
A β-D-glucose pentaacetate derivative as an efficient phase selective gelator for oil spill recovery†
Gang-Ren
Huang‡
a,
Xue-Wen
Shi‡
a,
Ya-Min
Wu
a,
Ban-Peng
Cao
 *a,
Hiroaki
Okamoto
b and
Qiang
Xiao
*a
*a,
Hiroaki
Okamoto
b and
Qiang
Xiao
*a
aJiangxi Key Laboratory of Organic Chemistry, Jiangxi Science and Technology Normal University, Nanchang, 330013, China. E-mail: caobanpeng@126.com; xiaoqiang@tsinghua.org.cn
bGraduate School of Sciences and Technology for Innovation, Yamaguchi University, 2-16-1 Tokiwadai, Ube, 755-8611, Japan. E-mail: oka-moto@yamaguchi-u.ac.jp
First published on 14th November 2022
Abstract
As a β-D-glucose derivative, compound 1 with a triazole group has been developed and used in oil spill recovery at room temperature within one minute. Compound 1 can efficiently solidify oil into oleogel, especially can solidify DMS at 0.09 wt%, and can exhibit solid mechanical strength and stability for a dozen days in different environments. Compound 1 can selectively solidify oil into opaque oleogel in less than a minute when it is dissolved in CHCl3, and the opaque oleogel can be easily scooped out of the aqueous surface with a spoon at room temperature. The advantages of easy-to-implement performance, highly efficient phase selectivity, and excellent oil recovery from oil–aqueous mixtures indicate that compound 1 has potential and promising prospects in applications in oil spill recovery.
1. Introduction
With the rapid development of industries and the increase in the necessities of life, crude oil, as the blood of industries, is becoming increasingly important in globalization.1 Oil extraction and transportation are indispensable for global crude oil operations, and the consequent emerging risks of oil spills can threaten the marine ecological environment and the human–environmental system.2 Oil spills can lead to the eutrophication of water bodies and health threats to humans through the food chain by disrupting the nutrient balance of the affected ecosystems. Furthermore, the PB oil spill accident has raised public concerns about the environmental hazards caused by oil transportation.3 Until today, many techniques, such as oil skimmers, anti-oil barriers, adsorbents, dispersants, microbial degradation, and oil gelling agents, have been widely employed to deal with oil spills.4 Although these techniques have played an important part in practical applications, they have some limitations which cannot be ignored, such as the incomplete recovery of the oil skimmers and anti-oil barriers, high cost and limited adsorption capacity of adsorbents, possible secondary pollution of chemical dispersants, and long remediation time of biodegradation.Phase selective organogelators (PSOGs), which can selectively solidify an organic solvent in a biphasic system consisting of both aqueous and non-aqueous solvents, have attracted considerable interest for their great potential in oil spill recovery.5 PSOGs are a feasible option for oil spill response as they can change the physical state of oil from a free-flowing state to a semi-solid or solid state, which can be easily removed and recovered from the environment. PSOGs can self-assemble molecules into a three-dimensional network structure for oil gelation through non-covalent intermolecular forces such as hydrogen bonding, π–π stacking, and van der Waals forces to solidify the oil phase to encapsulate the spilled oil. Due to the advantages of gelation with oils, easy separation and recovery, and biocompatibility, PSOGs have aroused intense interest of researchers, and different kinds of PSOGs have been developed rapidly.6
The first reported PSOG was N-lauroyl-L-alanine synthesized in 2001.7 Since then, significant advances have been made in PSOGs applied in oil spill recovery, and several previous main challenges are gradually being handled.8–11 For example, to improve the uniform dispersion of PSOGs in the oil layer, cosolvents are introduced into PSOGs to enhance gelation effectiveness.12 Moreover, powder PSOGs can be directly applied without cosolvents.13 Even though cosolvents and powder PSOGs are involved in deploying PSOGs, the gelator molecules self-assemble into a three-dimensional network structure mainly by hydrogen bonding as a driving force by which hydrogen donors usually coordinate with water molecules, leading to the formation of unstable oleogels in oil spill recovery.14 And then, some aprotic gelators short of hydrogen bonding have been reported.15–18
Carbohydrate-based low molecular weight gelators have been an intense subject of study over the last two decades.19–21 Not only do carbohydrates build biocompatible materials and renewable resources through self-assembling systems, but carbohydrates also have versatile structures available from naturally existing monosaccharides to construct gelators.22–27 As an example, a glucose ester derivative reported by Wang can solidify glycerol to oleogel at a high concentration of about 1.56 wt% (20.0 mg mL−1).28
PSOGs, as a promising oil spill recovery option, are not commercially available because some disadvantages still exist, including the complex synthesis process, effects in large-scale deployment, and a lack of application guidelines.29 As is known, copper-catalyzed azide–alkyne cycloaddition (CuAAC) is a reaction with high speed and convenient post-processing in the arsenal of organic chemistry possessing a broad range of applications.21,30–32 Based on our previous experience that an aromatic group can improve the gelation abilities of triazole derivatives,33 we designed an aprotic PSOG containing a triazole group that can be quickly and efficiently synthesized from a glucose derivative. The PSOG can solidify oil in 10 s at 2 wt% and then solidify oil even at 0.09 wt%.
2. Experimental
2.1 Materials and instruments
All the reagents were obtained from commercial suppliers and used unless otherwise noted. The Fourier-transform infrared (FT-IR) spectra of the xerogel and gelator were recorded on an FT-IR spectrometer from Nicolet, USA. The melting points were obtained with an X-4 micro-melting point apparatus (Beijing Tech Instrument Co., Ltd). The UV/vis spectra of the solution state and gel state were recorded on a Shimadzu UV-2501 spectrometer. Scanning electron microscopy (SEM) images of the xerogels were obtained using an SU8220 instrument. The rheological properties of a sample with high viscosity were measured using a Discovery hybrid rheometer DHR-3 equipped with a cone and plate geometry of 40 mm diameter. The mass spectra were recorded with a mass spectrometer (AB, SCIEX Triple TOF 4600, US).2.2 Synthesis
Compound 1 is synthesized through commercially available cheap β-D-glucose pentaacetyl and 4-bromophenol as raw materials. The synthetic route is shown in Scheme 1, and detailed synthetic procedures and analytical data are given in the ESI.†D-Glucose is brominated and then azidized at 1-position with β-D-glucose pentaacetyl through the SN2 reaction to yield compound 3. The aromatic terminal alkyne derivative is synthesized from 4-bromophenol through the Williamson ether synthesis reaction and then the Sonogashira coupling reaction to obtain compound 5. Compound 1 is afforded via the CuAAC reaction of compounds 3 and 5.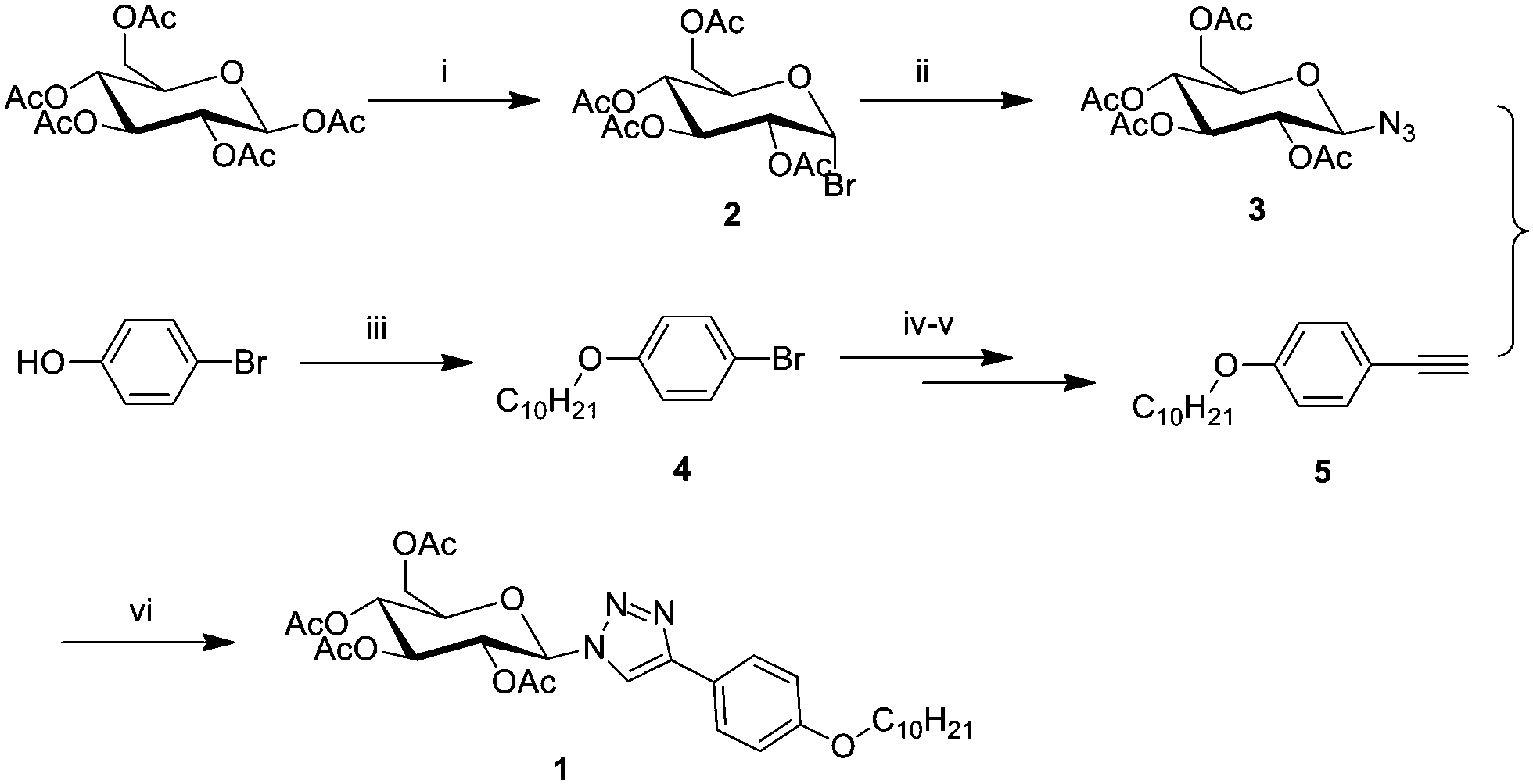 | ||
Scheme 1 Synthetic scheme for compound 1; reagents and conditions: (i) 30% HBr in CH3COOH, react overnight in the ice bath. (ii) NaN3, DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 4:1, react overnight at RT. (iii) C10H21Br, K2CO3, acetone, reflux for 18 hours. (iv) 3-Methyl butynol, CuI, Pd(Ph3P)2Cl2, PPh3, and NEt3, react at 80 °C for 24 hours in Ar. (v) NaOH, toluene, reflux for 2 h in Ar. (vi) Sodium ascorbate, CuSO4·5H2O, EtOH:H2O = 1:1, react at 60 °C for six hours. :H2O = 4:1, react overnight at RT. (iii) C10H21Br, K2CO3, acetone, reflux for 18 hours. (iv) 3-Methyl butynol, CuI, Pd(Ph3P)2Cl2, PPh3, and NEt3, react at 80 °C for 24 hours in Ar. (v) NaOH, toluene, reflux for 2 h in Ar. (vi) Sodium ascorbate, CuSO4·5H2O, EtOH:H2O = 1:1, react at 60 °C for six hours. | ||
2.3 Gelation experiments
The gelation abilities of compound 1 are investigated using the vial inversion method.34 Compound 1 (ca. 10 mg) is added to a tested solvent (19 times greater weight than that of compound 1, ca. 190 mg) in a glass tube which can be sealed through a ground glass stopper. The mixture is heated until compound 1 is dissolved or the tested solvent starts refluxing, and then the resulting solution is allowed to stand for two hours before being totally gelatinized at room temperature. The glass tube is inverted and the solution is allowed to flow down. If no solution flows down the wall of the glass tube, a gel is considered to have formed.The minimum gelation concentrations of compound 1 are determined using a dilution method.12 The formed oleogel system is stepwise diluted with a small amount of the tested solvent and the gelation abilities are investigated using the vial inversion method. The heating–cooling process is repeated until the oleogel system fails to resolidify. The last concentration at which the gel state remains without slipping after inverting the sample vial is recorded as the MGC value in the unit of wt%. This process is repeated more than three times for each tested solvent.
3. Results and discussion
3.1 Gelation ability
As shown in Table 1, compound 1 cannot solidify common organic solvents, whether in polar or non-polar solvents. Compound 1 can quickly dissolve in CH2Cl2, CHCl3, and THF at room temperature without heating while in DMF it dissolves on heating. The results suggest that CH2Cl2, CHCl3, and THF may be utilized as cosolvents to study the phase selectivity of compound 1. Compound 1 quickly precipitates from other polar solvents or high boiling solvents such as CH3CN, acetone, pentanol, and toluene after removing from heat while it is unable to dissolve in non-polar tested solvents and low boiling tested solvents.| Solvent | Status/MGC | T gel (°C) | Solvent | Status/MGC | T gel (°C) |
|---|---|---|---|---|---|
| a G = gel; S = soluble; P = precipitate; and I = insoluble; MGC values (wt%) are given behind status and tests started from a gelator concentration of 5 wt%. b MS, DMS, PEG-400, EA, and PE indicate methyl silicone oil, dimethyl silicone oil, polyethylene glycol-400, ethyl acetate, and petroleum ether, respectively. | |||||
| DMF | S | CH2Cl2 | S | ||
| THF | S | CHCl3 | S | ||
| CH3CN | P | Acetone | P | ||
| DMSO | P | PEG-400 | P | ||
| Ethanol | P | Pentanol | P | ||
| EA | P | Acetic acid | P | ||
| Toluene | P | Et3N | I | ||
| Hexane | I | CCl4 | I | ||
| Cyclohexane | I | PE | I | ||
| MS | G/0.90 | 77 | Diesel oil | G/0.80 | 69 |
| Paraffin oil | G/0.20 | 42 | Pump oil | G/0.15 | 64 |
| Mineral oil | G/0.125 | 38 | DMS | G/0.09 | 73 |
As expected, compound 1 shows an excellent ability to solidify the tested oils. The minimum gelation concentration (n) values are 0.125 wt%, 0.15 wt%, 0.2 wt%, 0.9 wt%, 0.8 wt%, and 0.09 wt% in mineral oil, pump oil, paraffin oil, MS, diesel oil, and DMS, respectively (Fig. S1, ESI†). A gelator, which can immobilize solvents to ≥100 times its own weight, is defined as a supergelator.35 From this conventional criterion, compound 1 is a supergelator for these tested oils. In Particular, compound 1 (10 mg) can solidify DMS (11110 mg) and immobilize DMS >1100 times its own weight.
As we know, the thermally reversible gel–sol transition temperature (Tgel) and mechanical stiffness of physical gels have an almost intimate relationship. The Tgel of compound 1 in oleogel at MGC is measured as shown in Table 1. Although the MGC value of compound 1 in DMS is low, which is 0.09 wt%, the Tgel of 73 °C is still high.
In order to gain more insight into the efficiency of gel formation, Tgel was measured from 5 to 1 wt% in mineral oil, paraffin oil, MS, and DMS, as shown in Table S1 (ESI†). Furthermore, the corresponding phase transition diagrams are presented in Fig. S2 (ESI†). From Table S1 (ESI†), Tgel in DMS at concentrations from 5 to 1 wt% are much higher than in other tested solvents. For example, Tgel is 153.7 °C at a concentration of 5 wt% in DMS, while it is 148.3 °C, 144.2 °C, and 136.9 °C in mineral oil, paraffin oil, and MS, respectively.
Gel formation can be assumed to resemble crystallization.36 From van’t Hoff plots, the thermodynamic parameters (ΔH, ΔS) associated with the gel phase to a quasi-sol phase (gel–sol) transition can be obtained using the following equation:
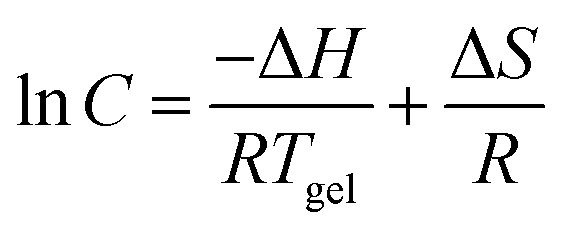
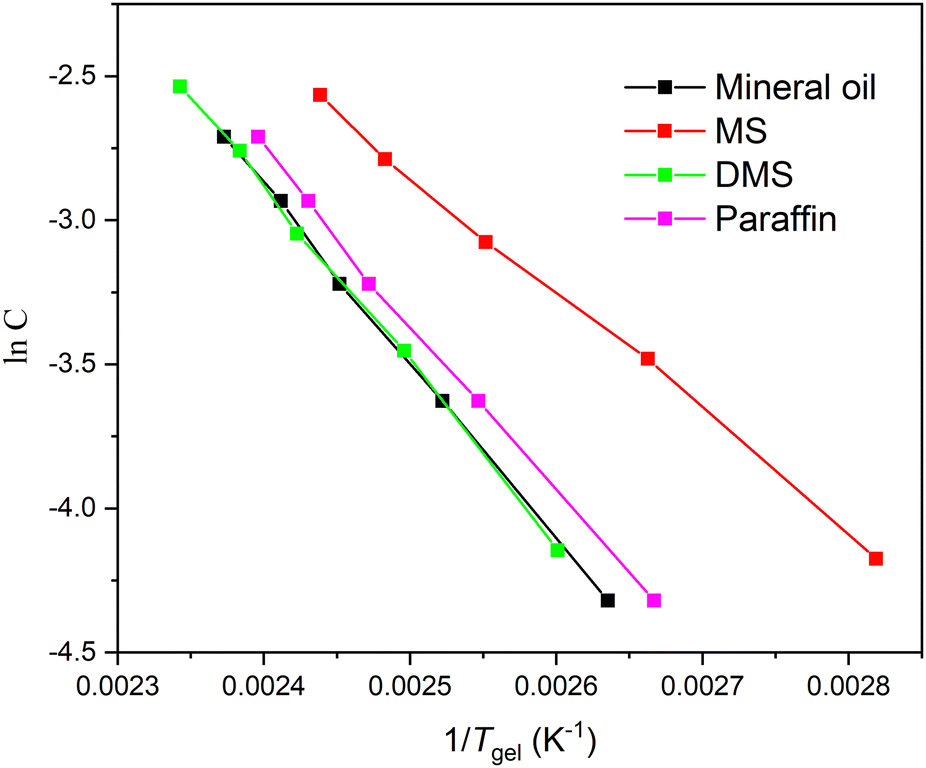 | ||
| Fig. 1 Van’t Hoff diagrams of InC and 1/Tgel. | ||
| Solvent | ΔH | ΔS | ΔG |
|---|---|---|---|
| MS | −34537.8 |
−62.7 | −15832.74 |
| Paraffin oil | −48975.0 |
−94.6 | −20770.1 |
| Mineral oil | −51007.5 |
−98.5 | −21645.4 |
| DMS | −51772.7 |
−100.3 | −21869.8 |
3.2 Rheological analysis
Gel strength is an important parameter that should be considered if materials are to be developed for practical applications. The storage modulus G′ of a gel is higher than its loss modulus G′′, indicating that a gel is a solid-like material with high strength and stiffness. The flow behavior and mechanical strength can be the best parameters to study the gel strength through rheological experiments. Therefore, rheological experiments are performed to understand the mechanical properties of compound 1 in mineral oil at 2 wt%, as shown in Fig. 2. Both moduli presented in the angular frequency sweep experiment shown in Fig. 2a are frequency-independent, as expected of solid gels. The storage modulus G′ of up to 12000 Pa and the loss modulus G′′ of 8000 Pa suggest that the mineral oil oleogel has a solid mechanical strength.
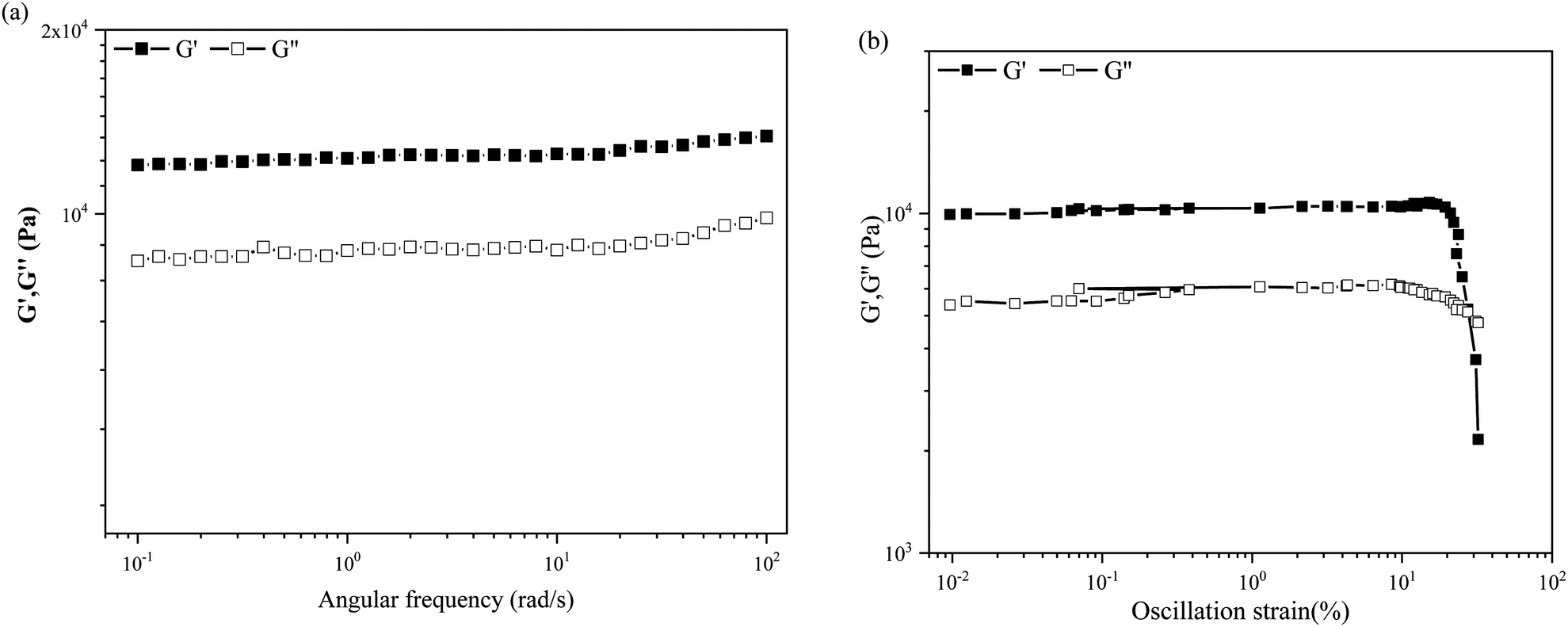 | ||
| Fig. 2 Rheological properties of the oleogel formed in mineral oil at 2 wt%: (a) angular frequency sweep and (b) stress sweep. G′ (closed symbols) and G′′ (open symbols). | ||
The oscillation stress sweep experiments are performed at a constant velocity of 10 rad s−1 on the gel states, as shown in Fig. 2b. G′ and G′′ of the mineral oil oleogels have a cross-over point (critical strain) at ca. 5100 Pa, which is detected to be 27.4% of strain for the mineral oil oleogel. The results of rheological experiments suggest that the oleogel has a solid mechanical strength and can withstand high pressure.
3.3 Phase-selective gelation
The excellent gelation ability in oils drives us curious to know whether compound 1 is a good candidate for efficient phase selectivity from oil–aqueous mixtures at room temperature for practical applications like oil spill recovery. The phase-selectivity of the oleogel solidified with compound 1 at 2 wt% is investigated using a conventional heating–cooling method. At first, water (2 mL) is added to the glass tube with oleogel to get a biphase mixture. Then, the mixture is heated until the oleogel phase turns into a liquid state. After cooling to room temperature, the oil in the liquid phase is resolidified into the oleogel phase as shown in Fig. S4 (ESI†), leaving water still in a free-flowing state.The weight-holding capacity of the oleogel is also determined to verify the solid mechanical strength. As shown in Fig. S5 (ESI†), 2 g of the mineral oil oleogel solidified with compound 1 at 2 wt% can hold 120 g of water above it against gravity. The solid mechanical strength and stability for a dozen days make compound 1 suitable for application in oil spill treatment.
To investigate the phase-selective gelation ability in different environments, the influence of both pH and salt concentration on aqueous solutions has been studied through the heating–cooling process. Compound 1 (10 mg) is added into a glass tube containing 0.5 g of mineral oil and 3 g of an aqueous solution. Compound 1 dissolves on heating and after that the mixture is cooled to room temperature and the phase state is determined using the vial inversion method. Fig. 3a shows that compound 1 can solidify mineral oil from an oil–aqueous mixture, irrespective of the pH conditions. Oil–aqueous mixtures containing different metal salt solutions (1 mol L−1) such as NaCl, KCl, ZnCl2, MgSO4, and CuSO4 have been investigated to determine the ionic effect on the phase-selective gelation ability. The result shown in Fig. 3b indicates that the tested metal salts do not influence the phase-selectivity of compound 1. Consequently, compound 1 can solidify oil to oleogel in different environments. The oleogel with acid and alkali resistance and salt tolerance can remain stable for more than a dozen days without collapsing, indicating its broad application potential in a realistic environment.
 | ||
| Fig. 3 Phase-selective gelation ability in different environments. From left to right (a) pH = 1, (b) pH = 3, (c) pH = 5, (d) pH = 7, (e) pH = 9, (f) pH = 11, (g) pH = 13, (h) NaCl solution, (i) KCl solution, (j) ZnCl2 solution, (k) MgSO4 solution, and (l) CuSO4 solution. | ||
However, the heating–cooling process is impractical under harsh field conditions for oil spill recovery. Therefore, quite a few methods carried out at room temperature, such as sonication, shaking, power, and cosolvent, have been reported.4 The method of using a solution of a gelator in a cosolvent is quite practicable in real-life applications to clean up an oil spill. CHCl3 is chosen as a cosolvent which might cause less adverse environmental impacts as it does not dissolve in water and solidifies with the oil layer instead.23 A concentrated solution of compound 1 (5 mg in 45 mg of CHCl3) is added by dropping into a glass tube containing 200 mg of mineral oil and 1 mL of water with stirring at room temperature. It is worth noting that the oil layer can be selectively solidified from oil–aqueous mixtures within 10 s after adding CHCl3 solution (Video S1, ESI†).
3.4 Recovery of gelators and oils
Oil spill recovery helps reduce possible damage to the environment and ecosystems. It is very important to have a gelator with a very low CGC for oil spills. In view of the phase selectivity and excellent mechanical properties of compound 1, with a CGC value of 0.09 wt% for mineral oil, this may be a practically viable method for oil-spill recovery.24 This study demonstrates the recycling process for collecting mineral oil and compound 1 from oil–aqueous mixtures (m:m = 2:6). The diagram is presented in Fig. 4. After adding a CHCl3 solution of compound 1 (10 wt%, 500 mg), the transparent mineral oil layer (2000 mg) is solidified into opaque oleogel, leaving the aqueous solutions in a free-flowing state. The oleogel is stiff enough to be scooped out of the aqueous surface using a spoon. After crushing, the oleogel is filtered and washed with petroleum ether (2 mL). The residue is dried to recycle compound 1 (40.6 mg) using a vacuum drying oven. The filtrate is evaporated under reduced pressure to yield a residue that is mineral oil (1860 mg, 93% recycled). The recycled compound 1 is used to restudy phase-selectivity and can recycle mineral oil ca. 92% from oil–aqueous mixtures, that is, the phase-selectivity of the recycled compound 1 is retained. The phase-selectivity of compound 1 at room temperature is also investigated in MS, paraffin oil, pump oil, and DMS using a similar process to that stated above. All of the oil recovery rates are about 90%.
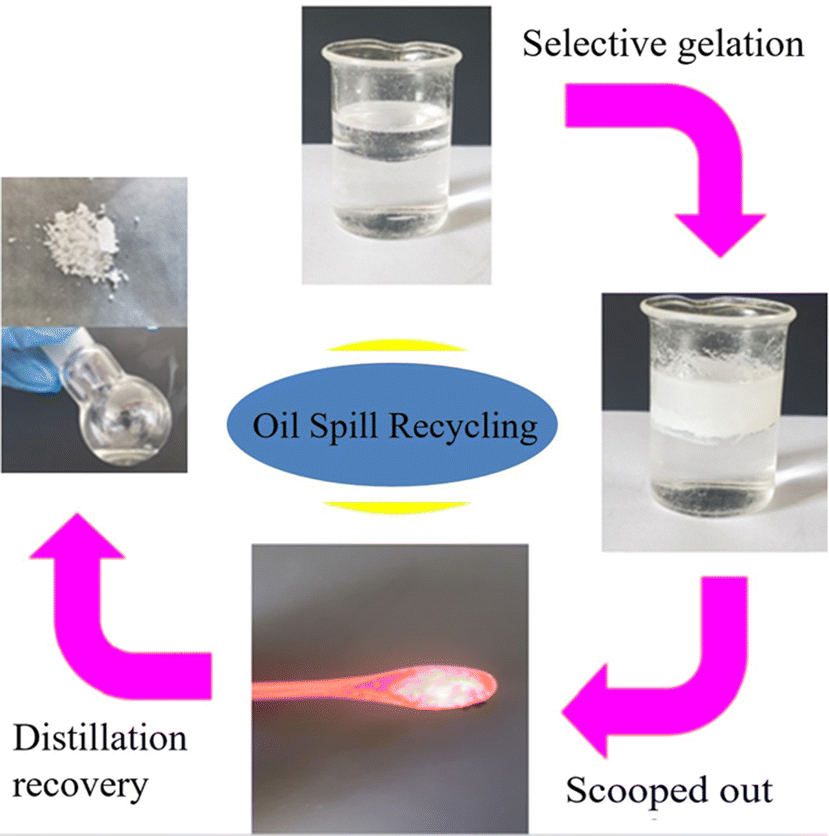 | ||
| Fig. 4 Phase-selective gelation of a mineral oil in the presence of water and its quantitative recovery through vacuum distillation. | ||
3.5 Investigation of the gelation mechanism
In order to gain insight into the morphology of compound 1 aggregates in the oleogel, the xerogel obtained from mineral oil is investigated by SEM. As seen in Fig. 5, the self-assembling molecules of the xerogel of compound 1 are twined around each other into three-dimensional networks through secondary bonds. | ||
| Fig. 5 SEM images of xerogels obtained from mineral oil. | ||
UV-vis absorption spectra have been extensively used to study π–π stacking.39 The absorption spectra of compound 1 in mineral oil solution and mineral oil oleogel are shown in Fig. S6 (ESI†). The absorption bands of compound 1 in mineral oil solution at 230 and 259 nm are attributed to the π–π* transition of the benzenoid rings, while in oleogel they are present at 230 and 250 nm. The maximum absorption band has an obvious blue shift from 259 nm to 230 nm, indicating that molecules of compound 1 may form H-type aggregates in the gel state.40
FT-IR spectroscopy is usually carried out to obtain structural information regarding self-assembled gelators, especially for hydrogen-bonding interactions.41 Two different states: a pure powder and a mineral oil xerogel, have been investigated by FT-IR spectroscopy. As displayed in Fig. S7 (ESI†), the spectra are also identical for both states. Based on the molecular structure of compound 1 and the experimental results discussed above, hydrogen-bonding interactions during self-assembly do not work.30
On the basis of the data presented above, a suggested packing process for compound 1 during gelation is described in Fig. 6. The main interactions contributing to the packing process are π–π stacking of the phenyl group and 1,2,3-triazole group and van der Waals interactions of long alkyl tails, and sugar segments. The molecules may be stacked in a face-to-face way driven by the strong π–π interaction. In the meantime, owing to the presence of effective chain interdigitation, molecules of compound 1 in oils can grow into long fibers and three-dimensional networks with ease.39
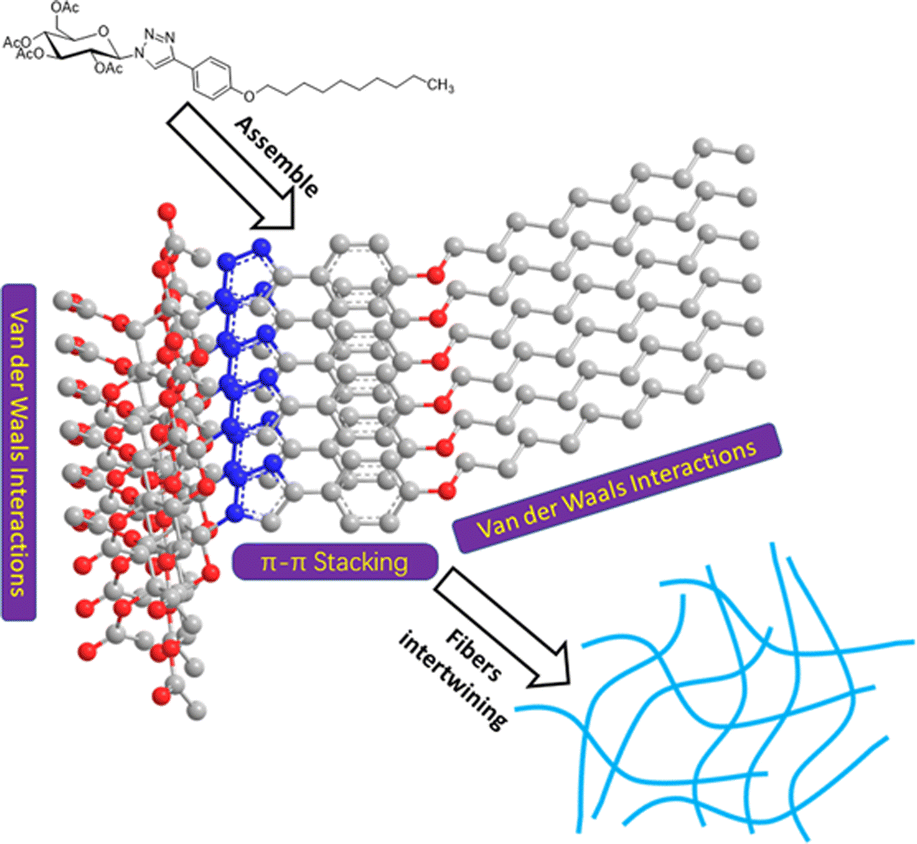 | ||
| Fig. 6 Schematic of possible packing of compound 1 during gelation. | ||
4. Conclusions
In summary, compound 1 as a β-D-glucose derivative with a triazole group was synthesized with high yields, and the gelation properties were investigated. Compound 1 can efficiently solidify the tested oil at ultralow concentrations. For instance, compound 1 can solidify DMS into oleogel at 0.09 wt%. What is more, compound 1 showed phase selectivity in oil–aqueous mixtures in different environments. In less than a minute, the CHCl3 solution of compound 1 can selectively solidify oil into opaque oleogel, which can be scooped out of the aqueous surface using a spoon at room temperature. After simply filtering and evaporating, both compound 1 and oil were efficiently recovered. The recycled compound 1 almost fully retained the phase-selectivity. With the ability to be stable for a dozen days without collapsing in different environments, compound 1 as a supergelator has promising prospects in future oil spill recovery applications.5. Procedure for synthesis
5.1 Synthesis of compound 2
β-D-Glucose pentaacetyl (10 g, 25.62 mmol) was added to 30% HBr in acetic acid (20 mL) in an ice bath and stirred overnight. The reaction was quenched with water and the product was extracted with CH2Cl2. The organic layer was dried over anhydrous sodium sulfate, and the solvent was removed by evaporation. The crude product was purified by silica gel column chromatography to obtain a white solid (compound 2, 6.43 g, 15.64 mmol, 61% yield). mp: 87–89 °C.1H NMR (400 MHz, CDCl3) δ 6.59 (d, J = 4.0 Hz, 1H), 5.53 (t, J = 9.6 Hz, 1H), 5.14 (t, J = 9.8 Hz, 1H), 4.85–4.78 (m, 1H), 4.33–4.25 (m, 2H), 4.10 (d, J = 11.2 Hz, 1H), 2.09–2.00 (m, 12H).
13C NMR (101 MHz, CDCl3) δ 170.6, 170.5, 169.9, 169.6, 169.5, 86.7, 72.3, 70.7, 70.3, 67.3, 61.1, 29.7, 20.7.
5.2 Synthesis of compound 3
Compound 2 (5 g, 12.16 mmol) was dissolved in DMSO (80 mL) and water (20 mL), and NaN3 (1.18 g, 18.24 mmol) was added with stirring at room temperature overnight. The reaction was quenched with water and the product was extracted with CH2Cl2. The organic layer was dried over anhydrous sodium sulfate, and the solvent was removed by evaporation. The crude product was purified by silica gel column chromatography to obtain a white solid (compound 3, 3.22 g, 8.63 mmol, 71% yield). mp: 127–129 °C.1H NMR (400 MHz, CDCl3) δ 5.20 (t, J = 9.4 Hz, 1H), 5.08 (t, J = 9.8 Hz, 1H), 4.93 (t, J = 9.2 Hz, 1H), 4.63 (d, J = 8.8 Hz, 1H), 4.28–4.22 (m, 1H), 4.14 (d, J = 12.4 Hz, 1H), 3.81–3.74 (m, 1H), 2.10–1.96 (m, 12H).
13C NMR (101 MHz, CDCl3) δ 170.7, 170.2, 169.4, 169.3, 88.0, 74.1, 72.7, 70.7, 68.0, 61.7, 20.8, 20.6, 20.6.
5.3 Synthesis of compound 4
1-Bromodecane (12.78 g, 57.80 mmol) and potassium carbonate (23.97 g,173.40 mmol) were added to an acetone solution (100 mL) of 4-bromophenol (10 g, 57.80 mmol) and the reaction mixture was refluxed for 18 hours. The solid in the reaction mixture was filtered off, and the filtrate was evaporated to give a crude product. The crude product obtained was purified by silica gel column chromatography to obtain a light yellow oil (compound 4, 15.57 g, 49.72 mmol, 86% yield).1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 8.8 Hz, 1H), 6.78 (d, J = 8.8 Hz, 1H), 3.91 (t, J = 6.4 Hz, 1H), 1.82–1.73 (m, 1H), 1.46–1.41 (m, 1H), 1.40–1.28 (m, 12H), 0.91 (t, J = 6.2 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 157.4, 131.3, 115.4, 111.7, 67.4, 31.0, 28.7, 28.5, 28.4, 28.3, 25.1, 21.8, 13.2.
5.4 Synthesis of compound 5
Compound 4 (10 g, 31.92 mmol), 2-methyl-3-butyn-ol (4.03 g, 4.88 mmol), bis(triphenylphosphine) palladium chloride (0.45 g, 0.64 mmol), triphenylphosphine (0.67 g, 2.55 mmol), and copper iodide (0.24 g, 1.28 mmol) were added in triethylamine (100 mL). The solution mixture was reacted in an argon atmosphere at 80 °C for 24 hours. The solvent was removed in vacuo, and the solid residue was dissolved in ethyl acetate, sequentially washed with water, saturated aqueous ammonium chloride and brine. The organic layer was dried over anhydrous sodium sulfate and evaporated to give a residue. The residue and potassium hydroxide (3.14 g, 78.5 mmol) was added to toluene (100 mL). The mixture was refluxed for two hours. The solid in the reaction mixture was filtered off, and the filtrate was evaporated to give a crude product. The crude product was purified by silica gel column chromatography to obtain a light yellow oil (compound 5, 3.45 g, 13.35 mmol, 43% yield).1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 3.95 (t, J = 6.6 Hz, 2H), 3.00 (s, 1H), 1.83–1.75 (m, 2H), 1.50–1.41 (m, 2H), 1.40–1.25 (m, 12H), 0.91 (t, J = 6.8 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 159.6, 133.6, 114.5, 114.0, 83.9, 75.8, 68.1, 32.0, 29.7, 29.5, 29.4, 29.2, 26.1, 22.8, 14.2.
5.5 Synthesis of compound 1
A mixture of compound 5 (1.04 g, 34.02 mmol), CuSO4·5H2O (0.02 g, 0.08 mmol), and sodium ascorbate (0.05 g, 0.26 mmol) in 10 mL of ethanol/water 1:1 was stirred vigorously. Compound 3 (1 g, 2.68 mmol) dissolved in 10 mL of ethanol/water 1:1 was added dropwise, and the resulting heterogenic mixture was kept at 60 °C for 6 h. After cooling, the suspension was poured into water (30 mL). The solid was filtered and washed with water. The crude product was purified by silica gel column chromatography to obtain a white solid (compound 1, 1.41 g, 2.23 mmol, 83% yield). mp: 185–187 °C.
MS-ESI: calculated for [M + Na]+ (C32H45N3O10): m/z 654.3003, found: m/z 654.2996.
1H NMR (400 MHz, CDCl3) δ 7.90 (s, 1H), 7.73 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 5.92 (d, J = 9.2 Hz, 1H), 5.52 (t, J = 9.4 Hz, 1H), 5.43 (t, J = 9.4 Hz, 1H), 5.26 (t, J = 9.6 Hz, 1H), 4.32 (dd, J = 5.2, 4.8 Hz, 1H), 4.15 (d, J = 11.6 Hz, 1H), 4.04–4.00 (m, 1H), 3.98 (t, J = 6.4 Hz, 2H), 2.07 (d, J = 2.8 Hz, 6H), 2.03 (s, 3H), 1.87 (s, 3H), 1.82–1.72 (m, 3H), 1.46–1.40 (m, 2H), 1.32–1.25 (m, 12H), 0.87 (t, J = 6.4 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 170.6, 170.0, 169.5, 169.1, 159.6, 148.6, 127.3, 122.5, 116.9, 115.0, 85.9, 75.3, 72.9, 70.3, 68.2, 67.9, 61.7, 32.0, 29.7, 29.6, 29.5, 29.4, 29.3, 26.1, 22.8, 20.8, 20.7, 20.6, 20.3, 14.2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 22066011), the Science Foundation of Jiangxi Province (no. 20192BBHL80017), the Department of Education of Jiangxi Province (no. GJJ211105), and Jiangxi Science & Technology Normal University (no. 2017BSQD020 and 2021QNBJRC002).References
- H. Mallick, M. K. Mahalik and M. Sahoo, Energy Econ., 2018, 69, 307–324 CrossRef.
- A. Lane and P. L. Harrison, Proceedings of the Ninth International Coral Reef Symposium, Bali, 2002, vol. 1, pp. 403–408 Search PubMed.
- Y. Shen, B. Li, X. Chen, S. Wang, Y. Guan and Y. Li, Environ. Sci.: Nano, 2022, 9, 489–498 RSC.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- Y. Ohsedo, Polym. Adv. Technol., 2016, 27, 704–711 CrossRef CAS.
- H. Bi, C. An, C. N. Mulligan, Z. Chen, K. Lee, J. Wen, Z. Qu and X. Chen, J. Mar. Sci. Eng., 2022, 10, 1111 CrossRef.
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185–186 RSC.
- A. M. Vibhute and K. M. Sureshan, ChemSusChem, 2020, 13, 5343–5360 CrossRef CAS.
- C. Raju, L. A. Mathew and K. M. Sureshan, Adv. Sustainable Syst., 2022, 6, 2100521 CrossRef CAS.
- J. Chen, C. E. Boott, L. Lewis, A. Siu, R. Al-Debasi, V. Carta, A. A. Fogh, D. Z. Kurek, L. Wang, M. J. MacLachlan and G. Hum, ACS Omega, 2020, 5, 18758–18765 CrossRef CAS.
- M. Colak, J. Mater. Sci., 2022, 57, 1044–1057 CrossRef CAS.
- X. Zhang, J. Song, W. Ji, N. Xu, N. Gao, X. Zhang and H. Yu, J. Mater. Chem. A, 2015, 3, 18953–18962 RSC.
- J. Liu, J. Li, P. Lin, N. Zhang, X. Han, B. Zhang and J. Song, Chem. Commun., 2016, 52, 13975–13978 RSC.
- T. Steiner and W. Saenger, J. Am. Chem. Soc., 1993, 115, 4540–4547 CrossRef CAS.
- B. Cao, Y. Kaneshige, Y. Matsue, Y. Morita and H. Okamoto, New J. Chem., 2016, 40, 4884–4887 RSC.
- T. Yoshida, T. Nakamura, Y. Morita and H. Okamoto, Chem. Lett., 2015, 44, 512–514 CrossRef CAS.
- T. Yoshida, T. Hirakawa, T. Nakamura, Y. Yamada, H. Tatsuno, M. Hirai, Y. Morita and H. Okamoto, Bull. Chem. Soc. Jpn., 2015, 88, 1447–1452 CrossRef CAS.
- T. Yoshida, A. Ohashi, Y. Morita and H. Okamoto, Mol. Cryst. Liq. Cryst., 2017, 647, 299–306 CrossRef CAS.
- C. Narayana, P. Kumari, G. Tiwari and R. Sagar, Langmuir, 2019, 35, 16803–16812 CrossRef CAS PubMed.
- J. Morris, J. Bietsch, K. Bashaw and G. Wang, Gels, 2021, 7, 24 CrossRef CAS PubMed.
- H. P. R. Mangunuru, J. R. Yerabolu, D. Liu and G. Wang, Tetrahedron Lett., 2015, 56, 82–85 CrossRef CAS.
- G. Wang, D. Wang, J. Bietsch, A. Chen and P. Sharma, J. Org. Chem., 2020, 85, 16136–16156 CrossRef CAS PubMed.
- A. Prathap and K. M. Sureshan, Chem. Commun., 2012, 48, 5250 RSC.
- A. M. Vibhute, V. Muvvala and K. M. Sureshan, Angew. Chem., 2016, 128, 7913–7916 CrossRef.
- A. Chen, I. S. Okafor, C. Garcia and G. Wang, Carbohydr. Res., 2018, 461, 60–75 CrossRef CAS.
- A. Prathap and K. M. Sureshan, Angew. Chem., Int. Ed., 2017, 56, 9405–9409 CrossRef CAS.
- I. Mattsson, M. Lahtinen, R. Sitdikov, B. Wank, T. Saloranta-Simell and R. Leino, Carbohydr. Res., 2022, 518, 108596 CrossRef CAS PubMed.
- A. Chen, L. P. Samankumara, C. Garcia, K. Bashaw and G. Wang, New J. Chem., 2019, 43, 7950–7961 RSC.
- H. Bi, C. An, C. N. Mulligan, Z. Chen, K. Lee, J. Wen, Z. Qu and X. Chen, J. Mar. Sci. Eng., 2022, 10, 1111 CrossRef.
- A. Chen, D. Wang, J. Bietsch and G. Wang, Org. Biomol. Chem., 2019, 17, 6043–6056 RSC.
- J. Huo, H. Hu, M. Zhang, X. Hu, M. Chen, D. Chen, J. Liu, G. Xiao, Y. Wang and Z. Wen, RSC Adv., 2017, 7, 2281–2287 RSC.
- K. Bozorov, J. Zhao and H. A. Aisa, Bioorg. Med. Chem., 2019, 27, 3511–3531 CrossRef CAS PubMed.
- B.-P. Cao, G.-R. Huang, X.-P. Tao, H. Okamoto and Q. Xiao, J. Nanomater., 2022, 2022, 1–4 CrossRef.
- J. Brinksma, B. L. Feringa, R. M. Kellogg, R. Vreeker and J. van Esch, Langmuir, 2000, 16, 9249–9255 CrossRef CAS.
- C. Ren, G. H. B. Ng, H. Wu, K.-H. Chan, J. Shen, C. Teh, J. Y. Ying and H. Zeng, Chem. Mater., 2016, 28, 4001–4008 CrossRef CAS.
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS.
- I. González Fernández, F. C. Gennari and G. O. Meyer, J. Alloys Compd., 2008, 462, 119–124 CrossRef.
- J.-P. Desvergne, T. Brotin, D. Meerschaut, G. Clavier, F. Placin, J.-L. Pozzo and H. Bouas-Laurent, New J. Chem., 2004, 28, 234 RSC.
- H. Wu, L. Xue, Y. Shi, Y. Chen and X. Li, Langmuir, 2011, 27, 3074–3082 CrossRef CAS PubMed.
- N. Yan, Z. Xu, K. K. Diehn, S. R. Raghavan, Y. Fang and R. G. Weiss, Langmuir, 2013, 13, 793–805 CrossRef.
- C. Bao, R. Lu, M. Jin, P. Xue, C. Tan, T. Xu, G. Liu and Y. Zhao, Chem. – Eur. J., 2006, 12, 3287–3294 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj04673c |
| ‡ These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |