
Tuning the annealing temperature to achieve heterostructured nanofibers for high performance lithium-ion batteries†
Shijin
Yu
 a,
Jiahao
Tong
a,
Ying
Wei
ab,
Tianrui
Chen
ab,
Xuannan
He
ab,
Huiqiang
Sui
a,
Cuiyun
Li
a,
Hua
Zhu
*a,
Qiuyun
Fu
*b and
LingBing
Kong
*c
a,
Jiahao
Tong
a,
Ying
Wei
ab,
Tianrui
Chen
ab,
Xuannan
He
ab,
Huiqiang
Sui
a,
Cuiyun
Li
a,
Hua
Zhu
*a,
Qiuyun
Fu
*b and
LingBing
Kong
*c
aSchool of Mechanical and Electrical Engineering, Jingdezhen Ceramic University, Jingdezhen 333001, Jiangxi, China. E-mail: zwchua@163.com
bSchool of Optical and Electronic Information, Engineering Research Center for Functional Ceramics of the Ministry of Education, Huazhong University of Science and Technology, Wuhan, 430074, Hubei, China. E-mail: fuqy@hust.edu.cn
cCollege of New Materials and New Energies, Shenzhen Technology University, Shenzhen 518118, Guangdong, China. E-mail: lbkongntunus@126.com
First published on 16th November 2022
Abstract
Heterointerfaces not only provide more sites for lithium ion storage, but also reduce the damage of electrode active materials, which are conducive to boosting the capacity and cycle stability of the electrode. This paper demonstrates for the first time that nanofibers with a NiO/Co3O4/NiCo2O4 heterostructure can be prepared by adjusting the annealing temperature. These heterostructured nanofibers are derived from as-spun Ni/Co nanofibers prepared by using electrospinning through thermal annealing at 500–800 °C. The phase formation and decomposition of spinel NiCo2O4 occur during heat treatment, and the composition is dependent on the annealing temperature. The electrochemical properties of the electrodes are correlated with the phase composition, particle size and morphology of the nanofibers. The nanofiber electrode annealed at 650 °C exhibits optimal electrochemical performances. Its specific capacity continues to increase at the initial stage of charge–discharge, and the activation cycle reached 61 cycles. The electrode has the largest boost value of 187.6 mA h g−1 (boost ratio, 21.08%), and its specific capacity reaches 1081.4 mA h g−1. The charge storage mechanism is analyzed by using cyclic voltammetry, electrochemical impedance spectroscopy and galvanostatic intermittent titration technique measurements. The contribution of electrode pseudo capacitance increases from 66.27% to 73.09% as the scan rate increases from 0.2 to 1.0 mV s−1, revealing that both the diffusion- and capacitive-controlled processes contributed. This is a facile and low-cost strategy to prepare high performance electrode materials for lithium-ion batteries, and stimulates the further development of phase-transition-induced synthesis of nanofibrous heterostructures.
1. Introduction
The increasingly serious global energy crisis has attracted the attention of governments and scientific researchers. Various high-performance energy conversion and energy storage devices have been developed. Lithium-ion batteries (LIBs) have the advantages of high energy/power density and long cycle life, and have been widely used in consumer electronics and electric vehicles (EVs).1–3 The application of commercial graphite anodes in energy storage systems is hindered by their low theoretical capacity.1,4–9 Transition metal oxides (TMOs) are considered as promising anode materials due to their low cost, high stability, and high theoretical capacity.10,11 A lithium intercalation reaction occurred at the carbonaceous electrodes, while TMO electrodes interacted with lithium through a phase inversion reaction (MOx + 2xLi ↔ M + xLi2O). Although TMO anodes exhibit a high specific capacity and impressive cycling stability, they suffer from poor electrical conductivity and large volume expansion during the charging–discharging process, which seriously limits their practical applications.5,12–17Compared with single phase metal oxide electrodes, binary metal oxide electrodes have the merits of higher structural stability and reversible capacity.6,18–20 For instance, compared with Co3O4 or NiO, spinel NiCo2O4 has higher theoretical capacity and conductivity.21 In addition, the elements in NiCo2O4 could have multiple oxidation states, thus offering multiple redox reaction electron pairs, such as Co3+/Co2+/Co and Ni2+/Ni. Therefore, the NiCo2O4 electrode exhibited enhanced electrochemical activity.22–24 NiCo2O4 based electrodes have been extensively studied, due to their synergistic effect as compared with cobalt oxide and nickel oxide based electrodes.21,25–27 Unfortunately, NiCo2O4 undergoes a large volume change during charging–discharging, resulting in electrode pulverization and consequent poor cycling performance.5,13
To address the above problems, developing one-dimensional nanostructures is an effective strategy.28–32 Electrospinning is a facile method to prepare nanofibers. The morphologies and surface functions of the nanofibers can be effectively adjusted by tuning the electrospinning parameters. It has been confirmed that NiCo2O4 nanofibers made by using electrospinning can be used as electrodes of LIBs, with alleviated volume expansion and improvement in conductivity.33–37 Zhang et al.13 reported polymer fibers by using electrospinning, which were carbonized in Ar to obtain NiCo2O4/NiO/carbon nanofibers. Li et al.18 reported a technique to prepare one-dimensional NiCo2O4 nanotubes.
Annealing temperature has a crucial effect on the crystallinity, crystal phase and morphology of nanoparticles.38–45 Siwatch et al.42 obtained three-dimensional flower-shaped NiCo2O4 nanoparticles by adjusting the annealing temperature. Do et al.46 demonstrated that the grain size of CoO increased nearly 5 times (from 6.59 to 31.5 nm) as the calcination temperature increased from 200 °C to 900 °C, and the activation period of the CoO electrode was also prolonged accordingly. Annealing treatment is an essential step for preparing anode materials with electrospinning, which removed high content polymers in the electrospun nanofibers and triggered the formation of nanocrystalline particles. Sundriyal et al.47 annealed as-spun Ca2Fe2O5 nanofibers at different temperatures. With an increasing annealing temperature, both the particle size and oxygen defect concentration are increased. In addition, the presence of a heterointerface provides more sites for Li-ion storage, which is beneficial to the specific capacity of the electrodes.48,49 Furthermore, the interaction of the multiphase interface can further suppress the damage of the active materials in the electrode.50
However, the effect of annealing temperature on the electrochemical performances of NiCo2O4 nanofibers has rarely been reported, while the formation of heterostructures during the high temperature annealing process has not been well studied. In this work, NiO/Co3O4/NiCo2O4 heterostructured nanofibers were prepared by using electrospinning and subsequent thermal annealing, and the formation mechanism of the heterostructures and the influence on the electrochemical performance were analyzed. It is found that NiCo2O4 decomposes at 550 °C, thus forming NiO/Co3O4/NiCo2O4 heterostructures. The particle size of the nanofiber increases as the annealing temperature becomes higher. The activation period (specific capacity continued to increase) of the electrode with NiO/Co3O4/NiCo2O4 heterostructured nanofibers annealed at 650 °C is 61 cycles and the specific capacity reached 1081.4 mA h g−1.
2. Experimental
2.1 Preparation of nanofibers
Polyvinylpyrrolidone (PVP) (K30) was added to a mixed solution of absolute ethanol and deionized water, and stirred for 2 h to form a clear solution. Then, cobalt nitrate hexahydrate and nickel nitrate hexahydrate (mass ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were slowly added to the solution and stirred to form a homogeneous precursor solution. It was then transferred to a needle cannula. The distance between the foil collector and the needle tip was adjusted to 20 cm, and a high voltage of 25 kV was applied while controlling the flow rate to 1 mL h−1. Finally, the precursor nanofibers were annealed to obtain nanofibers.
:1) were slowly added to the solution and stirred to form a homogeneous precursor solution. It was then transferred to a needle cannula. The distance between the foil collector and the needle tip was adjusted to 20 cm, and a high voltage of 25 kV was applied while controlling the flow rate to 1 mL h−1. Finally, the precursor nanofibers were annealed to obtain nanofibers.
2.2 Materials characterization
The phase compositions of the samples were examined by using an X-ray diffractometer (D8 Advance, Bruker Corporation). Field emission scanning electron microscopy (FESEM, JEOL JSM-7500F) and transmission electron microscopy (TEM, JEM-2100P) were used to observe the morphology and microstructure of the samples. Elemental compositions and valence states of the materials were analyzed by using X-ray photoelectron spectroscopy (XPS, Thermo K-Alpha).2.3 Electrochemical characterization
The active material, sodium carboxymethyl cellulose and conductive carbon black were mixed in deionized water with a mass ratio of 8:1:1 to form electrode slurry. This was coated on nickel foam and then dried in a vacuum oven at 80 °C for 12 h to obtain electrodes. The electrolyte was a mixture of 1 M LiPF6 in vinyl acetate, dimethyl carbonate and ethylene carbonate, with a volume ratio of 1:1:1. Microporous polymer membranes (Celgard 2400) and lithium foils were used as separators and counter electrodes, respectively, and coin cells (CR 2032) were assembled in a glove box. A LAND microcurrent test system (CT2001A) was used to test the charging–discharging performance of the cells. The charging–discharging voltages were in the range of 0.005–3 V and cells must be allowed to stand for 12 h before the testing. The galvanostatic intermittent titration technique (GITT) was performed by charging/discharging at 100 mA g−1 for 20 min and then standing for 120 min until the cut-off voltage was reached. An electrochemical workstation (CHI660e, Chenhua, China) was used to measure the electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV).
3. Results and discussion
The as-spun fibers of the Ni/Co compound with PVP as the plasticizer are shown in Fig. S1 (ESI†). The diameters of the Ni/Co compound fibers are 430 ± 40 nm and their surface is smooth. During the annealing process, after solvent volatilization and decomposition of PVP, nanofibers composed of nickel-cobalt oxides are formed.51 In order to identify the appropriate annealing temperature range, the as-spun fibers were heated to 1000 °C in air at a heating rate of 10 °C min−1, and the corresponding TGA curves are shown in Fig. 1a. The weight loss below 170 °C is 12.6%, which is mainly caused by the volatilization of the solvent. Then, the decompositions of cobalt nitrate and nickel nitrate occur between 170 °C and 330 °C, leading to a net weight loss of 6.8%. A sharp weight loss of 67.1% occurs in the temperature range of 330–500 °C due to the decomposition of PVP. It is worth noting that the weight change in the 4th temperature range (500–800 °C) shows an approximately flat curve, with a weight loss of only 0.26%. The weight loss related to the spinel NiCo2O4 is generally considered to be the decomposition of NiCo2O4 and the release of O2, which also leads to a change in the oxidation state of the metal cations.39,43 Therefore, the annealing temperature of the as-spun fibers is determined to be 500–800 °C.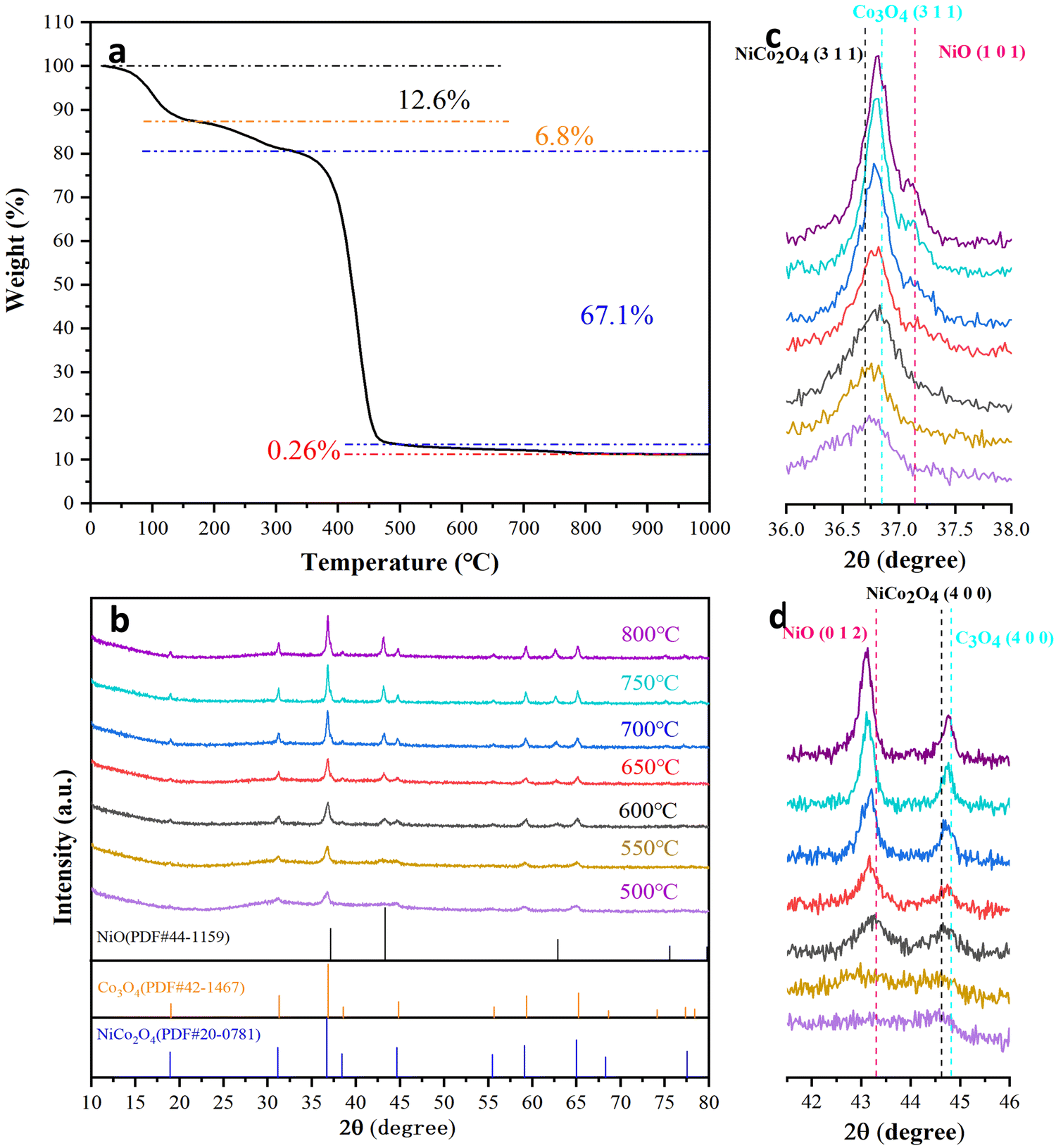 | ||
| Fig. 1 (a) TG curve of the as-spun Ni/Co compound fibers. (b) XRD patterns of the fibers annealed at different temperatures. The zoom-in region of 36°–38° (c) and 42°–46° (d). The standard Bragg peaks for NiO, Co3O4 and NiCo2O4 are marked. | ||
The controlled relationship between the NiCo2O4 spinel phase and the NiO/Co3O4/NiCo2O4 multiphase has been obtained, and the formation mechanism of the NiO/Co3O4/NiCo2O4 heterostructure is further explored. The samples annealed at 500, 550, 600, 650, 700, 750 and 800 °C are denoted as A-500, A-550, A-600, A-650, A-700, A-750 and A-800, respectively. With increasing annealing temperature, the diffraction peaks become stronger and the half-widths of the peaks become smaller (Fig. 1b). According to the Scherrer formula, crystal size increases with increasing annealing temperature.52 In the XRD pattern of the A-500 sample, only spinel NiCo2O4 is present (PDF#20-0781), which indicates that single phase spinel NiCo2O4 is formed firstly. When the annealing temperature is higher than 500 °C, NiO characteristic peaks are observed at 37.1°, 43.3° and 62.9° (Fig. 1c, d and Fig. S2, ESI†), which are sharper and sharper with increasing annealing temperature. The peaks of NiO in the A-650 sample shifted slightly to a lower angle than the characteristic peaks of the standard card. The magnitude of the left-shift is amplified with increasing annealing temperature. According to the Bragg law, an increase in interplanar spacing causes the diffraction peaks to shift to lower angles.53,54 It is necessary to emphasize that the molar ratio of Co:Ni in the precursor solution is 2:1. There are diffraction peaks of NiO in the XRD patterns of the samples annealed above 500 °C, which means that NiCo2O4 is decomposed into NiO and Co3O4 at high temperatures. Because the diffraction peaks of Co3O4 overlap with those of NiCo2O4, it is necessary to use the zoomed-in view to distinguish between them.39,55,56Fig. 1c shows a zoomed-in view near 36.7°, where the peak is a superposition of the NiCo2O4 and Co3O4 diffraction peaks. A slight right shift occurs with increasing annealing temperature. A similar phenomenon is observed around 44.6° (Fig. 1d). Therefore, the content of Co3O4 also increases with increasing annealing temperature.
The oxidation state of metal cation is closely related to the phase transition. Fig. 2 shows the XPS spectra and fitting curves of the samples, revealing the presence of Ni, Co and O in the nanofibers. As illustrated in Fig. 2b, the Co 2p spectra exhibit Co3+ peaks at 779.7 and 796.3 eV and Co2+ peaks at 781.1 and 795.9 eV. According to the relative intensities of the Co3+ and Co2+ peaks, Co3+ is dominant. Similarly, the peaks in Fig. 2c (Ni 2p spectra) at 874.0 and 855.4 eV belong to Ni3+, while those at 871.7 and 853.6 eV are assigned to Ni2+. The two satellite peaks at 861.1 and 879.6 eV are ascribed to the two nickel oscillation peaks on the high binding energy side of Ni 2p3/2 and Ni 2p1/2. The O 1s spectra present three oxygen contributions of O 1 (529.2 eV), O 2 (530.7 eV), and O 3 (532.1 eV), as observed in Fig. 2d, corresponding to the lattice oxygen (O(L)), oxygen deficiency and adsorbed water (H2O), respectively. The chemical states of Ni, Co and O basically confirm the presence of NiO, Co3O4 and NiCo2O4. The presence of Ni3+ can only be related to the formation of NiCo2O4, which strongly proves the incomplete chemical reaction between NiCo2O4, Co3O4 and NiO.55,57,58 As the annealing temperature increases, the proportion of Co2+ peak area decreases (Fig. 2b and c), i.e., the content of Co2+ in the samples decreases. However, the proportion of Ni2+ peak area in the fitted Ni peak remains unchanged, indicating that the chemical state of Ni is stable. Notably, the Ni 2p peak of the A-650 sample has a 0.4 eV (853.5 eV → 853.9 eV) shift relative to that of the standard card. In addition, the Co 2p peak of this sample also exhibits a slightly positive shift (779.5 eV → 779.7 eV). These results strongly suggest the presence of strong electronic interactions among NiCo2O4, NiO, and Co3O4, i.e., a NiO/Co3O4/NiCo2O4 heterostructure has been formed.25,54,59,60
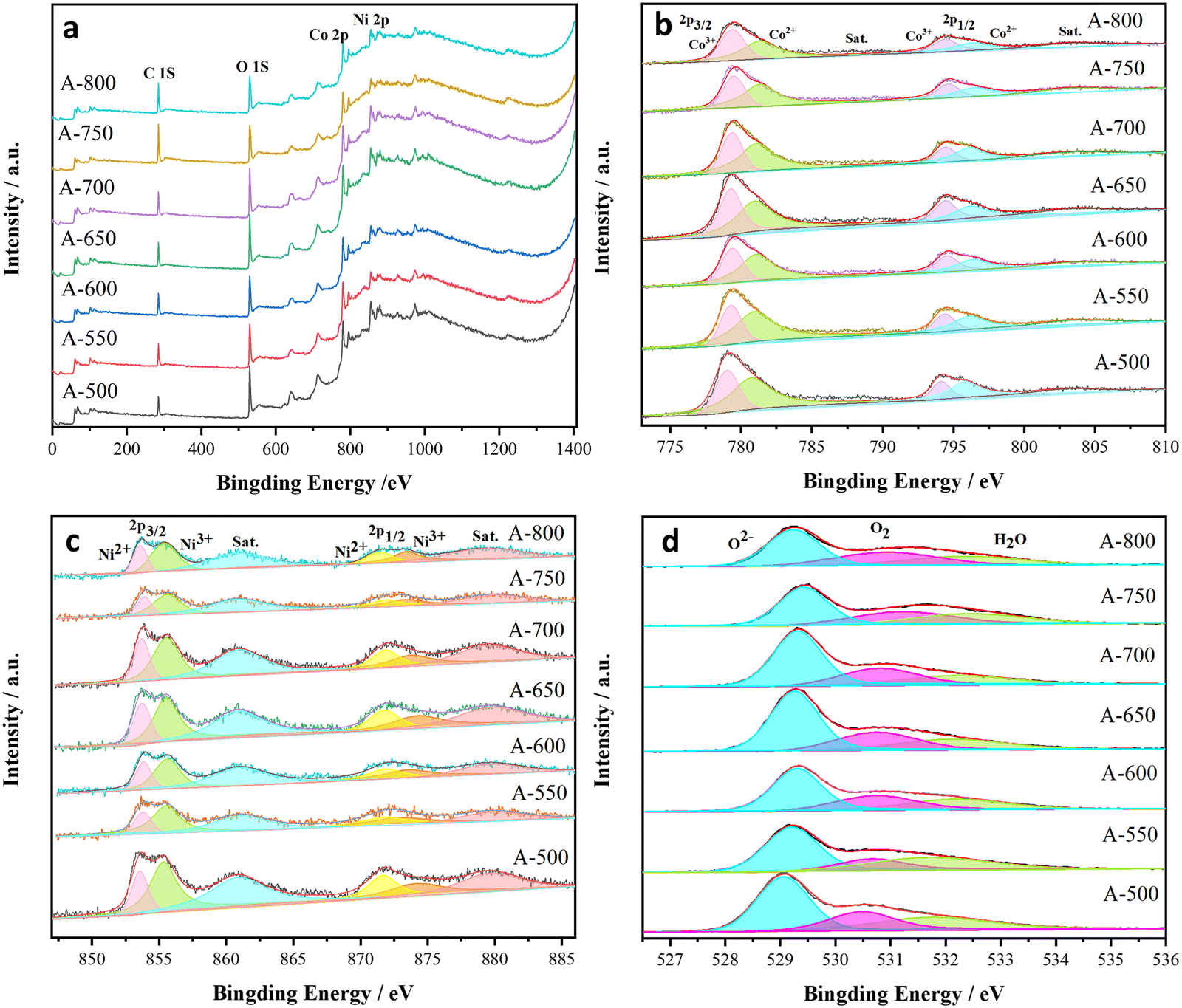 | ||
| Fig. 2 XPS spectra of the nanofibers annealed at different temperatures: survey spectra (a), and high-resolution of Co 2p(b), Ni 2p(c) and O 1s(d). | ||
Fig. 3 shows the morphologies of the nanofibers prepared at different annealing temperatures. A-500 nanofibers (Fig. 3a and d) consist of many small grains with diameters of about 40 nm. There are many voids between the grains, which may be the pores left after solvent evaporation and PVP decomposition.61 The grain size in the nanofibers is increased significantly as the annealing temperature is raised. Notably, both large particles with diameters of about 90 nm and small grains of about 10 nm are observed in A-600 nanofibers (Fig. 3c and f). This is mainly because the grains grow significantly at high annealing temperatures, consuming the surrounding grains. A-700 nanofibers composed of many larger particles in series (Fig. 3h and k) and have rough surfaces. Compared with A-500, the inter-grain voids of A-700 nanofibers disappeared and a small number of breakpoints appeared instead. Similar results were found in Ca2Fe2O5 nanofibers reported by Sundriyal et al.47 The morphology of A-650 nanofibers is similar to that of A-600 and A-700 samples, that is, there are large particles, small particles and a few breakpoints, which may provide a large specific surface area, abundant active sites and charge transfer channels. The grains of the A-800 sample (Fig. 3m and n) grow rapidly to about 220 nm, forming a cross-linked structure while the fiber morphology disappears. The variation trends of grain size observed in the SEM and TEM (Fig. 3a–n) images are consistent with the XRD spectra analysis results (Table S1, ESI†).
 | ||
| Fig. 3 Morphology of the nanofibers at different annealing temperatures. SEM images: (a) A-500, (b) A-550, (c) A-600, (g) A-650, (h) A-700, (i) A-750 and (m) A-800. TEM images: (d) A-500, (e) A-550, (f) A-600, (j) A-650, (k) A-700, (l) A-750 and (n) A-800. | ||
Fig. 4 shows the high-resolution TEM (HRTEM) analysis results of a representative sample. The elemental mapping profiles of the A-650 sample are shown in Fig. 4b–e, indicating that elements Ni, Co, and O are uniformly distributed throughout the nanofibers. Fig. 4f is an HRTEM image of the NiO/Co3O4/NiCo2O4 heterostructure in the A-650 sample. Three nanocrystals are closely packed with two clear interfaces (yellow dashed line). The interplanar spacings (0.457 nm, 0.209 nm, and 0.247 nm) can be calculated from the fast Fourier transform of the regions marked by the three dashed squares. They are well matched to the (111) plane of Co3O4 (No: 42-1467), the (012) plane of NiO (No: 44-1159) and the (311) plane of NiCo2O4 (No: 20-0781), respectively. Therefore, it is confirmed that there are heterogeneous interfaces of Co3O4/NiCo2O4 and NiO/NiCo2O4 in the nanofibers. The presence of such heterointerfaces would significantly promote electron transfer.
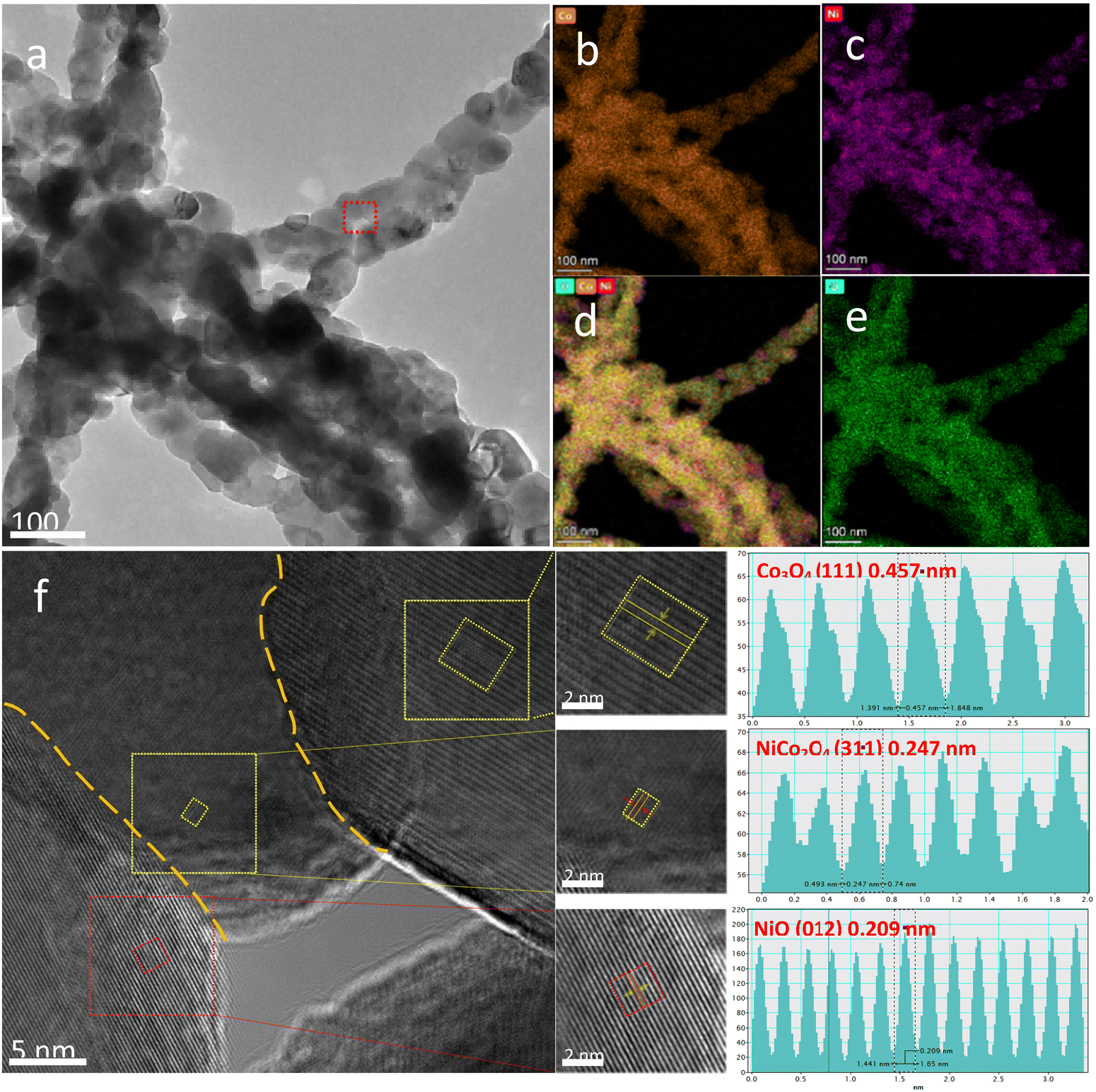 | ||
| Fig. 4 (a) TEM image of the A-650 sample. (b–e) Elemental mapping images. (f) HRTEM image and its lattice fringes. | ||
Fig. 5a–d and Fig. S3 (ESI†) present the initial three CV curves of the nanofiber electrodes. A weak shoulder and a strong reduction peak are observed at ∼0.5 V and ∼0.75 V, respectively, in the first cathodic sweep of the A-500 electrode. The shoulder originates from the formation of SEI films, while the strong peak is attributed to the reduction of NiCo2O4 to metallic Ni and Co.6,18,62 Fig. S3a (ESI†) depicts the CV curves of pure Co3O4 and pure NiO nanofiber electrodes. The reduction peaks of the first cycle for the Co3O4 and NiO electrodes are located at ∼0.76 V and ∼0.5 V, respectively. When the annealing temperature is ≥550 °C, the spinel NiCo2O4 partially decomposes and NiO is produced. The peak at ∼0.5 V may be associated with the formation of the SEI films and the reduction of NiO to metallic Ni.39,56 With increasing annealing temperature, more NiCo2O4 is decomposed, and the corresponding NiO content increases gradually. The contribution of the NiO reduction peak dominates, and the peak at ∼0.5 V is significantly higher. Interestingly, the change of the peak at ∼0.75 V with the annealing temperature is relatively complicated, that is, the peak height first decreases and then increases, and the peak width changes in the opposite trend. When annealed at relatively low temperatures (550–650 °C), the content of NiCo2O4 in the sample gradually decreases, while the proportion of Co3O4 increases accordingly. The heights of the superimposed peaks decrease and the peak widths increase accordingly. The reduction peaks of NiCo2O4 and Co3O4 are very close at about 0.75 V (difference is about 0.01 V). The superimposed peak height of the A-700 electrode increases and the peak width is obviously narrowed. Probably, the content of Co3O4 in the A-700 electrode has exceeded that of NiCo2O4, so that the reduction peak of Co3O4 is dominant.39 This observation is in agreement with the results shown in the XRD patterns (Fig. 1c and d). In addition, a slight shift in the NiCo2O4 reduction peak is observed in different samples, which may be related to the different contents of the spinel phase.43 In the first anodic sweep, the ∼1.6 V broad peak is attributed to the oxidation of metallic Ni to Ni2+, while the ∼2.1 V broad peak corresponds to the oxidation of metallic Co to Co2+.66 In the second cycle of all electrodes, the reduction peak at ∼0.5 V is merged because the SEI films are stable and don't consume more Li ions.63 On the other hand, the reduction peaks at ∼0.7 V shift to a high potential by ∼0.35 V, which may be related to the reversible reduction and the activation of the electrode material. Meanwhile, the oxidation peaks in the anodic scan did not change significantly.62,64 Compared with that of the A-500 electrode, the reduction peak of the A-650 electrode shifts to a lower potential, which indicates that the NiO/Co3O4/NiCo2O4 heterostructure increases the conductivity of the electrode and reduces the irreversible capacity. Therefore, the detailed electrochemical reactions can be expressed as follows:6
| NiCo2O4 + 8Li+ + 8e− → Ni + 2Co + 4Li2O | (1) |
| NiO + 2Li+ + 2e− ↔ Li2O + Ni | (2) |
| Co3O4 + 2Li+ + 2e− ↔ Li2O + 3CoO | (3) |
| CoO + 2Li+ + 2e− ↔ Li2O + Co | (4) |
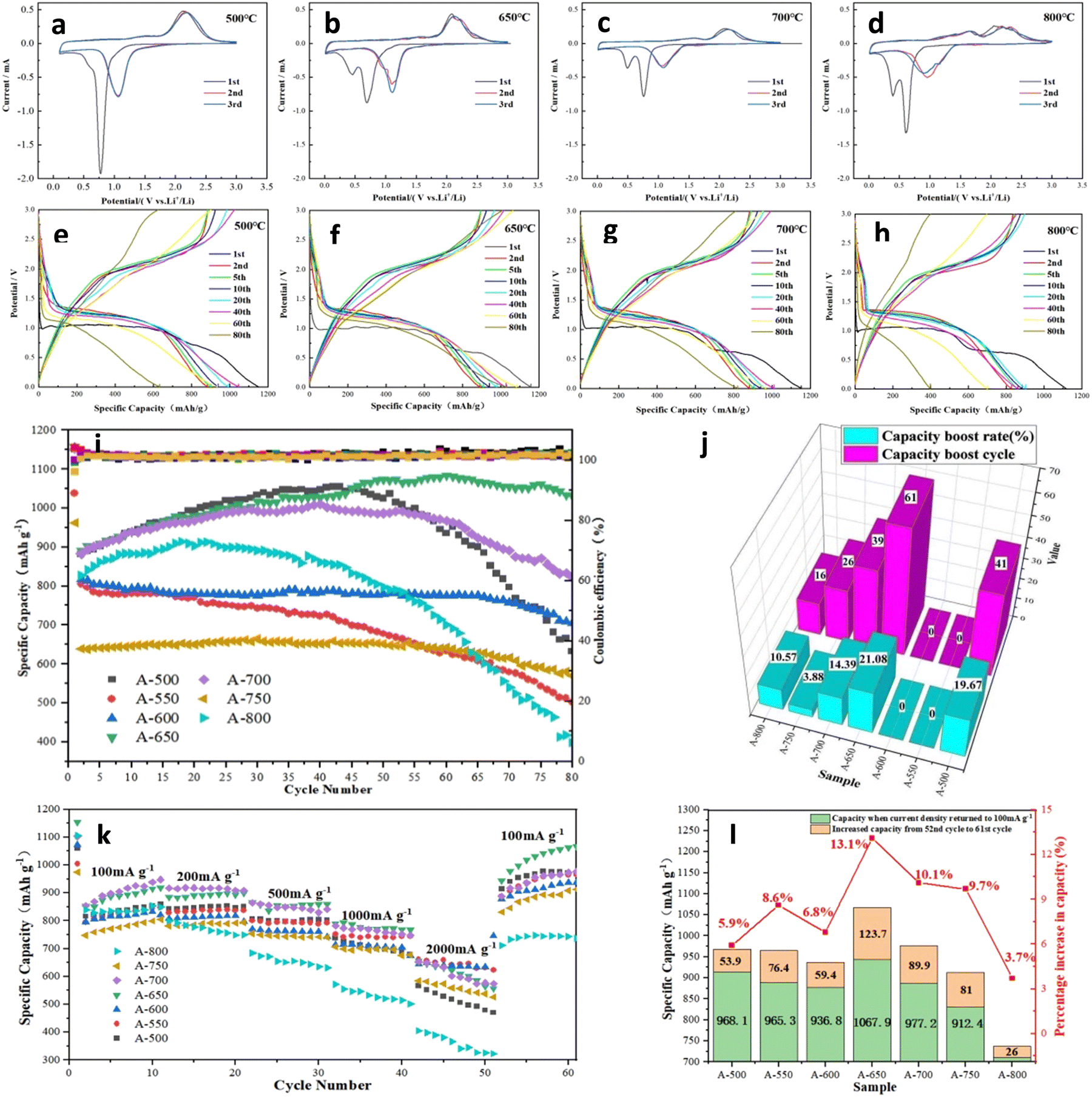 | ||
| Fig. 5 CV curves (a–d) and charge–discharge curves (e–h) of A-500, A-650, A-700, and A-800 nanofiber electrodes. (i) Cycling performance of nanofiber electrodes at 100 mA g−1. (j) Activation period cycles and capacity boost rates of electrodes. (k and l) Rate performance of nanofiber electrodes. | ||
Fig. 5 shows the electrochemical performance of the nanofiber electrodes. The galvanostatic charge–discharge curves were recorded at a current density of 100 mA g−1 in the potential range from 0.005 V to 3.0 V (V vs. Li/Li+), as shown in Fig. 5e–h. There is a significant capacity drop in the second cycle, and the irreversible capacity loss originates from the lithium and electrolyte being consumed during the SEI formation.65 A-650 electrode exhibits a stronger curve overlap than the other electrodes, indicating its excellent cycling stability. Fig. 5i shows the cycling performance at a current density of 100 mA g−1. Impressively, many electrodes exhibit an increase in specific capacity during the first few dozen cycles. As described in the open literature, the high catalytic activity of cobalt-based oxides may lead to the decomposition of the electrolyte into a gel-like electrolyte membrane. Different from the SEI film, the formation and decomposition of the gel electrolyte films are partially reversible, which also partially contribute to the capacity of the battery.37,40,46 The activation period and specific capacity boost value of the electrodes are compared in Fig. 5j and Table S2 (ESI†). Among them, A-650 and A-700 electrodes have the longest activation period, while the capacity improvement periods are 61 and 39 cycles, respectively. In terms of the boost in specific capacity, the A-650 electrode has the largest boost value of 187.6 mA h g−1 (boost ratio 21.08%), and its specific capacity reaches 1081.4 mA h g−1 (Fig. 5j). The long activation period and large capacity boost rate of the A-650 electrode may be ascribed to the presence of an appropriate amount of the heterostructures in the materials. Firstly, the uniformly distributed NiO/Co3O4/NiCo2O4 heterostructures in the electrode provides enough internal space to alleviate the volume expansion.13,50 Secondly, the heterostructures can modify the charge distribution of the electrode, resulting in a built-in electric field that provides channels for Li-ion storage and transport.66 Finally, the fragmentation of the large particles in the electrode leads to a larger specific surface area and more active sites of the active material, thereby increasing the specific capacity of the electrode. However, the A-800 electrode not only has a low initial capacity, but also decays rapidly. The specific capacity at the 80th cycle is only 387.8 mA h g−1, which is comparable to that of a commercial graphite electrode. This can be attributed to the excessively large particles of the active materials being very fragile during charge–discharge cycling and they move away from the current collector, resulting in active material loss and electrical insulation.67
Fig. 5k demonstrates the rate performances of A-500, A-550, A-600, A-650, A-700, A-750 and A-800 electrodes at current densities from 100 to 2000 mA g−1. The specific capacities of A-600, A-650 and A-700 electrodes remain relatively stable at different current densities, which may be related to the appropriate content ratio of NiO, Co3O4 and NiCo2O4 in the materials, so that they have higher activity and capacity stability.55,68 For the A-650 electrode, the specific capacities are 918.8, 900.1, 859.8, 767.3 and 556.3 mA h g−1 at current densities of 100, 200, 500, 1000 and 2000 mA g−1, respectively. In contrast, the specific capacity of the A-800 electrode dropped sharply, with a value of only 322.8 mA h g−1 at 2000 mA g−1. It is particularly noteworthy that the specific capacity of the A-650 electrode immediately recovers to 944.2 mA h g−1 at the current recovery of 100 mA g−1 and continues to boost to 1067.9 mA h g−1, which is much higher than that of the 10th cycle (918.8 mA h g−1). Fig. 5l and Table S3 (ESI†) show the capacity recovery of the electrodes when the current density is restored from 2000 to 100 mA g−1, and compare the capacity boost during the following 10 cycles. A-650 and A-700 electrodes have specific capacity recovery capabilities of 387.9 and 313.7 mA h g−1, respectively. The specific capacities of all electrodes continue to increase over the next 10 cycles. Among them, A-650 and A-700 electrodes have the larger specific capacity boost values, which are 123.7 and 89.9 mA h g−1, respectively, and specific capacity boost rates of 13.1% and 10.1%. This might be attributed to the structural reorganization and further activation of the material after high current charging and discharging.
The electrochemical properties of the NiCo2O4 based electrodes in this work and those reported in open literature are listed in Table S4 (ESI†). Some NiCo2O4 composite electrodes with carbonaceous materials (e.g., graphene) have high specific capacity. In contrast, the pristine NiCo2O4 electrodes usually deliver lower specific capacities (from 520 to 1198 mA h g−1). Obviously, the specific capacity of the NiO/Co3O4/NiCo2O4 heterostructure electrode in this work is higher than those of most pristine NiCo2O4 electrodes and some NiCo2O4 composite electrodes. Furthermore, the electrospinning method and annealing treatment used in this work are facile and low-cost.
The potential hysteresis of the electrode is closely related to the lithium-ion diffusion kinetics and energy efficiency, which is a crucial factor for practical applications.69 GITT was used to characterize the potential hysteresis of the electrodes in this work. Electrode potential versus time is shown in Fig. 6a and Fig. S4, S5 (ESI†). After charging/discharging for 20 min at 100 mA g−1, the cells were left to relax for 120 min. A long time is used for sufficient relaxation to allow lithium ions to reach equilibrium potential and minimize the self-discharge of the electrode. The current pulse polarization curves of A-500 and A-650 electrodes are shown in Fig. 6a. The GITT curves of all electrodes have similar variation trends. When the cells are charged to a higher voltage or discharged to a lower voltage, the polarization processes take a longer time to reach steady states. It is shown that the lithium-ion diffusion rate varies with the electrode potential. Overpotential is the voltage difference between the equilibrium potential at the end of relaxation and that at the end of current pulse (≈ΔEτ − ΔEs, ignoring the IR drop).70 The overpotentials of the A-500 and A-650 electrodes are listed in Table S5 (ESI†), where the overpotentials during charging are higher than those during discharging. The overpotential and potential hysteresis effect of the A-650 electrode is significantly lower than that of the A-500 electrode, which indicates that the A-650 electrode has a higher lithium ion diffusion rate and lower electrode polarization.
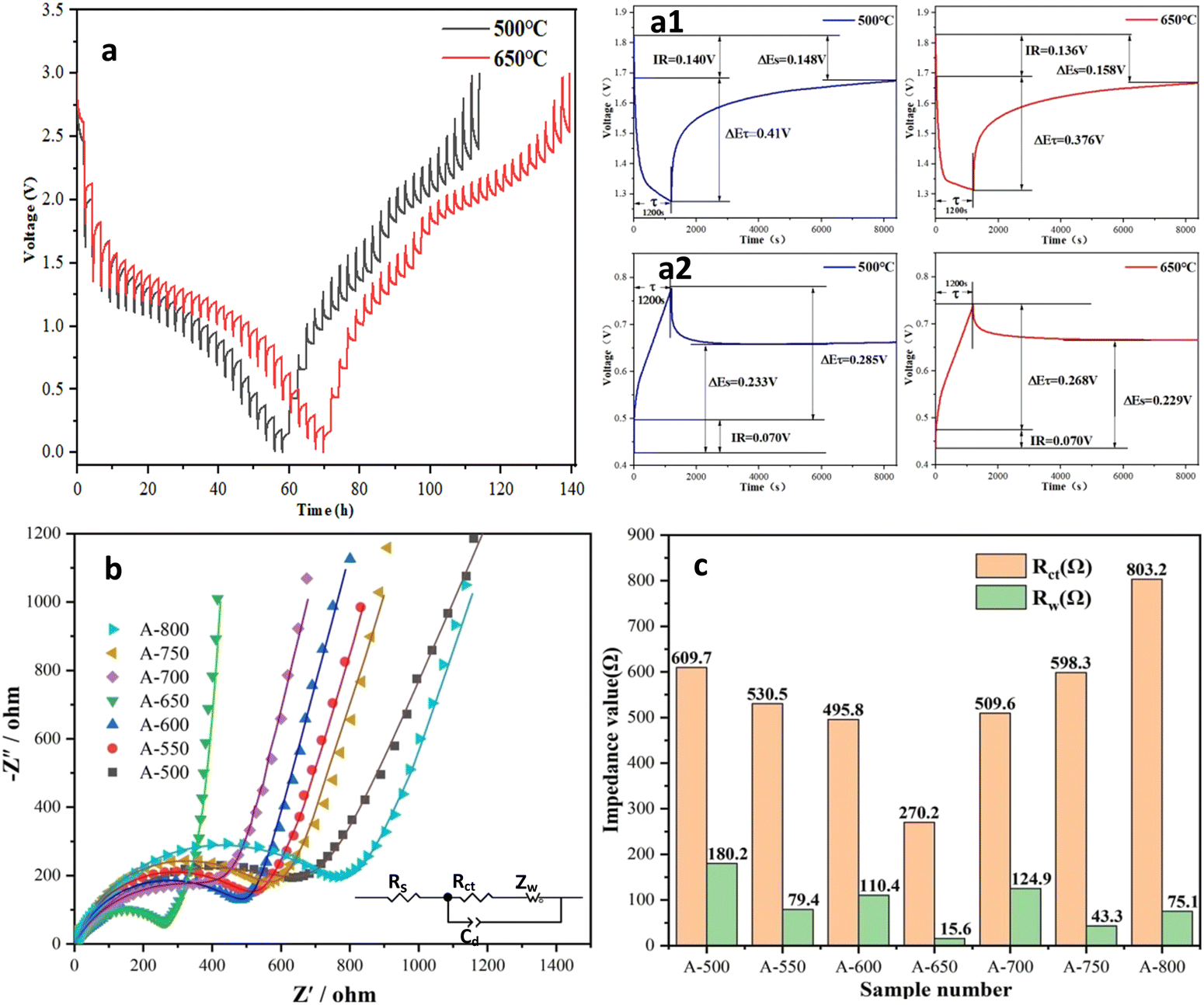 | ||
| Fig. 6 (a) GITT curves of A-500 and A-650 electrodes. EIS spectra of the nanofiber electrodes (b) and fitted Rct and Zw values (c). | ||
In this work, EIS was used to evaluate the reaction kinetics of the electrodes, and impedance spectra were recorded in the frequency range of 100 KHz–10 MHz. Fig. 6b shows the Nyquist plots of electrodes after standing for 12 h. The Nyquist plots consist of a recessed semicircle in the high frequency region and an oblique line in the low frequency region. Its semicircle is related to the charge transfer at the electrode/electrolyte interface. The oblique line shows the characteristic of the Warburg impedance, which is attributed to the diffusion of lithium ions in the electrode. The fitted equivalent circuits are shown in Fig. 6b (inset), the surface film resistance (Rsf) and their impedance components (CPEsf), electrolyte resistance (Re), charge transfer resistance (Rct) and the corresponding double layer (dl) CPE (Rct + CPEdl), and Warburg impedance (W) can be extracted from the Nyquist fitted curve. The fitting Rct and Zw values for the electrodes are listed in Fig. 6c and Table S6 (ESI†). The semicircle diameter of the A-650 electrode is smaller than that of other electrodes, while its Rct value is also much lower than that of all other electrodes. It is found that the A-650 electrode possesses a fast charge transfer capability and high electrical conductivity, which will contribute to faster lithiation/delithiation kinetics.71 This observation is consistent with that of the GITT test. Therefore, the EIS results also confirm that the A-650 sample has a high cycling performance (Fig. 5i) and rate capability (Fig. 5k).
To elucidate its charge storage mechanism, the CV measurements of electrodes were performed at scan rates from 0.2 to 1.0 mV s−1 (Fig. 7 and Fig. S6, ESI†) to distinguish diffusion-controlled and capacitive-controlled capacities. In order to obtain more accurate fitting data, the influences of Ohmic resistance and scan reversal residual current were eliminated successively by removing polarization and current residual.72 The relationship between the scan rate (v) and the current (i) can be described as:
| i = avb | (5) |
| log(i) = blog(v) + log(a) | (6) |
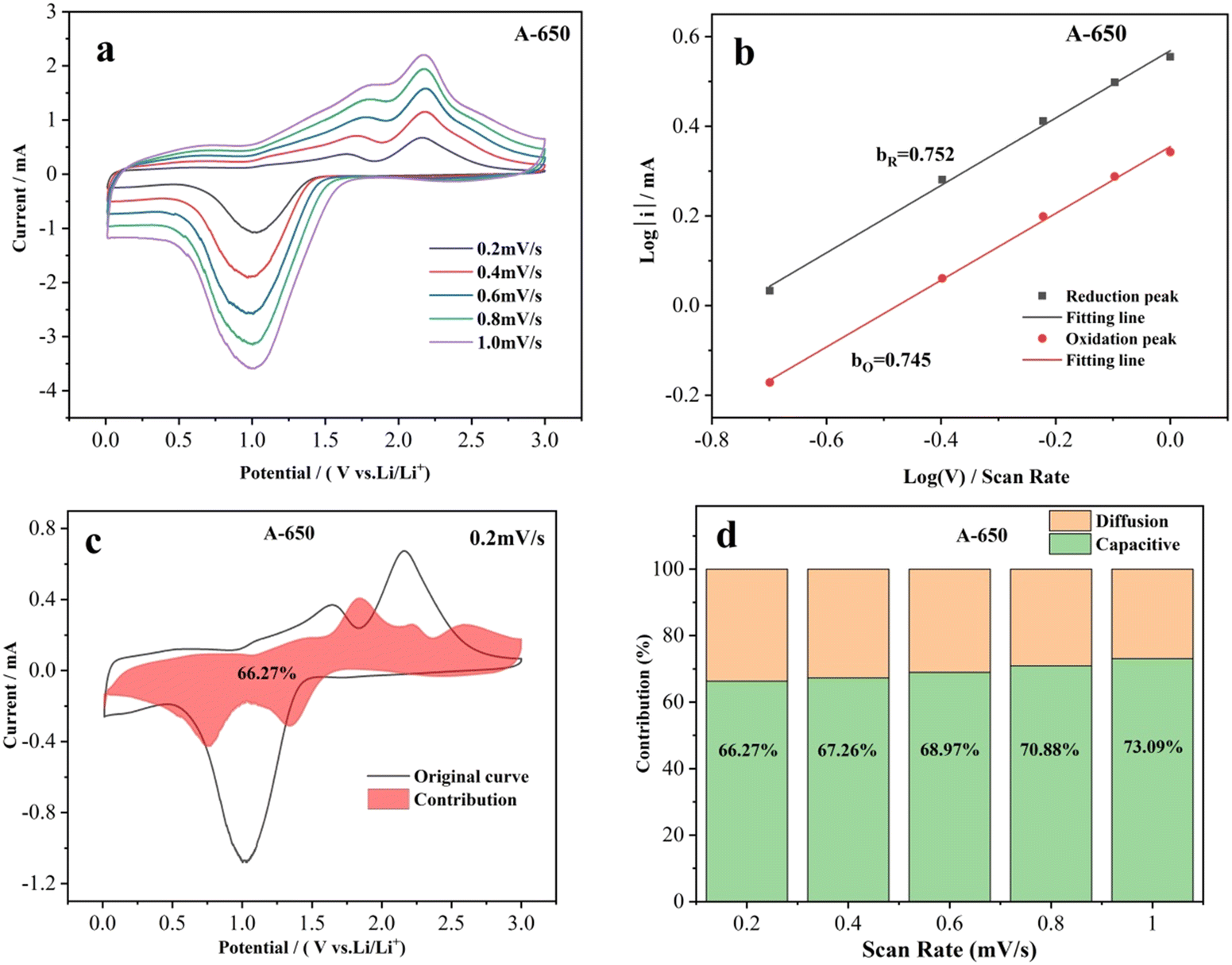 | ||
| Fig. 7 (a) CV curves of the A-650 electrode at scan rates from 0.2 to 2.0 mV s−1. (b) Peak current versus scan rate for the A-650 electrode. (c) CV curve of the A-650 electrode with the pseudocapacitive contribution at 0.2 mV s−1. (d) Pseudocapacitive contributions of the A-650 electrodes at different scan rates. | ||
During the charge-transfer/storage processes, the pseudocapacitive capacity (k1v) and diffusion-controlled capacity (k2v1/2) can be calculated with the following equations:77–80
| i = k1v + k2v1/2 | (7) |
| i/v1/2 = k1v1/2 + k2 | (8) |
As previously reported, the diffusion-controlled process of the electrode is affected by ion diffusion, while the capacitive-controlled process caused by surface redox reactions depends on the adsorption capacity of surface defects for lithium ions.74,83,84 In this work, the nanofibrous morphology of the A-650 electrode favors Li-ion diffusion and mass transport, thereby facilitating the diffusion-controlled process. The NiO/Co3O4/NiCo2O4 heterostructures and rough surface ensure high conductivity and abundant surface active sites on the electrode surface, which is responsible for the capacitive control process. As a result, the structural features and composition of the A-650 electrode ensure the combination of diffusion and capacitively controlled processes.
4. Conclusions
In this work, NiCo2O4 nanofibers were prepared by using electrospinning combined with subsequent heat treatment under an air atmosphere. With the increase of annealing temperature, the diameter and particle size of the nanofibers gradually increased. When the annealing temperature was ≥550 °C, NiCo2O4 was partially decomposed to Co3O4 and NiO, thus forming NiO/Co3O4/NiCo2O4 heterostructures and the decomposition ratio increased with increasing annealing temperature. The electrochemical performance of the electrode is dependent on the structural characteristics and composition of the nanofibers. The A-650 nanofiber electrode exhibits an optimal electrochemical performance, with the activation period reaching 61 cycles and a specific capacity of 1081.4 mA h g−1. The current density of the A-650 electrode recovered to 100 mA g−1 after high-current charge and discharge, and the specific capacity recovery value reached 387.9 mA h g−1, which continued to boost by 13.1% to 1067.9 mA h g−1. The charge storage mechanism is analyzed by using cyclic voltammetry, electrochemical impedance spectroscopy and galvanostatic intermittent titration technique measurements. The contribution of electrode pseudo capacitance increases from 66.27% to 73.09% as the scan rate increases from 0.2 to 1.0 mV s−1, revealing that both the diffusion- and capacitive-controlled processes have contributed. It is believed that this new strategy of constructing heterostructures through phase transition can serve as a reference to promote the development of anode materials for LIBs with excellent electrochemical performance.Author contributions
Shijin Yu: conceptualization, methodology, and writing – original draft. Jiahao Tong: investigation and data curation. Ying Wei: methodology. Tianrui Chen: investigation. Xuannan He: investigation. Huiqiang Sui: investigation. Cuiyun Li: methodology. Hua Zhu: methodology and writing – original draft. Qiuyun Fu: methodology, supervision, and writing – reviewing and editing. LingBing Kong: methodology, supervision, and writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 62041107, 61764007), the Natural Science Foundation of Jiangxi Province (No. 20181BAB202005), the Jingdezhen Science and Technology Project (20212KJHZ002, 2021GYD009-03), and the Special Fund for Graduate Innovation of Jingdezhen Ceramic University (YC2021-S546).References
- J. C. Zheng, Y. Y. Yao, G. Q. Mao, H. Z. Chen, H. Li, L. Cao, X. Ou, W. J. Yu, Z. Y. Ding and H. Tong, J. Mater. Chem. A, 2019, 7, 16479–16487 RSC.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- F. Wu, Q. Li, L. Chen, Z. Wang, G. Chen, L. Bao, Y. Lu, S. Chen and Y. Su, Acta Phys.-Chim. Sin., 2020, 0, 2007017 Search PubMed.
- Q. Wu, R. Xu, R. Zhao, X. Zhang, W. Li, G. Diao and M. Chen, Energy Storage Mater., 2019, 19, 69–79 CrossRef.
- Y. Ren, X. Li, Y. Wang, Q. Gong, S. Gu, T. Gao, X. Sun and G. Zhou, J. Mater. Sci. Technol., 2022, 102, 186–194 CrossRef.
- R. Li, H. Ke, C. Shi, Z. Long, Z. Dai, H. Qiao and K. Wang, Chem. Eng. J., 2021, 415, 128874 CrossRef CAS.
- B. Yuan, J. Li, M. Xia, Y. Zhang, R. Lei, P. Zhao and X. Li, Mater. Res. Express, 2020, 7, 115007 CrossRef CAS.
- J. Xu, Y. Xu, C. Lai, T. Xia, B. Zhang and X. Zhou, Sci. China: Chem., 2021, 64, 1267–1282 CrossRef CAS.
- H. Yang, T. Xiong, Z. Zhu, R. Xiao, X. Yao, Y. Huang and M. S. Balogun, Carbon Energy, 2022, 4, 820–832 CrossRef CAS.
- Y. Zhao, X. Li, B. Yan, D. Xiong, D. Li, S. Lawes and X. Sun, Adv. Energy Mater., 2016, 6, 1502175 CrossRef.
- S. Yu, V. M. Hong Ng, F. Wang, Z. Xiao, C. Li, L. B. Kong, W. Que and K. Zhou, J. Mater. Chem. A, 2018, 6, 9332–9367 RSC.
- H. Mou, S. Chen, W. Xiao, C. Miao, R. Li, G. Xu, Y. Xin and S. Nie, Ceram. Int., 2021, 47, 19945–19954 CrossRef CAS.
- W. Zhang, P. Cao, Z. Zhang, Y. Zhao, Y. Zhang, L. Li, K. Yang, X. Li and L. Gu, Chem. Eng. J., 2019, 364, 123–131 CrossRef CAS.
- C. Zhang and J.-S. Yu, Chem. – Eur. J., 2016, 22, 4422–4430 CrossRef CAS PubMed.
- D. Darbar, M. R. Anilkumar, V. Rajagopalan, I. Bhattacharya, H. I. Elim, T. Ramakrishnappa, F. I. Ezema, R. Jose and M. V. Reddy, Ceram. Int., 2018, 44, 4630–4639 CrossRef CAS.
- B. Li, J. Feng, Y. Qian and S. Xiong, J. Mater. Chem. A, 2015, 3, 10336–10344 RSC.
- X. Zhou, L. J. Wan and Y. G. Guo, Adv. Mater., 2013, 25, 2152–2157 CrossRef CAS.
- L. Li, Y. Ding, D. Yu, L. Li, S. Ramakrishna and S. Peng, J. Alloys Compd., 2019, 777, 1286–1293 CrossRef CAS.
- S. Peng, L. Li, H. B. Wu, S. Madhavi and X. W. D. Lou, Adv. Energy Mater., 2015, 5, 1401172 CrossRef.
- J. Cheng, Y. Lu, K. Qiu, H. Yan, J. Xu, L. Han, X. Liu, J. Luo, J.-K. Kim and Y. Luo, Sci. Rep., 2015, 5, 12099 CrossRef.
- X. Hou, H. Dang, M. Liu, X. Shang, Y. Fu and D. He, J. Alloys Compd., 2019, 810, 151736 CrossRef CAS.
- L. Li, Y. Cheah, Y. Ko, P. Teh, G. Wee, C. Wong, S. Peng and M. Srinivasan, J. Mater. Chem. A, 2013, 1, 10935–10941 RSC.
- F. Fu, J. Li, Y. Yao, X. Qin, Y. Dou, H. Wang, J. Tsui, K.-Y. Chan and M. Shao, ACS Appl. Mater. Interfaces, 2017, 9, 16194–16201 CrossRef CAS.
- S. Liu, J. Wu, J. Zhou, G. Fang and S. Liang, Electrochim. Acta, 2015, 176, 1–9 CrossRef CAS.
- Y. Li, W. Yang, X. Liu, S. Che, Z. Tu, L. Hou, C. Xu, H. Liu, G. Huang, Y. Zhou and Y. Li, Electrochim. Acta, 2021, 389, 138536 CrossRef CAS.
- L. Shen, L. Yu, X.-Y. Yu, X. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 1868–1872 CrossRef CAS PubMed.
- Y. Xia and H. Wang, Ionics, 2016, 22, 159–166 CrossRef CAS.
- A. Wang, Y. Hu, H. Wang, Y. Cheng, T. Thomas, R. Ma and J. Wang, Mater. Today Phys., 2021, 17, 100353 CrossRef CAS.
- J. Zhang, R. Chu, Y. Chen, H. Jiang, Y. Zhang, N. M. Huang and H. Guo, Nanotechnology, 2018, 29, 125401 CrossRef PubMed.
- J. Zhu, Z. Xu and B. Lu, Nano Energy, 2014, 7, 114–123 CrossRef CAS.
- J. Li, S. Lu, H. Huang, D. Liu, Z. Zhuang and C. Zhong, ACS Sustainable Chem. Eng., 2018, 6, 10021–10029 CrossRef CAS.
- W. Weng, J. Xu, C. Lai, Z. Xu, Y. Du, J. Lin and X. Zhou, J. Alloys Compd., 2020, 817, 152732 CrossRef CAS.
- M. Ma, H. Wang, X. Li, K. Peng, L. Xiong and X. Du, J. Eur. Ceram. Soc., 2020, 40, 5238–5246 CrossRef CAS.
- Z. Li, X. Hu, Z. Shi, J. Lu and Z. Wang, Appl. Surf. Sci., 2020, 531, 147290 CrossRef CAS.
- M. Hong, Y. Su, C. Zhou, L. Yao, J. Hu, Z. Yang, L. Zhang, Z. Zhou, N. Hu and Y. Zhang, J. Alloys Compd., 2019, 770, 116–124 CrossRef CAS.
- J.-M. Syu, M.-L. Hsiao and C.-T. Lo, J. Electrochem. Soc., 2017, 164, A3903–A3913 CrossRef CAS.
- M. L. Hsiao and C. T. Lo, Int. J. Energy Res., 2020, 44, 8606–8621 CrossRef CAS.
- L. Wang and X. Y. Qin, Bull. Mater. Sci., 2014, 37, 649–654 CrossRef CAS.
- B. Chi, Int. J. Hydrogen Energy, 2004, 29, 605–610 CrossRef CAS.
- S. Hwan, Oh, J.-S. Park, M. Su, Jo, Y. C. Kang and J. S. Cho, Chem. Eng. J., 2018, 347, 889–899 CrossRef.
- Y. Hoe Seon, Y. Chan Kang and J. S. Cho, Chem. Eng. J., 2021, 425, 129051 CrossRef CAS.
- P. Siwatch, K. Sharma and S. K. Tripathi, Electrochim. Acta, 2020, 329, 135084 CrossRef CAS.
- Y. Pei, C. Y. Xu, Y. C. Xiao, Q. Chen, B. Huang, B. Li, S. Li, L. Zhen and G. Cao, Adv. Funct. Mater., 2017, 27, 1604349 CrossRef.
- Y. Wang, J. Zhang, M. S. Balogun, Y. Tong and Y. Huang, Mater. Today Sustainability, 2022, 18, 100118 CrossRef.
- K. Ye, K. Li, Y. Lu, Z. Guo, N. Ni, H. Liu, Y. Huang, H. Ji and P. Wang, TrAC, Trends Anal. Chem., 2019, 116, 102–108 CrossRef CAS.
- J.-S. Do and C.-H. Weng, J. Power Sources, 2005, 146, 482–486 CrossRef CAS.
- S. K. Sundriyal and Y. Sharma, Appl. Surf. Sci., 2021, 560, 150055 CrossRef CAS.
- Y. Wu, G. Cheng, K. Katsov, S. W. Sides, J. Wang, J. Tang, G. H. Fredrickson, M. Moskovits and G. D. Stucky, Nat. Mater., 2004, 3, 816–822 CrossRef CAS.
- L. Cao, X. Gao, B. Zhang, X. Ou, J. Zhang and W.-B. Luo, ACS Nano, 2020, 14, 3610–3620 CrossRef CAS PubMed.
- H. Wu, N. Xu, Z. Jiang, A. Zheng, Q. Shi, R. Lv, L. Ni, G. Diao and M. Chen, Chem. Eng. J., 2022, 427, 131002 CrossRef CAS.
- Y. Li, M. Zhang, Q. Huang, P. Zhou, P. Xu, Z. Guo and K. Dai, Ionics, 2020, 26, 4351–4361 CrossRef CAS.
- Q. Wang, X. Qin, P. Jiang, J. Dai, W. Li and H. Gao, Mater. Res. Express, 2018, 5, 035503 CrossRef.
- G. M. Tomboc and H. Kim, Electrochim. Acta, 2019, 318, 392–404 CrossRef CAS.
- H. Wu, X. Chen, X. Zhang, Z. Jiang, Y. Dong, H. Li, L. Ni, G. Diao and M. Chen, Chem. Eng. J., 2022, 428, 131207 CrossRef CAS.
- J. Liu, C. Lin, H. Yao, S. Zhang, D. Fang, L. Jiang, D. Wang, Z. Zhang and J. Wang, J. Power Sources, 2021, 506, 230159 CrossRef CAS.
- F. Saeidpour and H. Ebrahimifar, Corros. Sci., 2021, 182, 109280 CrossRef CAS.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed.
- H. Zhu, X. Song, X. Han, X. Zhang, J. Bao, N. Zhang and G. He, Environ. Sci. Technol., 2020, 54, 8601–8611 CrossRef CAS PubMed.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6702–6707 CrossRef CAS.
- W. Jin, J. Chen, H. Wu, N. Zang, Q. Li, W. Cai and Z. Wu, Catal. Sci. Technol., 2020, 10, 5559–5565 RSC.
- L. Li, S. Peng, J. Wang, Y. L. Cheah, P. Teh, Y. Ko, C. Wong and M. Srinivasan, ACS Appl. Mater. Interfaces, 2012, 4, 6005 CrossRef CAS.
- F. Zou, Y. M. Chen, K. Liu, Z. Yu, W. Liang, S. M. Bhaway, M. Gao and Y. Zhu, ACS Nano, 2016, 10, 377–386 CrossRef CAS PubMed.
- Z. Zhang, Y. Huang, J. Yan, C. Li, X. Chen and Y. Zhu, Appl. Surf. Sci., 2019, 473, 266–274 CrossRef CAS.
- L. Guo, Q. Ru, X. Song, S. Hu and Y. Mo, J. Mater. Chem. A, 2015, 3, 8683–8692 RSC.
- D. Wang, W. Zhou, R. Zhang, X. Huang, J. Zeng, Y. Mao, C. Ding, J. Zhang, J. Liu and G. Wen, J. Mater. Chem. A, 2018, 6, 2974–2983 RSC.
- J. Wen, Y. Pei, L. Liu, D. Su, M. Yang, Q. Wang, W. Zhang, J. Dai, Y. Feng, T. Wu and X. Wang, J. Alloys Compd., 2021, 874, 159961 CrossRef CAS.
- Y. Yu, C.-H. Chen, J.-L. Shui and S. Xie, Angew. Chem., Int. Ed., 2005, 117, 7247–7251 CrossRef.
- Y. Lu, L. Yu, M. Wu, Y. Wang and X. W. Lou, Adv. Mater., 2018, 30, 1702875 CrossRef.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- Y. Zhu and C. Wang, J. Power Sources, 2011, 196, 1442–1448 CrossRef CAS.
- J. Guo, Q. Liu, C. Wang and M. R. Zachariah, Adv. Funct. Mater., 2012, 22, 803–811 CrossRef CAS.
- X. Pu, D. Zhao, C. Fu, Z. Chen, S. Cao, C. Wang and Y. Cao, Angew. Chem., Int. Ed., 2021, 60, 21310–21318 CrossRef CAS.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS.
- D. Meng, C. Zhang, Y. Liang, W. Qiu, F. Kong, X. He, M. Chen, P. Liang and Z. Zhang, J. Colloid Interface Sci., 2021, 599, 280–290 CrossRef CAS.
- L. Wang, M. Shi, C. Yang, Y. Liu, J. Jiang, K. Dai, Z. Guo and C. Yan, J. Alloys Compd., 2019, 804, 243–251 CrossRef CAS.
- T. Yuan, Y. Jiang, W. Sun, B. Xiang, Y. Li, M. Yan, B. Xu and S. Dou, Adv. Funct. Mater., 2016, 26, 2198–2206 CrossRef CAS.
- Y. Tang, Y. Zhang, O. I. Malyi, N. Bucher, H. Xia, S. Xi, Z. Zhu, Z. Lv, W. Li, J. Wei, M. Srinivasan, A. Borgna, M. Antonietti, Y. Du and X. Chen, Adv. Mater., 2018, 30, 1802200 CrossRef.
- Y. Liu, C. Ding, X. Yan, P. Xie, B. Xu, L. Chen, Y. Liu, C. Liu, Y. Yu and Y. Lin, Chem. Eng. J., 2021, 420, 129894 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597 RSC.
- Y. Wu, T. Ouyang, T. Xiong, Z. Jiang, Y. Hu, J. Deng, Z. Wang, Y. Huang and M. S. Balogun, Energy Environ. Mater., 2021, 5, 1251–1259 CrossRef.
- Z. He, H. Guo, J. D. LaCoste, R. A. Cook, B. Hussey, X. Zhang, D. D. Gang, J. Hao, L. Chen, P. Cooke, H. Yan and L. Fei, Sustainable Energy Fuels, 2021, 5, 166–174 RSC.
- Y. Wang, Y. Wang, W. Kang, D. Cao, C. Li, D. Cao, Z. Kang, D. Sun, R. Wang and Y. Cao, Adv. Sci., 2019, 6, 1801222 CrossRef PubMed.
- S. Li, J. Qiu, C. Lai, M. Ling, H. Zhao and S. Zhang, Nano Energy, 2015, 12, 224–230 CrossRef CAS.
- D. Li, L. Zhang, H. Chen, J. Wang, L. X. Ding, S. Wang, P. J. Ashman and H. Wang, J. Mater. Chem. A, 2016, 4, 8630–8635 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj05331d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |