DOI:
10.1039/D2NJ05206G
(Paper)
New J. Chem., 2023,
47, 156-166
Design, synthesis and biological evaluation of novel potent STAT3 inhibitors based on related heterocycle-fused naphthoquinones for cancer therapy†
Received
23rd October 2022
, Accepted 27th November 2022
First published on 28th November 2022
Abstract
Persistently activated signal transducer and activator of transcription 3 (STAT3) plays an important role in the development of multiple cancers, and therefore it is a potential therapeutic target for cancer treatment. Herein, we report the rational design, synthesis, and biological evaluation of novel potent STAT3 inhibitors based on iso-napabucasin. Among them, compound A14 exhibited the most potent in vitro tumor cell growth inhibitory activities against HepG2 and K562 cells with IC50 values as low as 0.88 μM and 2.16 μM, respectively. Compound A14 has a moderate percentage of plasma protein binding in vitro (88.6%), good plasma stability and solubility. After oral administration, the bioavailability of A14 was 44.22%. Finally, molecular docking and WB studies further clarified the binding mode of A14 in STAT3 SH2 domain.
1. Introduction
Naphthoquinones are natural pigments that are widely distributed as secondary metabolites among bacterial, fungal, and plant species.1 Several naphthoquinones were isolated and identified as biologically active compounds in the bark and heartwood of Tabebuia species.2 They are a commercial natural product obtained from the inner bark and heartwood of Tabebuia trees indigenous to the Amazonian rainforest.2 The stem bark of the tree shows a wide range of biological properties such as antibacterial, antifungal, and anti-inflammatory and, in particular, antitumor activities.3 Several naphthoquinones were isolated and identified as biologically active compounds in the bark and heartwood of Tabebuia species4. The most prominent ones are lapasol, dihydrobenzodiketone α-lapatone, its angular derivative β-lapatone and naphtho [2,3-b] furan-4,9-dione napacacin. Napbuccin was first discovered in 1982 as a plant-derived compound with anticancer activity.4 In 1998, napacacin was reported to have antipsoriatic, antibacterial, and antifungal activities.5 Despite the promising preliminary data on napacacin, there was no interest in its development for almost a decade until 2009 when it was re-discovered by Boston Biomedical, Inc. and given the name napabucasin, also known as BBI608.
Recent studies have shown that napabucasin is an effective inhibitor of stem cell6 activity in cancer cells by inhibiting the signal transducer and activator of transcription 3 (STAT 3).7
The STAT family is comprised of seven proteins including STAT1, STAT2, STAT3, STAT4, STAT5α, STAT5β and STAT6.8 STAT3 is a dual-functional protein with ability of signal transduction and activation of gene transcription. Under physiological conditions, it mediates cell proliferation, differentiation, apoptosis, survival, immune function, angiogenesis and other important cellular processes in response to stimulation of a large number of cytokines and growth factors.9,10 It plays a critical role in some cellular processes such as cell growth and apoptosis by mediating the expression of target genes.11 Abnormally activated STAT3 signaling is found in a variety of solid tumors and hematological malignancies, and can induce tumor angiogenesis and suppresses anti-tumor immune responses.12–15 In addition, aberrant STAT3 correlates with resistance to chemotherapy and poor prognosis.16,17 Abundant evidence suggests that STAT3 is an ideal target for cancer therapy.
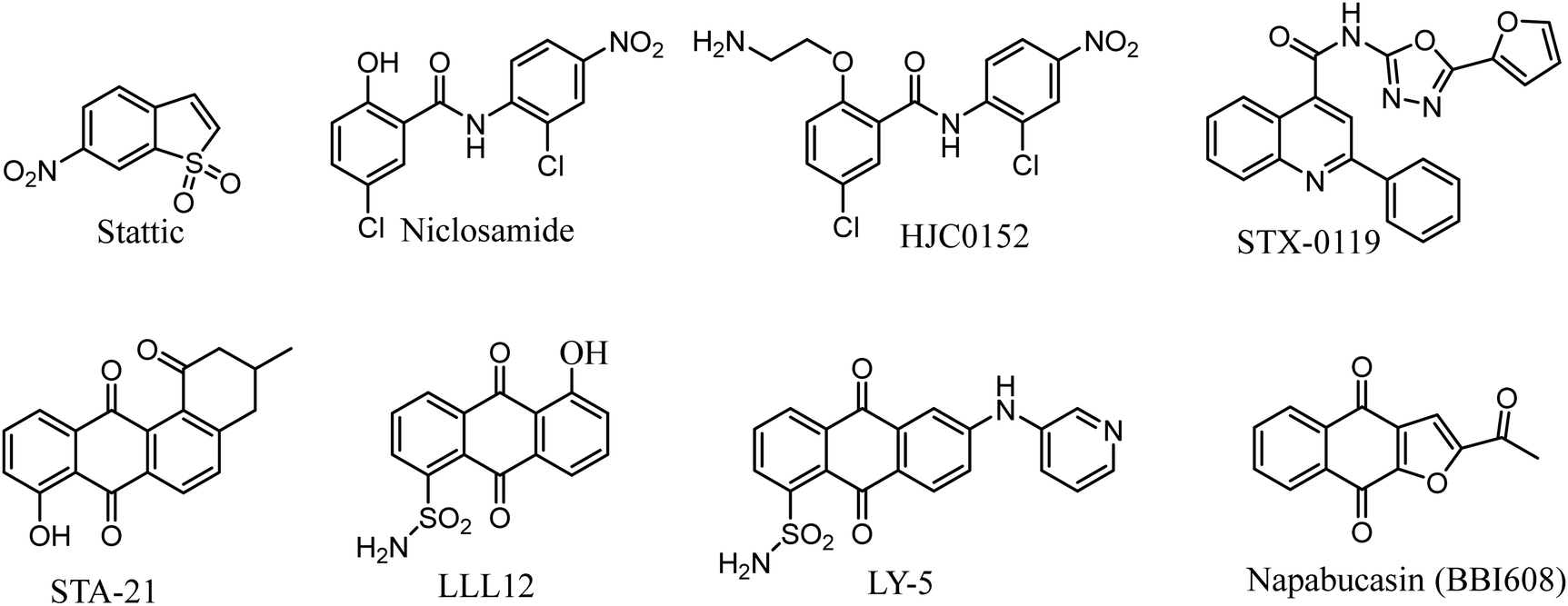
Researches on STAT3 inhibitors mostly stays in the initial stage, development of a potent inhibitor is imminent. In the past few years, significant progress has been made in the discovery of STAT3 inhibitors. Several effective small molecule inhibitors, such as Stattic,18 Niclosamide,19 HJC0152,20 STX-0119,21 STA-21,22 LLL12,23 LY-5,24 Napabucasin (Fig. 1).25
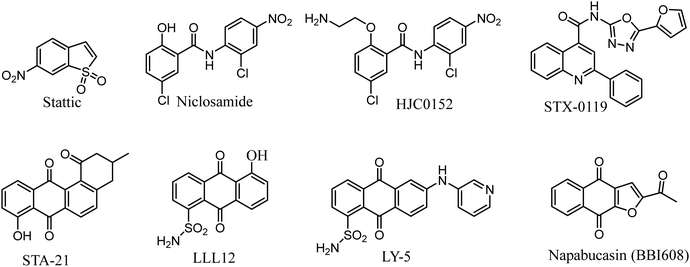 |
| | Fig. 1 Representative of known STAT3 inhibitors. | |
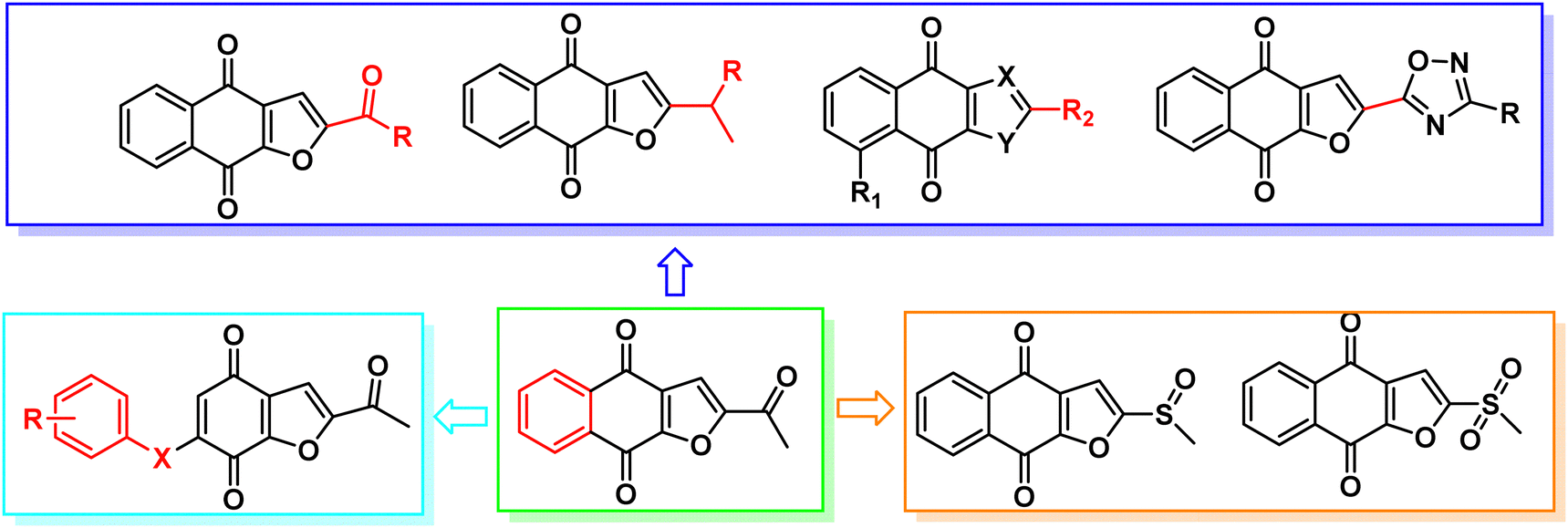
Napabucasin, an excellent STAT3 small molecule inhibitor by targeting and blocking cancer stem cell pathway activity, was evaluated in phase III clinical trials for the treatment of a variety of cancers such as metastatic colorectal carcinoma, pancreatic cancer, gastric cancer and non-small cell lung cancer.26–28 As far as we know, the previous structural modifications of BBI608 were mainly concentrated on the acetyl group at position 2 of the furan ring, without modification of the nucleus, and lack of structural diversity (Fig. 2).29–33
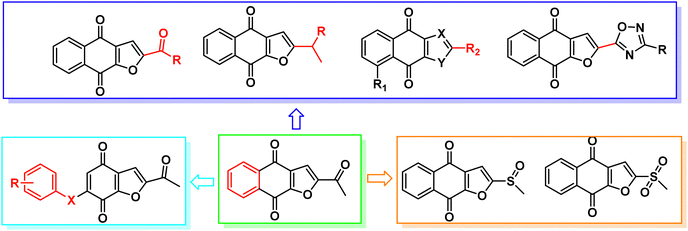 |
| | Fig. 2 Previous and our structure-based drug design of STAT3 inhibitors based on BBI608. | |
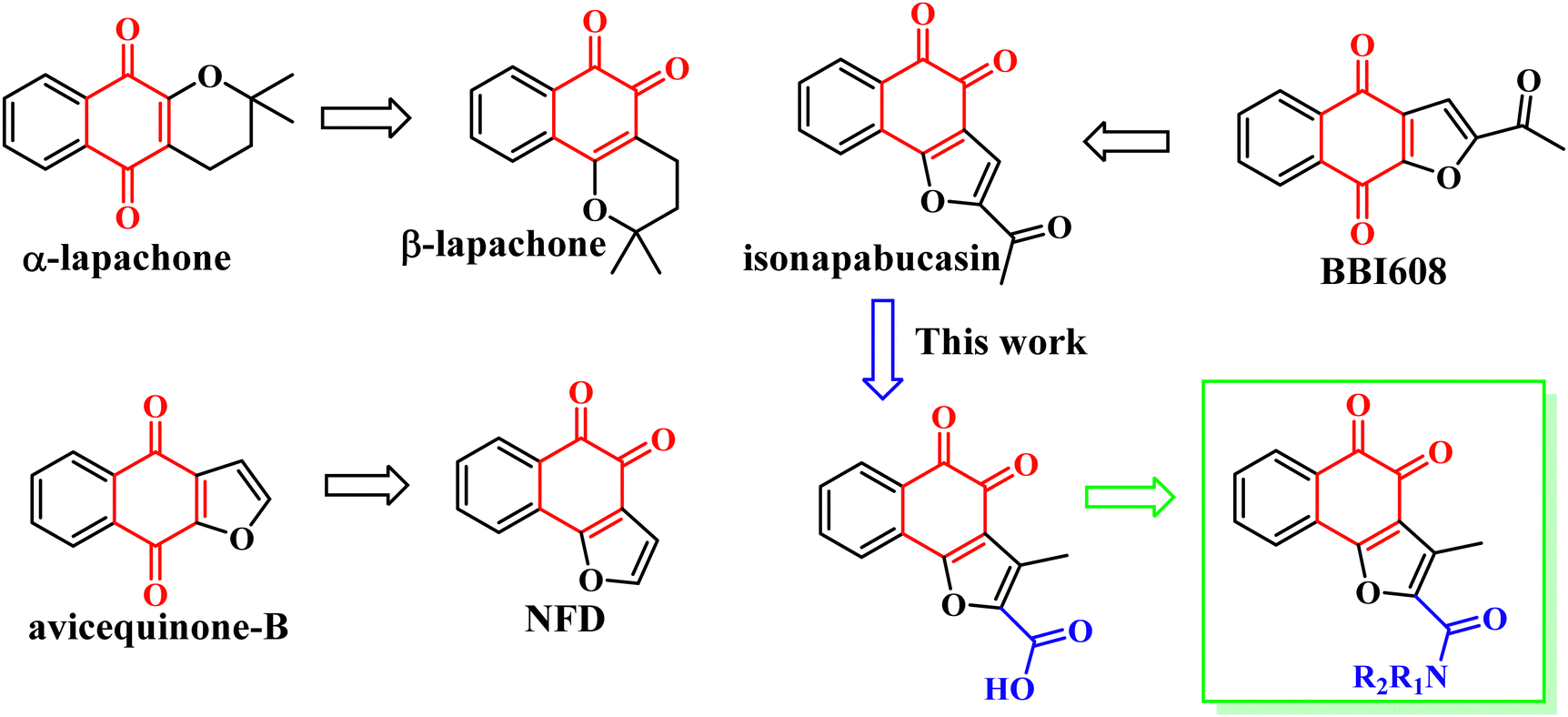
In 2018, Muller et al. found that the anti-tumor activity of β-Lapachone is better than that of α-lapachone. Their difference lies in the position of the carbonyl group. Continued research also found that the anti-tumor activity of naphtho[1,2-b]furan-4,5-dione (NFD) is better than that of naphtho[2,3-b]furan-4,9-dione (avicequinone-B). It is guessed that the activity of the carbonyl group at position 1, 2 is due to position 1, 4. In order to verify the conjecture, they synthesized 2-acetyl-naphtho[1,2-b]furan-4,5-dione (iso-napabucasin), and found that the anti-tumor activity of iso-napabucasin was three times than that of BBI608 (Fig. 3). Therefore, we intend to take the structure of iso-napabucasin as the lead compound to design novel derivatives. Due to the lack of hydrogen bond donor (HBD), we modified the side chain of isonapabucasin by means of mixing and bioisosteres, assisted by computer aided design and drawing (CADD), and introduced nitrogen atoms to improve the solubility and drug-like properties of the compounds, so as to find better lead compounds.
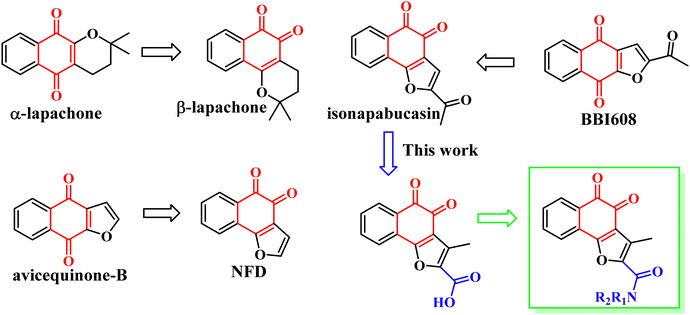 |
| | Fig. 3 Structure-based drug design of STAT3 inhibitors. | |
2 Chemistry
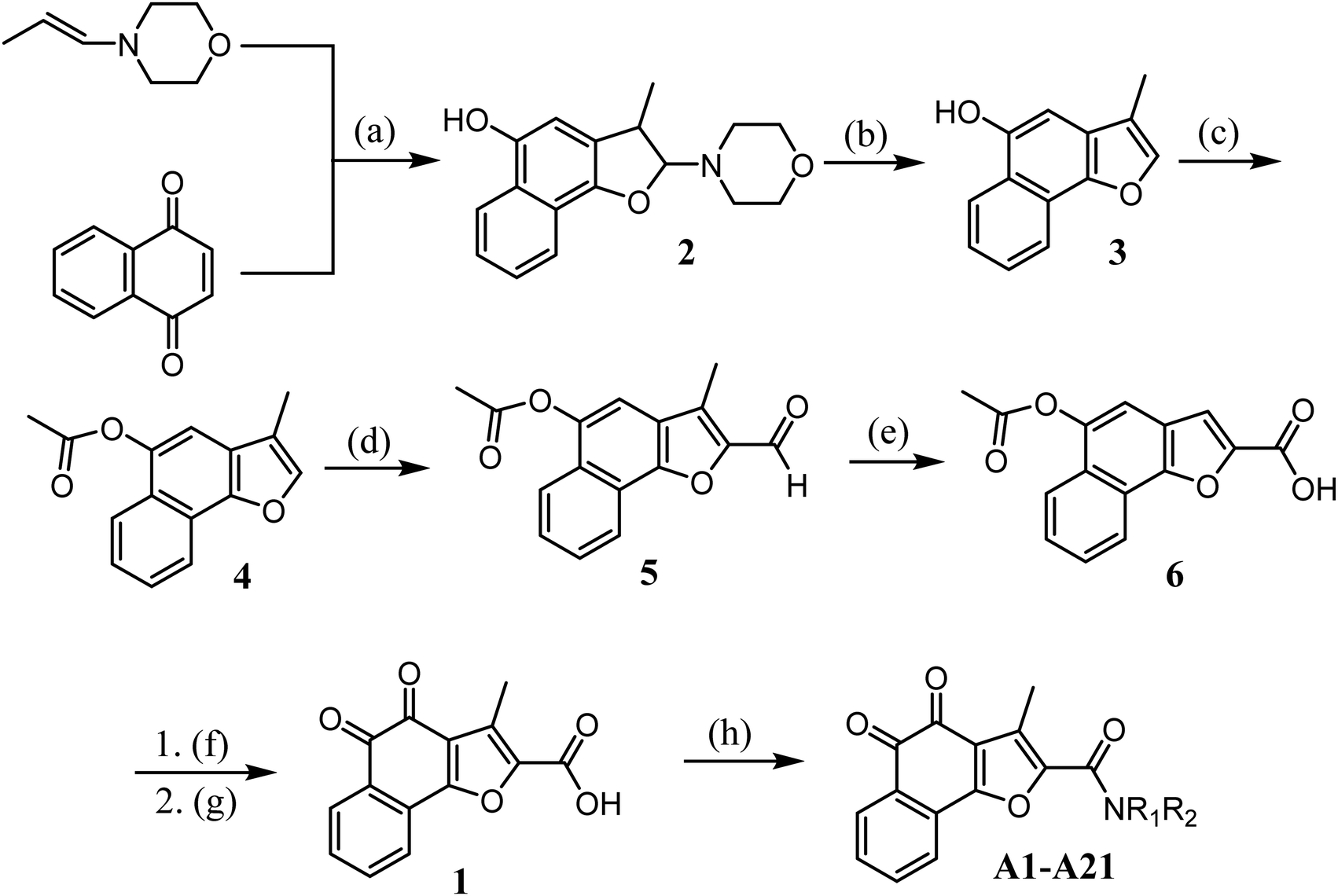
The synthetic route for designed compounds was described in Scheme 1. Treatment of 1,4-naphthoquinone with (E)-4-(prop-1-en-1-yl)morpholine provided the adduct product 2 in 30% yield. Compound 3 was obtained by heating intermediate 2 in the presence of diluted hydrochloric acid in 80.56% yield. Compound 3 is reacted with acetyl chloride to obtain compound 4. Compound 5 was obtained from compound 4 by Vilsmeier–Haack reaction. Compound 5 was oxidized to obtain compound 6. Compound 1 is obtained by hydrolysis and oxidation. Finally, the target compound can be easily prepared by using various aliphatic amines through the esterification reaction in the presence of 1-ethyl-3(3-dimethylpropylamine) carbodiimide (EDCI) and 1-hydroxybenzotriazole (HOBt) (Scheme 1).
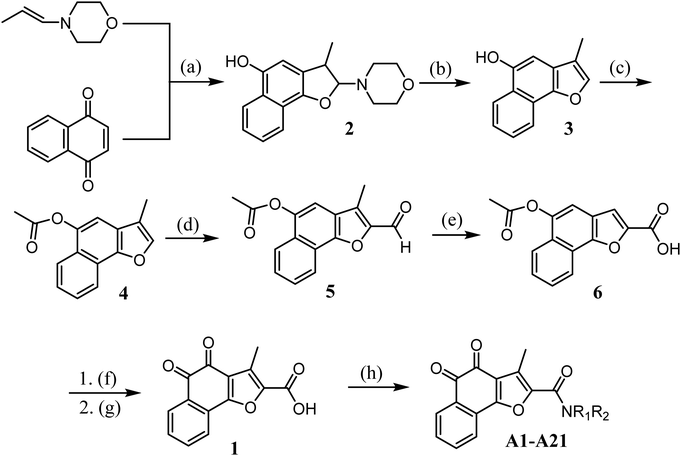 |
| | Scheme 1
Reagents and conditions: (a) dry DCM, Ar, 0 °C to rt, 12 h; (b) THF, 37% HCl, 75 °C, 3 h; (c) acetyl chloride, TEA, DCM, rt; (d) DMF, POCl3, DCM, 0 °C to reflux; (e) 30% H2O2, NaH2PO4, NaClO2, rt, 3 h; (f) NaOH, THF, H2O, rt; (g) IBX, acetone, H2O, rt, 3 h; (h) 1-ethyl-3(3-dimethylpropylamine) carbodiimide (EDCI), 1-hydroxybenzotriazole (HOBt), TEA, DCM, NR1R2, rt, 3 h. | |
3 Results and discussion
3.1 Biological assay
We firstly evaluated the inhibitory activities of target compounds against two STAT3 over-expressed human cancer cell lines (HepG2 and K562) via established CCK8 assay. The IC50 values were listed in Table 1 with BBI608 as positive control. The activity assay was started with the target compounds with small molecular chain amines on the branched chain. The preliminary structure–activity relationship shows that the inhibitory activity of the compound against HepG2 and K562 decreases with the extension of the carbon chain, and the activity of compound A2 against HepG2 is better IC50 = 2.90 μM Then the inhibitory activities of the target compounds with small molecule cyclic amines on the branched chain against HepG2 and K562 were evaluated. The results showed that the activities of cyclic small molecule amines is better than that of chain ones and also suggested that increasing carbon number of cyclic small molecule amine mildly decreased the inhibitory activity, with compound A8 (IC50 = 1.99 μM) among them showing the best activity. Next, the inhibitory activities of the target compounds with substituent on small molecule amines on the branched chain against HepG2 and K562 were further evaluated. The results showed that the substituent had some effect on their inhibitory activity and with the ester moiety at the 3-position (A14 = 0.88 μM) being the most active. The structure–activity relationship revealed that when a chain, such as a small molecule amine, was introduced, the activity of the compounds increased initially before decreasing with chain extension, with ethylamine having the highest activity. When the ring-shaped small molecule amine is present, tetrahydropyrrole has the highest activity of the compounds, with the activity decreasing with the number of carbons. When the substituent is added to the cyclic small molecule amine, position 3 is preferable to position 4; compound A14 has the best activity, and the substituent at position 3 is ethyl carboxylate. On the whole, the inhibitory activities of target compounds against K562 were a little better than against HepG2. Compound A14 was chosen for next investigation.
Table 1 The inhibition potencies of compounds in vitro against HepG2 and K562 cells
3.2 Toxicity testing of A-14 against L-02 cells
The cytotoxicity of compound A14 against human normal liver cell L-02 was evaluated. The IC50 against L-02 was 6.73 μM and is 7.6-fold of that against Hep G2. It showed that compound A14 has good selectivity and safety.
3.3 Microsomal stability
The microsomal stability of compound A14 was evaluated in human, Monkey, dog, rat, and mouse liver microsomes, respectively, which are widely used to determine the likely degree of primary metabolic clearance in the liver (Table 2). But the tested compounds displayed unsatisfactory stabilities in vitro microsomal stability.
Table 2 Half-life of the compound A14 in different species of liver microsomes
| Species |
Human |
Monkey |
Dog |
Rat |
Mouse |
|
T
1/2 (min) |
0.587 |
2.52 |
0.756 |
0.830 |
9.50 |
3.4 Plasma stability
The plasma stability of A14 compounds was evaluated in humans, monkeys, dogs, rats and mice. As shown in Table 3, compound A14 was relatively stable in human, monkey, and dog plasma at 37 °C, but less stable in rat and mouse plasma. The similar results were obtained under the room temperature.
Table 3 Plasma stability of the compound A14 in different species
| Compd |
Incubation conditions |
Species |
k (min−1) |
T
1/2 (min) |
Remaining after 60 min (%) |
| NA: not Applicable (not metabolized). |
|
A14
|
37 °C |
Human |
−0.00125 |
NA |
108 |
| Monkey |
0.00285 |
272 |
84.4 |
| Dog |
−0.00212 |
NA |
114 |
| Rat |
0.0954 |
7.27 |
0.332 |
| Mouse |
0.0872 |
7.98 |
0.562 |
| rt |
Human |
−0.00238 |
NA |
115 |
| Monkey |
0.00161 |
431 |
90.8 |
| Dog |
0.000103 |
6757 |
109 |
| Rat |
0.0891 |
7.77 |
0.476 |
| Mouse |
0.0670 |
10.4 |
1.85 |
3.5 Kinetic solubility
The kinetic solubility of compound A14 was tested at pH 2.0 and pH 7.4. The experimental results showed that compound A14 was significantly more soluble than Tamoxifen at pH 7.4. Even at pH 2.0, the solubility of compound A14 was better (Table 4).
Table 4 Kinetic solubility of compound A14 at pH 2.0 and pH 7.4
| Compd |
pH |
Solubility (μM) |
|
A14
|
2.0 |
69.5 |
| 7.4 |
39.6 |
| Tamoxifen |
7.4 |
0.124 |
3.6 Plasma protein binding rate
Plasma protein binding (%PPB) was measured in SD rats by a classical equilibrium dialysis device. Compound A14 showed a moderate rate of %PPB (88.6%). Since the therapeutic effect of a drug depends on the concentration of free drug, a moderate concentration of %PPB (88.6%) may facilitate the in vivo efficacy and thus the anti-tumor effect.
3.7 Pharmacokinetic study in vivo
To determine the metabolic stability of compound A14in vivo, the pharmacokinetic profile was therefore determined in Sprague-Dawley (SD) rats by oral (po) single dose of 50 mg kg−1 and intravenous (iv) single dose of 0.5 mg kg−1, and the pharmacokinetic parameters are shown in Table 5. After oral administration, blood levels were detected within 24 h. The maximum concentration reached after 0.25 h (1.32 μM); the area under the curve (AUC0–24h) was 3.64 (μM h); the plasma t1/2 for A14 was 0.68 h. The maximum concentration (3.78 μM) was reached 1.8 min after intravenous injection of compound A14; the area under the curve (AUC0–24h) was 0.082 μM h; the plasma t1/2 of A14 was 1.67 h. A14 had a moderate bioavailability of 44.22% laying a good foundation for the next in vivo study.
Table 5 Pharmacokinetics of A14 in rats following intravenous and oral administration (n = 6)
| Parameter |
A14 (n = 3) |
A14 (n = 3) |
| iv (0.5 mg kg−1) |
po (50 mg kg−1) |
|
T
max (h) |
0.03 |
0.66 |
|
C
max (μM) |
0.2 |
1.32 |
|
t
1/2 (h) |
1.67 |
0.68 |
| CL (L h−1 kg−1) |
0.66 |
1.17 |
|
V
Z (L kg−1) |
1.67 |
1.11 |
| AUC(0–24) (μM h) |
0.082 |
3.64 |
| AUC(0–∞) (μM h) |
0.225 |
3.97 |
|
F (%) |
|
44.22 |
3.8 WB analysis of compound A14 in Hep G2 cells with p-STAT3
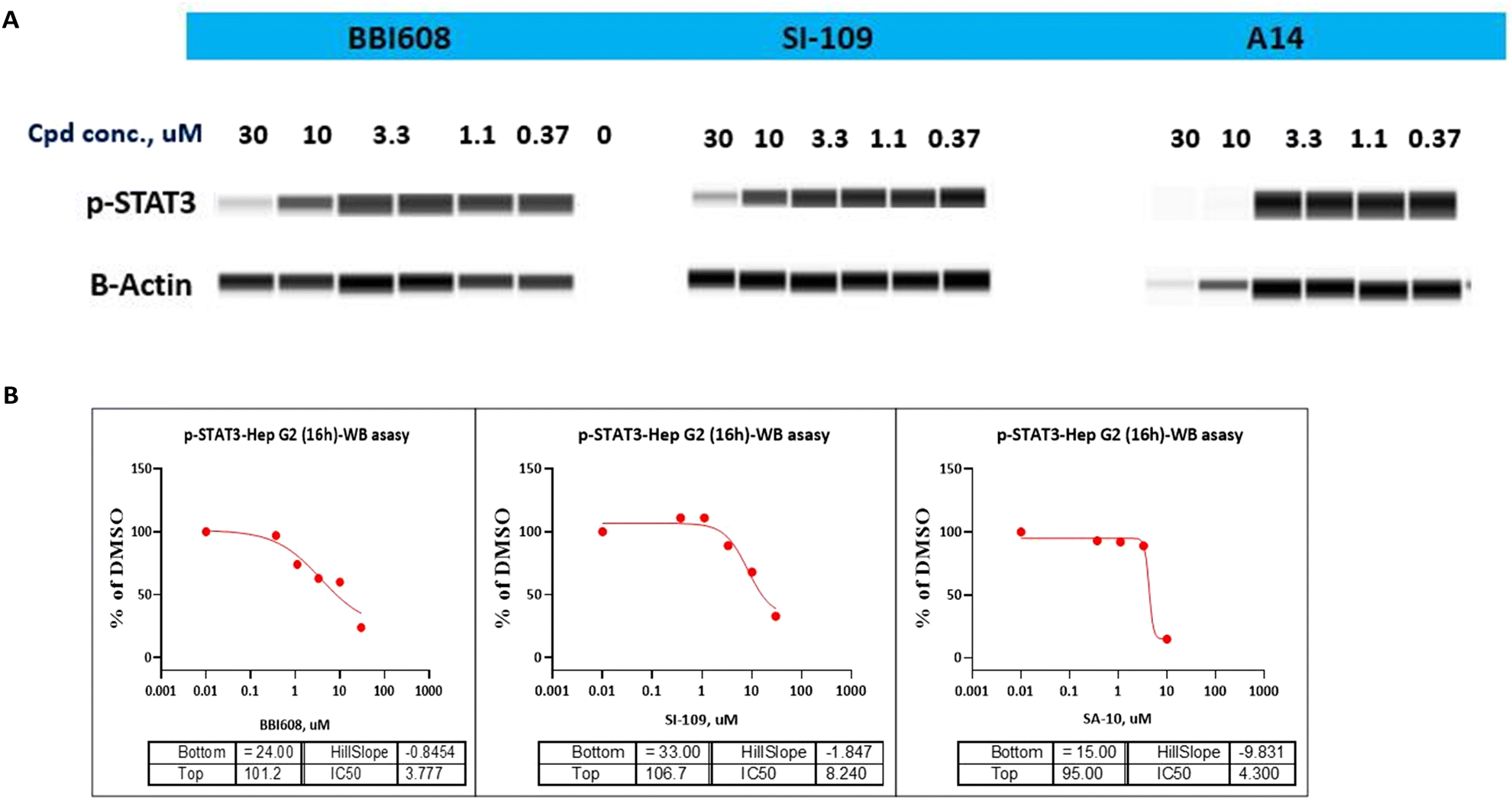
As STAT3 inhibitors can suppress the STAT3 phosphorylation on Tyr705 residue and thus restrain the expression of its downstream target proteins, then we turned our effort to the effects of compound A14 in Hep G2 cells adopting western blot analysis. As shown in Fig. 4A, compound A14 can significantly reduce the phosphorylation of STAT3 at 10 μM, which is superior to compounds BBI608 and SI-109. The IC50 values of A14, BBI608 and SI-109 inhibiting STAT3 phosphorylation in Molm-16 cells were 4.300, 3.777 and 8.240 μM (Fig. 4B).
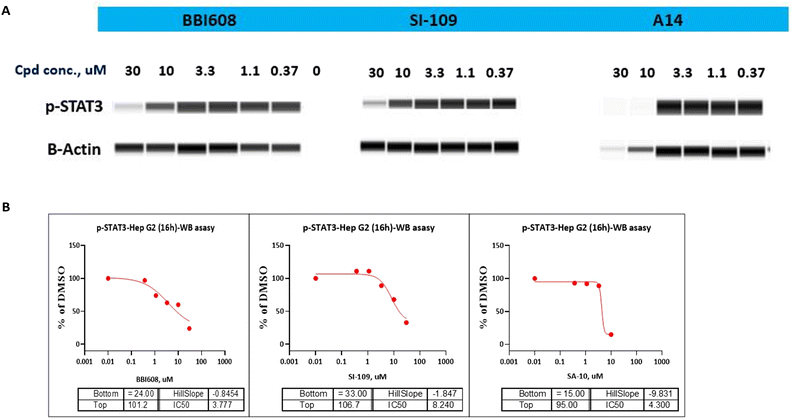 |
| | Fig. 4 (A) Western blot analysis of the inhibitory effect of compound A14, BBI608 and SI-109 on p-STAT3 in Hep G2 cell line. Cells were treated with A14, BBI608 and SI-109 for 16 h, and levels of p-STA T3 was probed by specific antibodies. β-Actin was used as the loading control. (B) IC50 of compound A14, BBI608 and SI-109 on p-STAT3 in Hep G2 cell line. | |
3.9 Molecular docking
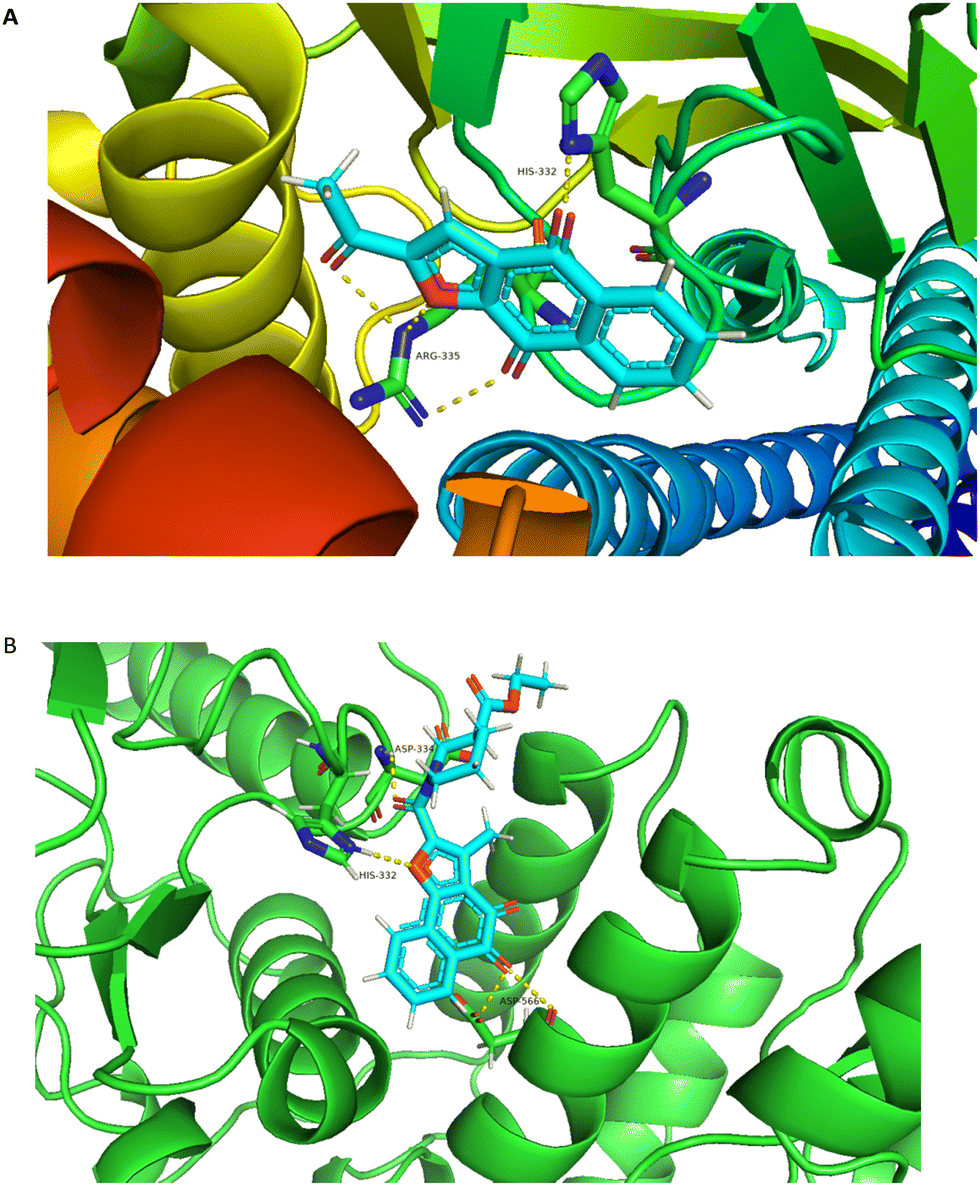
In an attempt to elucidate the binding mode of compound A14 with STAT3 SH2 domain, docking study was carried out on the basis of the crystal structure of STAT3 homo dimer (PDB code: 1BG1). For comparison, it is also compared with the combination mode of BBI608. As shown in Fig. 4A, BBI608 forms four key hydrogen bonds with HIS-332 and ARG-335. Compound A14 also forms four key hydrogen bonds with HIS-332, ASP-334 and ASP566. In general, all these interactions ensured the activities of compound A14 toward STAT3 protein (Fig. 5).
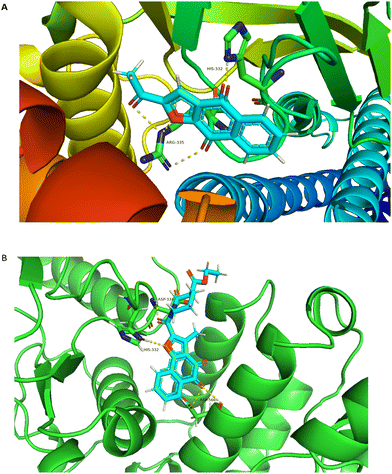 |
| | Fig. 5 Molecular modeling study of A14 and BBI608 in STAT3 SH2 domain (PDB code: 1BG1). (A) Superimposed pose of BBI608 within STAT3 SH2 domain. (B) Predicted interaction of A14 within STAT3 SH2 domain. The figures were generated using Pymol. | |
4 Conclusion
In summary, a novel series of isonapabucasin derivatives were designed, synthesized and biologically evaluated. Most of the designed compounds exerted promising inhibitory activity on HepG2 and K562 cells. The most potent compound A14 exhibited remarkable inhibitory activity on HepG2 and K562 cells with the IC50 values of 0.88 and 2.16 μM, respectively. Compound A14 has a moderate percentage of plasma protein binding in vitro (88.6%), good plasma stability and solubility. After oral administration, the bioavailability of A14 was 44.22%. Molecular docking and WB studies further clarified the binding mode of A14 in STAT3 SH2 domain. However, the clinical value of these compounds as novel antitumor agents can only be derived after further systematic evaluation of in vivo efficacy and toxicity profiles. We also recommend further studies of the compounds to assess their value in hepatocellular carcinoma.
5 Experimental
5.1 Chemistry
All reagents and starting materials were purchased from commercial suppliers, and used without further purification. Nuclear magnetic resonance (NMR) spectra were recorded on an Ascent 400 MHz spectrometer (1H frequency of 400 MHz; 13C frequency of 150 MHz). Chemical shifts (d) are reported in parts permillion (ppm) with Me4Si as internal standard. Melting points were determined with an X-4 melting point devices. High-resolution mass spectrometry (HRMS) analysis was performed on an Agilent Technologies 6530 Accurate-Mass Q-TOF MS instrument using electrospray ionization (ESI) as ionization source. Details of compound synthesis and characterization were provided in ESI.† The purity analysis of target compounds was carried out by high-performance liquid chromatograph (HPLC).
5.2 Chemistry
3-Methyl-2-morpholino-2,3-dihydronaphtho[1,2-b]furan-5-ol (2).
To a solution of 1,4 naphthoquinone (5.16 g, 32.65 mmol) in dry dichloromethane (90 mL) was added (E)-4-(prop-1-en-1-yl)morpholine (4.15 g, 32.65 mmol) at 0 °C. The reaction mixture was then warmed to room temperature and kept for 12 h. The precipitate was filtered, washed with dichloromethane (2 × 10 mL), and dried to give the titled compound as a while solid (2.79 g, 30%), m. p. 141–143 °C 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.94–7.90 (m, 2H), 7.82–7.73 (m, 2H), 6.03 (s, 1H), 3.73 (t, J = 4.6 Hz, 4H), 3.50 (t, J = 5.0 Hz, 4H) ESI-MS: m/z 244.0 [M + H]+.
3-Methylnaphtho[1,2-b]furan-5-ol (3).
3-Methyl-2-morpholinyl-2,3-dihydronaphtho[1,2-b]furan-5-ol (2) (6.20 g, 21.74 mmol), THF (31 mL), 3 N HCl (120 mL) were added to a 250 mL lask at room temperature, and the reaction solution was raised to 75 °C for 3 h. Pour the reaction solution into 500 mL ice water, filter, and dry to obtain a white solid compound 3 (3.46 g, 80.46%), m. p. 145–147 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.93(s, 1H), 8.23 (d, J = 8.3 Hz, 1H), 8.12(d, J = 8.2 Hz, 1H),7.79 (s, 1H), 7.61 (t, J = 7.8 Hz, 1H), 7.48 (t, J = 8.0 Hz, 1H), 6.96 (s, 1H), 2.23 (s, 3H).
3-Methylnaphtho[1,2-b]furan-5-yl acetate (4).
To a 100 mL flask was added 3-methylnaphthaleno[1,2-b]furan-5-ol (1.0 g, 5.05 mmol), dry DCM (20 mL), triethylamine (0.77 g, 7.57 mmol), and 5 mL of DCM solution of acetyl chloride (0.59 g, 7.57 mmol) was added slowly dropwise at room temperature for 10 min. The reaction solution is extracted with dichloromethane, dried with anhydrous magnesium sulfate, and concentrated to obtain a gray white solid 3-methylnaphthaleno [1,2-b] furan-5-acetate (4) (1.11 g, 92%), m. p. 73–75 °C 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 8.2 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.58–7.62 (m, 1H), 7.58 (d, J = 1.2 Hz, 1H), 7.49–7.53 (m, 1H), 7.31(s, 1H), 2.46 (s, 3H), 2.29 (d, J = 1.2 Hz, 3H).
2-Formyl-3-methylnaphtho[1,2-b]furan-5-yl acetate (5).
Add DMF (11.46 g, 20.08 mmol) and dry DCM (20 mL) to a 100 mL flask and add POCl3 (3.08 g, 20.08 mmol) 10 mL DCM solution slowly dropwise at −10 °C for 20 min. The reaction was carried out at room temperature for 0.5 h. (4) (2.27 g, 10.04 mmol) was added slowly dropwise at room temperature, the temperature was raised to 90 °C for 12 h. Pour the reaction solution into 400 mL ice water, filter, and dry to obtain a white solid compound (5) (2.28 g, 85.0%), m. p. 180–181 °C 1H NMR (400 MHz, CDCl3): δ 10.06 (s, 1H), 8.43–8.46 (m, 1H), 7.93–7.95 (m, 1H), 7.63–7.71 (m, 2H), 7.44 (s, 1H), 2.65 (s, 3H), 2.50 (s, 3H).
5-Acetoxy-3-methylnaphtho[1,2-b]furan-2-carboxylic acid (6).
2-Formyl-3-methylnaphthaleno[1,2-b]furan-5-acetate (5) (1.20 g, 4.47 mmol), acetone (30 mL), water (10 mL), 30% H2O2 (1.52 g, 13.43 mmol), NaH2PO4 (2.10 g, 13.43 mmol) were added to a 250 mL single-mouth flask at room temperature. Slowly add NaClO2 (1.52 g, 13.43 mmol) 20 mL aqueous solution dropwise for 30 min. Pour the reaction solution into 500 mL ice water, filter, and dry to obtain a white solid compound 6 (1.10 g, 87.0%), m.p. 230–232 °C. 1H NMR (400 MHz, CDCl3): δ 8.48 (d, J = 7.6 Hz, 1H), 7.93 (d, J = 7.6 Hz, 1H), 7.69–7.63 (m, 2H), 7.44 (s, 1H), 2.68 (s, 3H), 2.51 (s, 3H).
3-Methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxylic acid (1).
To a 100 mL single-mouth flask was added 5-acetoxy-3-methylnaphthaleno[1,2-b]furan-2-carboxylic acid (1.07 g, 3.76 mmol), THF (20 mL). NaOH (0.45 g, 11.28 mmol) was added slowly dropwise to 20 mL of aqueous solution at room temperature, and the dropwise addition was completed in 30 min. THF was removed from the reaction solution after 3 h of reaction at 60 °C. To the reaction was added acetone (25 mL), water (25 mL), IBX (1.12 g, 4.01 mmol) at room temperature, and the reaction solution was filtered after 3 h to give a red solid 3-methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxylic acid (0.77 g, 90.0%), m. p. 248–250 °C 1H NMR (400 MHz, DMSO-d6): δ (ppm) 13.57 (s, 1H), 8.02–7.94 (m, 2H), 7.80–7.79 (m, 2H), 2.53 (s, 3H).
General procedure for the synthesis of compounds A1–A21.
To a 25 mL vial was added 3-methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxylic acid (0.1 g, 0.39 mmol), DCM (5 mL), 1-ethyl-3(3-dimethylpropylamine) carbodiimide (EDCI) (0.11 g, 0.585 mmol), 1-hydroxybenzotriazole (HOBt) (0.08 g, 0.585 mmol), triethylamine (0.12 g, 0.585 mmol). Stir at room temperature for 1 hour, add amine (0.47 mmol), and extract the reaction solution with dichloromethane after 3 hours. Dry anhydrous magnesium sulfate, remove the organic solvent under vacuum, and purify the residue by silica gel column chromatography to obtain compound A1–A21.
N,3-Dimethyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A1).
Red solid (47 mg, 45%), m. p. 226–228 °C 1H NMR (400 MHz, CDCl3): δ (ppm) 8.12 (d, J = 7.8 Hz, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.70 (t, J = 7.4 Hz, 1H), 7.54 (t, J = 7.4 Hz, 1H), 6.40 (s, 1H), 3.30 (d, J = 4.9 Hz, 3H), 2.68 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 179.91, 175.26, 159.13, 158.88, 143.96, 135.39, 131.06, 130.79, 129.50, 127.91, 127.57, 122.38, 25.86, 9.89. HRMS (ESI) calcd for C15H12NO4 [M + H]+: 270.0688, found 270.0770.
N-Ethyl-3-methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A2).
Red solid (47 mg, 45%), m. p. 162–164 °C. 1H NMR (400 MHz, CDCl3): δ 8.11 (d, J = 7.7 Hz, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.69 (t, J = 7.5 Hz, 1H), 7.53 (t, J = 7.5 Hz, 1H), 6.36 (s, 1H), 3.56–3.49 (m, 2H), 2.67 (s, 3H), 1.30 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ 179.93, 175.28, 158.83, 158.44, 144.02, 135.38, 131.05, 130.75, 129.50, 127.90, 127.58, 122.56, 122.41, 34.14, 15.0, 9.90. HRMS (ESI) calcd for C16H14NO4 [M + H]+: 284.0845, found 284.0911.
N,N,3-Trimethyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A3).
Red solid (50 mg, 45%), m. p. 224–225 °C 1H NMR (400 MHz, CDCl3): δ (ppm) 8.09 (d, J = 7.2 Hz, 1H), 7.73(dd, J = 6.8 Hz, J = 0.8 Hz, 1H), 7.67 (dt, J = 7.5 Hz, J = 1.1 Hz, 1H), 7.51 (dt, J = 7.5 Hz, J = 1.1 Hz, 1H), 3.23(s, 3H), 3.15 (s, 3H), 2.49 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 180.14, 175.26, 160.46, 159.22, 145.08, 135.46, 130.80, 130.63, 129.34, 127.88, 126.22, 122.49, 121.59, 38.32, 35.90, 10.01. HRMS (ESI) calcd for C16H14NO4 [M + Na]+: 306.0845, found 306.0744.
N-Isopropyl-3-methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A4).
Red solid (50 mg, 43%), m. p. 117–119 °C 1H NMR (400 MHz, CDCl3): δ (ppm) 8.11 (dd, J = 7.8 Hz, J = 0.9 Hz, 1H), 7.80 (dd, J = 7.8 Hz, J = 0.9 Hz, 1H), 7.70 (dt, J = 7.6 Hz, J = 1.2 Hz, 1H), 7.53 (dt, J = 7.6 Hz, J = 1.2 Hz, 1H), 6.13(d, J = 7.4 Hz, 1H), 4.26–4.35 (m, 1H), 2.67 (s, 3H), 1.33 (s, 3H), 1.31 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 179.94, 175.29, 158.78, 157.76, 144.04, 135.37, 131.04, 130.74, 129.48, 127.87, 127.58, 122.62, 122.42, 41.39, 22.88, 9.92. HRMS (ESI) calcd for C17H16NO4 [M + Na]+: 320.3100, found 320.0903.
N-Isobutyl-3-methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A5).
Red solid (51 mg, 42%), m. p. 168 °C 1H NMR (400 MHz, CDCl3): δ (ppm) 8.11 (d, J = 7.7 Hz, 1H), 7.78 (dd, J = 7.7 Hz, 1H), 7.70 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 6.41(s, 1H), 3.30 (t, J = 6.6 Hz, 2H), 2.67 (s, 3H), 1.89–1.99 (m, 1H), 1.02 (s, 3H),1.01 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 179.93, 175.28, 158.33, 158.57, 144.05, 135.38, 131.06, 130.77, 129.51, 127.94, 127.59, 122.54, 122.45, 46.48, 28.76, 20.19, 9.93. HRMS (ESI) calcd for C18H18NO4 [M + Na]+: 334.1158, found 334.1071.
N-(2-Hydroxypropyl)-3-methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A6).
Red solid (46 mg, 38%), m. p. 210–211 °C. 1H NMR (400 MHz, CDCl3): δ 8.11 (d, J = 7.7 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.70 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 6.79 (s, 1H), 4.10–4.07 (m, 1H), 3.74–3.68 (m, 1H), 3.33–3.27 (m, 1H), 2.67(s, 3H), 2.24–2.20 (m, 1H), 1.30 (d, J = 6.2 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ 179.88, 175.26, 159.13, 159.06, 143.76, 135.43, 131.15, 130.78, 129.55, 128.44, 127.50, 122.66, 122.39, 67.41, 46.37, 21.22, 9.56. HRMS (ESI) calcd for C17H16NO35[M + H]+: 314.0950, found 314.1029.
N,N-Diethyl-3-methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A7).
Red solid (47 mg, 39%), m. p. 174–177 °C 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 7.7 Hz, 1H), 7.67–7.69 (m, 2H), 7.49–7.53 (m, 1H), 3.51–3.56 (m, 4H), 2.50 (s, 3H), 1.33 (s, 6H). 13C NMR (150 MHz, CDCl3): δ 180.10, 175.22, 172.70, 159.30, 159.23, 144.87, 135.45, 130.84, 130.64, 129.35, 127.83, 122.57, 121.61, 60.87, 27.40, 14.20, 9.94. HRMS (ESI) calcd for C18H18NO4 [M + H]+: 312.1158, found 312.1246.
3-Methyl-2-(pyrrolidine-1-carbonyl)naphtho[1,2-b]furan-4,5-dione (A8).
Red solid (52 mg, 43%), m. p. 209–210 °C 1H NMR (400 MHz, CDCl3): δ 8.11 (d, J = 7.7 Hz, 1H), 778 (d, J = 7.6 Hz, 1H), 7.69 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.41 (s, 1H), 6.68 (s, 1H), 6.35–6.37 (m, 2H), 4.65 (d, J = 5.7 Hz, 2H), 2.68 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 180.14, 175.29, 158.78, 158.40, 145.37, 135.41, 130.66, 129.45, 128.29, 127.92, 122.37, 121.86, 47.87, 46.97, 26.67, 23.76, 10.21.
N-Cyclopentyl-3-methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A9).
Red solid (45 mg, 36%), m. p. 224–226 °C. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 7.6 Hz, 1H), 7.78 (d, J = 7.6 Hz, 1 H), 7.70 (t, J = 7.3 Hz, 1H), 7.53 (t, J = 7.3 Hz, 1H), 6.25 (d, J = 7.0 Hz, 1H), 4.36–4.5 (m, 1H), 2.67 (s, 3H), 2.10–2.18 (m, 2H), 1.77–1.83(m, 2H), 1.65–1.71 (m, 2H), 1.51–1.59 (m, 2H).13C NMR (150 MHz, CDCl3): δ 179.92, 175.28, 158.77, 158.17, 144.07, 135.35, 131.02, 130.73, 129.47, 127.79, 127.56, 122.59, 122.41, 51.04, 33.26, 23.88, 9.91. HRMS (ESI) calcd for C19H18NO34[M + Na]+: 346.1158, found 346.1073.
N-(Furan-2-ylmethyl)-3-methyl-4,5-dioxo-4,5 dihydronaphtho[1,2-b]furan-2-carboxamide (A10).
Red solid (51 mg, 39%), m. p. 117–119 °C 1H NMR (400 MHz, CDCl3): δ 7.19 (d, J = 8.7 Hz, 2H), 6.86 (br, 2H), 3.12 (t, J = 5.2 Hz, 4H), 1.76–1.70 (m, 4H), 1.58–1.57 (m, 2H). 13C NMR (150 MHz, CDCl3): δ 179.86, 175.24, 159.07, 158.20, 150.71, 143.62, 142.54, 135.42, 131.16, 130.76, 129.52, 128.66, 127.46, 122.71, 122.36, 110.63, 108.11, 35.98, 9.95. HRMS (ESI) calcd for C19H14NO5 [M + H]+: 336.0794, found 336.0874.
3-Methyl-2-(piperidine-1-carbonyl)naphtho[1,2-b]furan-4,5-dione (A11).
Red solid (56 mg, 45%), m. p. 209–210 °C 1H NMR (400 MHz, CDCl3): δ (ppm) 8.09 (dd, J = 7.6 Hz, J = 0.6 Hz, 1H), 7.73 (dd, J = 7.6 Hz, J = 0.6 Hz, 1H), 7.67 (dt, J = 7.5 Hz, J = 1.1 Hz, 1H), 7.5 (dt, J = 7.5 Hz, J = 1.1 Hz, 1H), 3.64 (s, 4H), 2.4 (s, 3H), 1.70–1.75 (m, 6H). 13C NMR (150 MHz, CDCl3): δ 180.21, 175.29, 159.32, 159.15, 145.33, 135.45, 130.72, 130.61, 129.29, 127.96, 124.90, 122.53, 121.55, 48.15, 43.87, 25.95, 24.57, 9.85. HRMS (ESI) calcd for C19H18NO4 [M + Na]+: 346.1158, found 346.1064.
3-Methyl-2-(morpholine-4-carbonyl)naphtho[1,2-b]furan-4,5-dione (A12).
Red solid (52 mg, 41%), m. p. 189–191 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.97 (d, J = 7.4 Hz, 1H), 7.78 (d, J = 3.7 Hz, 2H), 7.58–7.62 (m, 1H), 3.66–3.68 (m, 8H), 2.36 (s, 3H).13C NMR (150 MHz, DMSO-d6): δ 179.54, 175.50, 158.59, 158.73, 144.14, 135.52, 131.17, 130.30, 129.80, 127.56, 125.50, 122.80, 121.53, 66.69, 40.53, 10.17. HRMS (ESI) calcd for C18H16NO5 [M + H]+: 326.0950, found 326.1032.
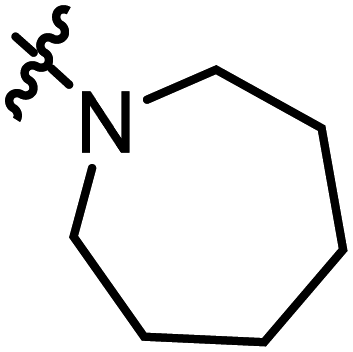
2-(Azepane-1-carbonyl)-3-methylnaphtho[1,2-b]furan-4,5-dione (A13).
Red solid (56 mg, 43%), m. p. 175–176 °C 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 7.7 Hz, 1H), 7.67–7.69 (m, 2H), 7.49–7.53 (m, 1H), 3.61–3.70 (m, 4H), 2.50 (s, 3H), 1.88 (s, 4H), 1.68 (m, 4H). 13C NMR (150 MHz, CDCl3): δ 180.21, 175.26, 160.35, 158.87, 145.73, 135.46, 130.68, 130.66, 129.33, 128.02, 126.03, 122.35, 121.72, 48.89, 46.99, 30.14, 27.50, 27.06, 26.91, 9.94. HRMS (ESI) calcd for C20H20NO4 [M + H]+: 338.1314, found 338.1402.
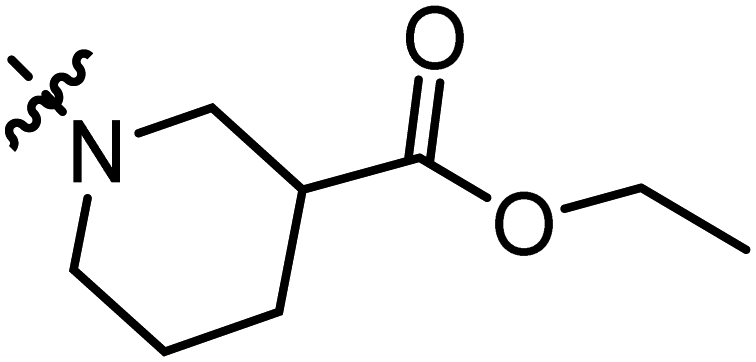
Ethyl 1-(3-methyl-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carbonyl)piperidine-3-carboxylate (A14).
Red solid (60 mg, 39%), m. p. 124–125 °C 1H NMR (400 MHz, CDCl3): δ (ppm) 8.10 (d, J = 7.7 Hz, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.68 (dt, J = 7.7 Hz, J = 0.9 Hz, 1H), 7.52 (dt, J = 7.7 Hz, J = 0.9 Hz, 1H), 4.13–4.19 (m, 4H), 3.33 (s, 1H), 3.16 (s, 1H), 2.62–2.69 (m, 1H), 2.47 (s, 3H), 2.16–2.20 (m, 1H) 1.82–1.92 (m, 2H), 1.60–1.70 (m, 1H), 1.23–1.26 (m, 3H). 13C NMR (150 MHz, CDCl3): δ 180.19, 175.22, 172.70, 159.30, 159.23, 144.87, 135.45, 130.84, 130.64, 129.35, 127.83, 122.57, 121.61, 60.87, 27.40, 14.20, 9.94. HRMS (ESI) calcd for C22H22NO6 [M + H]+: 396.1369, found 396.1459.
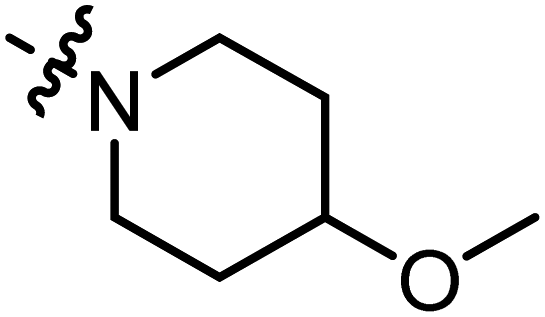
2-(4-Methoxypiperidine-1-carbonyl)-3-methylnaphtho[1,2-b]furan-4,5-dione (A15).
Red solid (58 mg, 43%), m. p. 177–179 °C 1H NMR (400 MHz, CDCl3): δ (ppm) 8.10 (d, J = 7.8 Hz, 1H), 7.71 (dd, J = 7.6 Hz, J = 0.8 Hz, 1 H), 7.67 (dt, J = 7.4 Hz, J = 1.0 Hz, 1H), 7.51 (dt, J = 7.4 Hz, J = 1.0 Hz, 1H), 3.89 (s, 2H), 3.52–3.57 (m, 3H), 3.40 (s, 3H), 2.45 (s, 3H), 1.94–1.99 (m, 2H), 1.71–1.79 (m, 2H). 13C NMR (150 MHz, CDCl3): δ 1180.15, 175.26, 159.34, 159.16, 145.05, 135.47, 130.81, 130.64, 129.32, 127.88, 125.50, 122.53, 121.58, 55.93, 30.38, 9.88. HRMS (ESI) calcd for C20H20NO5 [M + H]+: 354.1263, found 354.1356.
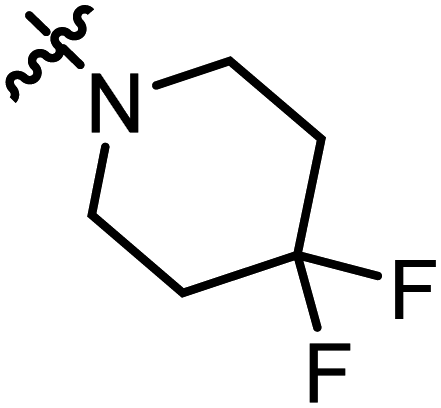
2-(4,4-Difluoropiperidine-1-carbonyl)-3-methylnaphtho[1,2-b]furan-4,5-dione (A16).
Red solid (63 mg, 45%), m. p. 213–214 °C. 1H NMR (400 MHz, CDCl3): δ 8.11 (d, J = 7.7 Hz, 1H), 7.71–7.69 (m, 2H), 7.56–7.52 (m, 1H), 3.84 (t, J = 5.0 Hz, 4H), 2.50 (s, 3H), 2.18–2.09 (m, 4H). 13C NMR (150 MHz, CDCl3): δ 179.91, 175.13, 159.38, 159.24, 144.26, 135.53, 131.08, 130.79, 129.42, 127.63, 127.32, 122.46, 121.65, 118.86, 34.38, 10.01. HRMS (ESI) calcd for C19H16F2NO4 [M + H]+: 360.0969, found 360.1052.
3-Methyl-2-(4-methylpiperazine-1-carbonyl)naphtho[1,2-b]furan-4,5-dione (A17).
Red solid (60 mg, 46%), m. p. 171–172 °C 1H NMR (400 MHz, CDCl3): δ 8.10 (dd, J = 7.7 Hz, J = 0.6 Hz, 1H), 7.66–7.74 (m, 2H), 7.49–7.54 (m, 1H), 3.77 (s, 4H), 2.54 (t, J = 4.4 Hz, 4H), 2.47 (s, 3H), 2.38 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 180.17, 175.22, 159.39, 159.14, 144.73, 135.48, 130.88, 130.66, 129.34, 127.79, 126.05, 122.56, 121.57, 55.06, 45.95, 9.96. HRMS (ESI) calcd for C19H19N2O4 [M + H]+: 339.1267, found 339.1362.
2-(4-(2-Hydroxyethyl)piperazine-1-carbonyl)-3-methylnaphtho[1,2-b]furan-4,5-dione (A18).
Red solid (61 mg, 39%), m. p. 176–178 °C 1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 7.72 Hz, 1H), 7.82–7.77 (m, 2H), 7.64–7.60 (m, 1H), 5.25 (s, 1H), 3.77 (s, 2H), 3.34–3.08 (m, 9H), 2.39 (s, 3H), 1.2 (t, J = 7.28, 1H). 13C NMR (150 MHz, CDCl3): δ 179.47, 175.47, 172.46, 158.85, 158.83, 143.66, 135.50, 131.31, 130.37, 129.81, 127.47, 126.67, 122.95, 121.58, 58.69, 56.07, 55.40, 51.96, 45.80, 10.28. HRMS (ESI) calcd for C20H21N2O4 [M + Na]+: 391.1372, found 391.1297.
3-Methyl-N-(2-morpholinoethyl)-4,5-dioxo-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A19).
Red solid (67 mg, 47%), m. p. 165–166 °C. 1H NMR (400 MHz, CDCl3): δ 8.12 (d, J = 7.7 Hz, 1H), 7.70–7.75 (m, 2 H), 7.13–7.17 (m, 1H), 7.0 (s, 1H), 3.78 (t, J = 4.4 Hz, 4H),3.57 (q, J = 5.7 Hz, 2H), 2.67 (s, 3H), 2.64 (d, J = 6.0 Hz, 2H), 2.57 (t, J = 3.9 Hz, 4H).13C NMR (150 MHz, CDCl3): δ 179.92, 175.27, 158.90, 158.46, 144.11, 135.46, 131.09, 130.79, 129.54, 127.77, 127.63, 122.37, 67.17, 56.76, 53.34, 35.26, 9.89. HRMS (ESI) calcd for C20H21N2O5 [M + H]+: 369.1372, found 369.1476.
3-Methyl-4,5-dioxo-N-(2-(piperidin-1-yl)ethyl)-4,5-dihydronaphtho[1,2-b]furan-2-carboxamide (A20).
Red solid (65 mg, 47%), m. p. 197–199 °C. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 7.7 Hz, 1H), 7.91 (s, 1H), 7.71 (t, J = 7.3 Hz, 1H), 7.52 (t, J = 7.3 Hz, 1H), 7.26 (s, 1H), 3.62 (d, J = 5.0 Hz, 2H), 2.73 (s, 2H), 2.67 (s, 7H), 1.76 (s, 4H), 1.56 (s, 2H). 13C NMR (150 MHz, CDCl3): δ 180.09, 175.36, 159.25, 158.68, 144.02, 135.60, 130.97, 130.57, 129.92, 123.04, 122.24, 58.47, 57.23, 54.33, 25.24, 23.72, 22.70, 9.89. HRMS (ESI) calcd for C21H23N2O4 [M + H]+: 367.1580, found 367.1655.
2-(4-Benzylpiperidine-1-carbonyl)-3-methylnaphtho[1,2-b]furan-4,5-dione (A21).
Red solid (69 mg, 43%), m. p. 195–197 °C. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 7.6 Hz, 1H), 7.73–7.66 (m, 2H), 7.51(td, J = 7.7 Hz, J = 1.2 Hz, 1H), 7.30 (t, J = 7.2 Hz, 2H),7.22 (t, J = 7.2 Hz, 1H), 7.15 (d, J = 7.2 Hz, 2H), 4.59 (s, 1H), 4.0 (s, 1H), 3.10–2.78 (m, 2H), 2.61 (d, J = 7.0Hz, 2H), 2.44 (s, 3H), 1.88–1.79 (m, 3H), 1.38–1.24 (m, 2H). 13C NMR (150 MHz, CDCl3): δ 180.18, 175.28, 159.34, 159.15, 145.22, 139.74, 135.45, 130.75, 130.63, 129.31, 129.07, 128.40, 127.92, 126.20, 125.14, 122.53, 121.55, 60.39, 38.31, 32.33, 9.89. HRMS (ESI) calcd for C26H24NO4 [M + H]+: 414.1627, found 414.1707.
5.3
In vitro cell growth inhibitory activity assays
All of these cancer cells and L-02 cells were seeded in 96-well culture plates at a density of 5000–7000 cells per well (100 μL per well) and incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 8 h. Next, the compounds were added in triplicate to the plates in 100 μL of fresh medium (total volume 200 μL, DMSO < 0.1%) at different concentrations and incubated at 37 °C for 48 h. After removing the cell culture medium, 10% Cell Counting Kit-8 (CCK-8, APExBIO, Houston, USA) solution (100 μL) was administered in the 96-well plates and re-incubated for 1 h (K562) and 1 h (HepG2). Finally, the absorbance was measured at the wavelength of 450 nm by a microplate spectrophotometer (MK3, Thermo, Germany). Each treatment was performed in triplicate. The half inhibitory concentration IC50 values were calculated by Prism 7.0 (GraphPad Software).
5.4 Microsomal stability
Human, Monkey, dog, rat, and mouse hepatic microsomes were purchased from Research Institute for Liver Diseases (Shanghai, China) Co., Ltd. The incubation mixture consisted of microsomal protein in PBS buffer (100 mM, pH = 7.4) and 100 μg mL−1A14 in a final volume of 100 mL. The concentration of human hepatic microsomal protein was 0.5 mg mL−1 (0.53 mg mL−1 of SD rat). A 0.83 mg mL−1 NADPH solution was prepared and added to the PBS buffer. After the addition of the NADPH-generating system, the resulting mixture was incubated at 37 °C for 0, 5, 15, 30, and 60 min. The reaction was terminated by the addition of methanol 300 mL containing diphenhydramine (0.02 ng mL−1). The mixture was vortexed for 1 min, centrifuged at 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 528 × g for 10 min at 4 °C. After centrifugation, 200 mL of the supernatant was transferred to 96-well plate, and 400 mL of purified water was added, and the solution was mixing with shaker at 500 rpm for 5 min, and the final concentration of compound A14 was analyzed by HPLC (Schimadzu LC-2010A with a Shimadzu Inertsil-SP C18 (5 μm; 150 mm × 4.6 mm).
528 × g for 10 min at 4 °C. After centrifugation, 200 mL of the supernatant was transferred to 96-well plate, and 400 mL of purified water was added, and the solution was mixing with shaker at 500 rpm for 5 min, and the final concentration of compound A14 was analyzed by HPLC (Schimadzu LC-2010A with a Shimadzu Inertsil-SP C18 (5 μm; 150 mm × 4.6 mm).
5.5 Plasma stability
Take 100 μL of human blank plasma, add A14 (1 μmol L−1), and incubate at 37 °C and 25 °C in a water bath for 0, 60 min. The reaction was terminated by adding ice-methanol and centrifuged at 4000 × g for 5 min. The supernatant was taken out 5 μL and the remaining amount of A14 in the sample was detected by API4000: LC–MS/MS-009.
5.6 Kinetic solubility of compound A14
Accurately aspirate 7.5 μL of 10 mM stock solution and add to 742.5 μL of phosphate buffer, vortex and mix for 2 min, fix on a shaker and shake for 1 h at room temperature. After shaking, 13000 rpm per centrifugation for 15 min. Aspirate 0.1 mL of supernatant into a new tube, shake and rinse for 5 min, then discard the liquid. Again, 0.5 mL of supernatant was transferred to a new tube, and the supernatant (or dilution) was taken at 13000 rpm per centrifuged for 15 min and analyzed by LC–MS injection. The sample concentration was quantified by fitting the calibration line (3–5 points).
5.7 Pharmacokinetics study in vivo
SPF-grade healthy male Wistar rats (3 females, 3 males, 8 weeks old), weighing 180 ± 20 g, were provided by Liaoning Changsheng Biotechnology Co., Ltd (Liaoning, China), license No. SCXK (Liao) 2015-0001. Wistar rats were housed under controlled environmental conditions at 22–24 °C, 50–60% relative humidity, natural circadian rhythm, free access to water and food, and acclimatized for one week. The experiments were approved by the Animal Management and Use Committee of Shenyang Pharmaceutical University, and the experiments conformed to the relevant experimental animal research guidelines of the Ethics Committee. 6 SD rats were randomly divided into two groups (half male and half female). Weigh before each dose to adjust the required volume. 3 SD rats were given 10 mg kg−1 of compound A14 by tail vein injection, and the other 3 SD rats were given 50 mg kg−1 of compound G1 by gavage. Blood samples (iv groups) were collected at different times (0 min, 2 min, 10 min, 15 min, 30 min, 60 min, 120 min, 240 min, 360 min, 480 and 1140 min). Blood samples (po groups) were collected at different times (0 min, 2 min, 10 min, 15 min, 30 min, 60 min, 120 min, 240 min, 360 min, 480 and 1140 min). Blood was collected from the orbital vein on an Eppendorf tube (1.5 mL) containing EDTA and then centrifuged at 4800 rpm for 10 minutes. Each plasma sample (50 μL) was mixed with 150 μL tolbutamide in acetonitrile (5 ng mL−1) and centrifuged at 13000 rpm for 10 min. Afterwards, 50 μL of supernatant was reconstituted with 200 μL of acetonitrile and the final concentration of compound A14 was analyzed by HPLC (Schimadzu LC-2010A with a Shimadzu Inertsil-SP C18 (5 μm; 150 mm × 4.6 mm).
5.8 Percentage plasma protein binding (%PPB)
The detailed experimental procedure followed the procedures reported of our previously work.34 The experimental assay was calibrated using warfarin of known %PPB: rat plasma (99.5%).
5.9 WB analysis of compound A14 in Hep G2 cells
Seeding Hep G2 cells into a 12 well plate. Add tested cmpds (final concetration was 30 μM, 1/3 diluent, 5 + 0 dose, final DMSO 0.3%) into the cell seeding plates, incubate at 37 °C & 5% CO2 incubator. Harvest cells and transfer into 1.5 mL microcentrifuge tube, centrifuge cells at 2000 rpm for 8 min at 4 °C. Wash cells once with ice-cold PBS, centrifuge at 4 °C. discard supernatant, lyse cells, incubate on ice for 30 min. Centrifuge at at 4 °C, gently transfer supernatant to microcentrifuge tube. Storage at −80 °C. Detect protein concentration by BCA kit. Load samples into gel and run the p-STAT3 detection by Jess.
5.10 Molecular docking
Molecular docking study was executed applying Schrodinger software package. Firstly, the X-ray crystal structure of STAT3 was retrieved from Protein Data Bank (PDB code: 1BG1) and prepared with the Protein Preparation Wizard model including the removement of one monomer, the addition of missing hydrogen atoms, the assignment of bond order, assessment of the correct protonation states, and a restrained minimization using the OPLS-2005 force field. Then, the receptor grid was generated at the centroid of selected residues (LYS591, ARG595) and the grid box size was set to 20 Å. After preparing the ligands, molecular docking was carried out using the standard precision (SP) with the default settings. Finally, the pictures were generated using pymol software.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 82273793), Major Basic Research Project of Natural Science Foundation of Shandong Province (ZR2018ZC1056, China), Taishan Industry Leading Talents Project, Project No: 2018TSCYCX-17, the Liaoning Revitalization Talents Program (XLYC1908031, China), Basic Research Project of Department of Education of Liaoning Province—natural sciences (2020LJC02, China).
References
- B. Petr, A. Vojtech, H. Ladislav and K. Rene, Curr. Pharm. Anal., 2009, 5, 47–68 CrossRef.
- J. R. Gómez Castellanos, J. M. Prieto and M. Heinrich, J. Ethnopharmacol., 2009, 121, 1–13 CrossRef PubMed.
-
(a) P. Arenas, J. Ethnopharmacol., 1987, 21, 279–295 CrossRef CAS PubMed;
(b) J. W. Bastien, J. Ethnopharmacol., 1983, 8, 97–111 CrossRef CAS PubMed;
(c) J. L. Hartwell, Lloydia, 1968, 31, 71–170 Search PubMed;
(d) M. L. S. Queiroz, M. C. Valadares, C. O. Torello, A. L. Ramos, A. B. Oliveira, F. D. Rocha, V. A. Arruda and W. R. Accorci, J. Ethnopharmacol., 2008, 117, 228–235 CrossRef CAS PubMed.
- M. M. Rao and D. G. Kingston, J. Nat. Prod., 1982, 45, 600–604 CrossRef CAS PubMed.
- K. Nagata, K. I. Hirai, J. Koyama, Y. Wada and T. Tamura, Antimicrob. Agents Chemother., 1998, 42, 700–702 CrossRef CAS PubMed.
- Y. Li, H. A. Rogoff, S. Keates, Y. Gao, S. Murikipudi, K. Mikule, D. Leggett, W. Li, A. B. Pardee and C. Li, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1839–1844 CrossRef CAS.
- C. Cafferkey and I. Chau, Expert Opin. Invest. Drugs, 2016, 25, 1023–1031 CrossRef CAS PubMed.
- N. Shahmarvand, A. Nagy, J. Shahryari and R. S. Ohgami, Cancer Sci., 2018, 109, 926–933 CrossRef CAS.
- E. J. Hillmer, H. Zhang, H. S. Li and S. S. Watowich, Cytokine Growth Factor Rev., 2016, 31, 1–15 CrossRef PubMed.
- M. Kujawski, M. Kortylewski, H. Lee, A. Herrmann, H. Kay and H. Yu, J. Clin. Invest., 2008, 118, 3367–3377 CrossRef CAS.
- S. Fletcher and J. Turkson, ChemMedChem, 2008, 3, 1159–1168 CrossRef CAS.
- M. McDaniel, K. E. Varley, J. Gertz, D. S. Savic, B. S. Roberts, S. K. Bailey, L. A. Shevde, R. C. Ramaker, B. N. Lasseigne, M. K. Kirby, K. M. Newberry, E. C. Partridge, A. L. Jones, B. Boone, S. E. Levy, P. G. Oliver, K. C. Sexton, W. E. Grizzle, A. Forero, D. J. Buchsbaum, S. J. Cooper and R. M. Myers, Oncotarget, 2017, 8, 8226–8238 CrossRef.
- H. L. Koskela, S. Eldfors, P. Ellonen, A. J. van Adrichem, H. Kuusanmaki, E. I. Andersson, S. Lagstrom, M. J. Clemente, T. Olson, S. E. Jalkanen, M. M. Majumder, H. Almusa, H. Edgren, M. Lepisto, P. Mattila, K. Guinta, P. Koistinen, T. Kuittinen, K. Penttinen, A. Parsons, J. Knowles, J. Saarela, K. Wennerberg, O. Kallioniemi, K. Porkka, T. P. Loughran, C. A. Heckman, J. P. Maciejewski and S. Mustjoki, N. Engl. J. Med., 2012, 336, 1905–1913 CrossRef PubMed.
- H. Yu, M. Kortylewski and D. Pardoll, Nat. Rev. Immunol., 2007, 7, 41–51 CrossRef CAS.
- A. V. Villarino, Y. Kanno, J. R. Ferdinand and J. J. O’Shea, J. Immunol., 2015, 194, 21–27 CrossRef CAS.
- D. E. Johnson, R. A. O’Keefe and J. R. Grandis, Nat. Rev. Clin. Oncol., 2018, 15, 234–248 CrossRef CAS PubMed.
- J. E. Darnell Jr., Nat. Rev. Cancer, 2002, 2, 740–749 CrossRef PubMed.
- J. Schust, B. Sperl, A. Hollis, T. U. Mayer and T. Berg, Chem. Biol., 2006, 13, 1235–1242 CrossRef CAS PubMed.
- X. Ren, L. Duan, Q. He, Z. Zhang, Y. Zhou, D. Wu, J. Pan, D. Pei and K. Ding, ACS Med. Chem. Lett., 2010, 1, 454–459 CrossRef CAS.
- K. Matsuno, Y. Masuda, Y. Uehara, H. Sato, A. Muroya, O. Takahashi, T. Yokotagawa, T. Furuya, T. Okawara, M. Otsuka, N. Ogo, T. Ashizawa, C. Oshita, S. Tai, H. Ishii, Y. Akiyama and A. Asai, ACS Med. Chem. Lett., 2010, 1, 371–375 CrossRef CAS PubMed.
- H. Chen, Z. Yang, C. Ding, L. Chu, Y. Zhang, K. Terry, H. Liu, Q. Shen and J. Zhou, ACS Med. Chem. Lett., 2013, 4, 180–185 CrossRef CAS.
- H. Song, R. Wang, S. Wang and J. Lin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4700–4705 CrossRef CAS.
- L. Lin, B. Hutzen, P. K. Li, S. Ball, M. Zuo, S. DeAngelis, E. Foust, M. Sobo, L. Friedman, D. Bhasin, L. Cen, C. Li and J. Lin, Neoplasia, 2010, 12, 39–50 CrossRef CAS.
- W. Yu, H. Xiao, J. Lin and C. Li, J. Med. Chem., 2013, 56, 4402–4412 CrossRef CAS PubMed.
- Y. Li, H. A. Rogoff, S. Keates, Y. Gao, S. Murikipudi, K. Mikule, D. Leggett, W. Li, A. B. Pardee and C. J. Li, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1839–1844 CrossRef CAS.
- S. Fiorito, F. Epifano, C. Bruyere, V. Mathieu, R. Kiss and S. Genovese, Bioorg. Med. Chem. Lett., 2014, 24, 454–457 CrossRef CAS PubMed.
- K. O. Eyong, P. S. Kumar, V. Kuete, G. N. Folefoc, E. A. Nkengfack and S. Baskaran, Bioorg. Med. Chem. Lett., 2008, 18, 5387–5390 CrossRef CAS.
- S. Bannwitz, D. Krane, S. Vortherms, T. Kalin, C. Lindenschmidt, N. Zahedi Golpayegani, J. Tentrop, H. Prinz and K. Muller, J. Med. Chem., 2014, 57, 6226–6239 CrossRef CAS.
- C. Li, C. Chen, Q. An, T. Yang, Z. Sang, Y. Yang, Y. Ju, A. Tong and Y. Luo, Eur. J. Med. Chem., 2019, 162, 543–554 CrossRef CAS.
- Q. Zhou, C. Peng, F. Du, L. Zhou, Y. Shi, Y. Du, D. Liu, W. Sun, M. Zhang and G. Chen, Eur. J. Med. Chem., 2018, 151, 39–50 CrossRef CAS.
- A. Reichstein, S. Vortherms, S. Bannwitz, J. Tentrop, H. Prinz and K. Muller, J. Med. Chem., 2012, 55, 7273–7284 CrossRef CAS PubMed.
-
T. Sone, W. Hirose, N. Shimada, C. J. Li, W. Li and D. Leggett, WO 2015/12030, 2015.
- K. R. Feng, F. Wang, X. W. Shi, Y. X. Tan, J. Y. Zhao, J. W. Zhang, Q. H. Li, G. Q. Lin, D. Gao and P. Tian, Eur. J. Med. Chem., 2020, 201, 112428 CrossRef CAS PubMed.
- P. Mehta, D. F. McAuley, M. Brown, E. Sanchez, R. S. Tattersall and J. J. Manson, Lancet, 2020, 395, 1033–1034 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a