
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural enzymology of iterative type I polyketide synthases: various routes to catalytic programming
Jialiang
Wang
 ,
Zixin
Deng
*,
Jingdan
Liang
* and
Zhijun
Wang
*
,
Zixin
Deng
*,
Jingdan
Liang
* and
Zhijun
Wang
*
State Key Laboratory of Microbial Metabolism, Joint International Research Laboratory of Metabolic & Developmental Sciences, School of Life Science & Biotechnology, Shanghai Jiao Tong University, Shanghai, China. E-mail: wangzhijun@sjtu.edu.cn; jdliang@sjtu.edu.cn; zxdeng@sjtu.edu.cn
First published on 15th August 2023
Abstract
Time span of literature covered: up to mid-2023
Iterative type I polyketide synthases (iPKSs) are outstanding natural chemists: megaenzymes that repeatedly utilize their catalytic domains to synthesize complex natural products with diverse bioactivities. Perhaps the most fascinating but least understood question about type I iPKSs is how they perform the iterative yet programmed reactions in which the usage of domain combinations varies during the synthetic cycle. The programmed patterns are fulfilled by multiple factors, and strongly influence the complexity of the resulting natural products. This article reviews selected reports on the structural enzymology of iPKSs, focusing on the individual domain structures followed by highlighting the representative programming activities that each domain may contribute.
 Jialiang Wang | Jialiang Wang studied structural and biochemical biology and received a PhD at Shanghai Jiao Tong University (SJTU) in 2021. Currently, he continues his research as a postdoctoral fellow in the group of Professor Zixin Deng and works with Professors Zhijun Wang and Jingdan Liang at SJTU. He is interested in visualizing and understanding nature's biosynthetic macromolecular machines including polyketide synthases and nonribosomal peptide synthetases that are central for the production of valuable natural products. Using cryo-electron microscopy and biochemical techniques, he is trying to resolve the architecture, catalytic mechanism and structural dynamics of these molecular factories. |
 Zixin Deng | Prof. Zixin Deng received his PhD in Microbial Genetics from the University of East Anglia (1987) while working in the Streptomyces group at the John Innes Centre. He is the Dean of the School of Life Sciences and Biotechnology, Shanghai Jiao Tong University and School of Pharmaceutical Sciences, Wuhan University and is a Member of the Chinese Academy of Sciences, Fellow of the American Academy of Microbiology, and Fellow of The Royal Society of Chemistry. His research interests center around synthetic biology, Streptomyces genetics, biochemistry and molecular biology of antibiotic biosynthesis, and phosphorothiolation of DNA. |
 Jingdan Liang | Jingdan Liang earned a PhD in Biology under the guidance of Prof. Zixin Deng at Shanghai Jiao Tong University in 2008. Afterward, she worked as an Assistant Professor at the State Key Laboratory of Microbial Metabolism at the same university from 2008 to 2012 and was promoted to Associate Professor in 2012. Following that, she spent two years as a visiting scholar at the University of California, Berkeley, studying the catalytic mechanisms of CMG. Currently, her research focuses on understanding the enzymology of proteins involved in DNA phosphorothioate modification and the biosynthesis of natural products. |
 Zhijun Wang | Dr Zhijun Wang received his PhD in 2006, studying protein interactions of polyketide synthases under the guidance of Professor Keqian Yang at the Institute of Microbiology, Chinese Academy of Sciences. He joined the faculty at the School of Life Sciences and Biotechnology, Shanghai Jiao Tong University in 2007. From 2011 to 2013, he was supported by the Tang scholarship to work with Professor Michael Botchan at the University of California, Berkeley, investigating the structure of CMG. Currently, Dr Wang is an associate professor specializing in the structural biology of natural product biosynthetic machinery and the enzymes responsible for DNA phosphorothioate modification. |
1. Introduction
Polyketides are naturally synthesized by utilizing a series of amazing enzymes called polyketide synthases (PKSs).1 They are multifunctional enzymes that produce numerous natural products with wide-ranging biological activities2,3 (Fig. 1), such as cholesterol-lowering drugs4 (e.g., lovastatin), antibiotics5 (e.g., erythromycin), carcinogens6 (e.g., aflatoxin B1) and toxins7,8 (e.g., cercosporin and T-toxin). PKSs construct these complex compounds via multiple extensions of mainly two- or three-carbon building blocks, with varied but controllable modification processes.9,10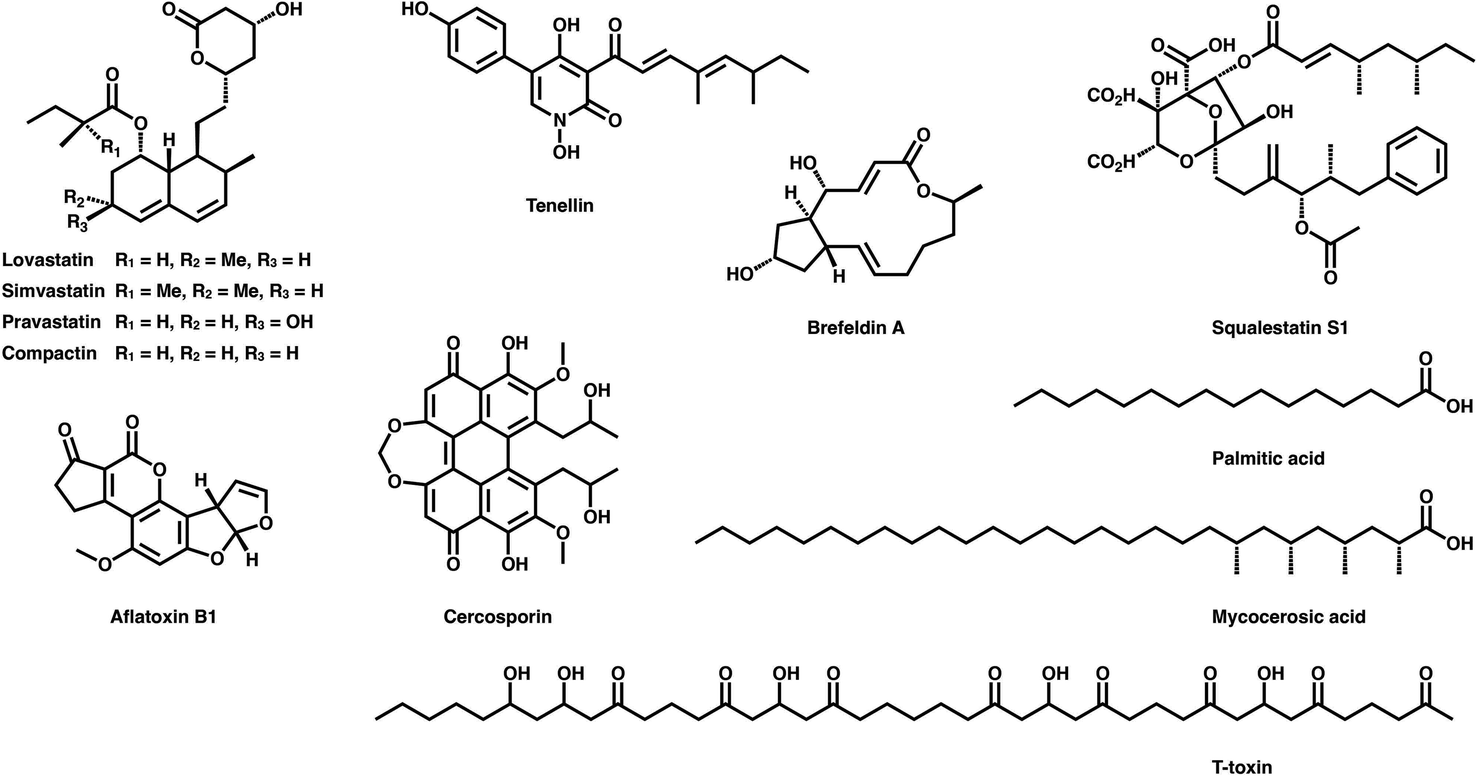 | ||
| Fig. 1 Representative polyketides synthesized using type I iPKSs and related mFAS. | ||
Based on domain organization, PKSs are usually divided into three types: type I PKSs, which are multidomain megaenzymes that act linearly through multiple modules (modPKSs) or iteratively through one module (iPKSs), type II PKSs, which contain several discrete stand-alone proteins, and type III PKSs, which contain a single domain.11 Among the most interesting yet least understood PKSs are the type I iterative PKSs, mostly found in fungi.10 Based on the various reductive degrees of the polyketide chain backbone, fungal type I iPKSs are further classified into highly reducing iPKSs (HR-iPKSs), nonreducing iPKSs (NR-iPKSs), and partially reducing PKSs (PR-iPKSs). Mycocerosic acid synthase (MAS), MAS-like PKSs and the related mammalian fatty acid synthase (mFAS), which generate fully reduced products, may also be viewed as subsets of type I iPKSs.12,13 In addition, a remarkable group of modular PKSs in bacteria such as aureothin PKSs obtained from Streptomyces thioluteus are observed to function iteratively.14
The hallmark of iterative type I iPKSs is that the usage of a single set of catalytic domains is repeated yet programmed. To realize the catalytic complexity of type I iPKSs, it is worth comparing their biosynthetic logic to those of the related mammalian fatty acid synthase (mFAS) and modPKSs (Fig. 2). In the representative case of the HR-iPKS LovBC complex involved in the biosynthesis of lovastatin15 (Fig. 2A), the biosynthetic cycle is initiated by loading malonyl-CoA catalyzed by a malonyl-acetyl transferase (MAT) domain, presumably followed by decarboxylation to generate the starter acetyl unit. Chain elongation is performed iteratively by eight rounds of Claisen condensation catalyzed by a β-ketosynthase (KS) domain and one Diels–Alder reaction. After each condensation reaction, the growing polyketide chain tethered to the phosphopantetheinyl (pPant) arm of the acyl carrier protein (ACP) domain and is subjected to modification processes, including α-carbon methylation by C-methyltransferase (CMeT), β-keto group reduction by the ketoreductase (KR) domain, β-hydroxyl group dehydration by the dehydratase (DH) domain and α,β-double bond reduction by the enoylreductase (ER) domain. Intriguingly, during the eight extension cycles, the CMeT-KR-DH-ER (LovC)-modifying domain usage is permutative. This is in stark contrast to either the mFAS in mammalian palmitic acid biosynthesis,16 where the elongated intermediate chain is faithfully modified in each extension cycle to finally form fully reduced C16 or C18 fatty acid (Fig. 2B), or the modPKSs, such as in erythromycin biosynthesis,17 where the modification steps are specified by each modular domain component (Fig. 2C). In addition, the modification processes of NR-iPKS PksCT18 (Fig. 3A) and PR-iPKS 6-MSAS19 (Fig. 3B) are less complicated than that of HR-iPKS LovBC.
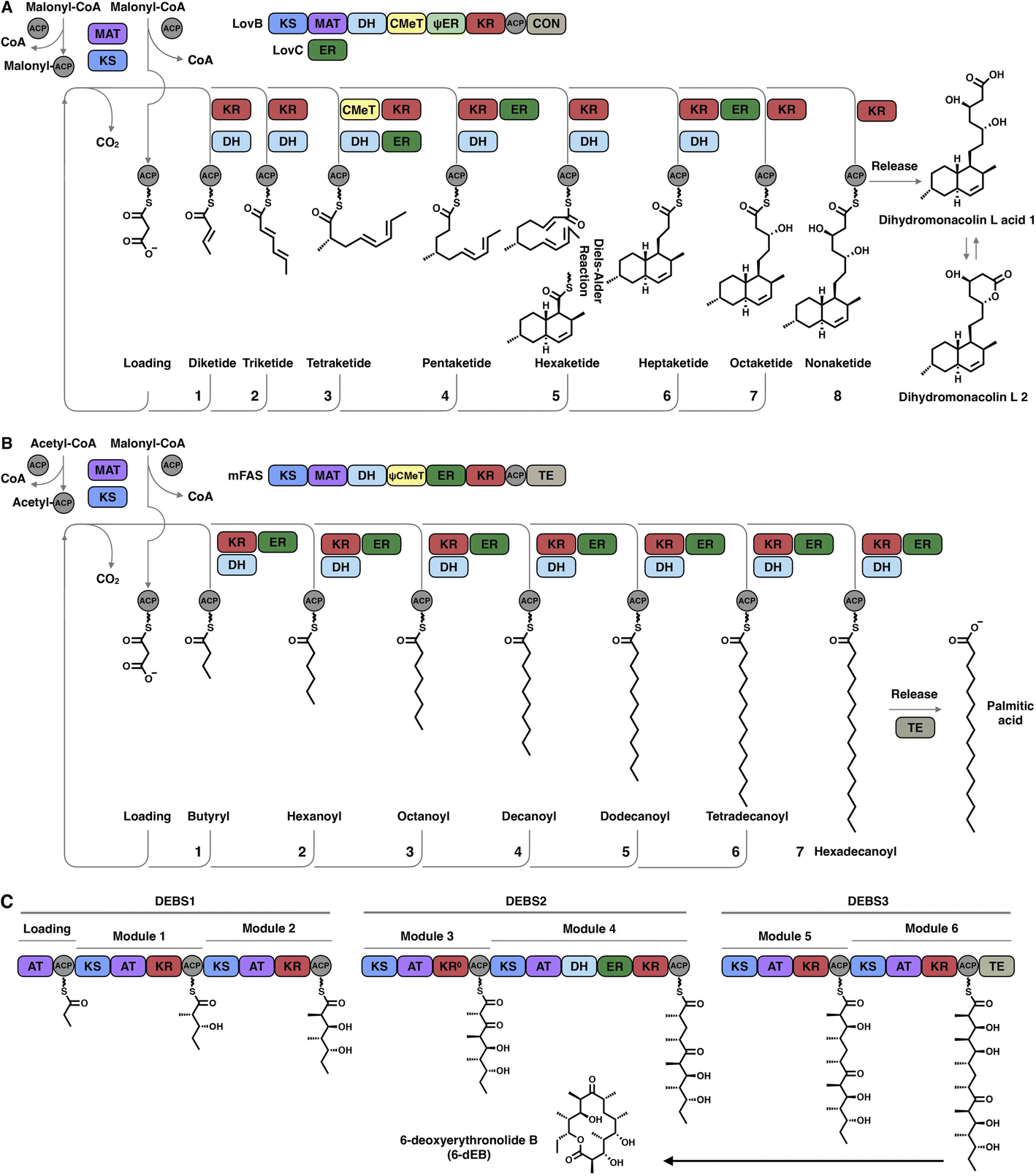 | ||
| Fig. 2 Comparison of the representative biosynthetic logic between (A) HR-iPKSs (LovBC complex in dihydromonacolin L acid biosynthesis), (B) mFAS in palmitic acid biosynthesis and (C) modPKSs (DEBS1-3 in 6-deoxyerythronolide B biosynthesis). Three subunits comprising one loading module and six extension modules, in which each module is used only once to elongate and specifically modify the polyketide chain in an assembly-line fashion to yield 6-deoxyerythronolide B (6-dEB). The inactive KR domain in DEBS2 module 3 is denoted as KR0. | ||
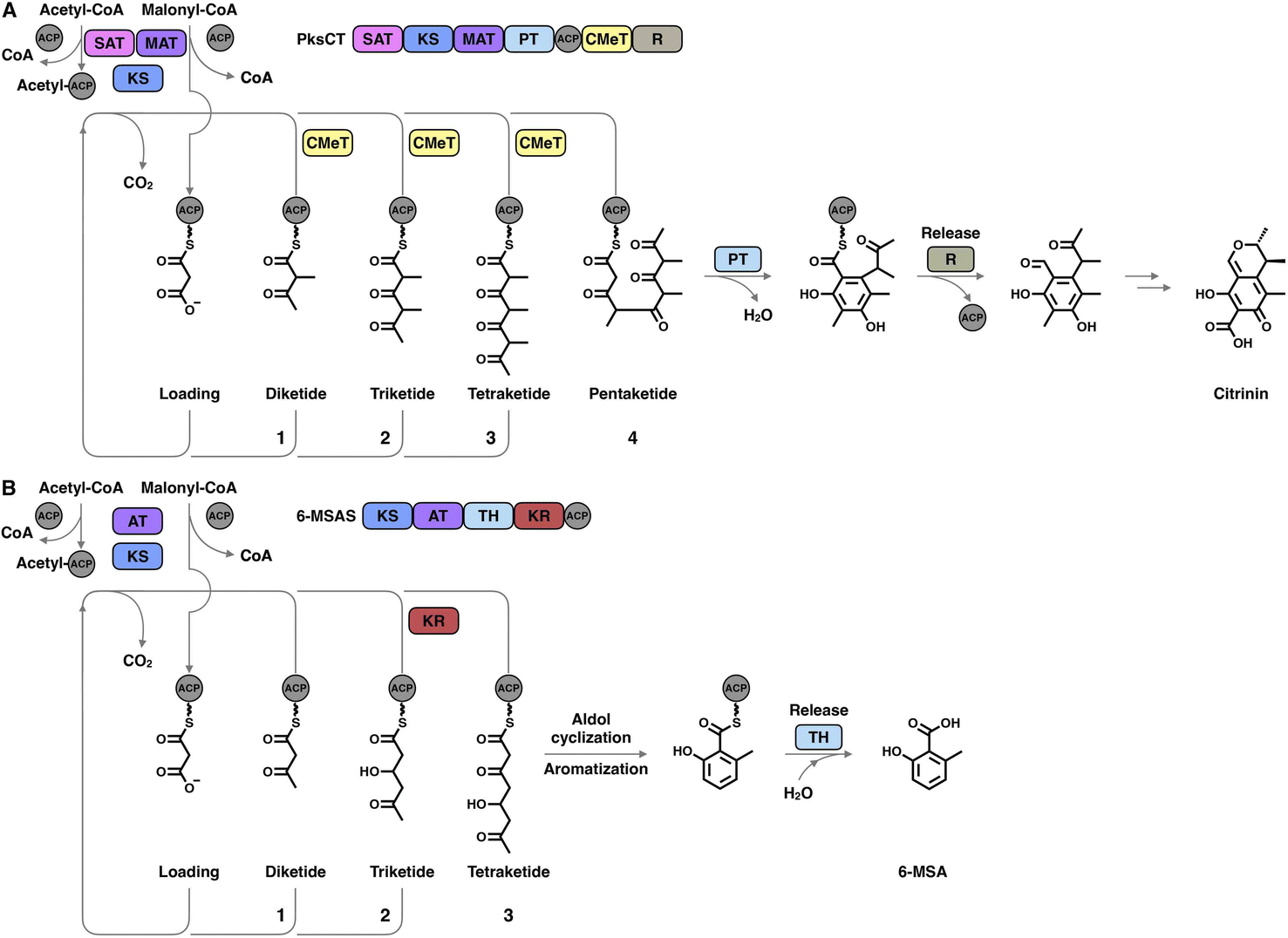 | ||
| Fig. 3 Representative biosynthetic logic of (A) NR-iPKSs (PksCT in citrinin biosynthesis) and (B) PR-iPKSs (6-methylsalicylic acid synthase (6-MSAS) in 6-methylsalicylic acid (6-MSA) biosynthesis). See also Fig. 2 for comparison. | ||
How does a single set of catalytic domains of type I iPKSs use different domain combinations to construct polyketides under specific program rules? The underlying programming is extremely complicated,20,21 intertwined with intrinsic and extrinsic contributions such as starter/extender unit selections, intermediate specificity, kinetic competition, gatekeeping and interdomain in trans interactions.
Fortunately, the biochemical activities established by in vitro reconstitution, domain swap and re-engineering have begun to unveil the program rules.20 Additionally, significant advances in obtaining structural information have been uncovered in the past decade.16 Looking into the architectures, the active sites and substrate tunnels of these molecular machineries will provide the necessary structural basis for the catalytic cycles of polyketide biosynthesis. This review summarizes the currently reported structures of type I iPKSs, either relatively complete regions or excised domains, and highlight the representative examples of programming mechanisms delicately contributed by catalytic domain(s).
2. Overall architectures of iterative type I PKSs
It was not until 2021 that the structural information of an HR-iPKS architecture became available. The cryo-EM structure of the 750 kDa LovB–LovC complex was reported at 3.60 Å resolution and the core LovB at 2.91 Å resolution22 (Fig. 4A). The domain organization of LovB is an X-shaped face-to-face dimer, and each monomer contacts each other extensively with an approximately 6197 Å2 interface mainly contributed by KS, ψER and DH domains. The LovB architecture is divided into two regions: lower condensing (KS-MAT) and upper modifying (DH–CMeT–ψKR–ψER–KR) regions. The lateral binding of LovC, the trans-acting ER, to the MAT domain allows the completion of an L-shaped catalytic chamber consisting of six active domains on each side. Further interface mutation experiments confirmed that the formation of the LovBC complex is essential for the correct programming fidelity of the DML acid synthesis.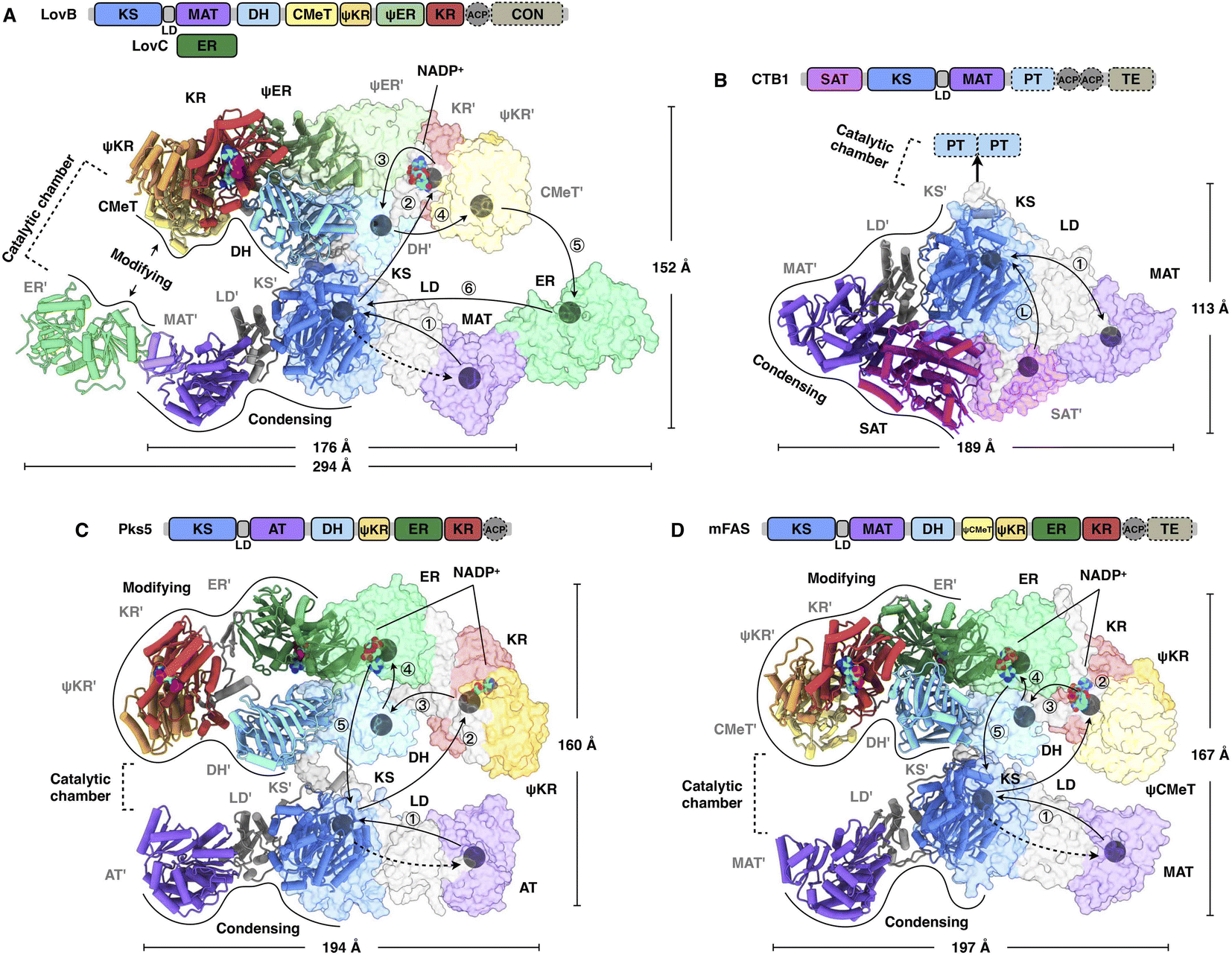 | ||
| Fig. 4 Overall architectures of type I iPKSs and mFAS. (A) HR-iPKS LovBC complex, PDB: 7CPY. (B) Loading/condensing region of NR-iPKS CTB1, PDB: 6FIJ. (C) Hybrid model containing the modifying and condensing regions of MAS-like PKS Pks5, PDB: 5BP4 and 5BP1, respectively. (D) mFAS (pig), PDB: 2VZ9. For each structure, the linear domain organization and dimensions are labeled. The hypothetical substrate shuttling trajectories (one side) within the catalytic chamber are shown as lines with each arrow pointing toward the next step; the locations of active site residues of each domain are marked as gray balls. The dashed arrows indicate the loading of the malonyl or acetyl starter unit. The double-sided arrow in (B) indicates a possible mechanism of NR-iPKSs in which the iterative polyketide extension is rapid and processive in the KS domain (PksA, discussed in Section 2.3). | ||
Great insights into the loading/condensing region architecture of an NR-iPKS have been provided by the 2.8 Å crystal structure of CTB1 comprising starter unit acyltransferase (SAT)-KS-MAT domains23 (Fig. 4B). The structure is particularly featured by the interactions between the SAT and MAT domains of opposite chains, covering an average of 957 Å2, which results in an overall rhomboid-shaped compact dimer. Furthermore, by using the mechanism-based crosslinker, the 7.1 Å cryo-EM structure traps the ACP docked to only one side of the KS dimer, which indicates that the asymmetric domain arrangements mediate polyketide biosynthesis.
A breakthrough in the understanding of a fully reducing PKS came from the hybrid model comprising the crystal structures of condensing and modifying regions of MAS-like PKS Pks5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 (Fig. 4C). The condensing region adopts a similar conformation to the known structures.25,26 In contrast, instead of being the V-shaped conformation in LovB22 and mFAS,26 the DH dimer of the Pks5-modifying region adopts a linear conformation at an angle of 222°, similar to that of modPKSs.27,28 The Pks5 hybrid model reveals a linker-based, rather than domain–domain interaction-based, architecture and establishes a framework for the modPKS domain organization.
24 (Fig. 4C). The condensing region adopts a similar conformation to the known structures.25,26 In contrast, instead of being the V-shaped conformation in LovB22 and mFAS,26 the DH dimer of the Pks5-modifying region adopts a linear conformation at an angle of 222°, similar to that of modPKSs.27,28 The Pks5 hybrid model reveals a linker-based, rather than domain–domain interaction-based, architecture and establishes a framework for the modPKS domain organization.
Compared to the well-studied mFAS structure26 (Fig. 4D), type I iPKSs exhibit a similar dimeric architecture; however, the condensing region of HR-iPKS LovB is rotated ∼180° opposite to that of mFAS when both modifying regions are superposed. Furthermore, the condensing and modifying regions of LovB clearly show contact at the “waist”, which is distinct from the central flexible linker observed in mFAS. This contact indicates that the condensing region of LovB may not be able to undergo large-scale rotation, as seen in the multiple conformations of mFAS analyzed by cryo-EM.29 Nevertheless, structural dynamics, either subtle or dramatic, are observed in all of these megaenzymes and believed to be necessary for creating asymmetric chambers during substrate shuttling.
2.1 Malonyl-acetyl transferase (MAT) domains
The MAT domain is responsible for selecting and loading the starter and extender building blocks to prime polyketide biosynthesis and required for further chain extension. Type I iPKSs use MAT domains to almost specifically select acetyl-CoA and malonyl-CoA, although several examples have shown that diverse acyl-CoAs can be the preferred substrates,30,31 which are more reasonably defined as acyltransferase (AT) domains. The MAT domain is composed of a large α/β-hydrolase-like subdomain and a ferredoxin-like small subdomain (Fig. 5), similar to the known AT of modPKS and MAT structures.25,32 The large subdomain is fairly stable, whereas the small subdomain can undergo flexible motion. This conformational flexibility is implied by the relatively weak integrity of the LovB cryo-EM map22 (Fig. 5C) and is clearly observed in the mFAS (murine) MAT domain33 and in the cryo-EM variability analysis of the modPKS Pik127 AT structure.34 How this small subdomain mobility is related to substrate selection and loading is not yet understood.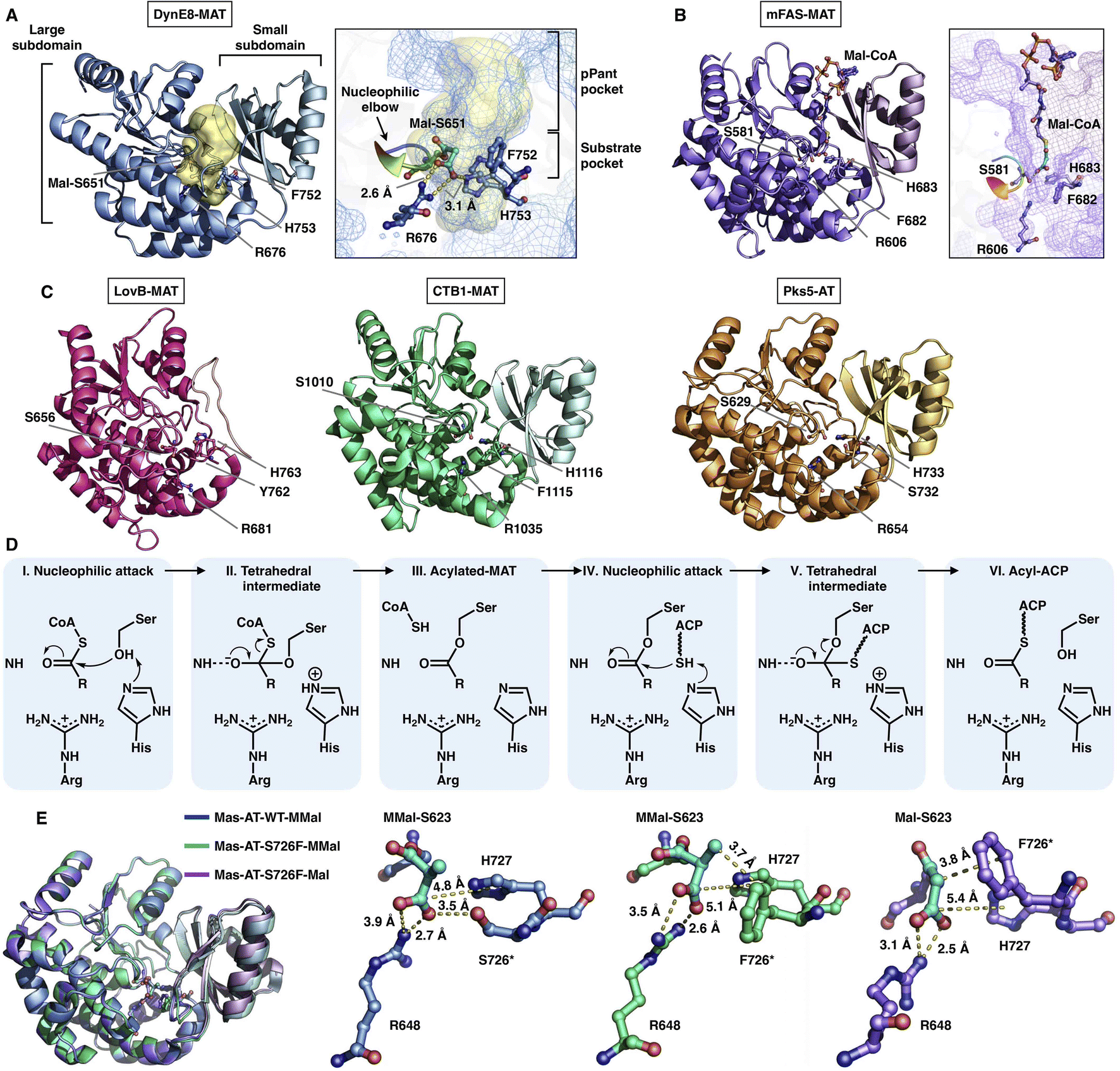 | ||
| Fig. 5 Structures of type I iPKS MAT and AT domains of DynE8 acylated with Mal ((A), PDB: 4AMP), mFAS (murine) in complex with Mal-CoA ((B), PDB: 5MY0, chain C), LovB, CTB1, Pks5 ((C), PDB: 7CPX, 6FIJ, 5BP1, respectively), and MAS WT and S726F mutant acylated with MMal-CoA, MMal-CoA and Mal-CoA ((E), PDB: 7AGS, 7AGU, 7AGT, respectively). The Ser-His catalytic dyads, key residues and interactions with substrates are labeled. The substrate pocket (yellow surface) is located between the two subdomains (shown as mesh), and the G-X-S-X-G motifs (nucleophilic elbows) are rainbow colored in (A) and (B). (D) Proposed catalytic mechanism of MAT. | ||
The deep substrate binding pockets are located between the two subdomains where the substrates are sandwiched (Fig. 5A and B). The two-subdomain formed interface harbors a Ser-His catalytic dyad (e.g., S651-H753 in DynE8-MAT35), in which Ser is located in the highly conserved G-X-S-X-G motif that is referred to as the “nucleophilic elbow”36 (Fig. 5A and B). The MAT-catalyzed transfer of building blocks to the ACP is conducted via a ping-pong bi–bi mechanism37 facilitated by the geometry of the nucleophilic elbow (Fig. 5D). The nucleophilic Ser attacks the thioester carbonyl carbon of malonyl-CoA to form a covalently Ser-acylated intermediate. Then, His acts as a general base mediating the deprotonation of the ACP pPant thiol group, and a subsequent nucleophilic attack results in malonyl-ACP. A key conserved Arg residue (e.g., R676 in DynE8-MAT) interacts with the malonyl carboxylate group by forming a salt bridge, which is responsible for holding the substrate. The specific substrate selectivity for malonyl-CoA (Mal-CoA) over α-substituted extender units such as methylmalonyl-CoA (MMal-CoA) is partly contributed by a bulk side chain containing phenylalanine (e.g., F752 in DynE8-MAT), one residue before the catalytic His. Recently, molecular-level evidence of substrate specificity in the AT crystal structures of iterative mycocerosic acid synthase (MAS) in complex with substrates has been provided38 (Fig. 5E). The M624V-S726F double mutants exhibit almost completely inverted specificity from natural MMal-CoA to Mal-CoA. Although the S726F mutant does not hinder the binding of MMal-CoA by a proposed steric clash with the additional methyl group, perhaps due to the motion of the entire ferredoxin-like small subdomain, the M624V-S726F double mutant structure completely abolishes the formation of the complex with MM-CoA. This study reinforces the influence of this Phe residue on Mal-CoA substrate specificity.
2.2 Starter-unit acyltransferase (SAT) domains
The SAT domain is a unique starter unit selector organized in the NR-iPKS.39 The substrate specificity is remarkably tolerant, ranging from acetyl-CoA (C2) to octanoyl-CoA (C8)39,40 and even various nonnative starter units, as exemplified by the PksA SAT domain,41 which is reminiscent of the loading AT domain of modPKSs. Interestingly, the SAT domain of the NR-iPKS is also often found to pair with an upstream HR-iPKS and transfer the synthesized polyketide intermediate to the NR-iPKS,21 further enhancing the flexibility of SAT in substrate selection.To date, two crystal structures have been reported: the SAT domain of CTB123 and CazM acylated with a hexanoyl substrate.42 Similar to the known MAT domains in both overall structure and catalytic mechanism, SAT comprises a large α/β-hydrolase-like subdomain and a ferredoxin-like small subdomain (Fig. 6A and B). The substrate pocket is located at the two-subdomain interface that contains a Cys-His catalytic dyad (e.g., C155-H277 in CazM-SAT). Acylated hexanoyl, the triketide substrate mimic, is stabilized by multiple hydrophobic interactions with H22, A156, A158, I191, I272 and I276, which may contribute to acyl substrate selection (Fig. 6A). The highly conserved substrate-holding Arg residue of the MAT domain is replaced by A187 in CazM-SAT, which lines at the bottom end of the pocket, resulting in a deeper L-shaped hydrophobic substrate pocket than that of MATs.
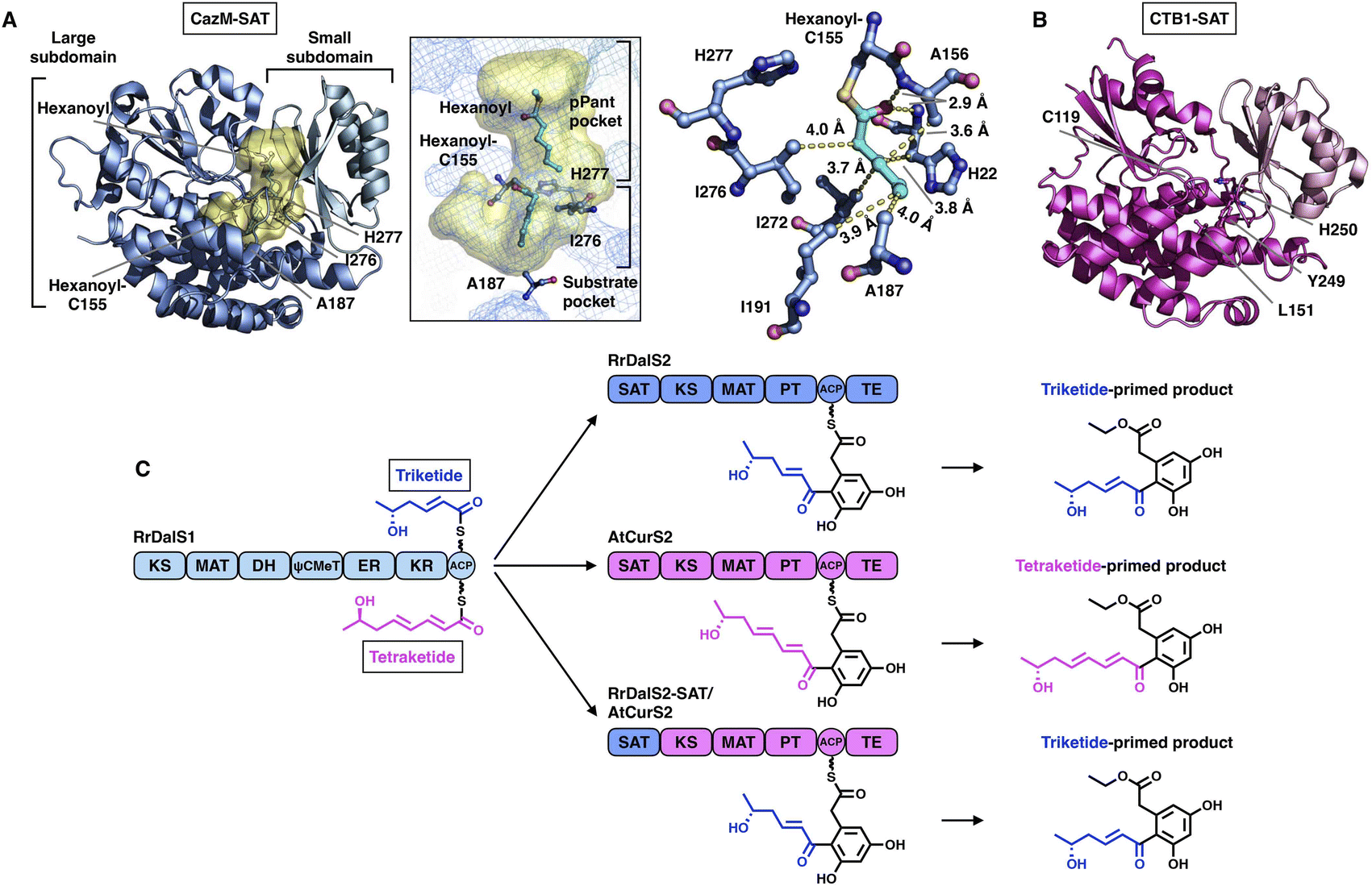 | ||
| Fig. 6 Structures of SAT domains of CazM ((A), PDB: 4RPM) and CTB1 ((B), PDB: 6FIJ). Cys-His catalytic dyads, key residues and hydrophobic interactions with the hexanoyl substrate mimic (cyan) are labeled. The substrate pocket (yellow surface) is located between the two subdomains (shown as mesh). (C) Programming mechanism of the RrDalS2 SAT domain revealed by chain length control. | ||
An excellent example of the programming of the SAT domain by chain-length control has been recently investigated by domain swapping43 (Fig. 6C). HR-iPKS RrDalS1 collaborates with downstream NR-iPKS RrDalS2 and AtCurS2 to produce triketide- and tetraketide-primed main products, respectively. By exchanging the SAT domains of the two NR-iPKSs, the chimeric RrDalS2-SAT/AtCuS2 intercepts and forces the HR-iPKS to offer the triketide, resulting in the dominance of the triketide-primed polyketide. This study shows that the SAT domain can, although rarely, be a proactive selector that acts in trans to strictly accept the preferred substrate based on the chain length (the triketide by the SAT of RrDalS2) from the upstream partner HR-iPKS to further polyketide biosynthesis.
2.3 β-Ketosynthase (KS) domains
The elongation KS domain of type I iPKSs catalyzes the central decarboxylative Claisen condensation reaction and needs to accommodate several distinct intermediates. Type I iPKSs that have been structurally determined include the KS domain of HR-iPKS LovB,22 the KS domain of NR-iPKS CTB1,23 the KS domain of MAS-like PKS Pks524 and mFAS KS.26 As the most highly conserved domain, dimeric KS adopts the typical thiolase fold, in which each monomer contains an αβαβα structure15,44 (Fig. 7). The KS homodimeric interface is extensive, with a surface area of nearly or more than 2000 Å2, and contributes to the largest homophilic interactions, maintaining the overall dimeric architecture of type I iPKSs and mFAS. The active site comprises the conserved Cys-His-His catalytic triad (e.g., C181-H320-H367 in LovB). It is proposed that when the ACP-tethered starter unit enters the KS tunnel, the nucleophilic Cys, which creates the oxyanion hole, attacks the thioester bond to form a covalently Cys-acylated starter unit. Then, the extender unit-ACP binds to KS, and two His promote the decarboxylation of the extender to form a carbon–carbon bond, leading to one round of polyketide chain elongation (Fig. 7F).
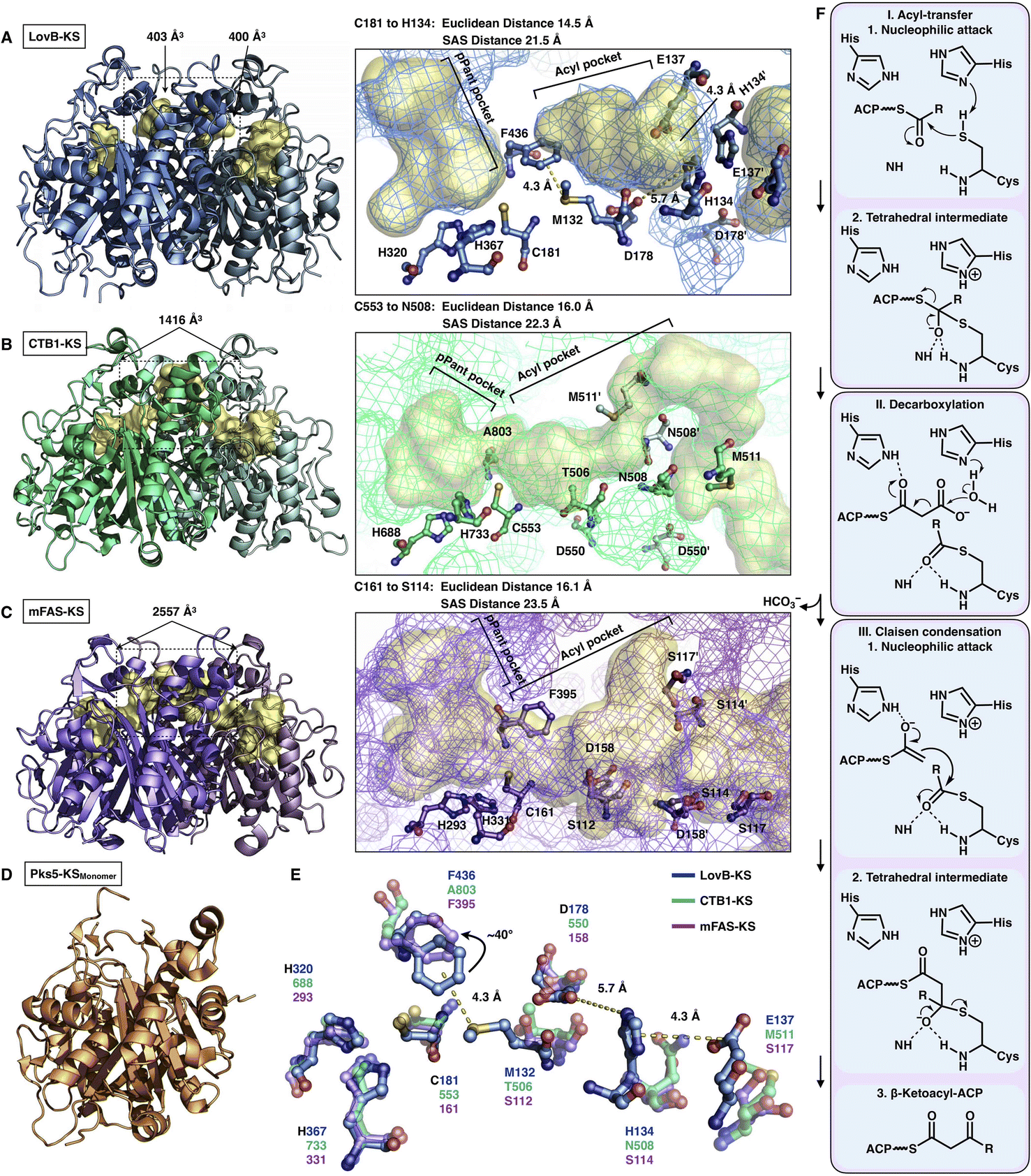 | ||
| Fig. 7 Structures of KS domains of type I iPKSs and mFAS. (A) KS of HR-iPKS LovB, PDB: 7CPX. (B) KS of NR-iPKS CTB1, PDB: 6FIJ. (C) KS of mFAS, PDB: 2VZ9. (D) Monomeric KS of MAS-like PKS Pks5, PDB: 5BP1. For each KS structure, the Cys-His-His catalytic triads, tunnel bottleneck residues, and substrate tunnels (yellow surfaces) are labeled. (E) Superposition of the catalytic triads and bottleneck residues reveals the potential side chain rotation of LovB-F436 of ∼40°. (F) Proposed catalytic mechanism of KS. | ||
An exceptional characteristic of the LovB KS domain is the substrate tunnel (Fig. 7A). In contrast to the end-to-end connected KS tunnels of CTB1 and mFAS (Fig. 7B and C), the LovB KS pPant pocket is disconnected from the acyl chain pocket by the F436-M132 hydrophobic interaction in the tunnel and truncated at the end by the putative ionic interactions formed by H134-E137-D178 (Fig. 7E). The acyl pocket volume is significantly smaller than that of CTB1 and mFAS. Both the Euclidean distance and the solvent-accessible surface (SAS) distance of LovB KS from the reactive Cys to the tunnel end are also shorter. The superposition of the three KSs shows that F436 of LovB exhibits an approximately 40° side-chain rotation (Fig. 7E) and perhaps suggests a “breathing motion” mechanism mediating the substrate entry process. A similar observation conducted by the corresponding F395 of mFAS (murine) has been postulated as a “gatekeeper” in which the side chain rotates approximately 120° after the loading with the C8 fatty acid chain substrate.33 The residues at this Phe residue-equivalent position of NR-iPKSs vary. Nevertheless, whether a gate-keeping or breathing motion mechanism is utilized, the potential conformational variability of F436 of the LovB KS domain needs to be further clarified by capturing the KS complex structure with Cys181-acylated intermediates inside the tunnel.
Although the chain length control of type I HR-iPKSs has not been deeply investigated, NR-iPKSs and type II HR-iPKSs have both offered valuable information. The clade II NR-iPKS16 PksA with the SAT-KS-MAT-PT-ACP-TE domain organization was interrogated by mass spectrometry.45 Interestingly, partially elongated intermediates covalently attached by ACP were not detectable, and only fully elongated octaketide could be observed. This observation suggests an unexpected yet efficient working mode of KS (Fig. 8A): since the β-carbon of the β-ketoacyl polyketide chain does not need to be modified, the intermediate chain elongation is processive and may never leave the KS substrate tunnel, shuttling back and forth expeditiously and progressively between the active site cysteine and the pPant-tethered extender unit (malonyl-ACP) until the correct chain length is achieved. The abovementioned gate-keeping residue Phe in LovB and mFAS is replaced by alanine in PksA without bulk side-chain restrictions, which may also contribute to the processive mechanism of clade II NR-iPKS. The evidence of chain length controlled by KS has also been shown by a domain swap of the two closely related NR-iPKS CoPKS1 and CoPKS4, in which it is the KS domain that determines whether the dominant product is hepta- or octaketide.46
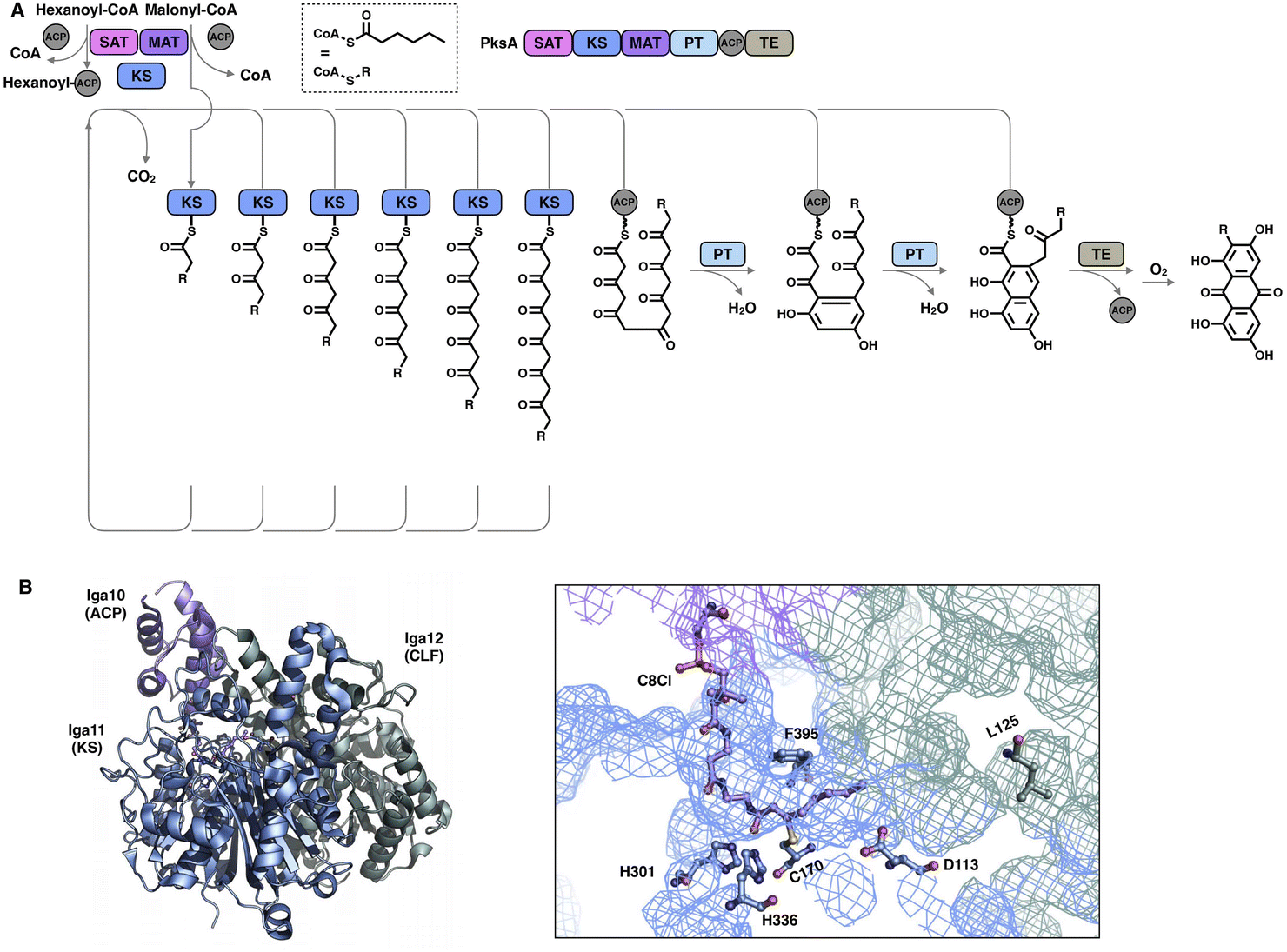 | ||
| Fig. 8 Representative programming mechanisms revealed by NR-iPKS PksA (A) which indicates a processive extension mechanism of KS, and type II HR-iPKS Iga11–Iga12 in complex with Iga10-tethered C8 β-chloracrylamide pantetheinamide (C8Cl), a mimic of the pPant arm ((B), PDB: 6KXF). The chain length is proposed to be restricted by L125 of Iga12. | ||
The second perception comes from the bacterial type II HR-iPKS Iga11-Iga1247 (Fig. 8B). The chain length controlled by Iga11 (KS) is observed with a non-active site-containing chain link factor (CLF, Iga12), which may be viewed as an inactive version of the KS monomer. A negative charge of the Asp113 side chain of KS promotes the release of the Iga10 (ACP)-tethered β-ketoacyl intermediate and drives the chain forward for further β-carbon modifications. The substrate tunnel is rigid, and Leu125 of CLF creates steric hindrance in the rigid tunnel, which prevents the acyl moiety from being transferred to Cys170, thus controlling the chain length up to C14.
2.4 C-Methyltransferase (CMeT) domains
The S-adenosyl-methionine (SAM)-dependent CMeT domain introduces a methyl group at the α-position of the β-ketoacyl intermediate after the KS-catalyzed polyketide chain elongation and before the KR-, DH-, ER- and PT-performed modifications. Methylation by CMeT occurs mostly once during each iteration of polyketide extension, although a rare case of the HR-iPKS Tv6-931 CMeT domain shows that the β-keto tetraketide intermediate, but not the triketide, could be consecutively methylated twice to form an α,α-gem-dimethyl product.48 Four structures of type I iPKS CMeT domains have been reported (Fig. 9A–D), including CMeT of HR-iPKS LovB,22 although this portion of the cryo-EM map suffers from relatively weak integrity; the crystal structure of CMeT of NR-iPKS PksCT in complex with the byproduct S-adenosyl-L-homocysteine (SAH);49 and the trans-acting CMeT of PsoF50 and CalH',51 both captured in complex with SAH. How such a stand-alone trans-acting CMeT interacts with megaenzymes is currently unknown.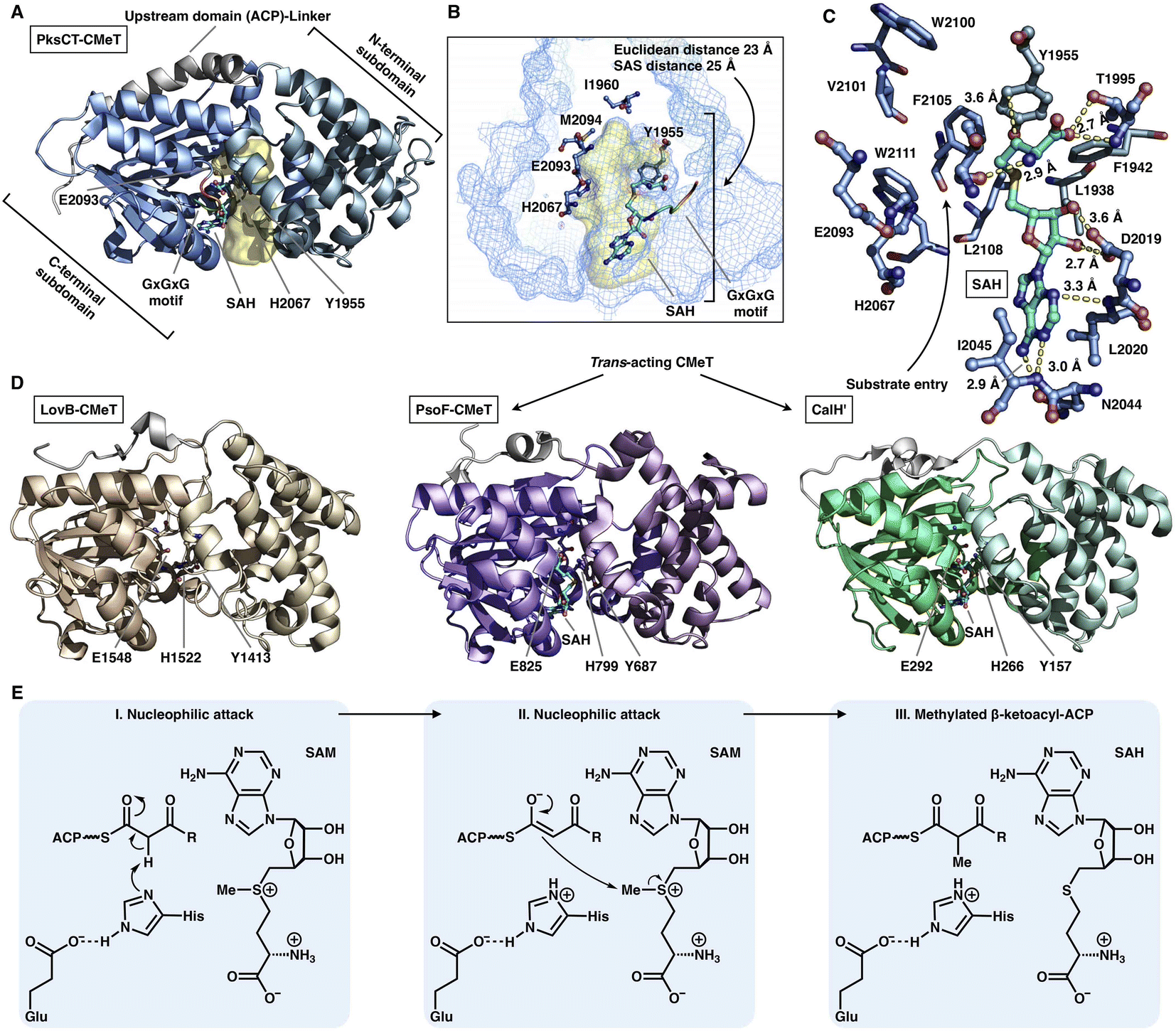 | ||
| Fig. 9 Structures and mechanism of CMeT domains. (A) CMeT structure of NR-iPKS PksCT, PDB: 5MPT. (B) H2067-E2093 catalytic dyad and pocket-end residues are labeled. The substrate pocket (yellow surface) is located between the two subdomains (shown as mesh), and the G-X-G-X-G SAM-binding motif is shown in rainbow colors. (C) Hydrogen bonding and hydrophobic interactions with the byproduct SAH. (D) Structures of HR-iPKS LovB CMeT and trans-acting PsoF CMeT and CalH'. PDB: 7CPX, 6KJI and 7DMB, respectively. (E) Proposed catalytic mechanism of CMeT. | ||
Both the cis- and trans-acting CMeTs exhibit similar overall monomeric architectures (Fig. 9A–D). For example, PksCT CMeT adopts a two-subdomain organization with a hydrophobic substrate pocket, and the conserved Tyr1955 and His2067-Glu2093 catalytic dyads are located at the interface (Fig. 9A). The core C-terminal subdomain displays a Rossmann-like fold and belongs to the typical class I methyltransferase superfamily,52 which is also responsible for binding the SAM cofactor. The N-terminal subdomain contains several helices that may be viewed as a large lid protecting the substrate entrance, whereas many HR-iPKSs contain an inactive version of CMeT in which the N-terminal subdomain is completely absent or severely truncated, similar to the ψCMeT domain of mFAS.26 The conserved G-X-G-X-G motif is responsible for binding the cofactor SAM. Hydrogen bonds and hydrophobic interactions stabilize SAH, and the SAH homocysteine moiety also assists in the formation of the funnel-shaped substrate pocket that has an ∼25 Å SAS distance from the protein surface to the I1960-M2094-defined end (Fig. 9B and C). The following methylation process has been proposed: activated by Glu, the His acts as a general base and abstracts the proton from the α-carbon of the ACP pPant-tethered β-ketoacyl intermediate to form an enolate, which can subsequently perform a nucleophilic attack on the methyl donor of SAM to complete the reaction. The Tyr may also facilitate the methyl transfer process (Fig. 9E).
Two layers of programming contributed by the CMeT domains have been significantly revealed. A kinetic competition experiment on LovB in lovastatin biosynthesis was performed in vitro53 (Fig. 10A). The LovB DH domain has been mutated previously, leaving only the CMeT- and KR-modifying domains to be analyzed. In the presence of NADPH and SAM cofactors, CMeT is exceptionally selective toward the natural tetraketide intermediate. This suggests that CMeT has a higher kinetic efficiency on this particular tetraketide intermediate and therefore outcompetes the downstream KR domain within the iteration. Furthermore, a rare case of a trans-acting CMeT domain of PsoF that acts as an essential checkpoint to maintain the correct acyl intermediate transfer has been reported54 (Fig. 10B). In azaspirene biosynthesis, the HR-iPKS-NRPS hybrid PsoA collaborates with the CMeT domain of PsoF to produce the aminoacylated polyketide. In the process, the programmed CMeT specifically methylates the tetraketide. Without the methylation pattern performed by trans-acting CMeT, the polyketide intermediate cannot be transferred to the NRPS, and PsoA only synthesizes and releases the shunt α-pyrone product.
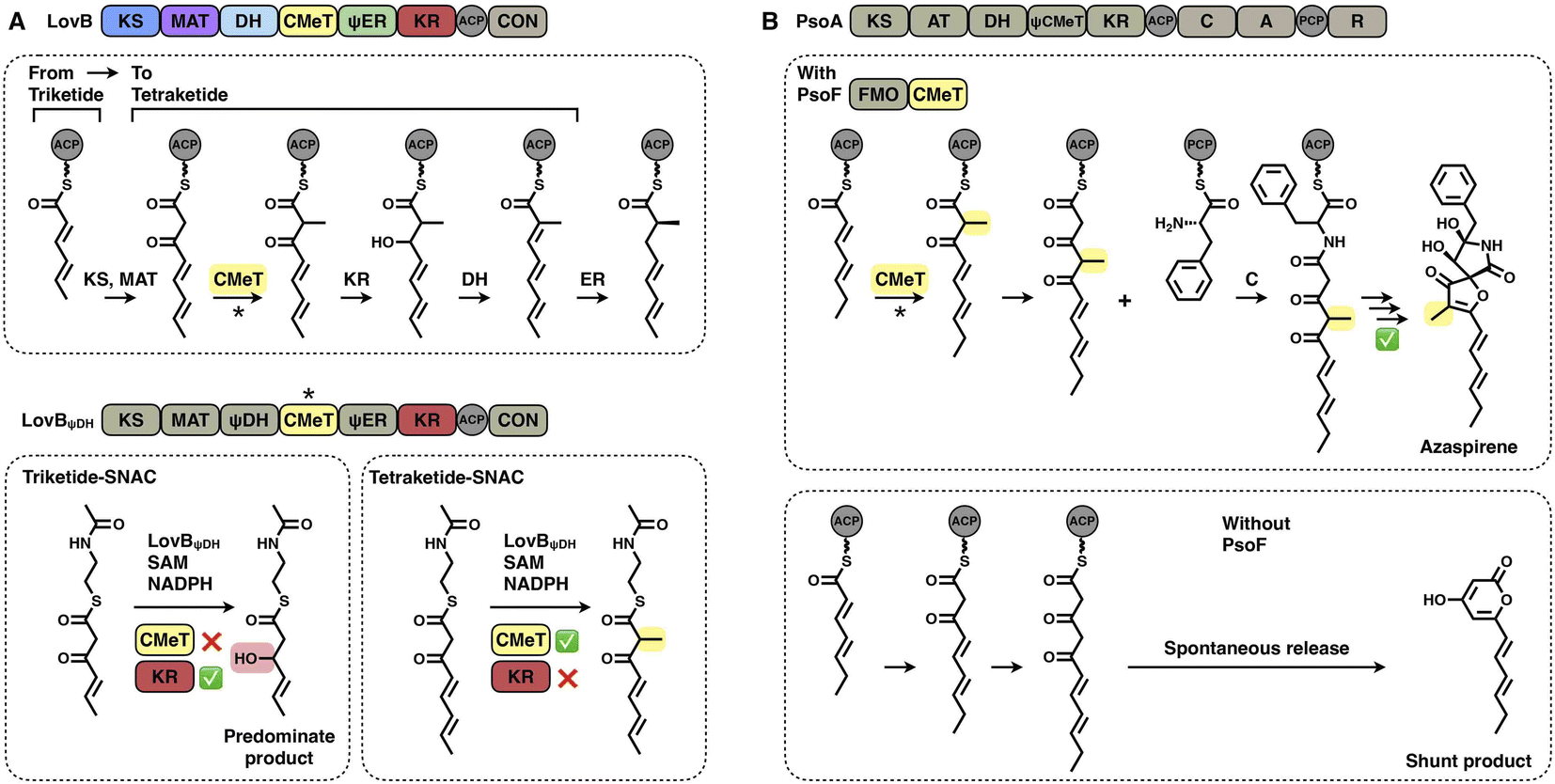 | ||
| Fig. 10 Representative programming mechanisms of CMeT domains. (A) CMeT program rule of LovB revealed by kinetic competition. (B) Correct chain transfer of the polyketide intermediate requires methylation catalyzed by trans-CMeT of PsoF. | ||
2.5 Ketoreductase (KR) domains
The NADPH-dependent KR domain stereoselectively reduces the β-keto group of the β-ketoacyl polyketide intermediate after the KS-catalyzed carbon–carbon bond-forming condensation reaction. The monomeric ψKR/KR domain serves as a central connector in the modifying regions of HR-iPKS (LovB),22 MAS-like PKS (Pks5)24 and mFAS26 (Fig. 4 and 11). It comprises an N-terminal structural subdomain (ψKR) and a C-terminal catalytic Rossmann-like subdomain (KR) that belongs to the short-chain dehydrogenase/reductase (SDR) superfamily55 and shares a common structure with its homologs. ψKR can be viewed as the inactive remnant of KR due to the truncation of nearly half of the Rossmann fold and is unable to bind NADPH (Fig. 11A).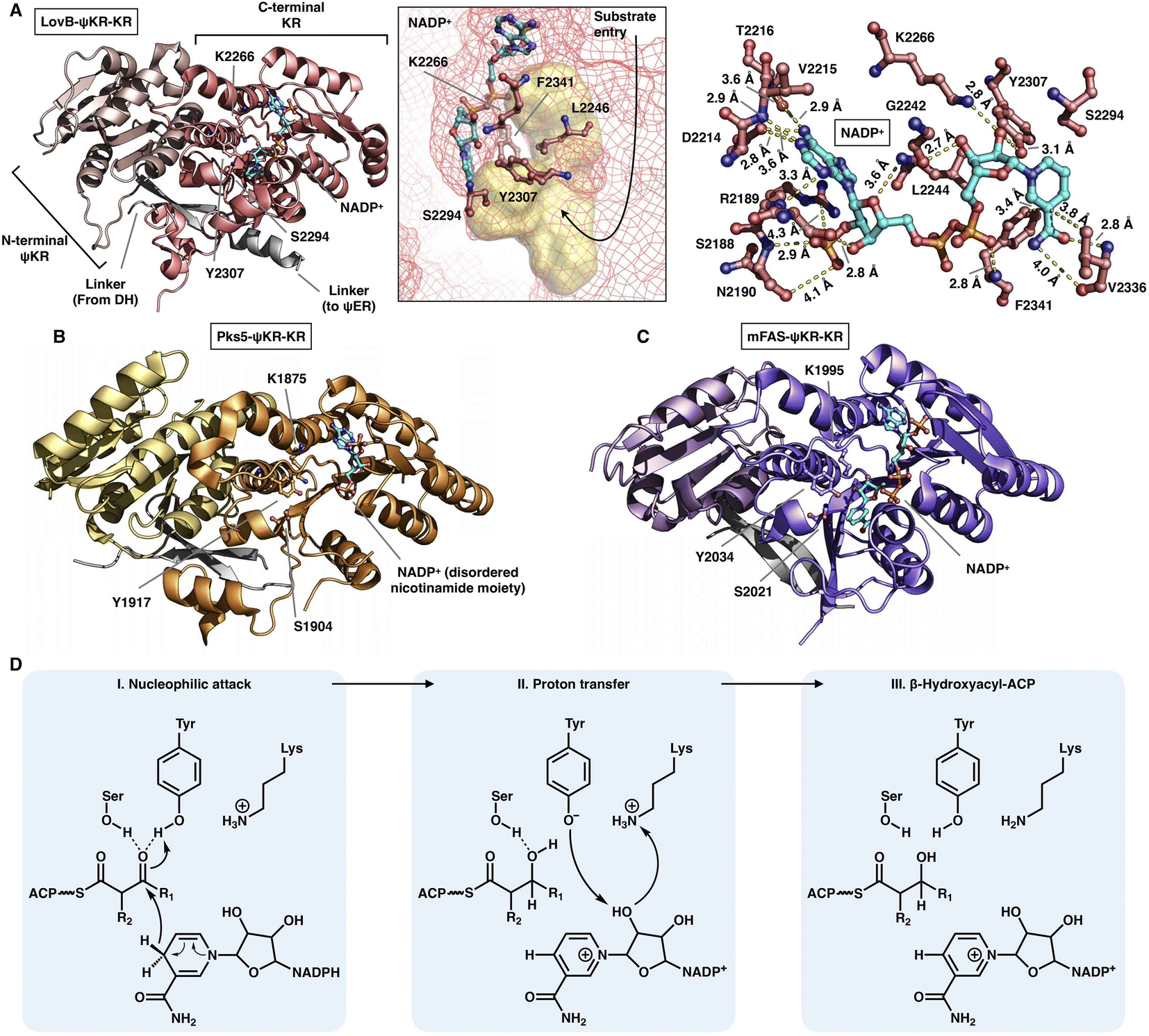 | ||
| Fig. 11 Structures of ψKR/KR domains of LovB ((A), PDB: 7CPX), Pks5 ((B), PDB: 5BP4) and mFAS ((C), PDB: 2VZ9). The Lys-Ser-Tyr catalytic triad below the substrate pocket (yellow surface) and the interactions with the cofactor NADP+ (cyan) are labeled in (A). (D) Proposed catalytic mechanism of KR. | ||
KR harbors the conserved Lys-Ser-Tyr catalytic triad (e.g., K2266-S2294-Y2307 in LovB-KR) and the substrate binding pocket. F2341 and L2246 of LovB narrow the pocket and constrict the substrate entry direction. The cofactor NADP+, located below the active site, is mainly stabilized by multiple hydrogen bonds, and F2341 and V2336 may also form hydrophobic interactions with the nicotinamide ring (Fig. 11A). It is suggested that the KR domain operates by a proton-relay mechanism56 (Fig. 11D): the hydride of NADPH nicotinamide ribose performs a nucleophilic attack on the β-carbon of the β-ketoacyl intermediate, activated by the hydroxyl groups of the Tyr and Ser side chains, and β-carbon oxygen abstracts a proton from Tyr. Proton transfer is also facilitated by Lys.
KRs are classified into A-type KRs that set a β-hydroxyl group product in the L-configuration, B-type KRs that set a hydroxyl in the D-configuration, and C-type KRs that are reduction-incompetent.57 Since a single KR domain is utilized in almost every iteration of the polyketide chain extension (e.g., LovB in Fig. 2A), it seems that type I iPKS KRs are expected to display a little substrate selectivity. However, an unusual example clearly shows that the KR domain can switch the stereochemical outcome, which is controlled by the substrate chain length58 (Fig. 12A–D). In hypothemycin biosynthesis, the KR domain of HR-iPKS Hpm8 reduces β-ketone of the diketide into the L-configuration, whereas it reduces β-ketone of the triketide exclusively to the common D-configuration (Fig. 12A). This also indicates that the diketide and triketide may enter the substrate tunnel from the opposite direction.58,59 A series of SNAC-substrates with various chain lengths were analyzed in vitro, which clearly shows that the tri-, tetra-, penta- and hexaketides are all reduced to the common D-configuration, except that the diketide is converted into the L-configuration (Fig. 12B). Furthermore, by a sequence swap between the Hpm8 and Rdc5 (reduces the diketide into the D-configuration in monocillin II biosynthesis)60 KR domains, the catalytic triad-containing α4β5α5α6 motif was pinpointed as the site responsible for this substrate-tuned stereospecificity alteration (Fig. 12C and D). Recently, similar stereochemistry alterations of HR-iPKS KR domains have also been reported, including ApmlA in phaeospelide A biosynthesis,61 MpmlA in phaseolide A biosynthesis62 and KU42 from the basidiomycete fungal species Punctularia strigosozonata.63
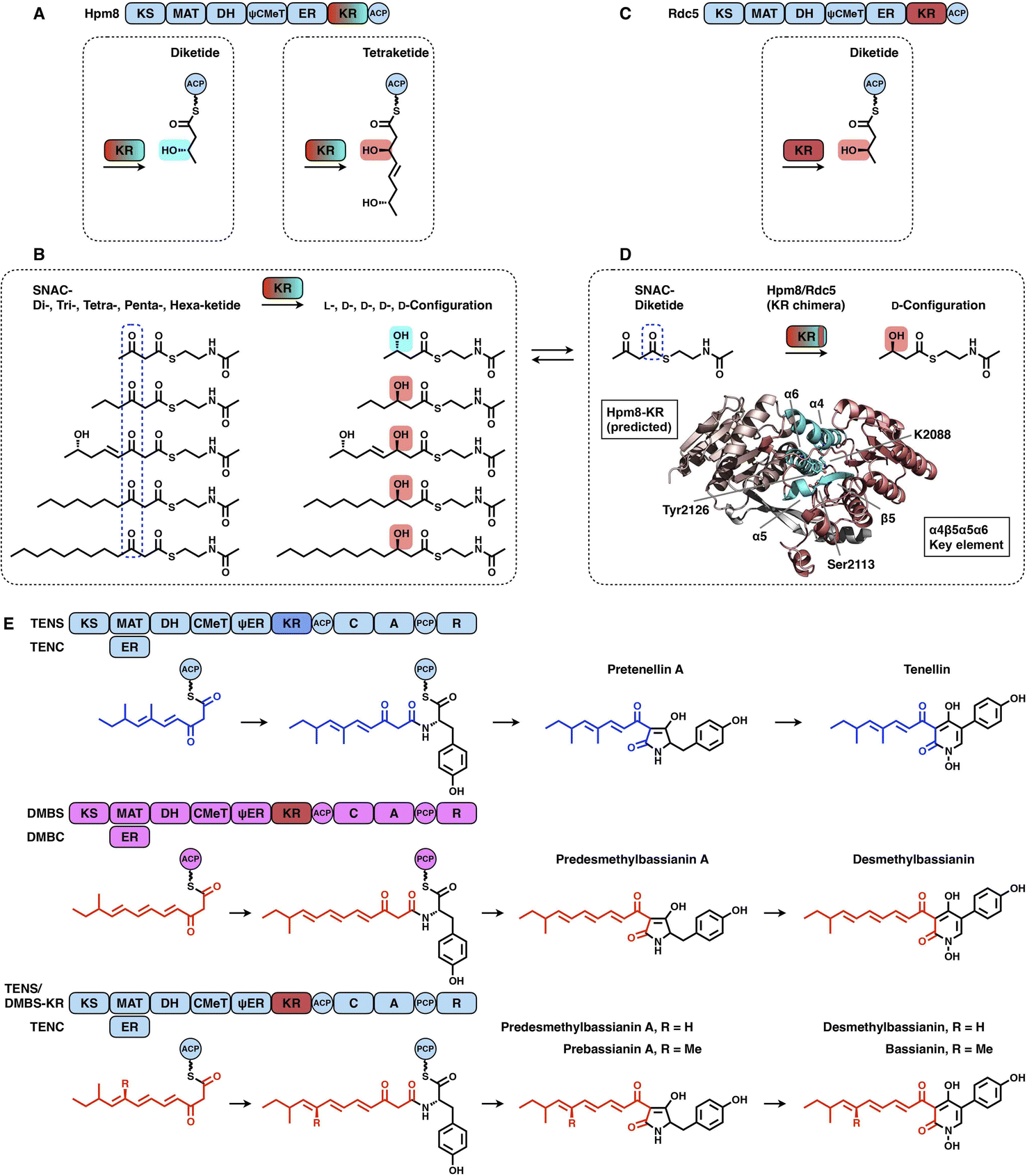 | ||
| Fig. 12 Representative programming mechanisms of KR domains. (A and B) Stereochemical alteration by the Hpm8 KR domain controlled by the substrate chain length. (C and D) Stereospecificity-determined α4β5α5α6 motif (cyan) revealed by the sequence swap experiment. The structure of the Hpm8 KR domain is predicted by Alphafold2. (E) Chain length control of DMBS KR revealed by the domain swap. | ||
Although the versatility and limited substrate specificity of KR domains suggest subtle contributions to programming, an insightful case of chain-length control by HR-iPKS KR was observed by rationally designed domain swaps64 (Fig. 12E). With the collaboration of the corresponding trans-acting ER domains, HR-iPKS-NRPS TENS and DMBS produce penta- and hexaketide-primed products, respectively. When the KR domains of the two PKS-NRPSs are exchanged, the reprogrammed TENS/DMBS-KR hybrid synthesizes the dominant hexaketide-primed product in which the chain length has been clearly altered.
2.6 Dehydratase (DH) domains
The DH domain of HR-iPKSs catalyzes the dehydration reaction on the β-hydroxyl-acyl polyketide intermediate to form a double bond between the α- and β-carbon, usually in the trans configuration. The dimeric DH of LovB22 is V-shaped, which is similar to that of mFAS26 (Fig. 13A and B), but in contrast to the relatively linear DH organization in MAS-like PKS Pks524 and modPKSs.27,28 Each monomer of the LovB DH domain displays a pseudodimeric hot-dog fold and contains the conserved His-Asp catalytic dyad (e.g., H985-D1174 in LovB-DH), which is contributed by both hot-dog folds. The substrate tunnel of LovB DH is truncated by hydrophobic residues, including I1181 and F1258, which are significantly shorter than the traverse tunnels of Pks5 and mFAS (Fig. 13B). It is proposed that the DH operates by an acid/base pair catalytic mechanism27 (Fig. 13C): His abstracts a proton from the α-carbon of the intermediate, followed by β-hydroxyl oxygen protonation by Asp, resulting in the syn elimination of water.
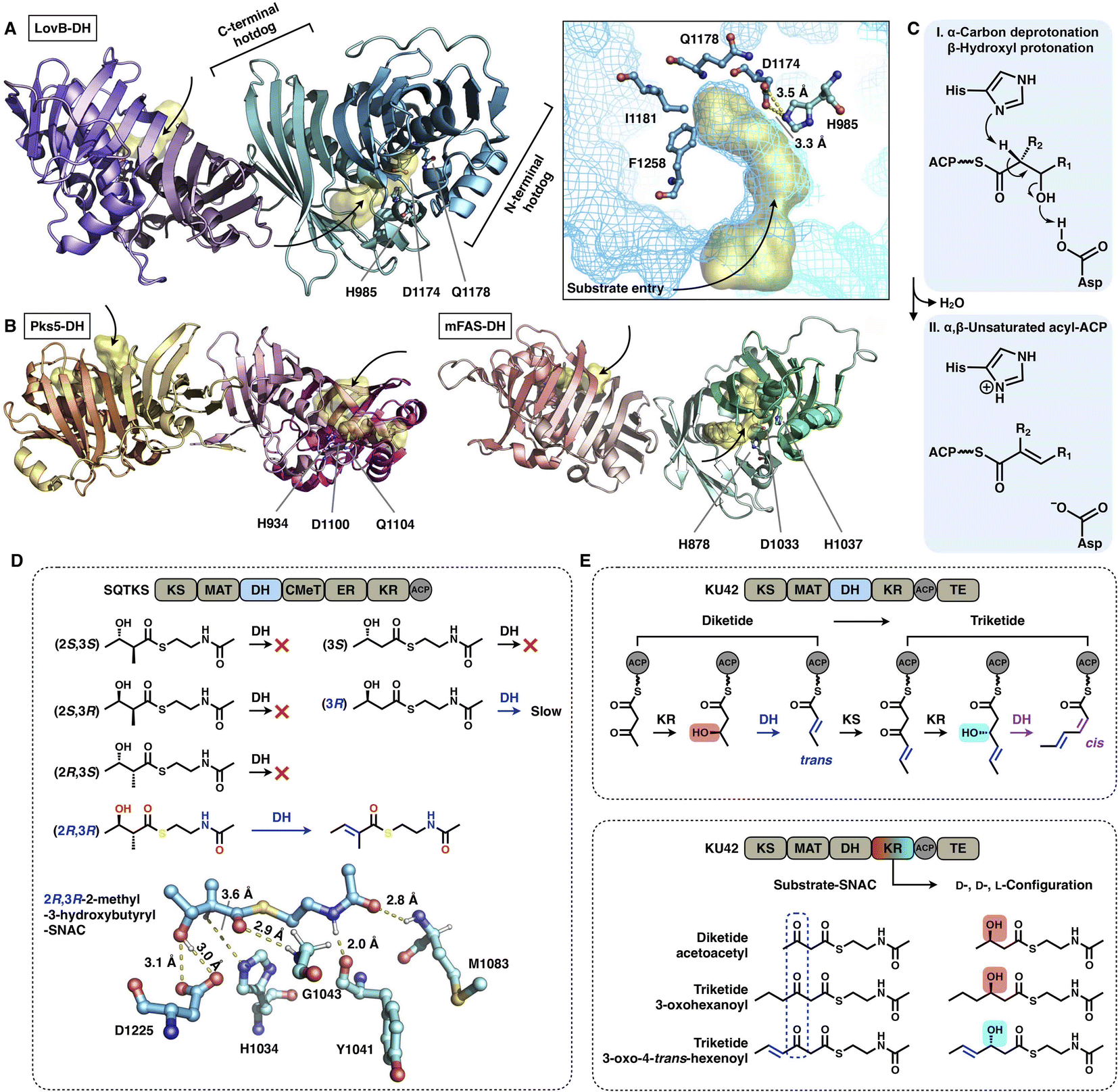 | ||
| Fig. 13 Structures and representative stereochemistry of DH domains. (A) HR-iPKS LovB DH domain, PDB: 7CPX. (B) MAS-like PKS Pks5 and mFAS DH domains, PDB: 5BP4 and 2VZ9, respectively. The 3.5 Å-distance-located His-Asp catalytic dyad and substrate pocket (yellow surface) are labeled in (A). H1037, which is crucial for the mFAS (pig) DH catalysis, and the equivalently positioned Q residues in LovB and Pks5 are also labeled. (C) Proposed catalytic mechanism of DH. (D) Substrate stereoselectivity of the SQTKS DH domain. (E) Stereochemistry determined by both the KR and DH domains. Note that the top scheme about the biosynthetic pathway for the conversion of diketides into triketides has been proposed, but not successfully characterized experimentally. | ||
The intrinsic substrate stereoselectivity of an HR-iPKS DH domain has been investigated in vitro.65 A total of six potential SNAC substrates (diketides) with opposite S and R configurations and various complexities were analyzed by the isolated DH from squalestatin tetraketide synthase (SQTKS) (Fig. 13D). SQTKS DH only efficiently catalyzes 2R,3R-2-methyl-3-hydroxybutyryl-SNAC to yield a trans-olefin product, which clearly reveals the strict stereoselectivity of the DH domain at both the α- and β-carbon positions. The successful molecular docking of the substrate into the DH shows a satisfactory geometry in which the α-proton is at a distance of 3.6 Å from catalytic H1034, and the β-hydroxyl group is ∼3.0 Å from D1225.
Several cases have been reported in which the structure of polyketides synthesized by type I iPKSs contains both common trans- and “less common” cis-α,β-double bonds.63,66,67 It is proposed that the stereochemistry of the DH domain is mainly determined by the β-hydroxyl group configuration reduced by the previous KR-catalyzed step, which is exemplified by HR-iPKS KU4263 (Fig. 13E). The diketide is reduced by the KU42 KR domain to generate the D-configuration β-hydroxyl group, and the following DH forms the product with the trans-double bond; however, when the triketide is reduced to give the L-configuration, the cis-double bond is formed by the same DH domain. Additionally, only the trans-double bond-containing diketide substrate is reduced to form the triketide with the L-configuration (Fig. 13E, bottom), which indicates that the stereoselectivity of KU42 KR is strongly influenced by the unsaturation degree of the substrate. This study reveals that the KU42 KR and DH domains are reciprocally related in determining the stereochemistry of the final product.
2.7 Product template (PT) domains
The PT domains housed by type I NR-iPKSs mediate the regioselective aldol cyclization reaction of the polyketide chain and, therefore, control the regioselectivity of the final product structures.68 Based on phylogenetic analysis, the PT domains are classified into five different groups69 (Fig. 14): group I and II, C2–C7 first-ring cyclization of a tetraketide and hexaketide, respectively; group III, C2–C7 first-ring cyclization of a longer polyketide chain (e.g., heptaketides and nonaketides); group IV, C4–C9/C2–C11 cyclization (first and second rings, respectively); and group V, C6–C11/C4–C13 cyclization (first and second rings, respectively). Currently, the most well-studied PksA PT domain that mediates C4–C9/C2–C11 cyclization during aflatoxin biosynthesis belongs to group IV.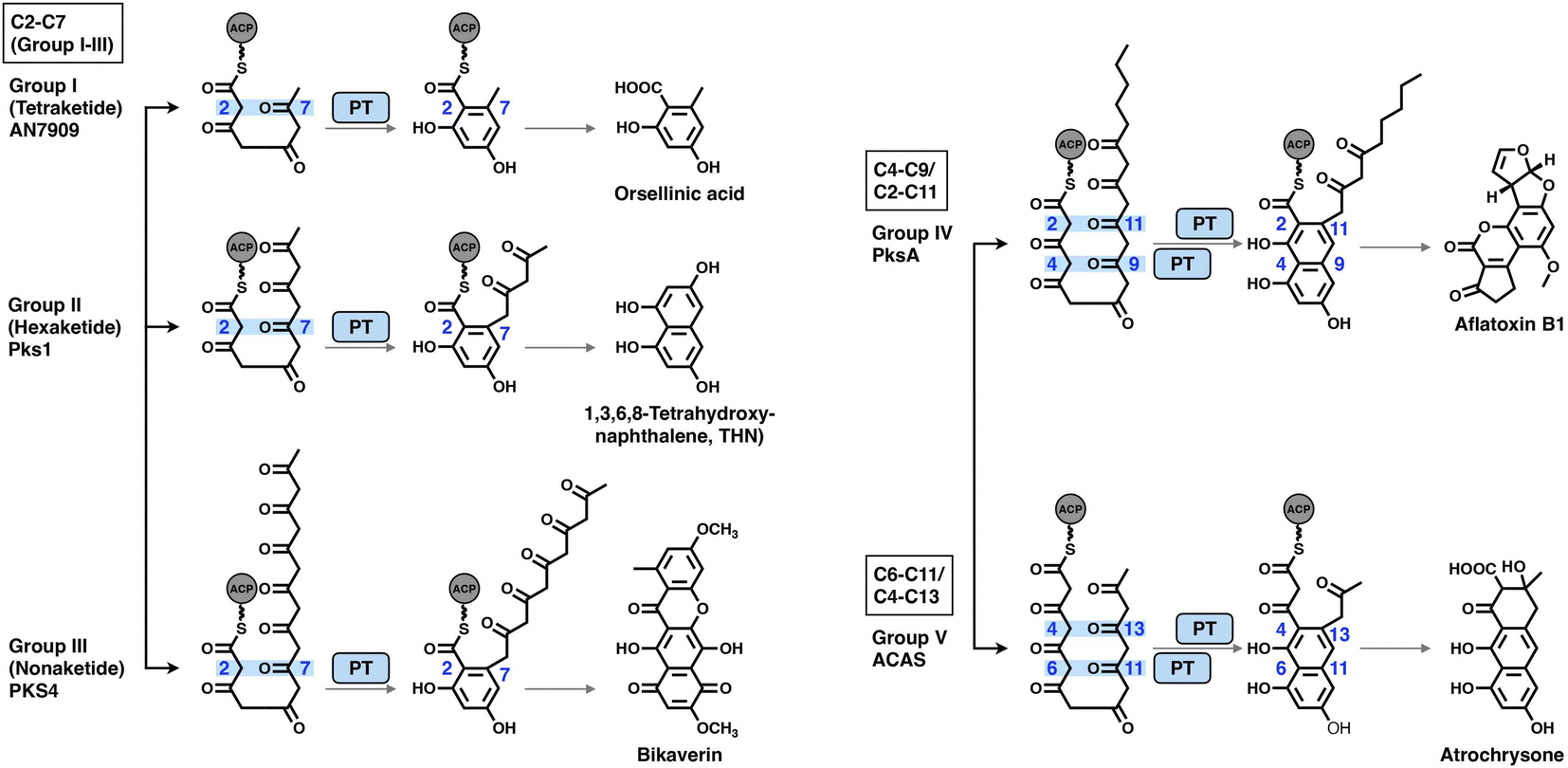 | ||
| Fig. 14 Regioselectivity of the aldol cyclization reactions catalyzed by five groups of NR-iPKS PT domains. | ||
The crystal structures of the PksA PT domain have been reported in complex with palmitate, a bicyclic substrate mimic or a bis-isoxazole heptaketide, which better mimics the natural poly-β-ketone intermediate70,71 (Fig. 15A–D). The dimeric PksA PT domain adopts a double hot-dog fold similar to that of the DHs (Fig. 15A, 13A and B). However, the two monomers of PT interact with each other via the C-terminal hot-dogs, which are directly opposite the DHs of modPKSs, HR-iPKSs and mFAS, which all interact via the N-termini. The approximately 30 Å (an SAS distance of 38 Å) substrate tunnel of PksA PT can be divided into three regions: an ∼14 Å linear region for binding the pPant arm of ACP, which delivers the intermediate polyketide chain; an ∼8 × 13.5 Å cyclization reaction chamber containing the His1345-Asp1543 catalytic dyad for the two regiospecific cyclization reactions; and an ∼6 × 6 Å hydrophobic hexyl binding region for accommodating the hexyl starter unit of the polyketide chain. The C16 palmitate captured in this deep tunnel is stabilized via multiple hydrophobic interactions (Fig. 15C). G1491 defines the end of the tunnel and is otherwise replaced by a bulk-side-chain hydrophobic residue in the nonhexyl binding PTs (Fig. 15B).
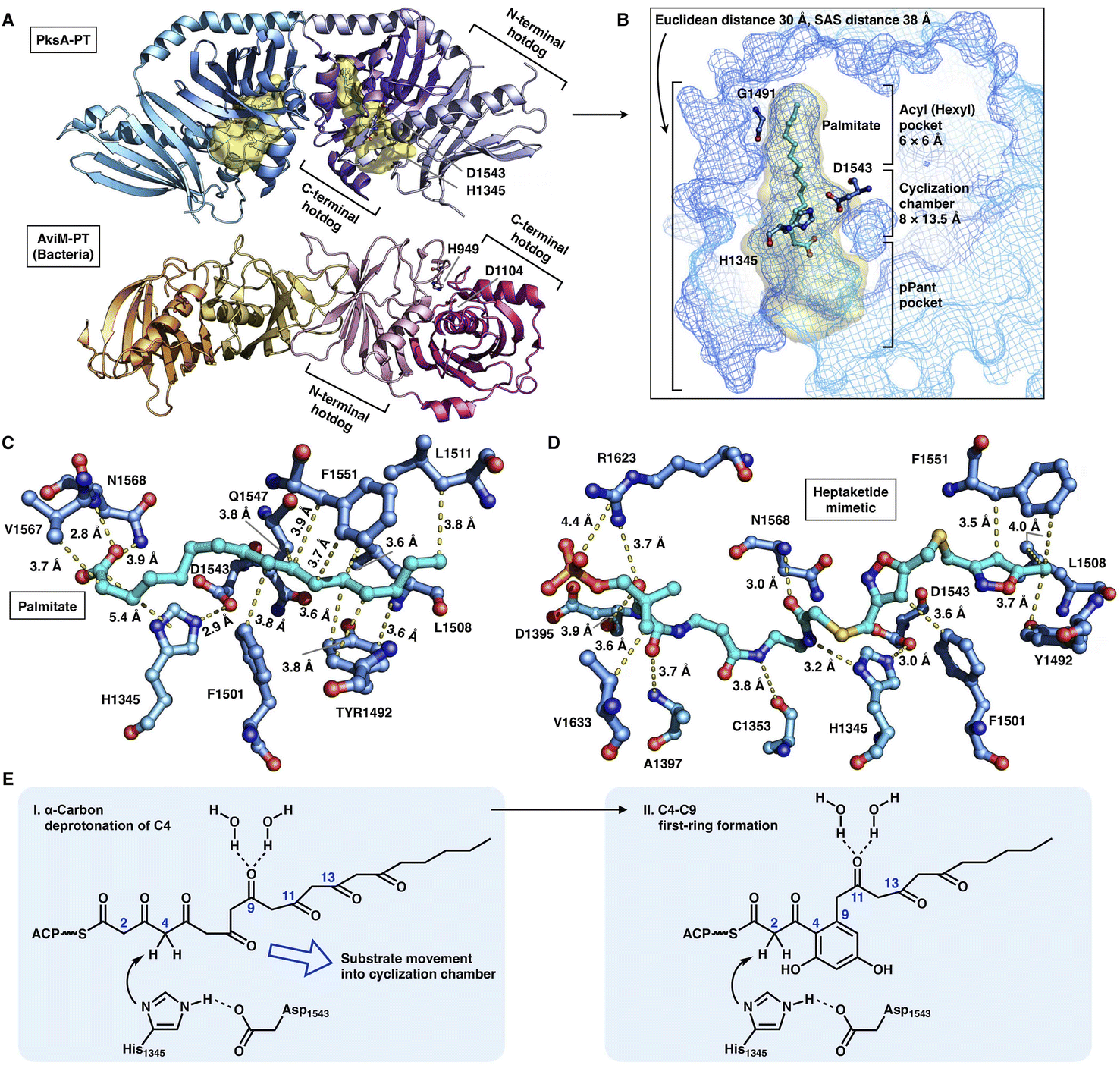 | ||
| Fig. 15 Structures and mechanism of PT domains. (A) Dimeric PT domains of NR-iPKS PksA and bacterial iterative AviM. PDB: 3HRQ and 7VWK, respectively. (B) Binding of palmitate (cyan) helps to identify the three-region-comprised substrate tunnel of ∼30 Å (yellow surface). The ∼3.0 Å distance-located H1345-D1543 catalytic dyad and G1491 are labeled. (C and D) Detailed views of the interactions between PksA PT active sites and palmitate and heptaketide substrate mimetic, respectively. The captured compounds are colored cyan. PDB: 3HRQ and 5KBZ, respectively. (E) Proposed catalytic mechanism of PksA-PT. | ||
Important insights into the catalytic mechanism have been uncovered by docking simulations70 and the structure of PT in complex with a C14 heptaketide (a substrate mimetic)71 (Fig. 15D). The mimetic is optimally positioned in an extended conformation, where pPant is strongly stabilized by R1623 via hydrogen bonding and a salt bridge, and the hexyl moiety forms hydrophobic interactions with Y1492, L1508 and F1551. In the cyclization reaction chamber, the critical heptaketide C4 is located precisely near the catalytic H1345 (3 Å), which catalyzes the regiospecific cyclization between C4 and C9 to form the first ring. The overall size and shape of the substrate tunnel are also important in chain length control.
Recently, the crystal structure of a bacterial PT domain of AviM that catalyzes C2–C7 cyclization in orsellinic acid synthesis has been reported72 (Fig. 15A, bottom). The overall structure, the dimeric interface and the active site catalytic dyad of the AviM PT domain are more similar to the canonical modPKS DHs than the fungal NR-iPKS PTs. Phylogenetic analysis showed that the bacterial AviM PT domain represents an evolutionary intermediate between the modPKS DH domains and NR-iPKS PT domains.
2.8 Enoylreductase (ER) domains
The NADPH-dependent ER domain stereoselectively reduces the α,β-double bond formed by the previous DH domain and belongs to the medium-chain dehydrogenase/reductase (MDR) superfamily.73 In type I iPKSs, the cis-acting ER domains form a homodimer74 and contribute the largest interface sticking the dimeric organization of the modifying region, as observed in the MAS-like PKS Pks524 and mFAS26 (Fig. 16B). In contrast, the trans-acting stand-alone ER domains are often found to collaborate with megaenzymes at the expense of the nonfunctional dimeric ψER domain, as exemplified by the LovB–LovC system in lovastatin biosynthesis17,75,76 (Fig. 16A).
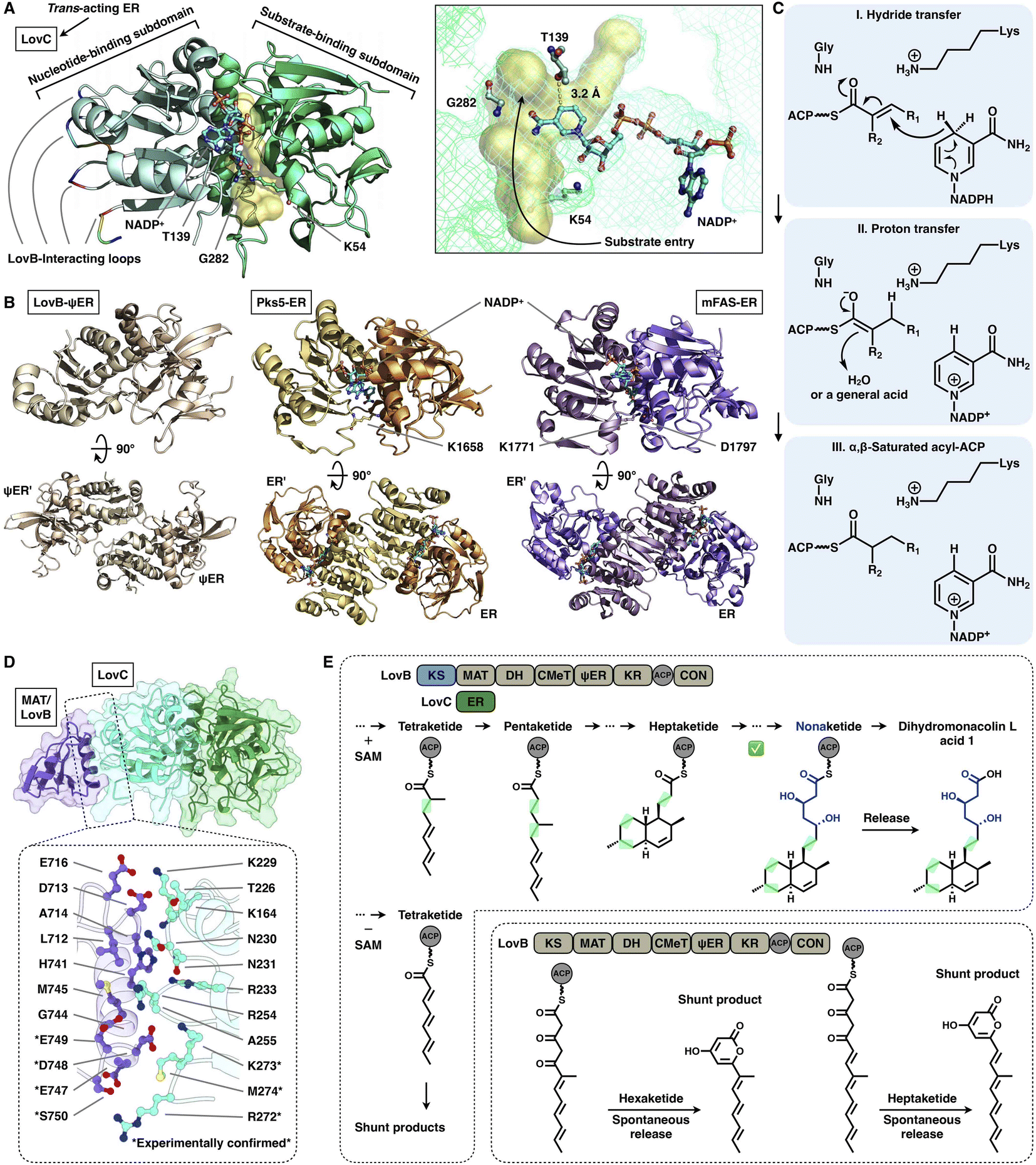 | ||
| Fig. 16 Structures and representative programming mechanism of ER domains. (A) Monomeric LovC, PDB: 3B70. The cofactor NADP+ (cyan), substrate tunnel (yellow surface) and active site residues are labeled. (B) Dimeric ψER of LovB, ERs of Pks5 and mFAS (PDB: 7CPX, 5BP4 and 2VZ9, respectively). (C) Proposed catalytic mechanism of ER (LovC). (D) LovB–LovC interface. (E) Gate-keeping function of trans-acting LovC. | ||
Despite the common dimeric architecture of MDR enzymes, LovC uniquely exists as a monomer either in the stand-alone state proven by the crystal structure and size-exclusion chromatography76 or in the complex state unveiled in the cryo-EM structure that LovC binds laterally to the LovB MAT domain through the C-terminal loops22 (Fig. 16A). LovC comprises of the substrate-binding subdomain and the nucleotide-binding subdomain. The hydrophobic substrate binding pocket is in the two-subdomain interface, which also binds the cofactor NADP+. The LovC substrate pocket is size-limited, which may contribute to the specific tetra-, penta- and heptaketide substrate selectivity, unlike the traverse tunnels of mFAS and Pks5. As indicated by substrate docking, the shorter di- or triketide may bind in the pocket in a nonproductive conformation, and the pocket is unable to accommodate polyketide intermediates longer than the heptaketide. K54 (highly conserved in the trans-type ER) and T139 are crucial for the reduction activity of LovC, in which T139 is at a distance of 3.2 Å from C4 of the NADPH nicotinamide ring, where the hydride is transferred. The enoylreduction mechanism has been proposed76 (Fig. 16C): when the ACP-tethered substrate enters the pocket, the hydride of NADPH nicotinamide ribose is transferred to the β-carbon of the α,β-unsaturated intermediate, followed by a proton transfer from an acidic residue or water to the α-carbon to form the α,β-saturated product. The enoyl reduction process may be facilitated by an oxyanion hole formed by the side chains of T139, K54 and G282.
The correct programming fidelity controlled by trans-acting ERs has been firmly established in multiple polyketide synthetic pathways.17,77–79 In the biosynthesis of dihydromonacolin L (the core of lovastatin), LovB faithfully constructs the nonaketide with the assistance of LovC, which has strict substrate selectivity for an α-methyl-substituted chain at the tetraketide stage17 (Fig. 16E). Without the partner LovC or eliminating the LovB–LovC interactions by interface mutation (Fig. 16D), LovB predominately produce the shunt pyrone products with shorter chain lengths (hexa- and heptaketides) (Fig. 16E) due to the highly reactive polyunsaturated polyketide structure. The highly substrate specificity of LovC toward the methylated tetraketide, not the unmethylated one, clearly provides evidence for the gate-keeping function of the trans-acting ER in maintaining the correct polyketide biosynthesis.
2.9 Polyketide chain release
The chain release of type I iPKSs has shown great diversity in different megaenzyme systems. In NR-iPKSs and mFAS, chain release is directly performed by a C-terminal TE domain similar to that in bacterial modPKSs. However, unlike the dimeric nature of modPKS TEs,80–82 the NR-iPKS PksA TE domain83 and hFAS TE domain84 are monomeric (Fig. 17). The PksA TE domain comprises two subdomains: the core region that displays the α/β-hydrolase fold and the inserted lid region, which contains two helices. The lid regions have been observed to exhibit various constituents and conformations across the α/β-hydrolase fold TE family.85 The substrate is suggested to enter the hydrophobic substrate pocket (closed conformation in the current structure) between the two subdomains, where the highly conserved Ser1937-Asp1964-His2088 catalytic triad is located. Instead of the typical hydrolysis reaction catalyzed by mFAS TE, which selects a water molecule as an intermolecular nucleophile and releases a linear carboxylic acid product, the NR-iPKS PksA TE can utilize a carbanion of the polyketide chain as an intramolecular nucleophile to form a C–C bond and produce a cyclic product. The mechanism of the PksA TE domain has been proposed: deprotonated by His, the nucleophilic Ser hydroxyl attacks the carbonyl carbon of the ACP pPant-tethered intermediate to form the tetrahedral intermediate. After the pPant arm leaves the active site, the substrate can undergo a conformational change to lock the substrate in a position near the catalytic His and undergo a nucleophilic attack to form the C–C bond (Dieckmann cyclization) and release the product.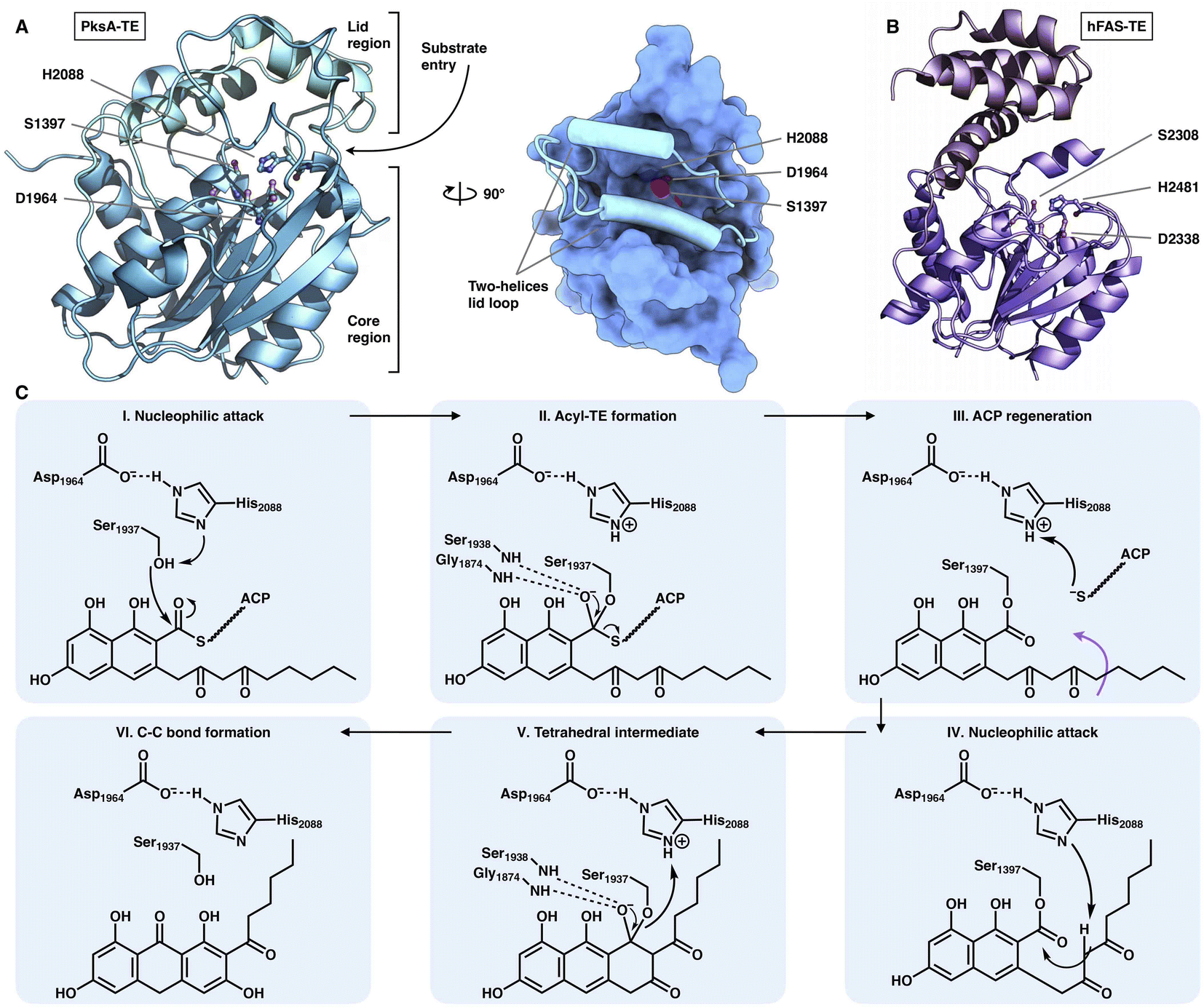 | ||
| Fig. 17 Structures of the C-terminus-fused TEs. (A) TE structure of NR-iPKS PksA shown in both cartoon and surface representations, PDB: 3ILS. The S1937-D1964-H2088 catalytic triad is labeled. (B) hFAS TE domain, PDB: 1XKT. (C) Proposed catalytic mechanism of NR-iPKS PksA TE. | ||
However, HR-iPKSs often do not contain a fused C-terminal TE domain and release the chain indirectly, although two rare cases of KU42-HR-iPKSs and KU43-HR-iPKSs show that the chains are released by their C-terminal TEs via thiolation and aminoacylation reactions,63 respectively. The polyketide chains are usually released by different stand-alone, trans-acting enzymes, including thioesterases (hydrolase fold and hot-dog fold), reductases, ATs, SATs, pyridoxal 5′-phosphate (PLP)-dependent enzymes (Fum8p)86 and even NRPSs.
Two trans-acting thioesterase crystal structures that display the α/β-hydrolase fold have been reported: DscB in complex with the substrate mimic during 10-membered lactone decarestrictine C1 biosynthesis87 and GrgF, which fuses the two different chain-length polyketides via C–C bond formation in gregatin A biosynthesis.88 Both of them are homodimeric in the asymmetric unit, and each monomer contains the Ser(Cys)-Asp-His catalytic triad (e.g., S114-D247-H276 in DcsB) (Fig. 18A–C). DcsB comprises the large α/β-hydrolase core region and the inserted small lid region containing three helices and two sheets. The capture of the pentaketide substrate mimic helps to identify an ∼151 Å3 substrate pocket located between the two regions. The mimic is stabilized by hydrogen bonds and hydrophobic interactions lining the pocket; however, the catalytic triad is 7.9 Å away from the thioester, which results in a nonproductive conformation. Subsequent docking simulations indicate that the substrate can be properly positioned near the triad in a ready-to-cyclize conformation, coordinated by hydrogen bonding interactions with the amides of F40 and F115.
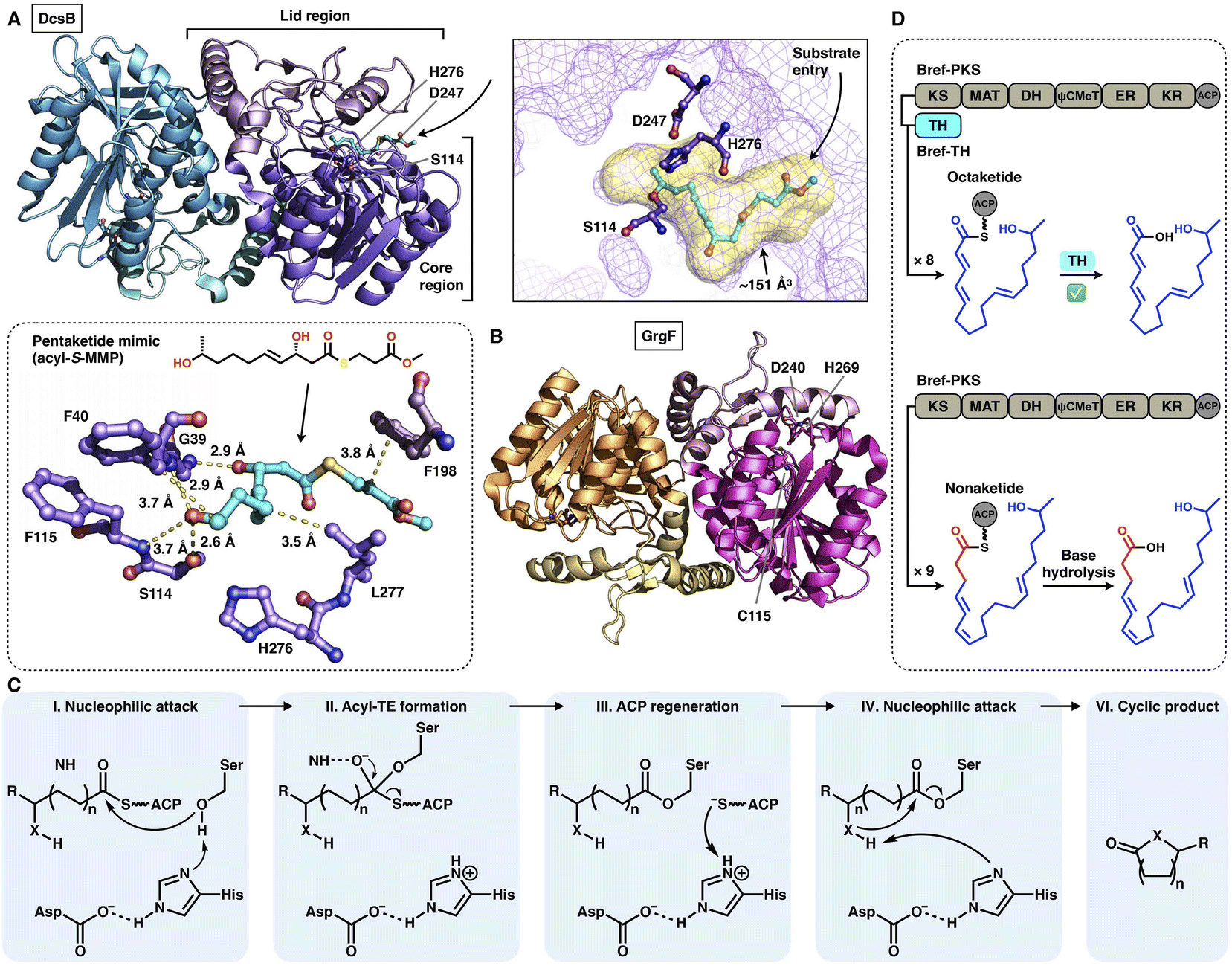 | ||
| Fig. 18 Structures and representative programming mechanism of α/β-hydrolase fold trans-acting TE. (A) Dimeric DcsB in complex with the pentaketide substrate mimic, PDB: 7D79. The substrate pocket (yellow surface) and Ser-Asp-His catalytic triad are labeled. A detailed view of the interaction between the active site and pentaketide mimic reveals a nonproductive conformation. (B) GrgF structure, PDB: 6LZH. Note that the conserved Ser in the triad is substituted by Cys (C115). (C) Proposed catalytic mechanism of α/β-hydrolase TE. (D) Chain length control of trans-acting TE revealed by Bref-PKSs and TH (thiohydrolase). | ||
The programming contributed by the hydrolase has been investigated. The chain-release of HR-iPKSs catalyzed by a trans-acting hydrolase is well demonstrated in the case of lovastatin biosynthesis.89 LovG, a serine hydrolase, is capable not only of releasing the correctly programmed dihydromonacolin nonaketide from LovB but also of proofreading by removing the incorrectly modified polyketide intermediates. Furthermore, in brefeldin A biosynthesis, how the product chain length of an HR-iPKS Bref-PKS is affected by a trans-acting hydrolase Bref-TH is elucidated90 (Fig. 18D). The Bref-PKS itself produces the acyclic nonaketide; however, in collaboration and specific interactions with Bref-TH, the octaketide product is released before the chain can be further elongated for an additional round. This hydrolase-mediated chain release indicates that Bref-TH contributes to the program rule by controlling the chain length of the final polyketide product. A similar chain-length control mechanism by the trans-acting releasing enzyme is exhibited by the pair of Fma-PKS and Fma-AT in the biosynthesis of fumagillin.91
The chain release catalyzed by trans-acting TEs is not limited to the α/β-hydrolase fold family. Three crystal structures of TE that adopt the hot-dog fold have been resolved (Fig. 19A and B): DynE7,92 CalE793 and SgcE10,94 which are involved in enediyne biosynthesis, including dynemicin, calicheamicin and C-1027, respectively. They form the homotetramer architecture, and each of the hot-dogs contains the conserved Arg catalytic residue (e.g., R35 in DynE7). The highly conjugated polyene, the product of the partner type I iPKS DynE8, is observed in the hot-dog B subunit of DynE7, which reveals an L-shaped traverse tunnel formed by both hot-dogs B and D. The tunnel can be divided into two regions: an ∼11 Å region for binding the pPant arm of ACP and a linear ∼19 Å acyl pocket for accommodating the extended polyketide chain in which the poly-carbon backbone is stabilized by multiple hydrophobic interactions. Intriguingly, although tetrameric, only one hot-dog's substrate tunnel is open and bound to the product, whereas the tunnels of the other three hot-dogs are closed, which support an induced-fit conformational change upon product binding. The following hydrolysis mechanism has been proposed (Fig. 19C): The Arg residue could serve as an oxyanion hole, facilitating the nucleophilic attack of the water molecule on the thioester bond of the ACP-tethered intermediate.
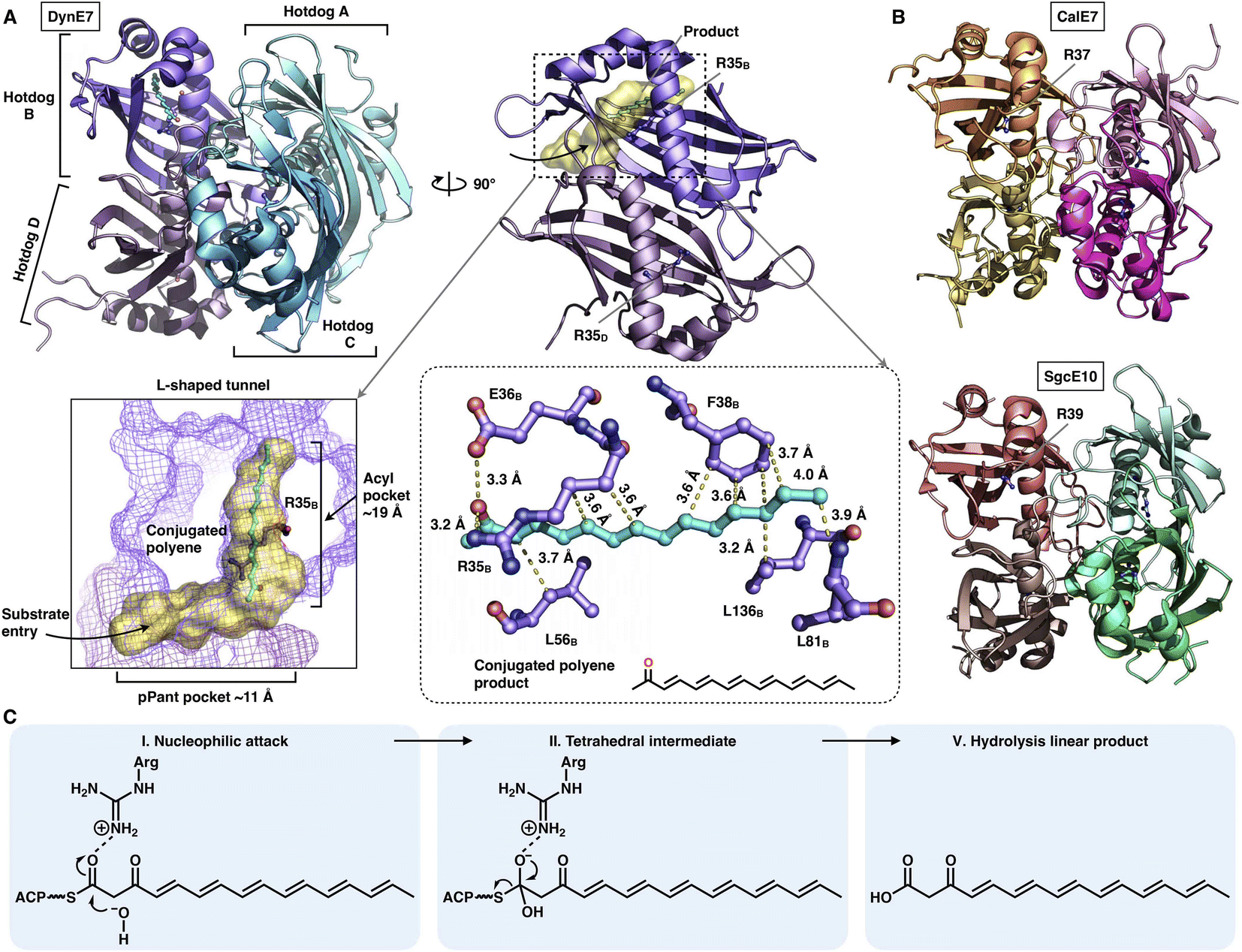 | ||
| Fig. 19 Structures of the tetrameric hot-dog fold trans-acting TE. (A) DynE7 in complex with the polyene product (cyan), PDB: 2XEM. The L-shaped substrate tunnel (yellow surface), residues interacting with the product and the Arg catalytic residue are labeled. (B) CalE7 and SgcE10 structures, PDB: 2W3X and 4I4J, respectively. (C) Proposed catalytic mechanism of hot-dog TE. | ||
A trans-acting acyltransferase can intercept and thus release the intermediate polyketide chain from an HR-iPKS, as also exemplified during lovastatin biosynthesis. The acyltransferase LovD is responsible for releasing the diketide synthesized by HR-iPKS LovF, and then transfers the chain to monacolin J acid to produce lovastatin95 (Fig. 20A). The chain transfer process essentially requires highly specific interactions between LovF-ACP and LovD. LovD exhibits broad substrate specificity toward various acyl-CoAs, acyl carriers, and variants of the monacolin J acceptor, and thus, can be used as an effective biocatalyst.96 A series of crystal structures of LovD have been reported,97,98 including wild-type (WT) LovD and directed evolution-improved LovD mutants (representative LovD G5, LovD6 and LovD9), which exhibit higher catalytic efficiency than the WT. LovD comprises a large α/β-hydrolase core region and a small lid region (Fig. 20B). The substrate pocket and Ser76-Lys79-Try188 are located at the two-region interface. Remarkably, the catalytic efficiency was enhanced by 1000-fold (LovD9) via directed evolution, considering that LovD WT has already been a broad substrate competent. By comparison of the WT with the evolved mutants, the dynamics of the lid region are observed, revealing a reduction of the pocket where the active sites are more deeply buried98 (Fig. 20C).
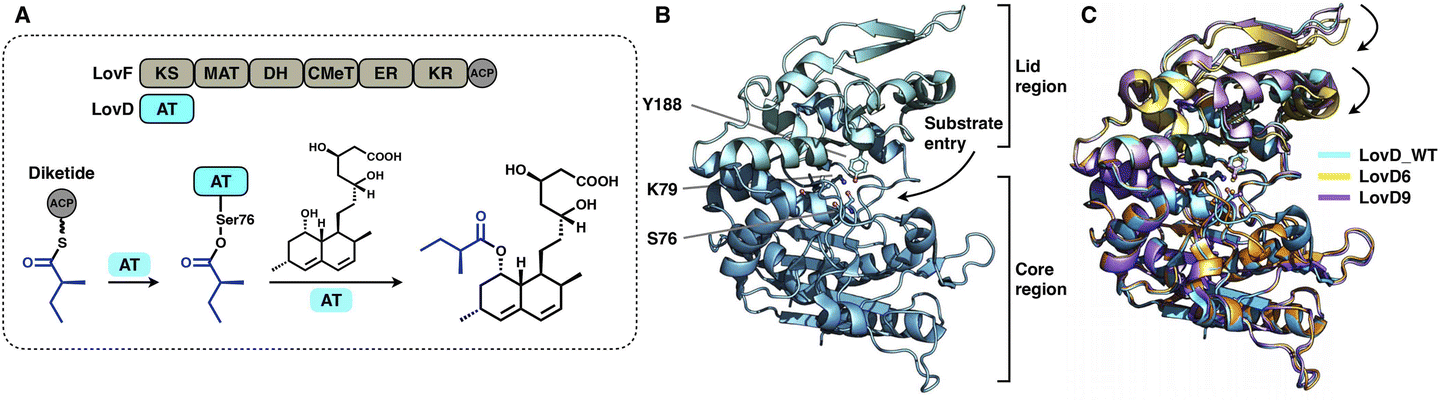 | ||
| Fig. 20 Structures of the α/β-hydrolase trans-acting AT. (A) Reaction of diketide release and transfer catalyzed by LovD. (B) LovD structure, PDB: 3HLB. The S76-K79-Y188 catalytic triad is labeled. (C) Superposition of the LovD WT and the evolved LovD6 and LovD9 on the core region reveals the dynamics of the lid regions, PDB: 3HLB, 4LCL and 4LCM, respectively. | ||
3. Conclusion and outlook
Type I iPKSs are elaborate nanomachines not only because of their multidomain architectures but also because of their complicated programming logic for the biosynthesis of complex polyketide products. Current evidence indicates that the overall programming of type I iPKSs may not be absolutely generalized but develops on a case-by-case basis with contributions from multiple intrinsic and extrinsic factors, including the relatively strict extension unit and broad starter unit selections performed by MAT and SAT domains, respectively; the substrate chain length controlled by SAT, KS, KR and trans-acting ER and TE domains; the stereochemical alterations of KR and DH domains; the intrinsic intermediate specificity of CMeT and KR domains, which combines the extrinsic kinetic competition between them; the acyl chain transfer checkpoint faithfully served by the trans-acting CMeT domain; and the gate-keeping function performed by trans-acting ER domains and specific domain–domain interactions.The interplay of catalytic domains is a key feature that has impacts on the programming pattern of type I iPKSs. A representative example of this is the biosynthesis of dihydromonacolin L acid (Fig. 21). The correct programming fidelity is achieved in the presence of the HR-iPKS LovB–LovC complex, substrate (malonyl-CoA) and cofactors (SAM, NADPH), leading to the nonaketide formation and released by LovG. LovB CMeT displays a higher kinetic efficiency at the tetraketide stage and thus outcompetes KR, and ER displays strict substrate selectivity toward this methylated tetraketide. If SAM, NADPH, and LovC are excluded (i.e., CMeT, KR, DH and ER function-disabled) in different combinations, the programming pattern goes off course and LovB can only produce pyrone shunt products. The production of pyrone shunt products is due to the highly reactive poly-β-carbonyl structure, which should be reduced by KR in the native context. Without the presence of LovC, the non-native tetraketide is elongated, but not further reduced by KR, which indicates that KR has also substrate selectivity. In addition, during the iterative LovB catalysis, LovG plays a proofreading role, removing the incorrectly modified polyketide intermediates from LovB, although it is not currently understood why the stalled aberrant intermediates cannot be further elongated by KS.
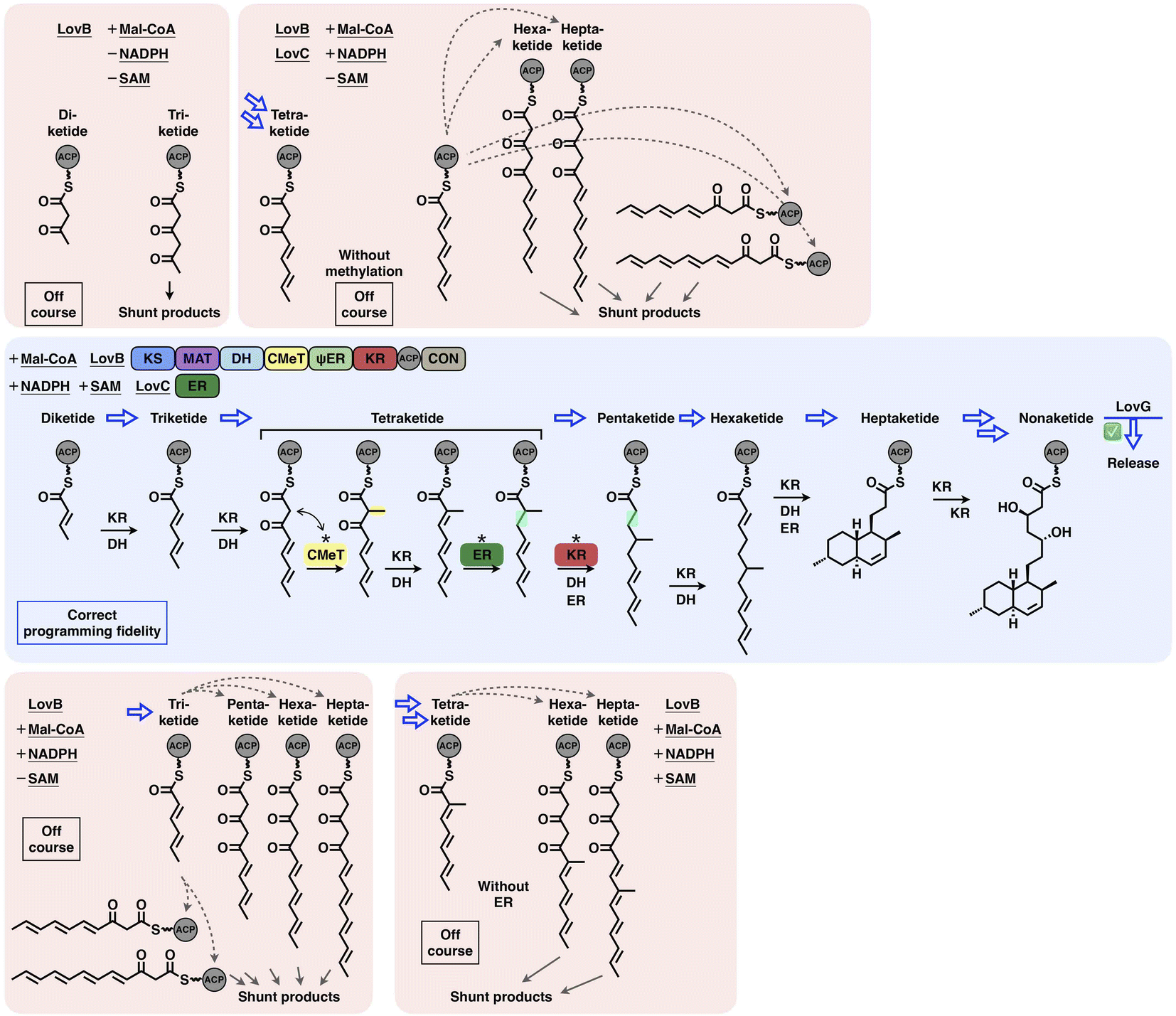 | ||
| Fig. 21 Overall view of the programming fidelity illustrated by the LovB–LovC system in dihydromonacolin L acid biosynthesis. | ||
The programming pattern of each catalytic domain can vary in the different types of I iPKS systems. In the case of kinetic competition between CMeT and KR, the TENS CMeT domain outcompetes KR at the triketide stage, resulting in the dimethylated polyketide (Fig. 22, left). However, in the closely related DMBS system, CMeT loses to KR and methylates the triketide only once. At the tetraketide stage (Fig. 22, right), CMeT of LovB is faster than its KR, whereas the TENS and DMBS KR domains beat their CMeT. With regard to the ER substrate selectivity, trans-acting LovC shows high specificity toward the methylated tetraketide (Fig. 16E). However, the cis-acting SQTKS ER is broadly selective toward a wide range of substrates that contain different chain lengths and methylation patterns.99
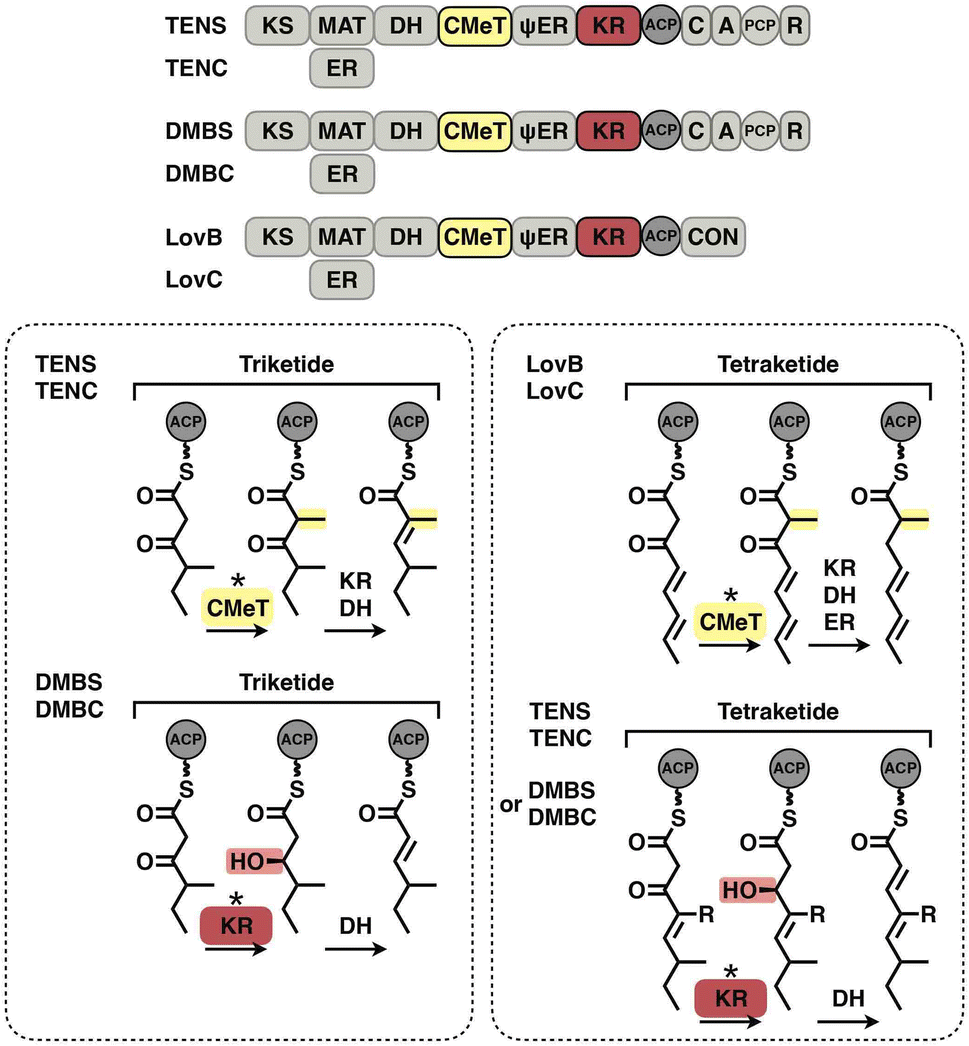 | ||
| Fig. 22 Kinetic competition between CMeT and KR in different systems. | ||
Although gradually revealed in recent decades, the overall programming of a type I iPKS is still very difficult to predict. However, the future is promising. With the development of the artificial intelligence-powered Alphafold2,100 one can now easily predict type I iPKS structures of either the excised domain or the complete architecture, which will rapidly provide fingerprints for rational engineering. Additionally, by using methods such as NMR, X-ray crystallography and cryo-EM single particle analysis, the capture of ACP pPant-tethered substrate binding in each domain will provide crucial information for scientists to uncover the catalytic mechanism, which might otherwise be difficult, as seen in many structures that adopt closed conformations without the binding substrate. Furthermore, using time-resolved cryo-EM101 combined with continuous heterogeneous cryo-EM reconstruction (such as cryoDRGN)102 will likely solve the transient, ever-changing intermediates inside the substrate tunnel of each domain during the time-ordered catalytic cycles. Visualizing these megaenzymes in a continuous action will provide invaluable molecular-level information, just as it is much clearer how a car engine works from a series of photographs, or even a video, than from just a single snapshot. With the further understanding of the programming mechanisms provided by the combinations of new strategies and technologies, it is eagerly anticipated that type I iPKSs may eventually be designed as programmable molecular machineries for the generation of desired products and novel pharmaceutical agents.
4. Author contributions
Z. W., J.L. and Z. D. supervised the study. All authors conceived the study. J. W. analyzed the structures and mechanisms and prepared the figures. All authors discussed and wrote the manuscript.5. Conflicts of interest
The authors declare no competing interests.6. Acknowledgments
This work was financially supported by the National Key R&D Program of China (2021YFA0910500 to J. L., 2018YFA0900700 to Z. W., 2019YFA0905400 to J. L.), the National Science Foundation of China (32270033 to Z. W., 32201035 to J. W., 32271302 to J. L., 32150013 and 32171252 to Z. D.), the China Postdoctoral Science Foundation (2022T150412 and 2022M720085 to J. W.) and the Shanghai ‘Super Postdoctoral’ Incentive Program (202112 to J. W.).7. Notes and references
- D. A. Hopwood, Chem. Rev., 1997, 97, 2465–2498 CrossRef CAS PubMed.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- J. A. Tobert, Nat. Rev. Drug Discovery, 2003, 2, 517–526 CrossRef CAS PubMed.
- J. Cortes, S. F. Haydock, G. A. Roberts, D. J. Bevitt and P. F. Leadlay, Nature, 1990, 348, 176–178 CrossRef CAS PubMed.
- R. E. Minto and C. A. Townsend, Chem. Rev., 1997, 97, 2537–2556 CrossRef CAS PubMed.
- A. G. Newman and C. A. Townsend, J. Am. Chem. Soc., 2016, 138, 4219–4228 CrossRef CAS PubMed.
- G. Yang, M. S. Rose, B. G. Turgeon and O. C. Yoder, Plant Cell, 1996, 8, 2139–2150 CAS.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2017 RSC.
- B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285–295 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2012, 29, 1050–1073 RSC.
- D. A. Herbst, C. A. Townsend and T. Maier, Nat. Prod. Rep., 2018, 35, 1046–1069 RSC.
- B. Busch, N. Ueberschaar, S. Behnken, Y. Sugimoto, M. Werneburg, N. Traitcheva, J. He and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 5285–5289 CrossRef CAS PubMed.
- S. M. Ma, J. W.-H. Li, J. W. Choi, H. Zhou, K. K. M. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. D. Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS PubMed.
- P. Paiva, F. E. Medina, M. Viegas, P. Ferreira, R. P. P. Neves, J. P. M. Sousa, M. J. Ramos and P. A. Fernandes, Chem. Rev., 2021, 121, 9502–9553 CrossRef CAS PubMed.
- C. Khosla, Y. Tang, A. Y. Chen, N. A. Schnarr and D. E. Cane, Annu. Rev. Biochem., 2007, 76, 195–221 CrossRef CAS PubMed.
- Y. He and R. J. Cox, Chem. Sci., 2015, 7, 2119–2127 RSC.
- T. Moriguchi, Y. Kezuka, T. Nonaka, Y. Ebizuka and I. Fujii, J. Biol. Chem., 2010, 285, 15637–15643 CrossRef CAS PubMed.
- R. J. Cox, Nat. Prod. Rep., 2023, 40, 9–27 RSC.
- E. Skellam, Nat. Prod. Rep., 2022, 39, 754–783 RSC.
- J. Wang, J. Liang, L. Chen, W. Zhang, L. Kong, C. Peng, C. Su, Y. Tang, Z. Deng and Z. Wang, Nat. Commun., 2021, 12, 867 CrossRef CAS PubMed.
- D. A. Herbst, C. R. Huitt-Roehl, R. P. Jakob, J. M. Kravetz, P. A. Storm, J. R. Alley, C. A. Townsend and T. Maier, Nat. Chem. Biol., 2018, 14, 474–479 CrossRef CAS PubMed.
- D. A. Herbst, R. P. Jakob, F. Zähringer and T. Maier, Nature, 2016, 531, 533–537 CrossRef CAS PubMed.
- Y. Tang, C.-Y. Kim, I. I. Mathews, D. E. Cane and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11124–11129 CrossRef CAS PubMed.
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 321, 1315–1322 CrossRef CAS PubMed.
- A. Keatinge-Clay, J. Mol. Biol., 2008, 384, 941–953 CrossRef CAS PubMed.
- D. L. Akey, J. R. Razelun, J. Tehranisa, D. H. Sherman, W. H. Gerwick and J. L. Smith, Structure, 2010, 18, 94–105 CrossRef CAS PubMed.
- E. J. Brignole, S. Smith and F. J. Asturias, Nat. Struct. Mol. Biol., 2009, 16, 190–197 CrossRef CAS PubMed.
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC.
- P. Mohr and C. Tamm, Tetrahedron, 1981, 37, 201–212 CrossRef.
- A. T. Keatinge-Clay, A. A. Shelat, D. F. Savage, S.-C. Tsai, L. J. W. Miercke, J. D. O'Connell, C. Khosla and R. M. Stroud, Structure, 2003, 11, 147–154 CrossRef CAS PubMed.
- A. Rittner, K. S. Paithankar, A. Himmler and M. Grininger, Protein Sci., 2020, 29, 589–605 CrossRef CAS PubMed.
- M. S. Dickinson, T. Miyazawa, R. S. McCool and A. T. Keatinge-Clay, Structure, 2022, 30, 1331–1339 CrossRef CAS PubMed.
- C. W. Liew, M. Nilsson, M. W. Chen, H. Sun, T. Cornvik, Z.-X. Liang and J. Lescar, J. Biol. Chem., 2012, 287, 23203–23215 CrossRef CAS PubMed.
- W. G. J. Hol, Prog. Biophys. Mol. Biol., 1985, 45, 149–195 CrossRef CAS PubMed.
- W. W. Cleland, Biochim. Biophys. Acta, Spec. Sect. Enzymol. Subj., 1963, 67, 173–187 CrossRef CAS.
- A. D. Grabowska, Y. Brison, L. Maveyraud, S. Gavalda, A. Faille, V. Nahoum, C. Bon, C. Guilhot, J.-D. Pedelacq, C. Chalut and L. Mourey, ACS Chem. Biol., 2020, 15, 3206–3216 CrossRef CAS PubMed.
- J. M. Crawford, B. C. R. Dancy, E. A. Hill, D. W. Udwary and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16728–16733 CrossRef CAS PubMed.
- J. M. Crawford, A. L. Vagstad, K. P. Whitworth, K. C. Ehrlich and C. A. Townsend, Chembiochem, 2008, 9, 1019–1023 CrossRef CAS PubMed.
- C. R. Huitt-Roehl, E. A. Hill, M. M. Adams, A. L. Vagstad, J. W. Li and C. A. Townsend, ACS Chem. Biol., 2015, 10, 1443–1449 CrossRef CAS PubMed.
- J. M. Winter, D. Cascio, D. Dietrich, M. Sato, K. Watanabe, M. R. Sawaya, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 9885–9893 CrossRef CAS PubMed.
- C. Wang, X. Wang, L. Zhang, Q. Yue, Q. Liu, Y. Xu, A. A. L. Gunatilaka, X. Wei, Y. Xu and I. Molnár, J. Am. Chem. Soc., 2020, 142, 17093–17104 CrossRef CAS PubMed.
- S. Smith and S.-C. Tsai, Nat. Prod. Rep., 2007, 24, 1041–1072 RSC.
- A. L. Vagstad, S. B. Bumpus, K. Belecki, N. L. Kelleher and C. A. Townsend, J. Am. Chem. Soc., 2012, 134, 6865–6877 CrossRef CAS PubMed.
- N. A. Löhr, M. C. Urban, F. Eisen, L. Platz, W. Hüttel, M. Gressler, M. Müller and D. Hoffmeister, Chembiochem, 2023, 24, e202200649 CrossRef PubMed.
- D. Du, Y. Katsuyama, M. Horiuchi, S. Fushinobu, A. Chen, T. D. Davis, M. D. Burkart and Y. Ohnishi, Nat. Chem. Biol., 2020, 16, 776–782 CrossRef CAS PubMed.
- L. Hang, M. Tang, C. J. B. Harvey, C. G. Page, J. Li, Y. Hung, N. Liu, M. E. Hillenmeyer and Y. Tang, Angew. Chem., Int. Ed., 2017, 56, 9556–9560 CrossRef CAS PubMed.
- P. A. Storm, D. A. Herbst, T. Maier and C. A. Townsend, Cell Chem. Biol., 2017, 24, 316–325 CrossRef CAS PubMed.
- S. Kishimoto, Y. Tsunematsu, T. Matsushita, K. Hara, H. Hashimoto, Y. Tang and K. Watanabe, Biochemistry, 2019, 58, 3933–3937 CrossRef CAS PubMed.
- H. Tao, T. Mori, X. Wei, Y. Matsuda and I. Abe, Angew. Chem., Int. Ed., 2021, 60, 8851–8858 CrossRef CAS PubMed.
- J. L. Martin and F. M. McMillan, Curr. Opin. Struct. Biol., 2002, 12, 783–793 CrossRef CAS PubMed.
- R. A. Cacho, J. Thuss, W. Xu, R. Sanichar, Z. Gao, A. Nguyen, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 15688–15691 CrossRef CAS PubMed.
- Y. Zou, W. Xu, Y. Tsunematsu, M. Tang, K. Watanabe and Y. Tang, Org. Lett., 2014, 16, 6390–6393 CrossRef CAS PubMed.
- K. L. Kavanagh, H. Jörnvall, B. Persson and U. Oppermann, Cell. Mol. Life Sci., 2008, 65, 3895–3906 CrossRef CAS PubMed.
- A. C. Price, Y.-M. Zhang, C. O. Rock and S. W. White, Structure, 2004, 12, 417–428 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Chem. Biol., 2007, 14, 898–908 CrossRef CAS PubMed.
- H. Zhou, Z. Gao, K. Qiao, J. Wang, J. C. Vederas and Y. Tang, Nat. Chem. Biol., 2012, 8, 331–333 CrossRef CAS PubMed.
- A. T. Keatinge-Clay and R. M. Stroud, Structure, 2006, 14, 737–748 CrossRef CAS PubMed.
- H. Zhou, K. Qiao, Z. Gao, J. C. Vederas and Y. Tang, J. Biol. Chem., 2010, 285, 41412–41421 CrossRef CAS PubMed.
- Y. Morishita, H. Zhang, T. Taniguchi, K. Mori and T. Asai, Org. Lett., 2019, 21, 4788–4792 CrossRef CAS PubMed.
- Y. Morishita, T. Sonohara, T. Taniguchi, K. Adachi, M. Fujita and T. Asai, Org. Biomol. Chem., 2020, 18, 2813–2816 RSC.
- M.-C. Tang, C. R. Fischer, J. V. Chari, D. Tan, S. Suresh, A. Chu, M. Miranda, J. Smith, Z. Zhang, N. K. Garg, R. P. St. Onge and Y. Tang, J. Am. Chem. Soc., 2019, 141, 8198–8206 CrossRef CAS PubMed.
- K. M. Fisch, W. Bakeer, A. A. Yakasai, Z. Song, J. Pedrick, Z. Wasil, A. M. Bailey, C. M. Lazarus, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 16635–16641 CrossRef CAS PubMed.
- E. Liddle, A. Scott, L.-C. Han, D. Ivison, T. J. Simpson, C. L. Willis and R. J. Cox, Chem. Commun., 2017, 53, 1727–1730 RSC.
- F. Kudo, Y. Matsuura, T. Hayashi, M. Fukushima and T. Eguchi, J. Antibiot., 2016, 69, 541–548 CrossRef CAS PubMed.
- R. Nofiani, K. de Mattos-Shipley, K. E. Lebe, L.-C. Han, Z. Iqbal, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Nat. Commun., 2018, 9, 3940 CrossRef PubMed.
- J. M. Crawford, P. M. Thomas, J. R. Scheerer, A. L. Vagstad, N. L. Kelleher and C. A. Townsend, Science, 2008, 320, 243–246 CrossRef CAS PubMed.
- Y. Li, I. I. Image, W. Xu, I. Image, Y. Tang and I. Image, J. Biol. Chem., 2010, 285, 22764–22773 CrossRef CAS PubMed.
- J. M. Crawford, T. P. Korman, J. W. Labonte, A. L. Vagstad, E. A. Hill, O. Kamari-Bidkorpeh, S.-C. Tsai and C. A. Townsend, Nature, 2009, 461, 1139–1143 CrossRef CAS PubMed.
- J. F. Barajas, G. Shakya, G. Moreno, H. Rivera, D. R. Jackson, C. L. Topper, A. L. Vagstad, J. J. L. Clair, C. A. Townsend, M. D. Burkart and S.-C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4142–E4148 CrossRef CAS PubMed.
- Y. Feng, X. Yang, H. Ji, Z. Deng, S. Lin and J. Zheng, Commun. Biol., 2022, 5, 508 CrossRef CAS PubMed.
- H. Riveros-Rosas, A. Julián-Sánchez, R. Villalobos-Molina, J. P. Pardo and E. Piña, Eur. J. Biochem., 2003, 270, 3309–3334 CrossRef CAS PubMed.
- D. M. Roberts, C. Bartel, A. Scott, D. Ivison, T. J. Simpson and R. J. Cox, Chem. Sci., 2017, 8, 1116–1126 RSC.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS PubMed.
- B. D. Ames, C. Nguyen, J. Bruegger, P. Smith, W. Xu, S. Ma, E. Wong, S. Wong, X. Xie, J. W.-H. Li, J. C. Vederas, Y. Tang and S.-C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11144–11149 CrossRef CAS PubMed.
- W. Xu, X. Cai, M. E. Jung and Y. Tang, J. Am. Chem. Soc., 2010, 132, 13604–13607 CrossRef CAS PubMed.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS PubMed.
- Z. Song, W. Bakeer, J. W. Marshall, A. A. Yakasai, R. M. Khalid, J. Collemare, E. Skellam, D. Tharreau, M.-H. Lebrun, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Sci., 2015, 6, 4837–4845 RSC.
- S.-C. Tsai, L. J. W. Miercke, J. Krucinski, R. Gokhale, J. C.-H. Chen, P. G. Foster, D. E. Cane, C. Khosla and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14808–14813 CrossRef CAS PubMed.
- S.-C. Tsai, H. Lu, D. E. Cane, C. Khosla and R. M. Stroud, Biochemistry, 2002, 41, 12598–12606 CrossRef CAS PubMed.
- J. B. Scaglione, D. L. Akey, R. Sullivan, J. D. Kittendorf, C. M. Rath, E. Kim, J. L. Smith and D. H. Sherman, Angew. Chem., Int. Ed., 2010, 49, 5726–5730 CrossRef CAS PubMed.
- T. P. Korman, J. M. Crawford, J. W. Labonte, A. G. Newman, J. Wong, C. A. Townsend and S.-C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6246–6251 CrossRef CAS PubMed.
- B. Chakravarty, Z. Gu, S. S. Chirala, S. J. Wakil and F. A. Quiocho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15567–15572 CrossRef CAS PubMed.
- M. Nardini and B. W. Dijkstra, Curr. Opin. Struct. Biol., 1999, 9, 732–737 CrossRef CAS PubMed.
- R. Gerber, L. Lou and L. Du, J. Am. Chem. Soc., 2009, 131, 3148–3149 CrossRef CAS PubMed.
- D.-W. Gao, C. S. Jamieson, G. Wang, Y. Yan, J. Zhou, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2021, 143, 80–84 CrossRef CAS PubMed.
- W.-G. Wang, H. Wang, L.-Q. Du, M. Li, L. Chen, J. Yu, G.-G. Cheng, M.-T. Zhan, Q.-F. Hu, L. Zhang, M. Yao and Y. Matsuda, J. Am. Chem. Soc., 2020, 142, 8464–8472 CrossRef CAS PubMed.
- W. Xu, Y. Chooi, J. W. Choi, S. Li, J. C. Vederas, N. A. D. Silva and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 6472–6475 CrossRef CAS PubMed.
- A. O. Zabala, Y.-H. Chooi, M. S. Choi, H.-C. Lin and Y. Tang, ACS Chem. Biol., 2014, 9, 1576–1586 CrossRef CAS PubMed.
- H.-C. Lin, Y.-H. Chooi, S. Dhingra, W. Xu, A. M. Calvo and Y. Tang, J. Am. Chem. Soc., 2013, 135, 4616–4619 CrossRef CAS PubMed.
- C. W. Liew, A. Sharff, M. Kotaka, R. Kong, H. Sun, I. Qureshi, G. Bricogne, Z.-X. Liang and J. Lescar, J. Mol. Biol., 2010, 404, 291–306 CrossRef CAS PubMed.
- M. Kotaka, R. Kong, I. Qureshi, Q. S. Ho, H. Sun, C. W. Liew, L. P. Goh, P. Cheung, Y. Mu, J. Lescar and Z.-X. Liang, J. Biol. Chem., 2009, 284, 15739–15749 CrossRef CAS PubMed.
- T. Annaval, J. D. Rudolf, C.-Y. Chang, J. R. Lohman, Y. Kim, L. Bigelow, R. Jedrzejczak, G. Babnigg, A. Joachimiak, G. N. Phillips and B. Shen, ACS Omega, 2017, 2, 5159–5169 CrossRef CAS PubMed.
- X. Xie, M. J. Meehan, W. Xu, P. C. Dorrestein and Y. Tang, J. Am. Chem. Soc., 2009, 131, 8388–8389 CrossRef CAS PubMed.
- X. Xie, K. Watanabe, W. A. Wojcicki, C. C. C. Wang and Y. Tang, Chem. Biol., 2006, 13, 1161–1169 CrossRef CAS PubMed.
- X. Gao, X. Xie, I. Pashkov, M. R. Sawaya, J. Laidman, W. Zhang, R. Cacho, T. O. Yeates and Y. Tang, Chem. Biol., 2009, 16, 1064–1074 CrossRef CAS PubMed.
- G. Jiménez-Osés, S. Osuna, X. Gao, M. R. Sawaya, L. Gilson, S. J. Collier, G. W. Huisman, T. O. Yeates, Y. Tang and K. N. Houk, Nat. Chem. Biol., 2014, 10, 431–436 CrossRef PubMed.
- D. M. Roberts, C. Bartel, A. Scott, D. Ivison, T. J. Simpson and R. J. Cox, Chem. Sci., 2016, 8, 1116–1126 RSC.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- J. Frank, J. Struct. Biol., 2017, 200, 303–306 CrossRef CAS PubMed.
- E. D. Zhong, T. Bepler, B. Berger and J. H. Davis, Nat. Methods, 2021, 18, 176–185 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |