters ranging from 500 nm to 2 μm (Fig. 1c and Fig. S2, ESI†). Fig. 1d and Fig. S3 (ESI†) confirm that there is no deformation in the shape of these microspheres after treating with 3 M KOH solution, i.e., on the formation of 1_OH. This indicates their textural stability under the harsh corrosive environment. Thus, 1_OH could be suited for operating under concentrated alkaline conditions. In the energy dispersive X-ray (EDX) analysis, the drop in the atomic percentage of Br− ions from 1.74% to 0.21% confirms the effective exchange of Br− ions with OH− to form 1_OH (Fig. S4 and S5, ESI†). The K+ ion traces in 1_OH is negligible, which confirms that the hydroxide ions are counterbalanced by the cationic centers of the polymer. The high-resolution transmission electron microscopy (HRTEM) images of 1 and 1_OH are displayed in Fig. 1e and f, respectively. The HRTEM showed that the spherical balls in 1_OH are continuously connected, which could aid the ionic transport across grains and spheres. After OH− ion exchange, we noticed a slight decrease in the surface diameter of 1_OH spheres. This could be due to more compacting from OH− compared to Br−. However, the spherical morphology is still intact (Fig. S6 and S7, ESI†).
O (1690 cm−1) feature from the monomer VBrBA with the polymerization confirms the C–C bond formation between the aldehyde and the phenolic moieties (Scheme 1). Furthermore, the presence of CN (1620 cm−1) assures that the building blocks are intact in 1 and 1_OH (Fig. 1b). The field emission scanning electron microscopy (FESEM) images of 1 indicate that the particles are either connected or intergrown microspheres of different diameters ranging from 500 nm to 2 μm (Fig. 1c and Fig. S2, ESI†). Fig. 1d and Fig. S3 (ESI†) confirm that there is no deformation in the shape of these microspheres after treating with 3 M KOH solution, i.e., on the formation of 1_OH. This indicates their textural stability under the harsh corrosive environment. Thus, 1_OH could be suited for operating under concentrated alkaline conditions. In the energy dispersive X-ray (EDX) analysis, the drop in the atomic percentage of Br− ions from 1.74% to 0.21% confirms the effective exchange of Br− ions with OH− to form 1_OH (Fig. S4 and S5, ESI†). The K+ ion traces in 1_OH is negligible, which confirms that the hydroxide ions are counterbalanced by the cationic centers of the polymer. The high-resolution transmission electron microscopy (HRTEM) images of 1 and 1_OH are displayed in Fig. 1e and f, respectively. The HRTEM showed that the spherical balls in 1_OH are continuously connected, which could aid the ionic transport across grains and spheres. After OH− ion exchange, we noticed a slight decrease in the surface diameter of 1_OH spheres. This could be due to more compacting from OH− compared to Br−. However, the spherical morphology is still intact (Fig. S6 and S7, ESI†).
Thermogravimetric analysis in Fig. 1g revealed that the framework 1 is stable at temperatures up to 280 °C. We attribute this to the strong C–C bond formed between the monomers, resembling Bakelite. The cationic polymer treated with 3 M (1_OH) and 6 M KOH (1_6OH) display slightly different TGA profiles (Fig. 1g and Fig. S8a, ESI†), but their thermal stabilities are comparable. The contact angle measurements pointed at the surface of the as-made polymer powder, 1 being super-hydrophobic. However, the 1_OH prepared by anion exchange in an aqueous alkaline solution yielded a final powder that was hydrophilic (Fig. 1h and Fig. S8b, S8c, ESI†). This surface wettability is crucial for the material's interaction with electrolytes. We investigated the intrinsic porosity with water sorption isotherms. The water adsorption isotherms of 1 and 1_OH showed a very similar profile, indicating that there were no major changes to the pore environment or structure. The overall water capacity of 1_OH is higher than that of 1, suggesting improved intrinsic wettability. Thus, the near super-hydrophobic (148° ± 3°) material with Br− containing framework becomes much more hydrophilic when exchanged with OH−. Notably, there was no noticeable swelling of the polymer or significant change in density.
The density functional theory-based model fitted to the 77 K N2 isotherm shows the majority of the pores being 15 to 18 Å in size, which are still intact after the ion exchange (Fig. S9, ESI†). However, the N2 uptakes are nominal, which is expected as the pores are permanently occupied by the counter-balancing anions. Still, there is sufficient openness to support the transport of OH− ions. Furthermore, unlike the crystalline frameworks, these polymers exhibit structural flexibility with changing humidity, opening up spaces for dynamic interactions between the framework atoms and the free hydroxide ions. This impacts the electrolyte's wetting and hydroxide ion transport characteristics (Fig. 1h).
Electrochemical studies
Electrochemical impedance analysis was performed in the frequency range of 106 to 0.1 Hz with an AC amplitude of 10 mV using a Solartron impedance analyzer. The impedance was recorded at variable temperatures from 30 °C to 80 °C with a constant relative humidity of 95%, and the data are presented in Fig. 2a. To evaluate the intrinsic conductivity of 1_OH, the measurements were performed on a densely pressed pellet of the same. The equivalent circuit was employed for fitting all of the Nyquist curves, using the Zview-4 software. At a constant relative humidity of 95%, the OH− ion conductivity value increased from 2.7 × 10−4 S cm−1 at 30 °C to 1.4 × 10−2 S cm−1 at 80 °C (Fig. 2a and Table S1, ESI†). In order to check the humidity-dependent conductivity of 1_OH, we measured the ac-impedance at 80 °C by varying the humidity (60% RH, 80% RH and 95% RH) (Fig. S10a, ESI†). The conductivity values are 3.6 × 10−5, 3.8 × 10−4 and 1.4 × 10−2 S cm−1 at RH values of 60%, 80% and 95%, respectively. The increase in hydroxide ion conductivity at 80 °C with increasing temperature suggests lowering of the activation energy, while the increase in conductivity with increasing humidity suggests that water could be playing a crucial role via hydrogen bonding.42 Fitting an Arrhenius equation to this temperature-dependent impedance data yields an activation energy of 0.52 eV (Fig. 2b), suggesting a mixed Grotthuss–Vehicular diffusion mechanism.43 The observed activation energy is comparable to some of those reported for hydroxide ions in other nanoporous structures.44 An equivalent circuit fit to the most promising Nyquist plots (80 °C, 95% RH) reveals an Ohmic resistance of 3.2 Ω (neat pellet) (Fig. 2c). In the fitted equivalent circuit, we confirmed the need for a CPE component, by measuring the CV of 1_OH, which clearly indicated the presence of capacitive behavior (Fig. S11, ESI†). In our case, considering that the hydroxide ion is moving in channels lined with hydroxyl groups and triazine, both of which might provide some hydrogen bond assistance resulting in the Grotthuss mechanism. Simultaneously, under the applied ac-potential, there could also be sufficient force for vehicular motion. Due to the absence of OH− ions in the pristine polymer (1), it shows a poor ionic conductivity value of 7.39 × 10−5 S cm−1 at 80 °C and 95% RH. This is expected as the Br− ions are heavier and will be even less effective for a hydrogen-bond-assisted Grotthuss-type movement. Furthermore, the comparison of the equivalent circuit fitted Nyquist plots of 1vs.1_OH (Fig. 2d) suggests that the series resistance and charge-transfer resistance are both substantially higher for 1 (R1 = 352 Ω, R2 = 509 Ω) compared to the 1_OH (R1 = 3.2 Ω, R2 = 11.8 Ω). The large semicircle in the low-frequency regime in the case of 1 suggests limited or restricted diffusion even in the same time-scale of measurements as employed for the 1_OH. Meanwhile, 1_OH has a typical semi-infinite linear diffusion; that is, diffusion in one dimension, which is only bound by a large planar electrode on one side (45° angle with Z'-axis), a typical Warburg behavior. A CPE-P value close to 0.5 for the 1_OH pellet is suggestive of pseudocapacitive behavior, which is expected for the polymer with porous structure (Fig. 2c). Remark: A transmission line model mimicking the Fickian diffusion, wherein the movement of ions through the pores is coupled to the movement of electrons through the pore walls, was not chosen. This is considering that the polymer does not have a crystalline structure with highly ordered pores, and also the electronic conductivity in larger length scales is limited.45 Linear sweep voltammetry (LSV) was conducted to check the electronic conductivity of 1_OH in the potential window of −0.6 to 0.6 V with a scan rate of 10 mV s−1 (Fig. S12, ESI†). 1_OH shows negligible electronic conductivity, which suggests that the observed conductivity is predominantly ionic, arising from the OH− ions. To assess the ability of 1_OH in delivering a stable hydroxide-ion conductivity, we measured it at 80 °C and 95% RH for 15 h. There is a build-up of a small charge transfer resistance over 15 h. Furthermore, 1_OH retains >95% of its conductivity (Fig. 2e and Fig. S10b, ESI†). This hydroxide ion conductivity is reproducible across different batches of the sample (Fig. 2e inset). To check the cyclic stability of 1_OH for OH− transport, the pellet was dried overnight at 100 °C in a vacuum oven. Impedance analysis was then carried out for both the forward cycle (increasing the temperature from 30 to 80 °C at 95% RH) and the backward cycle (decreasing the temperature from 80 to 30 °C at 95% RH) (Fig. 2f). The conductivities remained the same during the cycling, reflecting the electrochemical stability of the polymer. Furthermore, the shape/profile of the Nyquist plots remained the same, suggesting that the pathway and the mechanism of conduction remained unchanged. We calculated the diffusion coefficient for the hydroxide ions inside the cationic polymer framework using the Z' vs. ω−1/2 plot. The low-frequency spectrum of the impedance plot yielded the characteristic linear fits (Fig. S13, ESI†), from which the diffusion coefficients were estimated to be 2.9 × 10−5 cm2 s−1 at 80 °C with 95% RH. The diffusion coefficient increased systematically with an increase in temperature (Fig. 2f). The calculated diffusion coefficients matched well with the theoretical values reported in the literature.46,47 However, we could not find any experimentally calculated diffusion coefficients for hydroxide ions inside a porous material. However, for Cl−, the diffusion coefficient is on the order of 10−8 cm2 s−1.48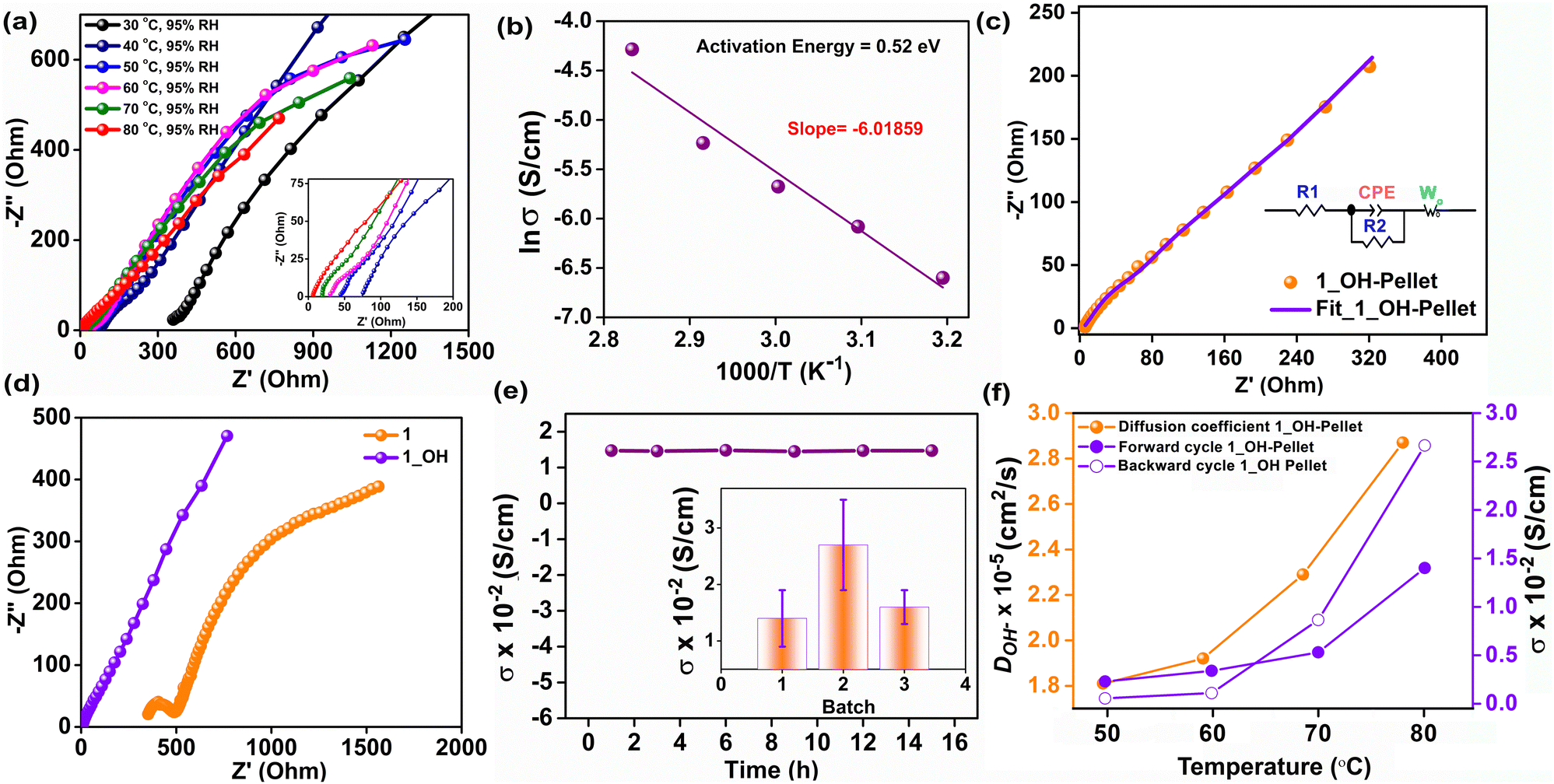 | ||
| Fig. 2 (a) Nyquist plots from the temperature-dependent conductivity measurements of 1_OH; inset presents its zoomed-in view. (b) Arrhenius plot of 1_OH. (c) Equivalent circuit fit to the Nyquist plot of 1_OH at 80 °C and 95% RH. (d) Comparison of the Nyquist plots of 1 containing Br− as the counter ion vs.1_OH. (e) Conductivity of 1_OH at 80 °C and 95% RH recorded for 15 h; inset shows that the conductivity is reproducible across different batches of the sample. (f) Hydroxide ion diffusion coefficient and forward–backward cycling plot for the OH− conductivity of 1_OH at different temperatures and constant relative humidity of 95%. | ||
Conductivity characteristics of 1_OH coated on Filter Paper (1_OH@FP)
Encouraged by the appreciable hydroxide ion conductivity and stability exhibited by this viologen polymer, we set out to investigate its scope as a solid-state electrolyte in a zinc–air battery. It is hugely advantageous to coat the polymer on a porous filter paper, which provides a bulk wettable microporous-macroporous separator-cum-electrolyte membrane. However, the composite needs to retain the high levels of OH− ion conductivity. To investigate this, 1_OH was devised into a separator-cum-electrolyte by coating it onto a research-grade Whatman filter paper (1_OH@FP). The commonly available filter paper was chosen owing to its advantageous features, such as low cost, lightweight, flexibility and easy accessibility.49 The dip-coating method was used to deposit the polymer by soaking cellulose paper in a dispersed solution of 1_OH in water. The process is scalable and 25 cm21_OH@FP was prepared in a few hours. The fluorescence microscopy images illustrated in Fig. 3a show the loss of fluorescence of the FP upon loading with the polymer. Its uniform loading on paper is evident. Typically, the filter paper has entangled cellulose fibers (Fig. 3b). These fibers are rich in terminally placed binding groups, which are likely to be compatible with our hydroxyl-functionalized polymer backbone. The solid-state UV-Vis absorption spectra of neat FPvs.1_OH@FP showed lowering of the absorbance intensity (@280 nm) of the FP upon coating with the polymer (Fig. S14, ESI†). This is in agreement with the results from the fluorescence microscopy. These suggest that the polymer directly contacts the electrolyte and the electrodes, while the paper is acting as a separator. From the FESEM images, it is evident that the polymer composite is deeply buried within the cellulose matrix of the paper (Fig. 3b, c and Fig. S15, S16, ESI†). This could be crucial to providing an intimate contact between the two entities in the composite. The cross-sectional SEM analysis of 1_OH@FP indicated that the distribution of 1_OH is uniform in FP. The fibers of the FP are intimately blended to a good thickness (not merely coating the surface) with small granules of 1_OH. (Fig. S16, ESI†). Even a strong thumb-pressure does not make the polymer particles lose its adhesion with the paper. The hydroxide ion conductivity of 1_OH@FP was analyzed at 80 °C and 95% RH, and the Nyquist plot is presented as Fig. 3d and Fig. S17 (ESI†). 1_OH@FP shows a OH− conductivity value of 4.5 × 10−3 S cm−1 at 80 °C and 95% RH (R1 = 10.4 and R2 = 18.2 Ω), which is quite comparable to the values obtained with the pellets at similar operating conditions. The same equivalent circuit fits extremely well. From the fitted data, we observe that the resistances of the 1_OH@FP is relatively higher. This is expected as more interfaces are generated and the paper is much less conductive. Notably, the CPE value of 1_OH (0.45) increases to 0.72 for 1_OH@FP, indicating the increase in double layer capacitive character. Interestingly, the ion diffusion is noticeably improved when the material is coated on the FP (Fig. S18 ESI†). Comparing the Warburg tail of the 1_OH pellet vs. the 1-OH@FP brings some noteworthy features. At lower frequencies, the diffusion tail of 1_OH@FP starts to deviate more from making an angle of 45° with the x-axis. This indicates deeper penetration of the ions into the material even at these low frequencies. This is expected, as the active material is coated on a paper to form a much thinner electrolyte membrane compared to the pellet. This points at the advantage of making this paper-derived electrolyte-cum-separator unit. Importantly, the overall conductivity still remains very much in an acceptable regime for a hydroxide-ion conducting electrolyte. Thus, the 1_OH@FP provides an appropriate electrolyte material with a micro-macroporous structure that still inhibits the direct zincate ion crossover through the separator. These properties are crucial for the battery performance.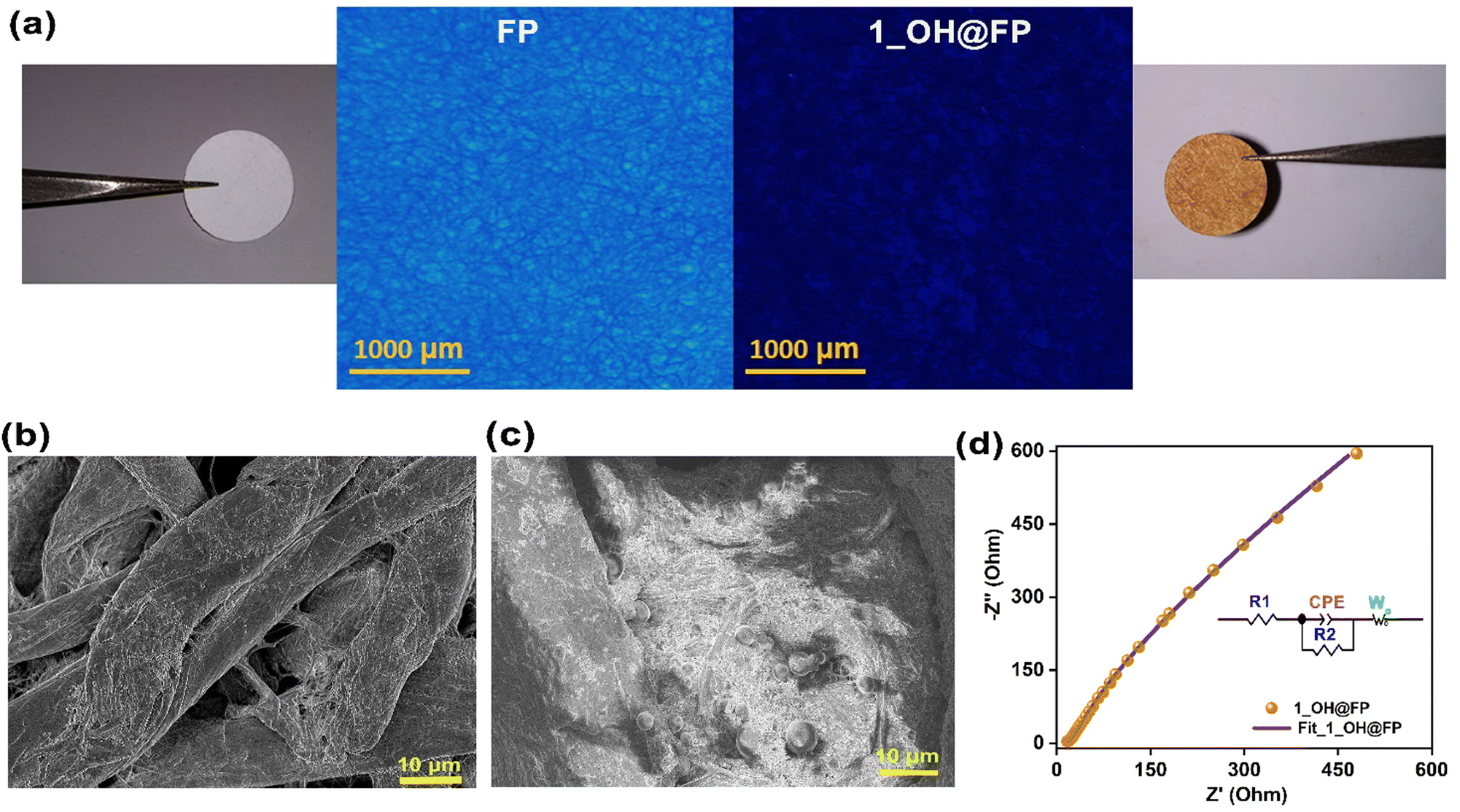 | ||
| Fig. 3 (a) Macro zoom fluorescence microscope images of neat FP and 1_OH@FP with the corresponding photographs on the side. (b, c) FESEM image of neat FP and 1_OH@FP. (d) Equivalent circuit fit to the Nyquist plot of 1_OH@FP at 80 °C and 95% RH. | ||
1_OH@FP as solid-state electrolyte in ZAB
The excellent ionic conductivity of 1_OH@FP motivated us to use it for zinc–air battery applications. For this, 1_OH@FP was employed as the solid-state separator-cum-electrolyte by sandwiching between the electrodes. Fig. 4a depicts the schematic of the zinc–air battery assembly with its components (Fig. S19, ESI†). During discharge, oxygen electrocatalytically reduces to form hydroxide ions at the cathode.50 At the anode, zinc would oxidize to Zn2+. The zinc ions would react with the hydroxide ions coming from the cathode through the separator to form zincate ions and further to ZnO. To assess the performance of 1_OH@FP in ZAB, LSV was performed at a scan rate of 10 mV s−1, and is shown in Fig. 4b. The polarization analysis presented in Fig. 4b shows a maximum power density of 115 mW cm−2 for the 1_OH@FP based battery compared to 38 mW cm−2 obtained for the neat filter paper-based counterpart. The resistances of the 1_OH@FPvs. neat FP were determined from impedance analysis performed on their respective ZAB assemblies. The Ohmic drop in 1_OH@FP is notably lower compared with neat FP (1.55 vs. 2.1 Ω) (Fig. S20 and S21, ESI†). Furthermore, the resistance increases almost by 0.85 Ω during the use of the neat FP as the electrolyte support in the battery, which was run for about 2 h. In contrast, the 1_OH@FP, maintains almost the same resistance even after 8 h under a comparable battery set-up. It is worth mentioning that even a difference of 0.55 Ω (between FPvs.1_OH@FP) hugely impacts the power density (Fig. 4b), as well as the run time of the ZAB derived out of these electrolyte supports. Accordingly, the 1_OH@FP containing battery exhibits a higher specific capacitance of 435 mA h g−1 compared to 397 mA h g−1 obtained for the neat FP based battery. Furthermore, the Fig. 4c inset shows that the 1_OH@FP containing battery delivers four times more run time compared with FP based ZAB. Flexible solid-state ZAB was fabricated to show the practical applicability of the as-prepared 1_OH@FP (details provided in the experimental section). The fabricated solid-state battery offers an OCV value of 1.44 V in an air atmosphere (without purging any additional O2 gas). Three flexible devices connected in series yielded an output of 4.22 V (Fig. S22, ESI†). The output voltage was stable to bending and twisting (Fig. 4d) owing to the liquid-free nature of 1_OH@FP. By combining the batteries in series, we could power three light-emitting diodes (LEDs) connected in parallel, resembling the traffic signal lights (Fig. 4e). Furthermore, 10 months after fabrication, we were able to regenerate the OCV of 1.10 V on wetting the device with just 50 μL of water (Fig. 4f). Overall, the results indicate the feasibility of employing 1_OH@FP as the hydroxide ion-conducting material for flexible batteries.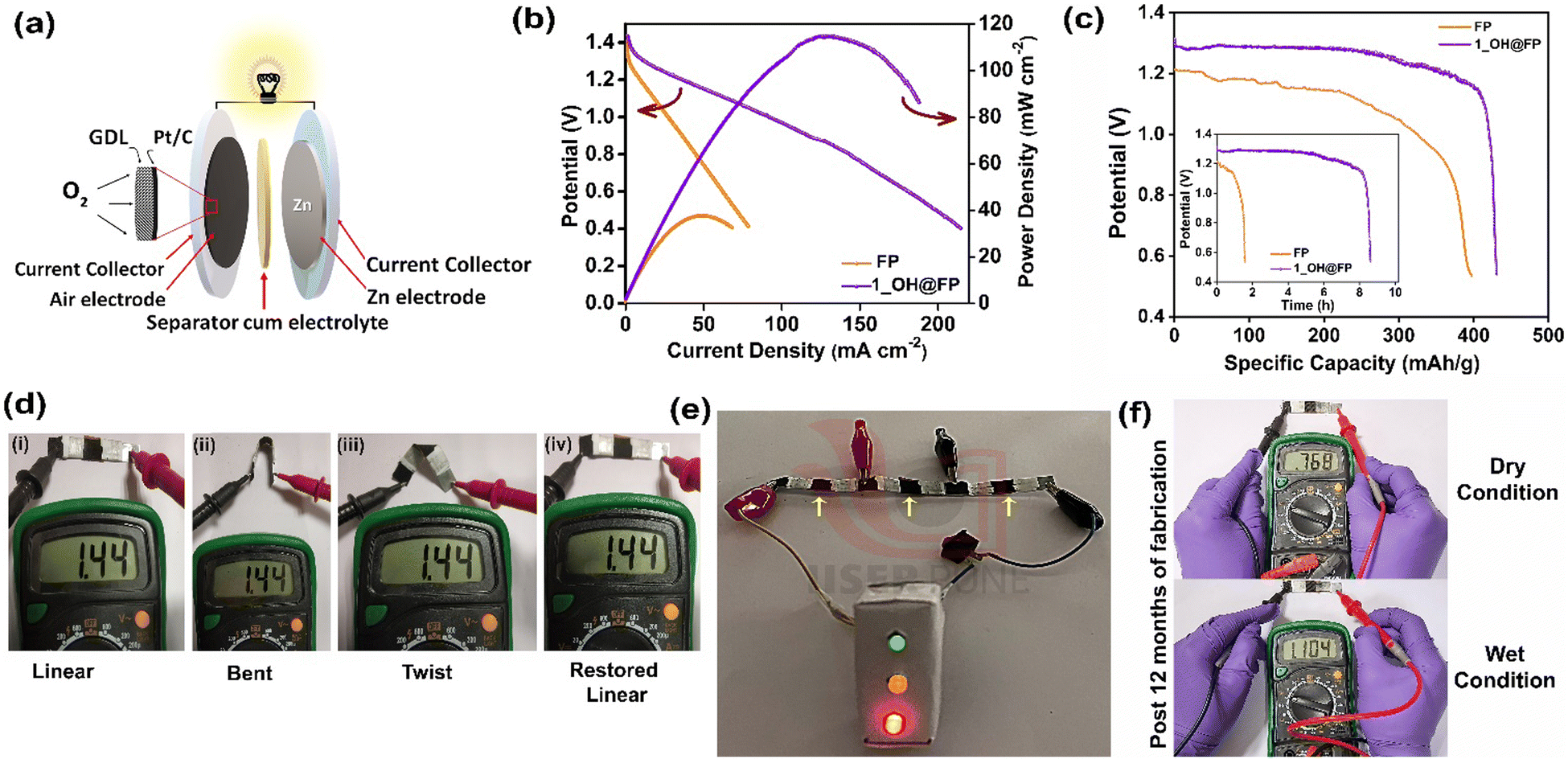 | ||
| Fig. 4 (a) Schematic of the ZAB with its components. (b) Polarization plot of neat FP and 1_OH@FP-based ZAB. (c) Comparative specific capacity of neat FP and 1_OH@FP based batteries; inset shows the durability of the ZAB over time. (d) Photographs showing constant OCV at different flexible modes of the solid-state ZAB. (e) Image of the flexible solid-state ZAB powering a demo traffic signal with yellow arrows indicating the air-breathing cathode. (f) OCV regeneration of solid-state ZAB after 10 months of fabrication on wetting with water. | ||
Post ZAB usage stability analysis of 1_OH@FP
The stability of 1_OH@FP when employed as separator-cum-electrolyte in the ZAB was investigated. PXRD and IR analysis indicated that the polymer remained intact on the FP even after the complete discharge cycle (8 h) (Fig. S23 and S24, ESI†). Then, this composite electrolyte in the ZAB, 1_OH@FP, was subjected to high-frequency impedance analysis after a complete discharge cycle and compared with the neat FP's performance (Fig. S20 and S21, ESI†). The resistance remains the same for the 1_OH@FP, but increases for the neat FP. This is due to the relatively higher loss of KOH (aqueous solution) from the neat FP, creating a passivation layer that is unfavorable for the conduction. This problem is not observed in the case of 1_OH@FP. This is most likely due to the intrinsic tendency of the polymer to hold onto the OH− species driven by its charge-balance requirements, and their pores trap water more efficiently (Fig. 1h). Furthermore, in the case of 1_OH@FP, the conduction pathway and diffusion mechanism remain the same, as seen from the equivalent circuit fits. The post-ZAB microscopy analysis of the 1_OH@FP further confirms the homogeneous adherence of the active material on the FP (Fig. S25 and S26, ESI†).Push–pull electronics in the triazine-resorcinol-viologen framework portrayed by frontier orbitals
To investigate the potential role of the cationic polymers in interacting with the hydroxide ion movement in this viologen functionalized spongy polymer, we looked at the potential/electron distribution within the polymer backbone. As a coherent step, we replaced the heteroatoms within the covalently linked modules and monitored the impact it brings to the HOMO–LUMO distribution. The local electronics is likely to be dictating the hydroxide-framework (heteroatom) interactions. For this, we developed a two-repeat unit polymer using the Random Copolymer module in the Materials Studio. This initial model was then geometry-optimized using the DMOL3 package.Geometry optimizations were performed for each of the individual configurations that were generated with and without heteroatoms. Important demarcation arises from the position of the HOMO and the LUMO. The 1_OH compounds with conjugated triazine-resorcinol core covalently linked to the viologen units have the HOMO residing on the resorcinol ring and the triazine nitrogen, and to some extent on the hydroxyl groups that are located proximal to the viologen (Fig. 5a). Meanwhile, the LUMO resides mostly on the viologen rings. For the as-made polymer, the HOMO–LUMO gap turned out to be 0.03 eV. However, when the triazine rings are replaced with phenyl moieties, the all-carbon backbone does not demarcate much between the HOMO–LUMO levels, leading to a gap of 0.002 eV.
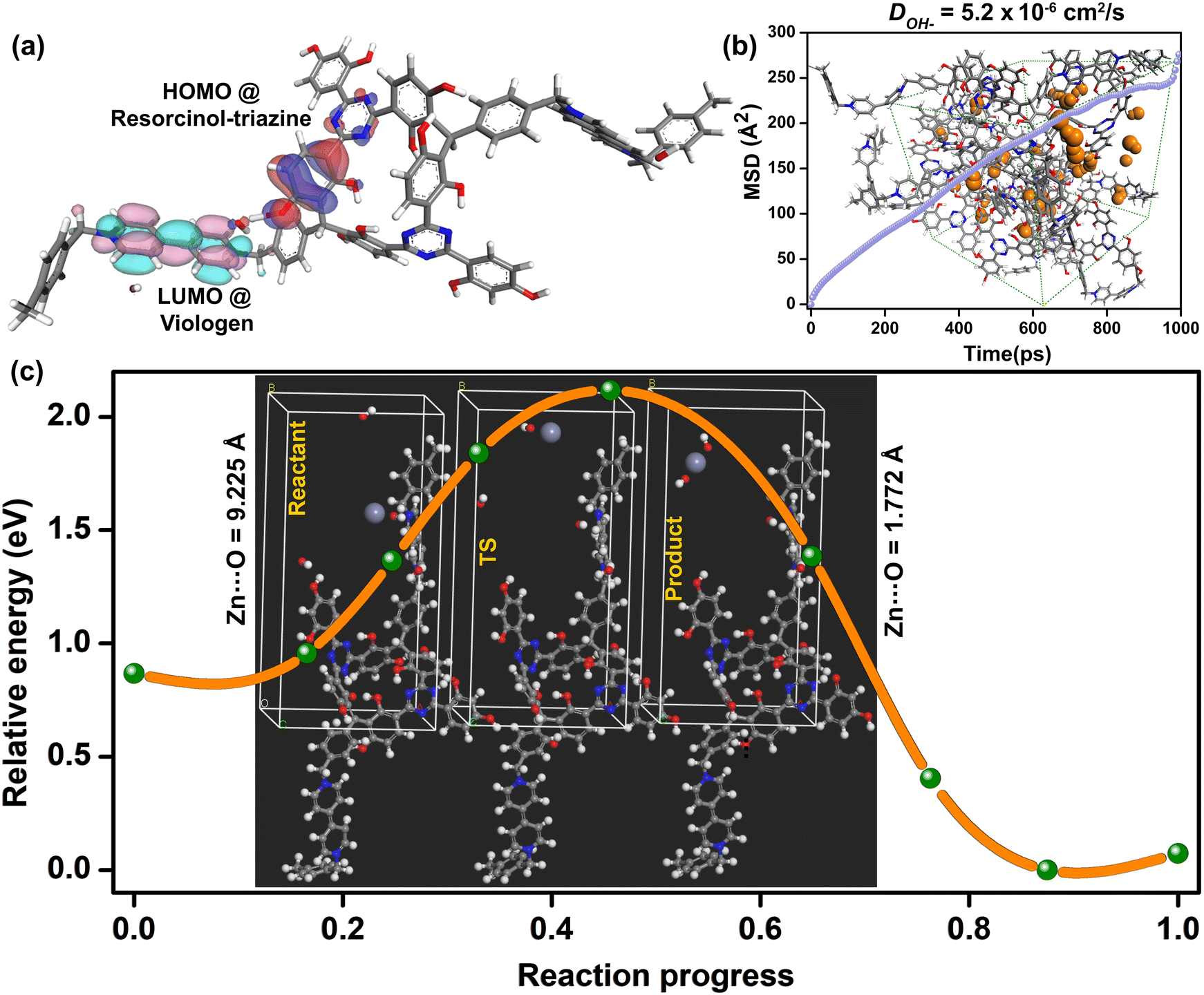 | ||
| Fig. 5 Computational modeling: (a) Frontier orbital locations within the polymer backbone. The resorcinol-triazine can set up a push–pull with the e− deficient viologen units. (b) Mean square displacement (MSD) vs. time plot calculated using MD simulations and the derived hydroxide ion self-diffusion coefficient. Inset shows a snapshot of an optimized structure with hydroxide ions shown as space fills. (c) Representative minimum energy path (MEP) for Zn2+ ion movement within the hydroxyl-containing polymer assessed from NEB. | ||
This removes the push–pull characteristics within the framework backbone. This points at how crucial the triazine nitrogens are in setting up the push–pull characteristics between the triazine-resorcinol unit and the viologen rings. Remarkably, this is only for the small section of the polymer. As the polymer grows into an amorphous extended cross-linked structure, there is bound to be a breakdown of the long-range conjugation assisted electronic transport. The overall bandgap will be much larger, and typically in the semiconducting regime.31 However, the redox active localized regions are sufficient to set-up a push–pull effect, which would govern the movement of the hydroxide ions.
Diffusion coefficient of hydroxide ions in the viologen polymer
We resorted to MD simulations for garnering insights into the transport of hydroxide ions within the polymer framework. All structural dynamics were carried out using the Forcite module in the Materials Studio. An amorphous cell was constructed for a structure encompassing cross-linked chains. These were filled with hydroxide ions in an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32 ratio per unit cell. A rigorous geometry optimization was performed until excellent convergence was achieved. The optimized structure was further equilibrated using an NPT ensemble, and the average lattice parameters were employed to the output structure. The migration of the hydroxide ions in the cationic framework was explored under the NVT conditions. The final trajectory was obtained from a 1 ns microcanonical NVE dynamics with a large 50 Å cut-off distance performed at 300 K. The Mean Square Displacement (MSD) vs. time plots were used to calculate the average diffusion coefficient for the hydroxide ions, which turned out to be 5.2 × 10−6 cm2 s−1 (Fig. 5b). However, the computed diffusion coefficient is lower than the experimental one (2.9 × 10−5 cm2 s−1). This is because the hydration was not considered in the MD simulations.51 Importantly, the observed and calculated diffusion coefficients for the hydroxide ions were comparable with those available from the recent computational works.52–55 From the fits to the MSD plots, we estimated the activation energy for the diffusion of the hydroxide ions in the cationic polymer framework to be 0.22 eV (21 kJ mol−1, Fig. S27, ESI†), which is comparable to the experimental activation energy (0.153 eV) (14.77 kJ mol−1) calculated using the Warburg Bode plot (Fig. S27, ESI†).56
:32 ratio per unit cell. A rigorous geometry optimization was performed until excellent convergence was achieved. The optimized structure was further equilibrated using an NPT ensemble, and the average lattice parameters were employed to the output structure. The migration of the hydroxide ions in the cationic framework was explored under the NVT conditions. The final trajectory was obtained from a 1 ns microcanonical NVE dynamics with a large 50 Å cut-off distance performed at 300 K. The Mean Square Displacement (MSD) vs. time plots were used to calculate the average diffusion coefficient for the hydroxide ions, which turned out to be 5.2 × 10−6 cm2 s−1 (Fig. 5b). However, the computed diffusion coefficient is lower than the experimental one (2.9 × 10−5 cm2 s−1). This is because the hydration was not considered in the MD simulations.51 Importantly, the observed and calculated diffusion coefficients for the hydroxide ions were comparable with those available from the recent computational works.52–55 From the fits to the MSD plots, we estimated the activation energy for the diffusion of the hydroxide ions in the cationic polymer framework to be 0.22 eV (21 kJ mol−1, Fig. S27, ESI†), which is comparable to the experimental activation energy (0.153 eV) (14.77 kJ mol−1) calculated using the Warburg Bode plot (Fig. S27, ESI†).56
Large energy barrier for zinc-ion incorporation into the cationic polymer
To estimate the energy barriers associated with the process of incorporation of the Zn2+ ions into the polymer and its interaction with the OH− ion, we used the Nudged Elastic Band calculations. In the “reactant” model, the Zn2+ ions were positioned relatively farther from the hydroxide ions present within the polymer's unit cell. Meanwhile, we configured the “product” to have the Zn2+ ions at a bonding distance to the hydroxide ions (Fig. 5c). The framework atoms of the “reactant” and “product” were made indistinguishable to sequester only the effect of Zn-ion incorporation. Then, a transition state confirmation for the bound state was performed using the climbing-image NEB method. We included multiple intermediate configurations between reactant–TS and TS–product to obtain a good extrapolation of the minimum energy path (MEP) and optimized under the NEB. The Ea for the Zn2+ ions incorporation was calculated using as much as 7 intermediate configurations.Most importantly, both the reactants and the product configurations showed stable structures. Furthermore, the incorporation of the Zn2+ ions goes through an acceptable intermediate, but with a significant energy barrier (2.11 eV). We observe that the energy barriers for incorporating the zincate ion into the polymer were even higher, and the zincate species goes through some unacceptable geometry deformations due to repulsive interactions with the polymer's framework atoms. This reveals the non-compatibility of the Zn2+ ions and zincates in the polymer, which is a desirable feature in preventing zinc-dendrimer formation or cell short-circuiting.
Conclusions
This work describes a cationic polymer with counterbalancing hydroxide anions as a solid-state electrolyte for a zinc–air battery. The uniqueness of the work is its simplicity, despite delivering high performance. We couple Bakelite and viologen chemistry to make this polymer, which is exceptionally stable, easy to make, and affordable. The organic nature of this ion-conducting polymer (1.4 × 10−2 S cm−1 at 80 °C and 95% RH) enables it to integrate well with filter paper to yield an electrolyte requiring minimal KOH wetting. 1_OH@FP delivers excellent performance as the separator-cum-electrolyte for the zinc–air battery with a power density of 115 mW cm−2, which is three times more than the neat filter paper-based counterpart. The fabricated device easily withstands bending and twisting operations. A gradual and constant increment in OCV was observed for devices upon connecting them in series, enabling them to light up a power-demanding traffic signal for a long time. Computational modeling reveals the importance of heteroatoms in providing an electronic push–pull across the framework, which aids in transporting the hydroxide ions. At the same time, the MD simulations confirm the favorable diffusion of hydroxide ions and the high barrier for the undesirable penetration of Zn2+ ions into the polymer. Thus, our findings assert the readily accessible C–C-linked cationic polymer's capacity as solid-state electrolytes for ZAB and any anion conducting membrane.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge IISER Pune for support and funding by the “DST for material for energy storage (DST/TMD/MES/2k17/103), Air Force Office of Scientific Research under Award Number FA2386-21-1-4022, MHRD-STARS [STARS1/278] and SERB (CRG/2021/008250) program. We thank DST-Nanomission for technical support under the Thematic Unit Program (SR/NM/TP-13/2016). We thank the biology microscopy facility, IISER Pune. D. R. and R. I. acknowledge IISER Pune and MHRD-STARS for financial support. We thank Dr Sreekumar Kurungot, CSIR-NCL, Pune, for the materials support.References
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- N. Chen, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, C. Hu, J. Y. Bae, E. S. Sim, Y.-C. Chung, J.-H. Jang, S. J. Yoo, Y. Zhuang and Y. M. Lee, Nat. Commun., 2021, 12, 2367 CrossRef CAS PubMed.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- S. Tao, H. Xu, Q. Xu, Y. Hijikata, Q. Jiang, S. Irle and D. Jiang, J. Am. Chem. Soc., 2021, 143, 8970–8975 CrossRef CAS PubMed.
- K.-D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- Y. Meng, J. Gao, Z. Zhao, J. Amoroso, J. Tong and K. S. Brinkman, J. Mater. Sci., 2019, 54, 9291–9312 CrossRef CAS.
- L. Malavasi, C. A. J. Fisher and M. S. Islam, Chem. Soc. Rev., 2010, 39, 4370–4387 RSC.
- S. S. Shinde, C.-H. Lee, A. Sami, D.-H. Kim, S.-U. Lee and J.-H. Lee, ACS Nano, 2017, 11, 347–357 CrossRef CAS PubMed.
- Z. Li, W. Niu, Z. Yang, N. Zaman, W. Samarakoon, M. Wang, A. Kara, M. Lucero, M. V. Vyas, H. Cao, H. Zhou, G. E. Sterbinsky, Z. Feng, Y. Du and Y. Yang, Energy Environ. Sci., 2020, 13, 884–895 RSC.
- S. Haldar, D. Kaleeswaran, D. Rase, K. Roy, S. Ogale and R. Vaidhyanathan, Nanoscale Horiz., 2020, 5, 1264–1273 RSC.
- R. Kushwaha, S. Haldar, P. Shekhar, A. Krishnan, J. Saha, P. Hui, C. P. Vinod, C. Subramaniam and R. Vaidhyanathan, Adv. Energy Mater., 2021, 11, 2003626 CrossRef CAS.
- J. Fu, R. Liang, G. Liu, A. Yu, Z. Bai, L. Yang and Z. Chen, Adv. Mater., 2019, 31, 1805230 CrossRef PubMed.
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu and C. Wang, Science, 2021, 371, 46–51 CrossRef CAS PubMed.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- Q. Liu, Y. Wang, L. Dai and J. Yao, Adv. Mater., 2016, 28, 3000–3006 CrossRef CAS PubMed.
- M. Yu, Z. Wang, C. Hou, Z. Wang, C. Liang, C. Zhao, Y. Tong, X. Lu and S. Yang, Adv. Mater., 2017, 29, 1602868 CrossRef PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- N. S. V. Narayanan, B. V. Ashokraj and S. Sampath, J. Electrochem. Soc., 2009, 156, A863 CrossRef CAS.
- A. R. Mainar, E. Iruin, L. C. Colmenares, A. Kvasha, I. De Meatza, M. Bengoechea, O. Leonet, I. Boyano, Z. Zhang and J. A. Blazquez, J. Energy Storage, 2018, 15, 304–328 CrossRef.
- P. Chen, K. Zhang, D. Tang, W. Liu, F. Meng, Q. Huang and J. Liu, Front. Chem., 2020, 8, 372 CrossRef CAS PubMed.
- H. Miao, B. Chen, S. Li, X. Wu, Q. Wang, C. Zhang, Z. Sun and H. Li, J. Power Sources, 2020, 450, 227653 CrossRef CAS.
- J. Park, M. Park, G. Nam, J.-s Lee and J. Cho, Adv. Mater., 2015, 27, 1396–1401 CrossRef CAS PubMed.
- D. Chen and M. A. Hickner, ACS Appl. Mater. Interfaces, 2012, 4, 5775–5781 CrossRef CAS PubMed.
- Y. A. Elabd, Mol. Syst. Des. Eng., 2019, 4, 519–530 RSC.
- Z. Cao, H. Hu, M. Wu, K. Tang and T. Jiang, J. Mater. Chem. A, 2019, 7, 17581–17593 RSC.
- J. Fu, J. Zhang, X. Song, H. Zarrin, X. Tian, J. Qiao, L. Rasen, K. Li and Z. Chen, Energy Environ. Sci., 2016, 9, 663–670 RSC.
- S. Nandi, V. M. Dhavale, S. Shalini, U. Werner-Zwanziger, H. Singh, S. Kurungot and R. Vaidhyanathan, Adv. Mater. Interfaces, 2015, 2, 1500301 CrossRef.
- R. Illathvalappil, V. M. Dhavale, S. N. Bhange and S. Kurungot, Nanoscale, 2017, 9, 9009–9017 RSC.
- M. T. Tsehaye, F. Alloin, C. Iojoiu, R. A. Tufa, D. Aili, P. Fischer and S. Velizarov, J. Power Sources, 2020, 475, 228689 CrossRef CAS.
- D. J. Arnot, M. B. Lim, N. S. Bell, N. B. Schorr, R. C. Hill, A. Meyer, Y.-T. Cheng and T. N. Lambert, Adv. Energy Mater., 2021, 11, 2101594 CrossRef CAS.
- Y. Kim, M. Künzel, D. Steinle, X. Dong, G.-T. Kim, A. Varzi and S. Passerini, Energy Environ. Sci., 2022, 15, 2610–2618 RSC.
- Y. Zeng, P. Gordiichuk, T. Ichihara, G. Zhang, E. Sandoz-Rosado, E. D. Wetzel, J. Tresback, J. Yang, D. Kozawa and Z. Yang, Nature, 2022, 602, 91–95 CrossRef CAS PubMed.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2011, 23, 1818–1824 CrossRef CAS.
- R. Paul, S. C. Shit, T. Fovanna, D. Ferri, B. Srinivasa Rao, G. T. K. K. Gunasooriya, D. Q. Dao, Q. V. Le, I. Shown, M. P. Sherburne, Q. T. Trinh and J. Mondal, ACS Appl. Mater. Interfaces, 2020, 12, 50550–50565 CrossRef CAS PubMed.
- R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 RSC.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- S. Nandi, U. Werner-Zwanziger and R. Vaidhyanathan, J. Mater. Chem. A, 2015, 3, 21116–21122 RSC.
- D. Chakraborty, S. Nandi, R. Kushwaha, D. Kaleeswaran and R. Vaidhyanathan, Mater. Res. Bull., 2021, 146, 111614 CrossRef.
- D. Crespy, M. Bozonnet and M. Meier, Angew. Chem., Int. Ed., 2008, 47, 3322–3328 CrossRef CAS PubMed.
- E. Breitmaier and W. Voelter, Carbon-13 NMR spectroscopy, VCH Publishers Inc, United States, 1987, 26, 351 Search PubMed.
- S. Haldar, D. Rase, P. Shekhar, C. Jain, C. P. Vinod, E. Zhang, L. Shupletsov, S. Kaskel and R. Vaidhyanathan, Adv. Energy Mater., 2022, 12, 2200754 CrossRef CAS.
- L. Feng, X. Zhang, C. Wang, X. Li, Y. Zhao, X. Xie and Y. Lv, Int. J. Hydrogen Energy, 2016, 41, 16135–16141 CrossRef CAS.
- G. A. Ludueña, T. D. Kühne and D. Sebastiani, Chem. Mater., 2011, 23, 1424–1429 CrossRef.
- D. W. Kang, M. Kang, H. Yun, H. Park and C. S. Hong, Adv. Funct. Mater., 2021, 31, 2100083 CrossRef CAS.
- U. Tröltzsch and O. Kanoun, Electrochim. Acta, 2012, 75, 347–356 CrossRef.
- J. Chen, P. Li, N. Zhang and S. Tang, J. Mater. Chem. A, 2022, 10, 7146–7154 RSC.
- S. H. Lee and J. C. Rasaiah, J. Chem. Phys., 2013, 139, 124507 CrossRef PubMed.
- R. Vedalakshmi, V. Saraswathy, H.-W. Song and N. Palaniswamy, Corros. Sci., 2009, 51, 1299–1307 CrossRef CAS.
- H. Zhang, B. Zhang, Y. Yang, D. Ye, R. Chen, Q. Liao and X. Zhu, Chem. Commun., 2021, 57, 1258–1261 RSC.
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- V. Dubey and S. Daschakraborty, J. Phys. Chem. B, 2022, 126, 2430–2440 CrossRef CAS PubMed.
- J. Chen, P. Li, N. Zhang and S. Tang, J. Mater. Chem. A, 2022, 10, 7146–7154 RSC.
- P. Prakash, A. Shylendran, B. Fall, M. J. Zdilla, S. L. Wunder and A. Venkatnathan, J. Phys. Chem. C, 2022, 126, 4744–4750 CrossRef CAS.
- T. Zelovich, L. Vogt-Maranto, C. Simari, I. Nicotera, M. A. Hickner, S. J. Paddison, C. Bae, D. R. Dekel and M. E. Tuckerman, Chem. Mater., 2022, 34, 2133–2145 CrossRef CAS.
- C. Chandana, H. D. Singh, L. S. Leo, P. Shekhar, D. Rase, D. Chakraborty, C. P. Vinod and R. Vaidhyanathan, J. Mater. Chem. A, 2022, 10, 15647–15656 RSC.
- P. Prakash, J. Aguirre, M. M. Van Vliet, P. R. Chinnam, D. A. Dikin, M. J. Zdilla, S. L. Wunder and A. Venkatnathan, J. Mater. Chem. A, 2018, 6, 4394–4404 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00455k |
| ‡ D. R and R. I contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |