
Two dimensional materials are non-nanotoxic and biocompatible towards cyclotides: evidence from classical molecular dynamics simulations†
Anupam
Ghosh
,
Titas Kumar
Mukhopadhyay
and
Ayan
Datta
 *
*
School of Chemical Sciences, Indian Association for the Cultivation of Science, Jadavpur – 700032, West Bengal, India. E-mail: spad@iacs.res.in; Tel: +91-33-24734971
First published on 22nd November 2022
Abstract
Cyclotides are backbone-cyclized peptides of plant origin enriched with disulfide bonds, having exceptional stability towards thermal denaturation and proteolytic degradation. They have a plethora of activities like antibacterial, antifungal, anti-tumor and anti-HIV properties predominantly owing to their selective interaction with certain phospholipids, thereby leading to the disruption of cellular membranes. On the other hand, low-dimensional materials like graphene and hexagonal boron nitride (h-BN) are also known to show membrane-proliferating activities through lipid extraction. A plausible and more effective antibacterial, anti-tumor and antifungal agent would be a composite of these 2D materials and cyclotides, provided the structures of the peptides remain unperturbed upon adsorption and interaction. In this study, classical molecular dynamics simulations are performed to understand the nature of adsorption of cyclotides belonging to different families on graphene and h-BN and analyze the resulting structural changes. It is revealed that, due to their exceptional structural stability, cyclotides maintain their structural integrity upon adsorption on the 2D materials. In addition, the aggregated states of the cyclotides, which are ubiquitous in plant organs, are also not disrupted upon adsorption. Extensive free energy calculations show that the adsorption strength of the cyclotides is moderate in comparison to those of other similar-sized biomolecules, and the larger the size of the aggregates, the weaker the binding of individual peptides with the 2D materials, thereby leading to their lower release times from the materials. It is predicted that graphene and h-BN may safely be used for the preparation of composites with cyclotides, which in turn may be envisaged to be probable candidates for manufacturing next-generation bionano agents for agricultural, antibacterial and therapeutic applications.
1. Introduction
In recent years, cyclotides, a topologically fascinating family of cyclic peptides, have gathered increasing attention from structural biologists, chemists, and biochemists for their unique structural features.1,2 Cyclotides are backbone-cyclized disulfide-rich circular peptides containing 28 to 37 amino acid residues which are the first class of gene-expressed cyclic peptides found in plants.3 The structure of all of the cyclotides involves a characteristic head-to-tail cyclized peptide backbone and three interlocking disulfide bridges built from six conserved cystine residues, where one of the disulfide bonds penetrates via a macrocycle formed from the two disulfide bonds. This topology is known as the cyclic cystine knot (CCK) motif (Fig. 1(a–c)), which gives the cyclotides a highly rigid structural framework, thereby resulting in exceptional thermal stability and resistance to chemical denaturation and enzymatic degradation.4,5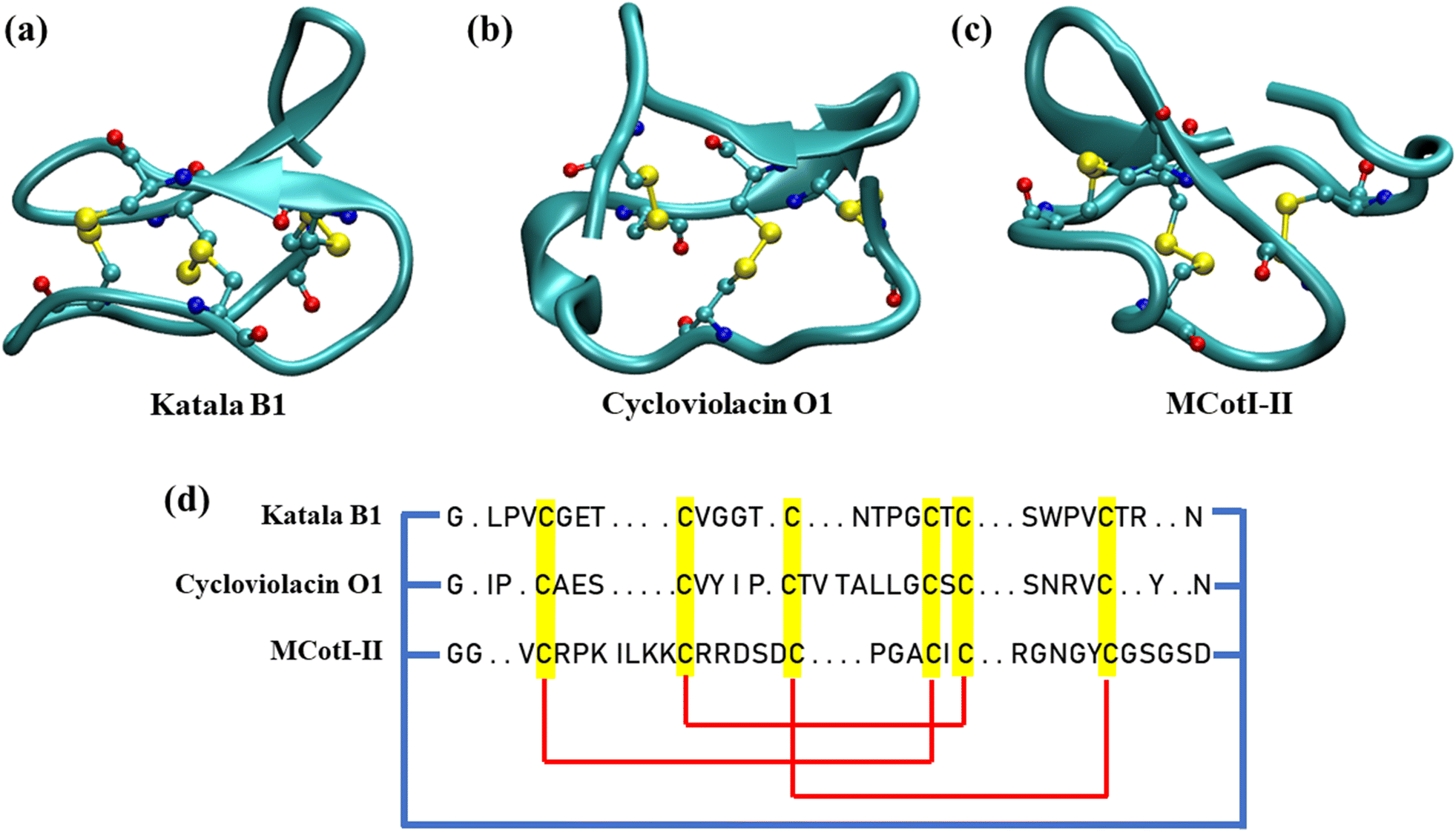 | ||
| Fig. 1 Secondary structures of the cyclotides considered in this study: (a) Katala B1 (the Möbius family), (b) cycloviolacin O1 (the bracelet family), and (c) MCoTI-II (the cyclic knottin family). The ball-and-stick representation is used for the depiction of the cyclic cystine knots (CCK), and the hydrogen atoms are removed for clarity. (d) Schematic representations of the sequence of amino acids in the three cyclotides. The cystine residues are highlighted in yellow, the cyclic knots are represented by red lines, and the cyclic connection between the amino acid residues are shown by bold blue lines. | ||
Hundreds of cyclotides have been obtained from plants in the Rubiaceae, Violacea, Apocynaceae, Cucurbitaceae, and Fabaceae families.1 Nevertheless, it has been predicted that ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cyclotides might exist. Based on the similarities in sequences and topological differences, the cyclotide family members found so far have been classified into two main subdomains depending on the absence or presence of a cis-proline (Pro) residue in the fifth loop.6,7 Cyclotides that contain this Pro residue are referred to as Möbius cyclotides (Fig. 1(a)), while those in which the Pro residue is absent are referred to as bracelet cyclotides (Fig. 1(b)).6 Recently, a third category of cyclotides, known as chimeric cyclotides, has been discovered which carries the structural properties of both the Möbius and bracelet families.7 Also, another subdomain of cyclotides, the trypsin inhibitor cyclotides, has been found recently which is different from the Möbius and bracelet families and shares more similarities with knottins, linear peptides bearing the cystine knot motif, and therefore, trypsin inhibitor cyclotides are also known as cyclic knottins (Fig. 1(c)).8 However, the last subclass of cyclotides has similar biosynthetic pathways as observed for Möbius and bracelet cyclotides, thereby justifying their classification. Cyclotides remain in the aggregated state in all plant parts where they are produced; however, each of the plant organs and tissues has its unique distribution of different categories of cyclotides having different amounts and varieties of their amino acid sequences.9,10 In fact, cyclotides expressed in different plant organs and tissues depend on the surrounding, climate and hurdles faced by the species such as insects, pathogens, and extreme environmental conditions among many others.
000 cyclotides might exist. Based on the similarities in sequences and topological differences, the cyclotide family members found so far have been classified into two main subdomains depending on the absence or presence of a cis-proline (Pro) residue in the fifth loop.6,7 Cyclotides that contain this Pro residue are referred to as Möbius cyclotides (Fig. 1(a)), while those in which the Pro residue is absent are referred to as bracelet cyclotides (Fig. 1(b)).6 Recently, a third category of cyclotides, known as chimeric cyclotides, has been discovered which carries the structural properties of both the Möbius and bracelet families.7 Also, another subdomain of cyclotides, the trypsin inhibitor cyclotides, has been found recently which is different from the Möbius and bracelet families and shares more similarities with knottins, linear peptides bearing the cystine knot motif, and therefore, trypsin inhibitor cyclotides are also known as cyclic knottins (Fig. 1(c)).8 However, the last subclass of cyclotides has similar biosynthetic pathways as observed for Möbius and bracelet cyclotides, thereby justifying their classification. Cyclotides remain in the aggregated state in all plant parts where they are produced; however, each of the plant organs and tissues has its unique distribution of different categories of cyclotides having different amounts and varieties of their amino acid sequences.9,10 In fact, cyclotides expressed in different plant organs and tissues depend on the surrounding, climate and hurdles faced by the species such as insects, pathogens, and extreme environmental conditions among many others.
Cyclotides have been extensively studied for their several biological and biochemical activities such as haemolytic, antifouling, anti-HIV, and antibacterial properties and most importantly, cytotoxicity toward human cancer cells.11–14 While cyclotides belonging to both the main categories, namely the Möbius and bracelet families, show antibacterial activities, the bracelet cyclotides particularly cycloviolacins (cyO 1/2/3/19) demonstrate stronger activities compared to Möbius.11 In addition, cyclotides are found to be active against a variety of fungi, the activity again being sensitive towards the composition of the amino acid residues and they might be useful as an eco-friendly way to protect crops.15,16 It has been revealed that the antibacterial activities of cyclotides are related to their selective membrane binding and penetration properties and resulting membrane disruption. In fact, researchers have explored the means to boost the antibacterial and anti-tumour activities of cyclotides for therapeutic applications and the structural motifs as templates for drug design.17–19
On the other hand, two dimensional (2D) systems have gained importance in biomedical research recently owing to their remarkable structural properties and chemical stability along with an exceptionally high surface area and can behave like a flat platform for the adsorption and interaction of a variety of biomolecules.20–24 However, contemporary bio-nano usage of such 2D systems has caused concerns regarding their nanotoxicity effects.20,25–30 Graphene and its oxides have been found to act as effective antibacterial and antifungal agents, destroying the bacterial cell membrane and disrupting their cellular functions.31,32 Also, graphene can destroy the secondary and tertiary structures of proteins and enzymes, thereby affecting their biological activities.33,34 Hexagonal boron nitride (h-BN), an analogue of graphene, shows similar membrane penetrating properties, and both graphene and h-BN are capable of extracting lipid molecules from membranes and inducing phase transitions in lipid membranes, affecting the membrane dynamics.35,36 Both computational and experimental studies reveal that graphene and h-BN perturb the structure of several nucleic acids including single and double-stranded DNA as well as guanine quadruplexes (GQs).37,38
Since the flat surfaces of 2D materials are efficient in adsorbing proteins, it is possible to prepare bio-nanocomposites of these materials with cyclotides, which may retain their individual antifungal, antibacterial and anti-tumor properties and might even show an enhanced efficacy cooperatively. Nevertheless, to envisage such possibilities, it must be ensured that the structural properties of the cyclotides adsorbed on the two-dimensional materials remain intact, maintaining their secondary structures. In addition, the adsorption strength between the peptides and the materials should be moderate so as to facilitate the interaction between lipid molecules and the peptides upon insertion of the 2D material–cyclotide composites onto lipid membranes. However, to date, no study has addressed this possibility and evaluated the potential of the state-of-the-art 2D materials for the preparation of such composite materials. In this context, “in silico” techniques can predict the outcome of such intricate molecular-level interactions, providing crucial details regarding the subtle balance of intra- and intermolecular interactions.39–41 In this regard, in this article, we have studied the interaction of different cyclotides with graphene and h-BN using all-atom classical molecular dynamics simulations. We have selected three cyclotides, one from each of the three unique families, viz. Möbius (Katala B1), bracelet (cycloviolacin O1), and cyclic knottins (MCoTI-II). The structures of these cyclotides are depicted in Fig. 1(a–c) and the amino acid sequences are schematically represented in Fig. 1(d). The structural evolution of the peptides during and after the adsorption is studied and compared with that without any 2D material. Cyclotides often exist in the form of aggregates in different plant organs and penetrate the lipid membranes in that aggregated state.42,43 Therefore, we allowed cyclotide aggregates to be adsorbed on these 2D materials and checked the stability of the aggregates. Free energy calculations are performed to shed light on the aspects of the peptide adsorption and release from the surfaces. It is revealed that 2D materials form stable heterostructures with cyclotides without perturbing their structural properties, and sustained release of adsorbed cyclotide aggregates can be achieved by controlling the aggregate size. The current study provides a microscopic understanding of the molecular-level details of the interaction of cyclotides with 2D materials, thereby providing the possibility of the fabrication of 2D material–cyclotide composites for the development of next-generation antibacterial, antifungal, and anti-tumour agents for agricultural, biomedical, and therapeutic applications.17
2. Computational details
The coordinates of the three different cyclotides were extracted from the Protein Data Bank (PDB ID: 1NB1 for Katala B1, 1NBJ for cycloviolacin O1, and 1IB9 for MCoTI-II). All of these peptides were solvated in cubic boxes of the dimension 50 × 50 × 50 Å3 and the charges resulting from the charged amino acid residues were neutralized by the addition of suitable numbers of sodium/chloride ions and then NaCl was incorporated to make the concentration 0.15 M. These solvated systems were subjected to a multistep minimization-equilibration protocol as reported in our earlier studies (see the ESI† for further details).37,38,44 The equilibrated systems were used for the 500 ns production simulations in the isothermal–isobaric (NPT) ensemble at 300 K. The structures of the cyclotides were extracted from the last frames of the production simulations and used as initial configurations for adsorption simulations. Although the adsorption simulations could have been started from the crystal structure of the cyclotides, without equilibrating them in a solvated state, solvation followed by equilibration and production simulations for 500 ns were carried out for each of the cyclotides to observe if the MD parameters used to model the cyclotides have any adverse effect on the dynamics of the structures. However, the structural changes during the equilibration of the cyclotides in water were negligible and judged in terms of the root mean squared displacement (RMSD) with respect to the initial crystal structure. The insignificant change in the crystal structure upon solvation, as evident from the RMSD plot (Fig. S1†), provided us additional confidence regarding the quality of the MD parameters used in this study. Graphene and h-BN sheets of dimensions 60 × 60 Å2 were constructed along the XY plane using the VMD nanotube builder plugin and for each of the 2D nanosheets, three separate systems were prepared, keeping the three peptides at a noninteracting distance of 15 Å from the surfaces along the Z axis. Water and salt were added to each of these systems following the same protocol mentioned above and the final size of all the systems was 60 × 60 × 60 Å3 before starting the simulations. We applied the periodic boundary conditions in all directions and all of the prepared systems were subjected to a similar multistep minimization followed by the equilibration process as adopted for the peptides in water. The equilibrated systems were subsequently subjected to production simulations for 500 ns using the canonical ensemble at 300 K. All the atoms of the graphene and h-BN sheets were constrained harmonically with a force constant of 10 kcal mol−1 nm−2 throughout the minimization and equilibration processes. To ensure the reproducibility of the results, we prepared two different initial configurations for each of the systems, and for each initial configuration, the production simulations were performed for 500 ns. The harmonic constraint was employed for the 2D materials in order to avoid their movement (if any) relative to the peptide molecules.Next, we prepared medium-sized cyclotide aggregates taking four of each of the cyclotides in simulation boxes of dimensions 80 × 80 × 80 Å3. The boxes were solvated and ions were added following the protocol as mentioned above and similar minimization-equilibration methodologies were followed as used for the single cyclotides. These equilibration simulations were followed by production simulations for 500 ns at 300 K using the isothermal–isobaric (NPT) ensemble. At the end of 500 ns, the structures of the aggregates were extracted and further used for the preparation of the initial structures for adsorption simulations. Larger graphene and h-BN sheets of dimensions 80 × 80 Å2 were prepared for the adsorption of aggregated cyclotides and a similar protocol was followed for adsorption simulations as done before. 500 ns of simulations were performed for each of the aggregates.
Isothermal conditions were maintained in all of the simulations using Langevin dynamics by employing a damping coefficient of 5 ps−1, and we have used the Langevin piston method to maintain a constant pressure of 1 atm.45–47 To introduce the constant pressure coupling, a 100 fs piston period, a 50 fs damping time constant and a 300 K piston temperature were used. The particle mesh Ewald (PME) method having a 1 Å grid was used to calculate the electrostatic interactions using the periodicity of the systems and a 2 fs time step was used to integrate the classical equations of motion according to the Velocity Verlet algorithm.48 Covalent bonds involving hydrogen atoms were made rigid using the SHAKE algorithm.48 Non-bonded interactions were calculated employing a cut-off distance of 1.2 nm and the atomic coordinates were continuously stored after every 20 ps for visualization and trajectory analysis. Each of the initial configurations was prepared with the Packmol code. The NAMD 2.12 package was used for the classical molecular dynamics simulations, the VMD for the trajectory visualization and the in-house Tcl scripts for the statistical analysis of the simulation data.49,50 The TIP3P model was used for water and CHARMM27 parameters were used for the peptides.51 To computationally model the graphene and h-BN sheets, we have used the same parameters as those used in our previous investigations to model their liquid-phase exfoliation and the small molecular and biomolecular interactions.37,38,52–55 For analysis, a contact between the peptide and the 2D materials was considered only if any of the peptide atoms were within a cut-off distance of 0.6 nm with respect to the 2D materials, while the contact surface area (CSA) was calculated as half of the difference between the solvent accessible surface area (SASA) of the cyclotide–2D material hybrid and the sum of the SASAs of the material and cyclotide. We defined a hydrogen bond using the criteria: D–A distance ≤0.35 nm and D–H–A angle ≥120° (D and A being the donor and the acceptor atoms, respectively), which have been utilized previously for related systems.37,38,44
The changes in free energies associated with peptide adsorption were calculated in terms of the potential of mean forces (PMF) utilizing the adaptive biasing force (ABF) method implemented in NAMD 2.12.56,57 The details of the PMF calculations for single cyclotides and cyclotide aggregates are provided in the ESI.†
3. Results and discussion
Since the first stage of the fabrication of cyclotide–2D material composites involve adsorption of the peptides on the 2D materials, in the present study, we first focus on the mechanism of adsorption of all three cyclotide molecules on both graphene and h-BN as discussed below. Furthermore, we performed detailed analyses of their structural changes throughout the simulations.3.1. Adsorption of cyclotides on 2D materials
Fig. 2(a–f) shows the snapshots of the adsorbed structures of the three cyclotides on both graphene and h-BN at the end of the 500 ns simulations, while Fig. 3(a–l) represents the time evolution of various dynamical quantities during the entire course of simulations. Initially, all of the cyclotides are found to wander randomly over both the 2D materials for the first few nanoseconds and then rapidly build interactions with them, as clearly suggested by a steep decrease in the distance (Fig. 3(a–c)) along with a rapid increase in the number of contacts (Fig. 3(d–f)) and contact surface area (CSA) (Fig. 3(g–i)). The building of contacts between the peptides and the surfaces continues for a few hundred nanoseconds and after nearly 300 ns, they reach a stable magnitude. Moreover, the number of contacts and CSA follow the order MCoT > cycloviolacin > Katala B1 and the total interaction energy is nearly commensurate with this trend as observed from Fig. 3(j–l). It is worthwhile to mention that the interaction energy with graphene is of van der Waals type only as the carbon atoms do not bear any partial charge. On the other hand, due to the partial charges over the B and N atoms in h-BN, the interaction energy is expressed as a sum of the electrostatic and van der Waals interactions. The decomposition of the peptide⋯h-BN interaction energy shows that the predominant contribution arises due to the van der Waals forces of interaction and the electrostatic interactions are just additions to the same (Fig. S2(a–c)†). Therefore, it may be contemplated that, during the initial stages of the adsorption on h-BN, the long-range electrostatic forces operate to bring the peptide close to the surface and once the distance of the peptide with the surface is reduced, the van der Waals forces become predominant and carry out the rest of the adsorption. It is due to the same reason that even though h-BN is isomorphous and iso-electronic to graphene, the interaction energy of all of the three peptides with h-BN is stronger than the former (Fig. 3(j–l)). For instance, the equilibrated interaction energies between cycloviolacin and MCoT with graphene at the end of 500 ns are observed to be between −100 to −150 kcal mol−1 while that on h-BN reaches a magnitude of nearly −250 kcal mol−1. On the other hand, Katala B1 has much lower interaction energies compared to the other peptides and for both graphene and h-BN, the interaction energy appears to be between −100 and −125 kcal mol−1, with a late adsorption on graphene. Therefore, it may be concluded that Katala B1 has inherently lesser affinity towards adsorption on 2D materials, irrespective of the nature of the surfaces. In addition, a comparison of the temporal evolution of the contact numbers and CSA with the interaction energies clearly shows that even though there are fluctuations in the former quantities, the reduction in interaction energy is significantly smooth. Thus, the time evolution of the contacts proceeds such that the interaction energy increases and there exists a dynamic equilibrium between the adsorption of the different residues, keeping the interaction energy decreasing and the equilibrium proceeding towards the adsorption of residues with successively higher adsorption energies with the 2D materials.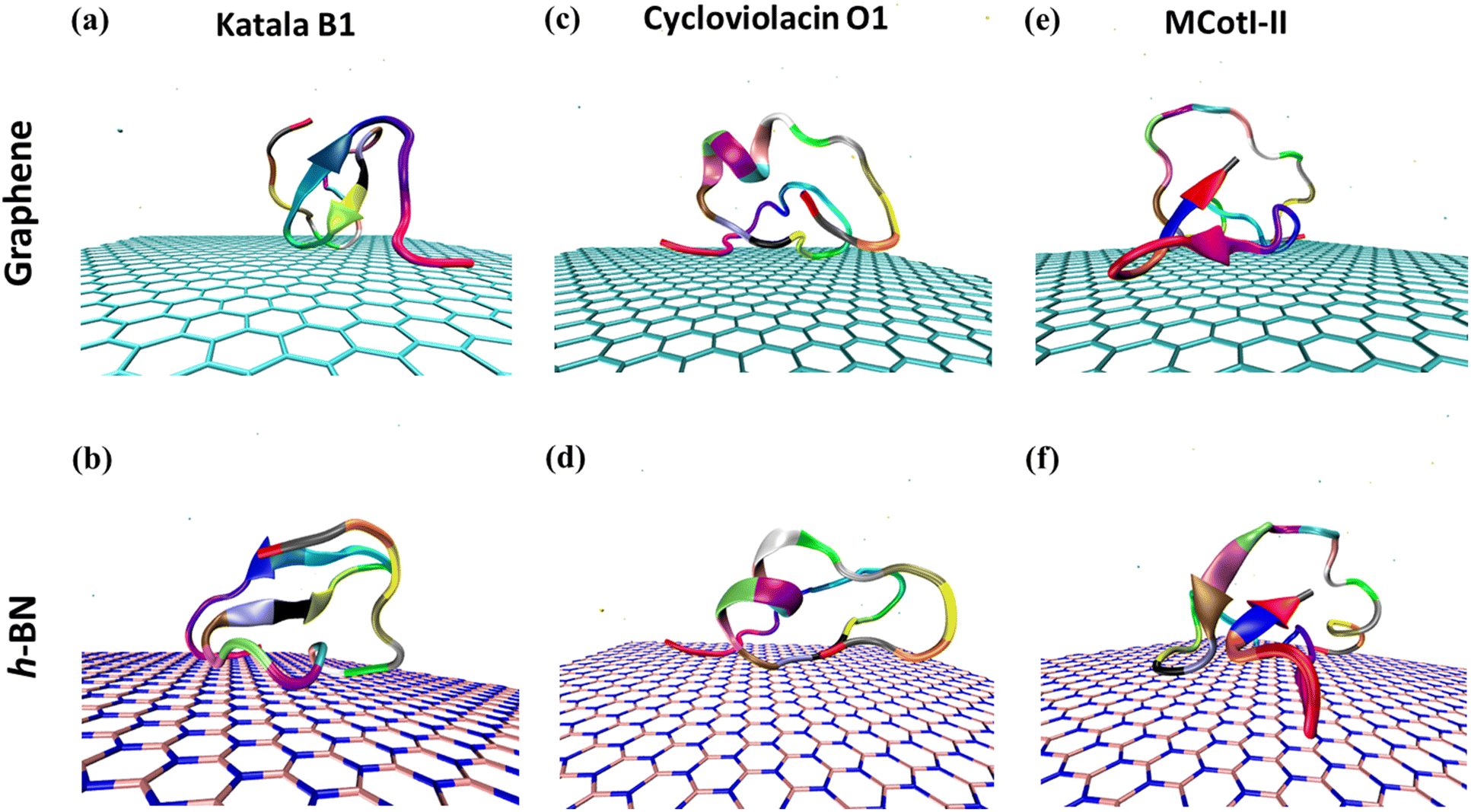 | ||
| Fig. 2 Snapshots from the 500 ns adsorption simulations showing the final adsorbed configurations of the cyclotides on graphene and h-BN for (a and b) Katala B1, (c and d) cycloviolacin O1, and (e and f) MCoTI-II, respectively. The peptide molecules are depicted through the VMD ‘Cartoon’ representation. Visual inspection clearly shows that the secondary structures of the cyclotides are nearly intact after adsorption on both graphene and h-BN. The different amino acid residues are coloured according to the residue type and the same colour code is maintained for all three cyclotides. | ||
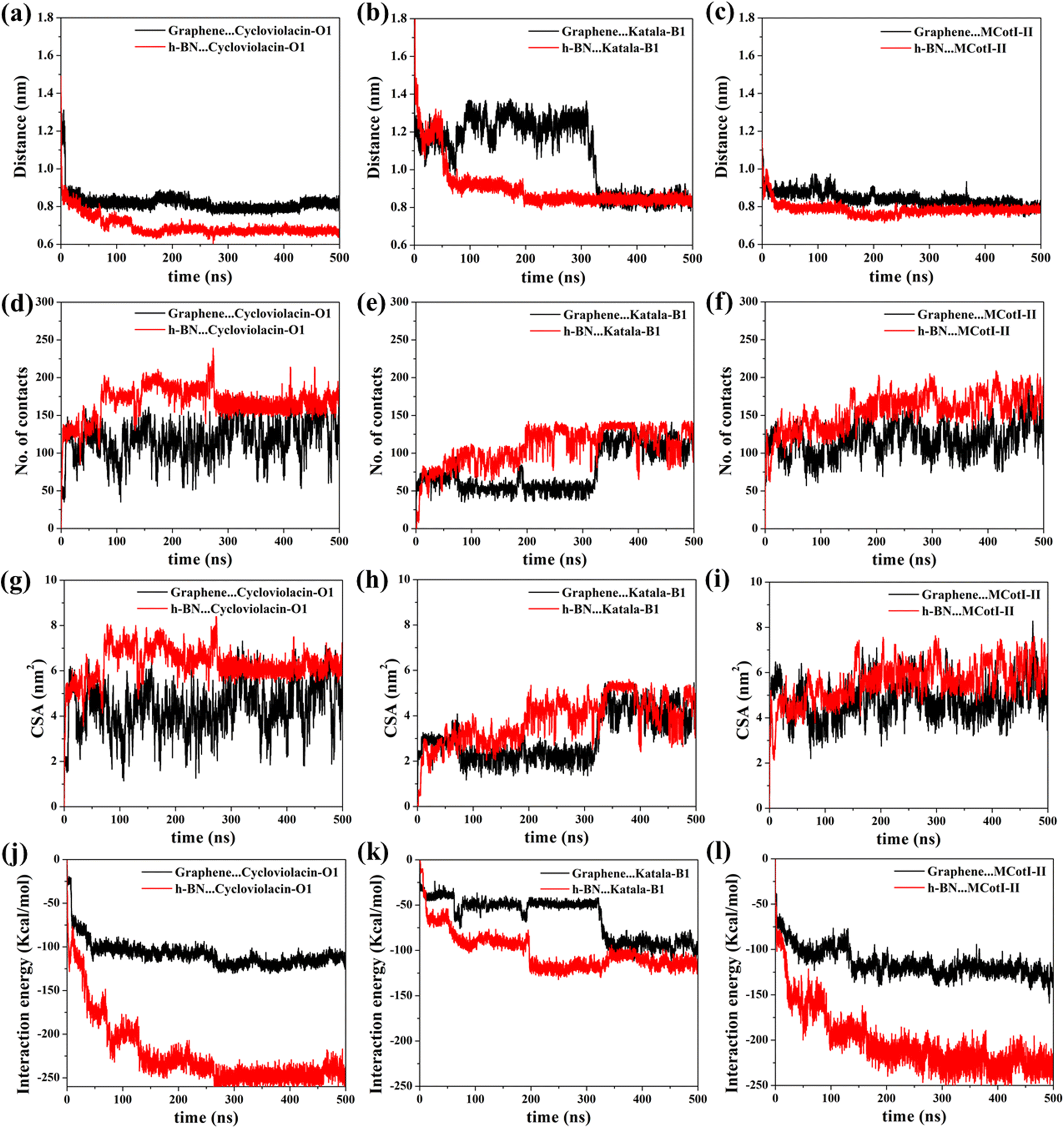 | ||
| Fig. 3 Time evolution of the dynamic properties showing the adsorption of different cyclotides on graphene and hexagonal BN at 300 K for the entire 500 ns production simulations: (a–c) center-to-center distance, (d–f) contact number, (g–i) contact surface area, and (j–l) interaction energy. Similar time evolution of the dynamical quantities is observed in both sets of simulations, suggesting that the nature of the adsorption of small peptide molecules is independent of the initial configuration. | ||
One of the most significant consequences of the adsorption of biomolecules onto 2D materials is their structural disruption, primarily due to strong interaction with the material underneath. Therefore, judging the biocompatibilities of graphene and h-BN towards cyclotides warrant the evaluation of the structural integrity of the peptides during and after adsorption. In this regard, we first evaluate the root mean squared displacement (RMSD) of all three cyclotides for the entire duration of the adsorption simulations, taking the initial structures as the reference. It is evident from Fig. 4(a–c) that the magnitude of the RMSD is significantly low in each case and the maximum RMSD is around ∼3 Å (0.3 nm) only, thereby clearly suggesting minimum structural perturbation upon adsorption on the 2D materials and eliminating the possibility of structural disruption.
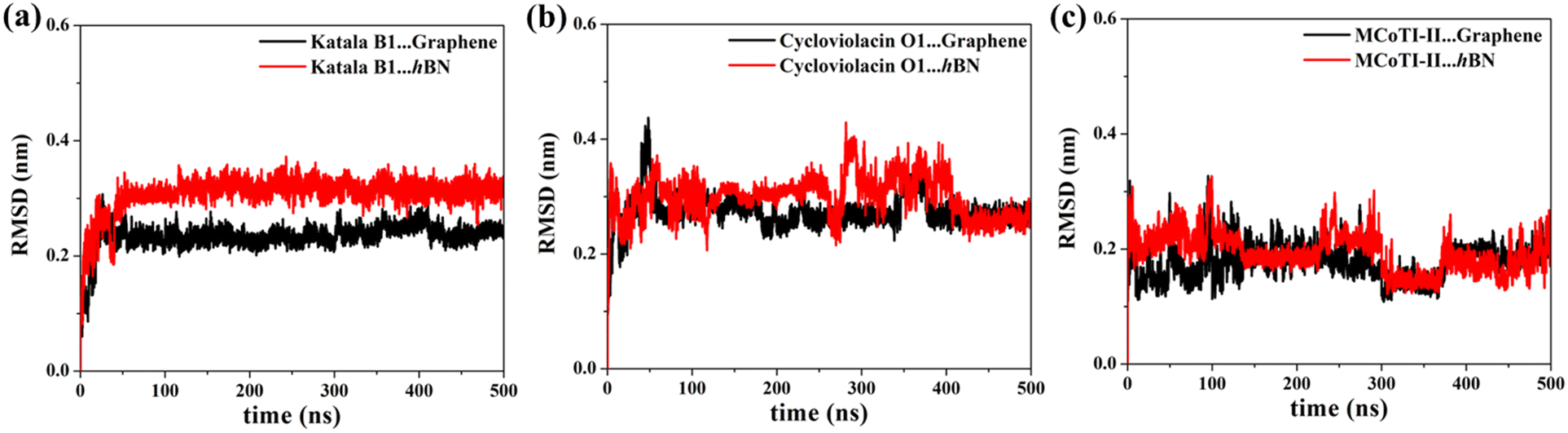 | ||
| Fig. 4 Root-mean-squared displacement (RMSD) plots for the adsorption simulations of (a) Katala B1, (b) cycloviolacin O1, and (c) MCoTI-II for the entire duration of the adsorption simulations. The heavy atoms of the peptides are considered for the calculations of RMSD and the initial structure of the cyclotides were taken as references. | ||
To deduce the evolution of the secondary structure after the adsorption of the cyclotides on the 2D materials, we first developed residue–residue contact maps (Fig. 5(a–c)) for the adsorbed cyclotides after 500 ns of the adsorption simulations on both graphene and h-BN and compared them with those obtained for the peptides in the absence of any 2D material (blank). The contact maps show that all the residues that are in contact with each other have a maximum contacting distance of 10 Å. The contacts belonging to different distances are represented through different colours. A contact distance of 0 Å represents a self-contact and is coloured black, while the other colour codes and the corresponding distances are explained in Fig. 5. Above an inter-residue distance of 10 Å, the residues are assumed to be non-contacting. From Fig. 5(a–c), it is evident that the contact maps of all the three cyclotides in the blank simulations are very similar to that observed at the end of 500 ns when adsorbed on graphene and h-BN. Therefore, it is inferred that due to the adsorption on the 2D materials, significant perturbation is not inflicted upon the peptides and the native inter-residue contacts are nearly preserved, as also observed from the visual analyses of the snapshots provided in Fig. 2(a–f).
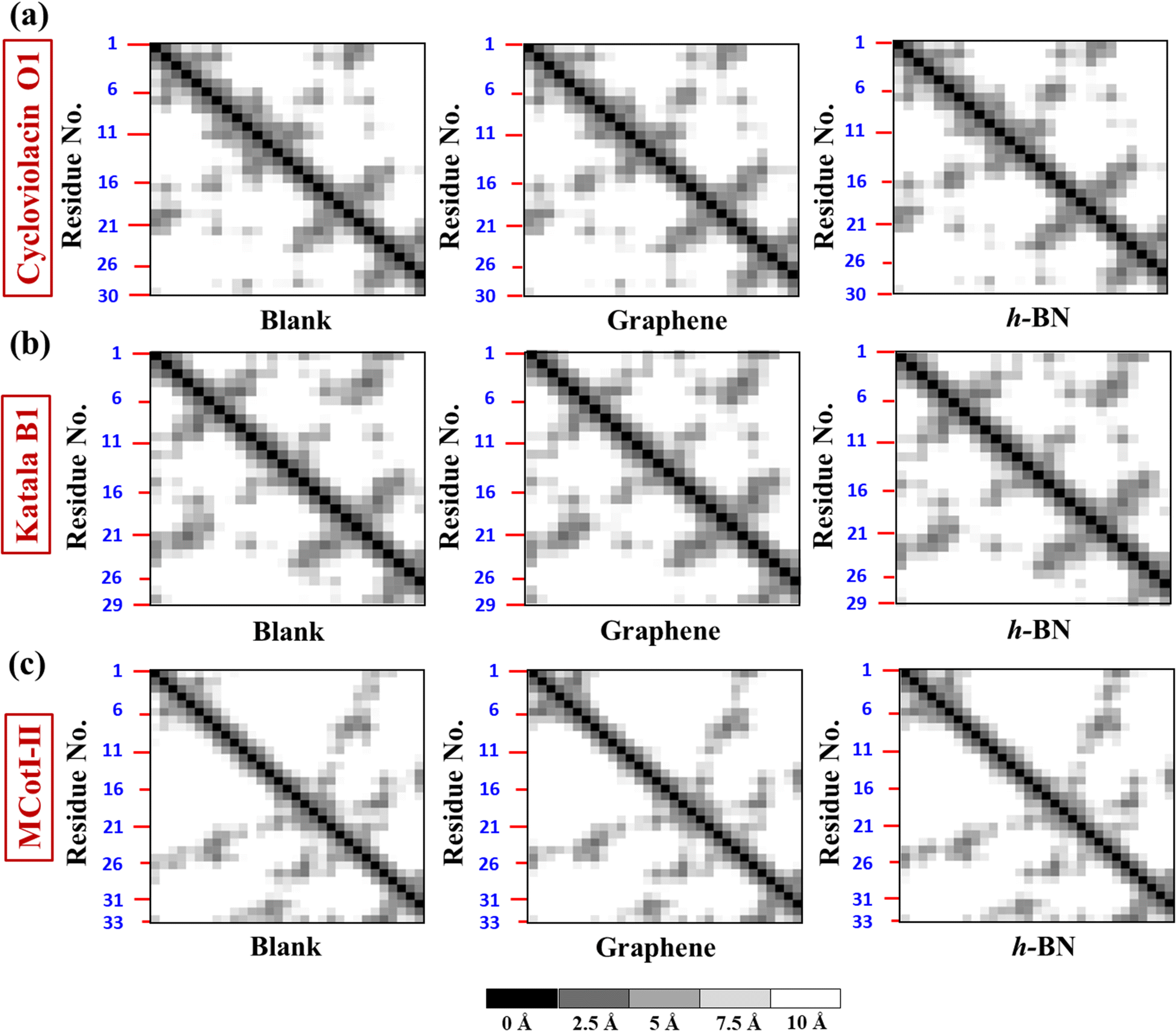 | ||
| Fig. 5 Residue–residue contact map at the end of 500 ns for free cyclotides in water (blank) and the adsorbed structure on graphene and h-BN for (a) cycloviolacin O1, (b) Katala B1, and (c) MCoTI-II. The colour code at the bottom indicates the corresponding distances between different residues. | ||
To gain further insights, we evaluated the time evolution of the secondary structures of the cyclotides (Fig. 6(a and b)), and similar to the previous discussion, we compared the secondary structures during and after adsorption on the 2D materials with those evaluated for the blank simulations. It appears that there are insignificant changes in the secondary structures of all of the three peptides at the end of 500 ns. For example, the structure of Katala B1 consists of two patches of extended beta sheets (colour code yellow, abbreviation E) between the residues 14 and 25, while cycloviolacin O1 forms the alpha helical conformation between the residues 7 and 15. These structural features are almost maintained both during and after adsorption on both graphene and h-BN. However, from Fig. 6(a and b), it is clear that the extent of perturbation inflicted upon the peptides is more in the case of h-BN as compared to graphene, which is again in harmony with the much higher interaction energy of the peptides on the former 2D material. It is noteworthy that even if there are some fluctuations and perturbations in the secondary structures of all three cyclotides on h-BN during the adsorption simulations, nevertheless, they tend to disappear near the ends of the trajectories, which clearly suggests that the changes in the secondary structures are reversible, and as soon as the adsorptions are nearly complete, the peptides reorganize themselves so that they can regain their native internal interactions. Apart from some specific perturbations, the rest of the structural fluctuations observed are present in both the blank and the adsorption simulations and they probably originate due to the small structures of the peptides compared to large proteins, which gives them more flexibility.
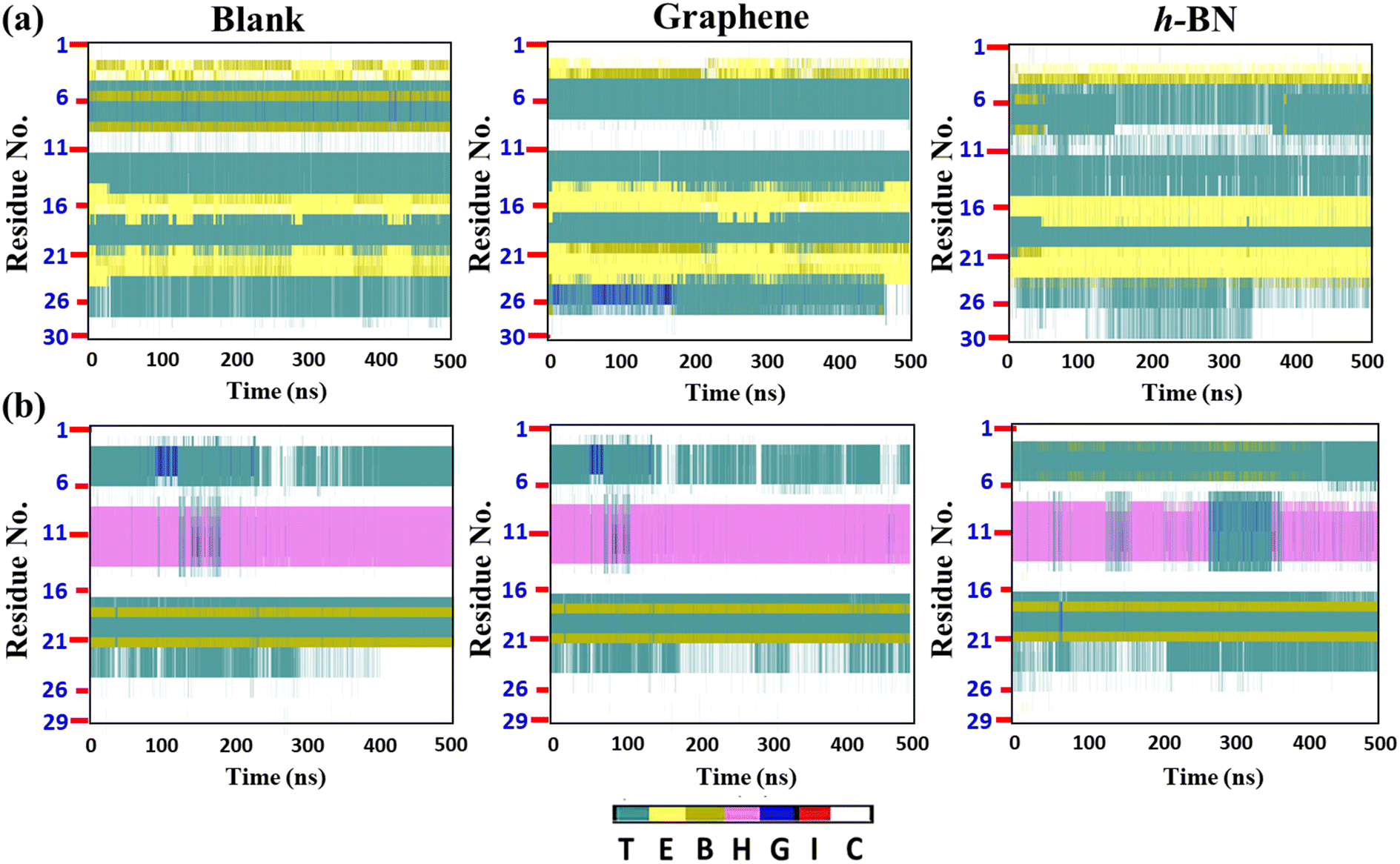 | ||
| Fig. 6 Time evolution of the secondary structures of the cyclotides in the absence of any 2D material (blank) and during as well as after adsorption on graphene and h-BN for (a) Katala B1 and (b) cycloviolacin O1. Color codes are used to indicate various secondary structures: T = turn, B = isolated β-bridge, H = α-helix, E = extended β-sheet, I = π-helix, G = 3–10 helix, and C = random coil. | ||
To shed light on the nature of the interaction between the cyclotides and the 2D materials, we decomposed the total interaction energies into residue-wise interaction energies. The interaction energy profiles (Fig. 7(a–c)) were obtained for the last 100 ns of the adsorption simulations and by averaging over two independent simulation trajectories having different initial configurations of the cyclotides. As observed from Fig. 7(a), the predominantly interacting residues are mostly common for both graphene and h-BN for all three cyclotides and they exist as clusters of 3 to 7 residues. For the adsorption of Katala B1, three common residue-clusters are observed, one between Cys5 and Gly8 followed by two groups of interacting residues between Thr16 and Trp19 and Leu27 and Val29. Similarly, for the adsorption of cycloviolacin O1, two clusters (Fig. 7(b)) of interacting residues are observed between Cys15 and Arg20 and Ile26 and Glu30, while for MCoTI-II, we found four clusters (Fig. 7(c)) between residues between Ser1 and Ser3, Cys15 and Asp18, Pro22 and Ala24, and Ile26 and Gly31. The residues having an average interaction energy equal to or more than −15 kcal mol−1 are found to be Tyr, Trp, Arg, and Asn, which contribute the maximum towards the total adsorption energies. The next category of residues is those having interaction energy between −10 kcal mol−1 to −15 kcal mol−1, and consist of Cys, Ser, Leu, Pro, and Ile residues. In addition, few unique interacting residues are observed for each of the cyclotides; however, the contributions of these residues towards the total interaction energies are substantially low. It is worthwhile to mention that the calculated interaction energies between the common interacting residues and the 2D materials are much higher with h-BN as compared to graphene, which is also in agreement with the results presented in this article regarding the analysis of adsorption. This result also corroborates with our previous findings of higher strength of adsorption of various nucleic acids with h-BN compared to graphene, which can be attributed to the polar nature of h-BN, in conjunction with the greater van der Waals interaction of biomolecules with the B and N atoms compared to only carbon in graphene.38,58 Furthermore, since cystine residues are one of the most important characteristic features of cyclotides, we compared the interaction between the Cys residues and 2D materials with the total interaction energies for all three cyclotides as shown in Fig. 7(d). These analyses also allow us to decipher the interaction of the 2D materials with the cyclic knots. Each of the cyclotides contains 29 to 34 amino acid residues, among which there are only six Cys residues and as Fig. 7(d) clearly shows the interaction energy contribution from the Cys residues towards the total adsorption energy is between only 13% to 17%, which is certainly not a large share. Therefore, cystine residues only add a favorable contribution during the adsorption of cyclotides on 2D materials but do not dictate the outcome of the process, which in turn, suggests that the cystine residues are relatively free to protect the structure. It should be noted that aromatic moieties have extra affinity towards their adsorption on graphene and h-BN due to the aromaticity of the 2D materials and their capability of efficient π–π stacking with adsorbed aromatic rings. In fact, both graphene and h-BN have been identified to partially or completely disrupt the structures of nucleic acids, the driving force behind such events being the same π–π stacking interactions, as observed in previous studies.38,53,58 Therefore, the presence of aromatic motifs is one of the pivotal contributing factors to the structural disruption of biomolecules. On a similar note, the presence of a greater population of aromatic amino acids may lead to the structural perturbation of proteins and peptides. However, cycloviolacin O1 has two aromatic residues (Tyr4, and Tyr23) and both Katala B1 as well as MCoTI-II have only one aromatic residue each (Trp19 and Tyr32). Most of these residues have interactions with the 2D materials with interaction energy of more than −15 kcal mol−1; however, the overall contribution towards the total interaction energy is not significant. Therefore, in addition to cystine knots, the present study identifies the absence of multiple aromatic residues in the cyclotides as an additional reason for the maintenance of structural integrity upon adsorption on common 2D materials such as graphene and h-BN. Hughes et al. determined the adsorption free energies of all naturally occurring amino acids on graphene and found Arg to have the strongest affinity towards the 2D material followed by Trp, Tyr, and Asn and the magnitude of the free energies for all these amino acids were found be greater than −15 kJ mol−1, while those of Cys, Ser, Leu, and Pro amino acids were between −7 and −15 kJ mol−1.59 In addition, they observed that all the aromatic amino acids, viz. Tyr, His, and Phe, are strongly adsorbed on graphene through π–π stacking interactions, each of them having adsorption free energies equal to or more than −15 kJ mol−1. These results are in complete coherence with the interaction patterns of amino acid residues observed in the present study and also provide additional confidence to the accuracy of the results and suitability of the MD parameters used to model the peptides as well as the 2D materials. It should be noted that the higher interaction energy observed in this study compared to the free energies observed by Hughes et al. is due to the entropic considerations when calculating free energies unlike the interaction energies and is a common observation in the molecular dynamics literature.37,58
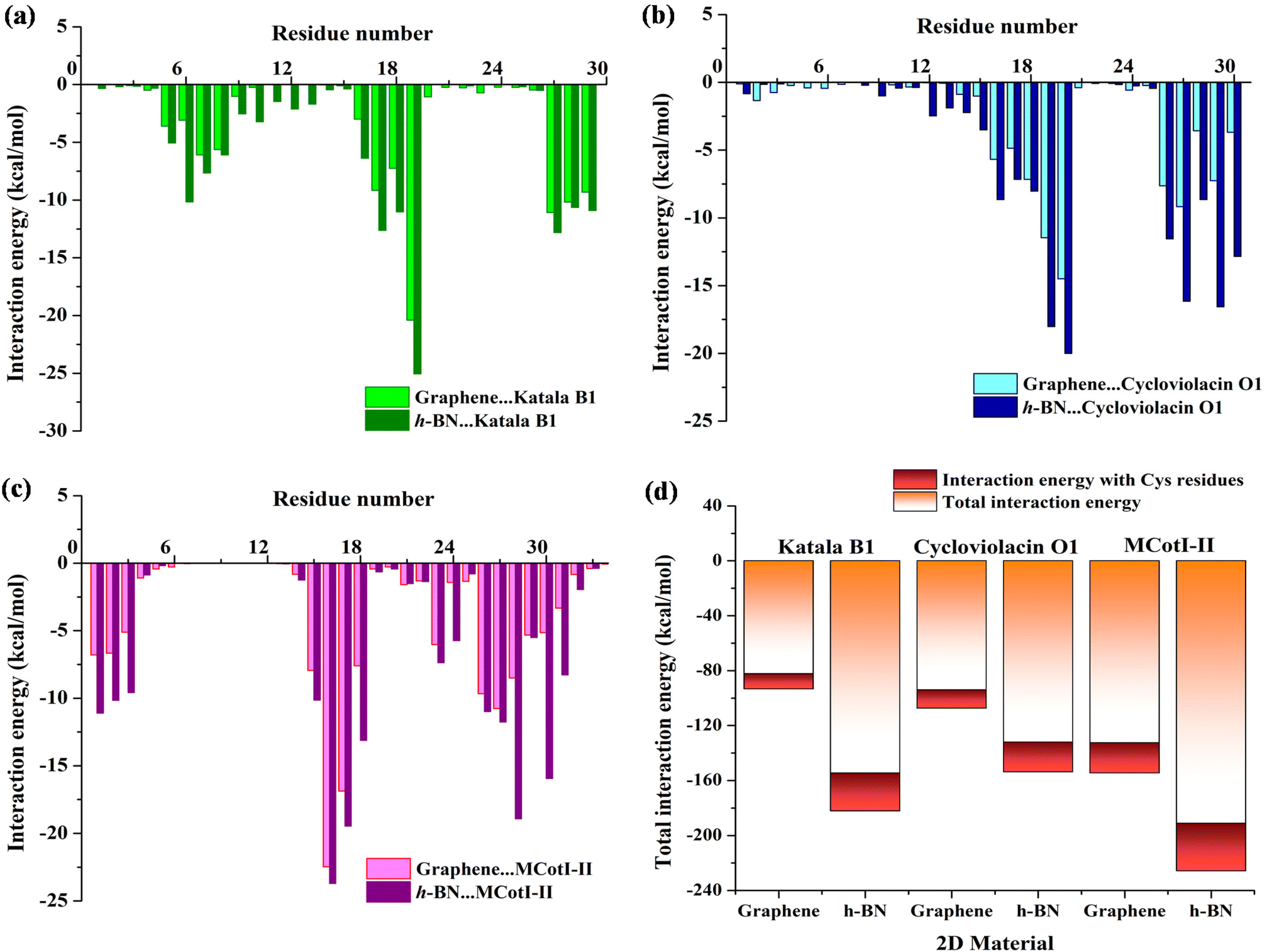 | ||
| Fig. 7 Residue-wise interaction energies (in kcal mol−1) among (a) Katala B1, (b) cycloviolacin O1, and (c) MCoTI-II on graphene and h-BN for the last 100 ns of the adsorption simulations showing clusters of common interacting residues and few residues unique to each of the materials. For most of the amino acid residues, the average interaction energies are much higher for h-BN as compared to graphene. The interaction energy profiles are obtained by taking the mean over two independent simulation trajectories having different initial configurations of the cyclotides. (d) Total interaction energy between cyclotides and 2D materials and the contribution from the interaction with six Cys residue in each of the cyclotides. | ||
It is well-known that adsorbed molecules on 2D materials might undergo longitudinal diffusion across the surfaces. To examine the mobility of the cyclotides after adsorption, we tracked the X–Y positions of the center of masses (COMs) of the peptides for the last 300 ns of the adsorption simulations (Fig. 8(a–c)), which clearly shows that the COM position is significantly scattered for all three peptides on both graphene and h-BN. Therefore, the adsorbed cyclotides move significantly freely on the 2D materials, and they “crawl” on the surfaces even if the adsorption free energy of the peptides is relatively high. This observation is in harmony with previous reports on the in-plane movement of nucleic acids on graphene and h-BN and occurs due to the presence of symmetric hexagonal rings, which isotopically interact with the peptide molecules during their in-plane translation.38,44,58 Moreover, graphene is nonpolar and each of the hexagonal rings of h-BN consists of polar B and N atoms related by local three-fold symmetry, which as a whole does not produce any long-range polarity. Therefore, the interaction energies of different configurations with the surfaces related by in-plane translational symmetry are expected to be identical, thereby drastically lowering the translational energy barrier and favouring the movement of the peptides on them.
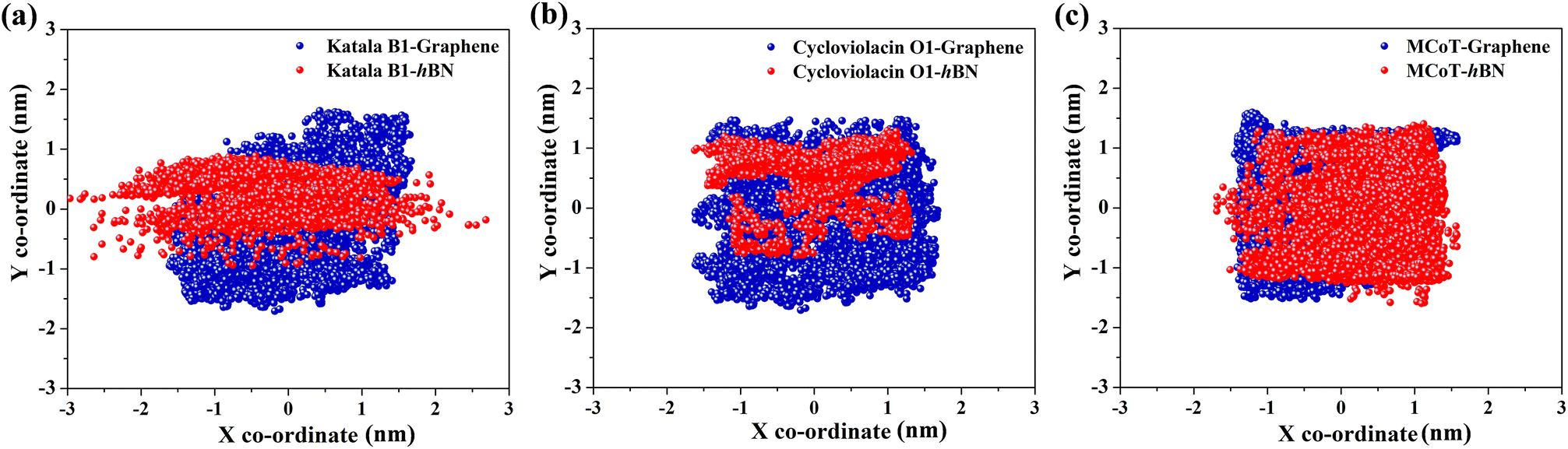 | ||
| Fig. 8 X and Y coordinates of the center-of-mass of (a) Katala B1, (b) cycloviolacin O1, and (c) MCoTI-II for the last 300 ns of the adsorption simulations on graphene (blue dots) and h-BN (red dots), representing the mobility of the adsorbed peptides on the 2D materials. The scattered nature of the dots clearly suggest that the molecules move significantly freely and “crawl” on the surfaces. | ||
Often cyclotides remain as aggregates in different parts of plants, usually where they are produced. In many instances, the antibacterial and antifungal activities of the cyclotides depend on the stability of the aggregated structures. Therefore, in addition to the adsorption of single cyclotides on graphene and h-BN, it is imperative to judge the nature of adsorption and the stability of the adsorbed aggregates on these 2D materials. In this regard, we first prepared aggregates of four cyclotide molecules for all three types of cyclotides in water and devoid of any 2D material (Fig. 9(a and b)). Cyclotides aggregate through inter-peptide hydrogen bonding, van der Waals, and electrostatic interactions, which have been discussed in the ESI (Fig. S3(a–d)†). These aggregates are then used to study their adsorption on both graphene and h-BN through 500 ns of adsorption production simulations. Fig. 9(c and d) shows the representative snapshots of the initial and final conformations of cycloviolacin O1 before and after adsorption on graphene. All of the other simulations produce very similar adsorbed structures of the aggregates. Since the structural integrity of single cyclotides is not lost upon adsorption on graphene and h-BN, it is most unlikely that upon adsorption of the cyclotide aggregates, the secondary structures of the constituent individual cyclotides would get perturbed. Therefore, the most important structural quantity that needs to be investigated is the integrity of the aggregate which can be conveniently judged by tracking the average inter-peptide distances during and after the adsorption of the aggregates and comparing them with those obtained in the blank simulations. As observed from Fig. 9(e–g), the average distance between the units of the aggregate do not change appreciably upon adsorption on 2D materials and remain nearly the same as observed in the last 200 ns of the blank simulation. In fact, in some of the adsorption simulations, the inter-cyclotide distance reduces compared to the blank simulations, suggesting that the aggregates remain unperturbed and even undergo further compaction upon adsorption, with the 2D materials behaving as the supporting platform for the latter.
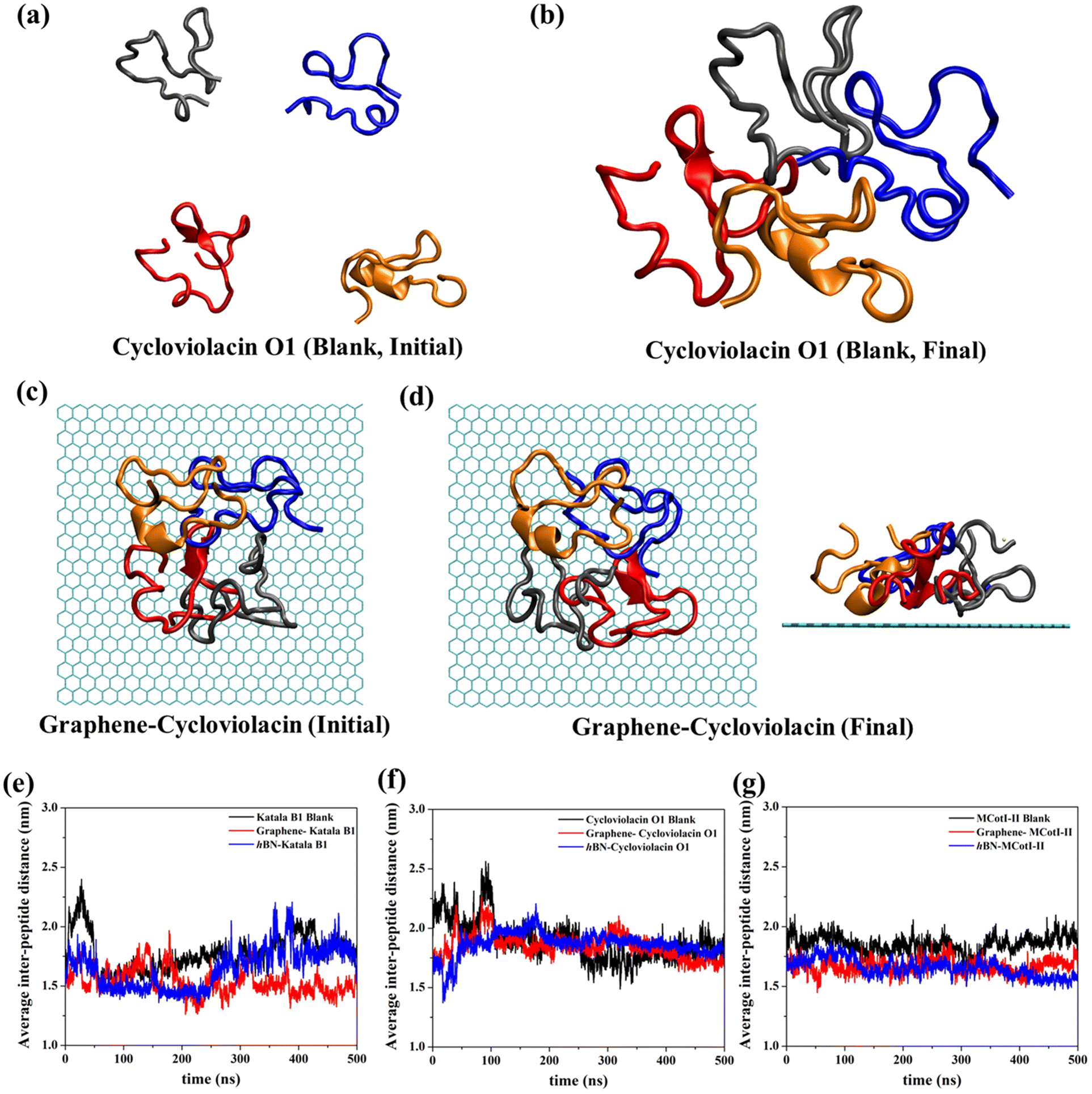 | ||
| Fig. 9 Representative snapshots of the (a) initial and (b) final structures (after 500 ns) of cycloviolacin O1 before and after the formation of stable aggregates in the blank simulations. (c) The initial (top view) and (d) final structures (top and side views) of cycloviolacin O1 aggregates before and after adsorption on graphene, respectively. The average inter-peptide distance for the blank simulations and the adsorption on both graphene and h-BN for (e) Katala B1, (f) cycloviolacin O1, and (g) MCoTI-II. The intricate details of the secondary structures are not shown for clarity. | ||
3.2. Thermodynamics of cyclotide adsorption
To shed light on the energetics involved in the adsorption of cyclotides on the 2D materials, we calculated the adsorption free energies as the potential of mean forces (PMF) for all three cyclotides considered in this study at 300 K. Fig. 10(a) and (b) show the same on graphene and h-BN, respectively. During the PMF calculations, the z-component of the distance between the center of masses of the cyclotide and the 2D material is taken as the reaction coordinate. From the PMF profiles it is clear that the trend of the free energies of adsorption is Katala B1 < cycloviolacin O1 < MCoTI-II, which is common for both of the 2D materials and the same as that found for the interaction energies and discussed in the previous section. Next, we compared the adsorption free energies on graphene and h-BN as shown in Fig. 10(c–e) for the three cyclotides. For each of the cyclotides, the adsorption free energy is observed to be higher on h-BN as compared to graphene. We define the quantity, ΔGprefad = ΔGsingle,h-BNad − ΔGsingle,graphenead, with ΔGprefad being the adsorption free energy preference of the cyclotides towards h-BN compared to graphene and ΔGsingle,h-BNad and ΔGsingle,graphenead being the adsorption free energies of single cyclotides on h-BN and graphene, respectively. For instance, Katala B1 has adsorption free energies of −25.3 kcal mol−1 and −31.9 kcal mol−1 on graphene and h-BN, respectively, and therefore produces a free energy inclination (ΔGadpref) of −6.6 kcal mol−1 towards h-BN. Similarly, the magnitudes of ΔGprefad for cycloviolacin O1 and MCoTI-II are calculated to be −3.8 kcal mol−1 and −9.1 kcal mol−1, respectively. The adsorption preference for h-BN as compared to graphene is in complete accordance with previous reports on the adsorption and interaction of small organic molecules, nucleobases, and nucleic acids with 2D materials. However, the magnitudes of the adsorption free energies are much smaller compared to similar sized biomolecules and organic molecules as has been reported in previous studies.60,61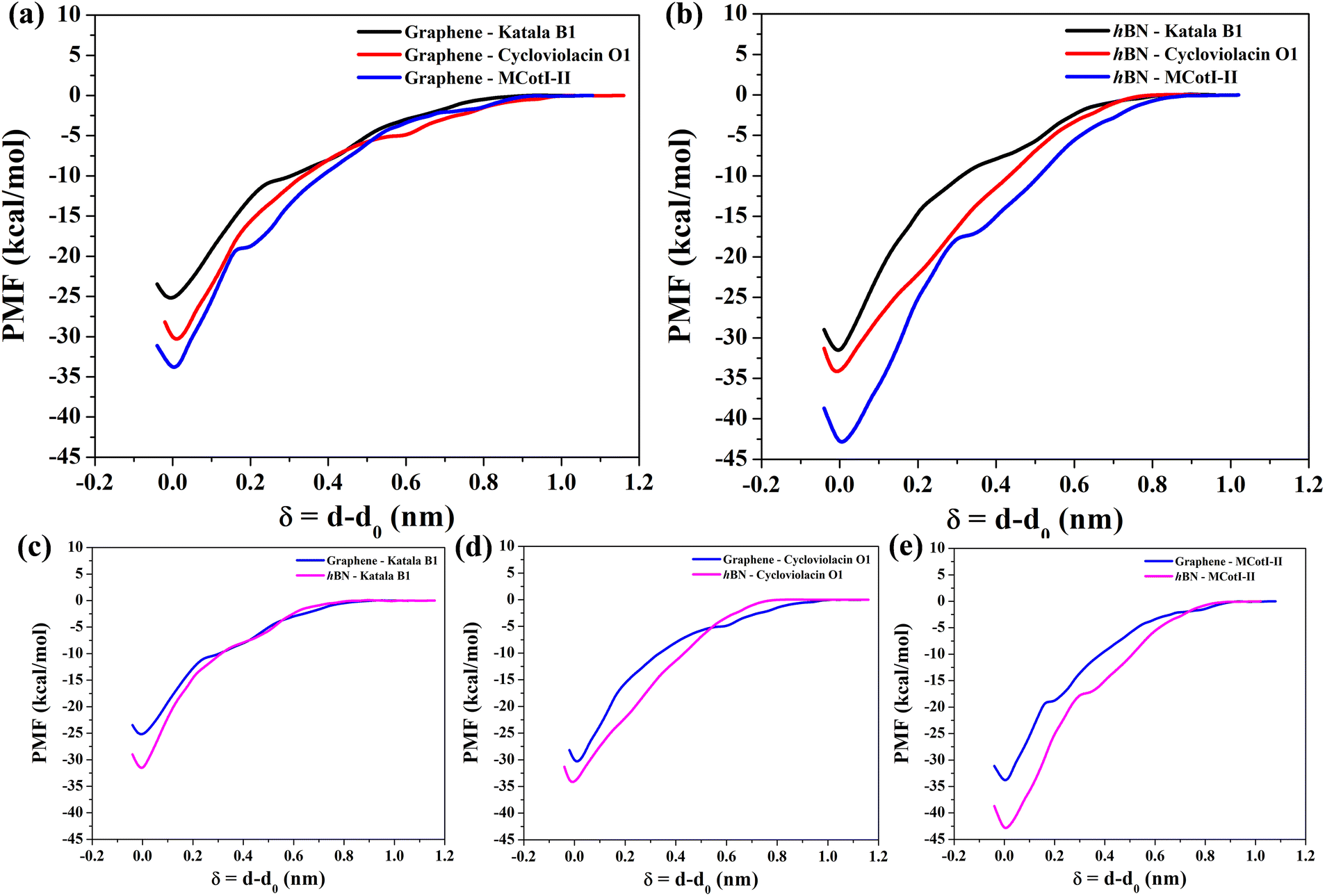 | ||
| Fig. 10 Free energy of adsorption (in kcal mol−1) of the three cyclotides Katala B1 (black curve), cycloviolacin O1 (red curve), and MCoTI-II (blue curve) on (a) graphene and (b) h-BN in terms of potential of mean forces (PMFs). Comparison of the PMF profiles on graphene (light blue curve) and h-BN (magenta curve) of (c) Katala B1, (d) cycloviolacin O1, and (e) MCoTI-II, clearly suggesting the higher strength of adsorption of each of the cyclotides on h-BN as compared to graphene. The PMF profiles are obtained at 300 K and at least two independent PMF simulations are carried out to check the reproducibility and produce an average profile. Since cyclotides are adsorbed at different COM distances with respect to the 2D materials, we substituted the reaction coordinate with δ = (d − d0), with d0 and d being the COM distances initially and at any instant during the PMF calculations, respectively. Free energy of the completely desorbed structure is set to zero, where the cyclotide is not interacting with the surface. | ||
For the integration of heterostructures with 2D materials and cyclotides, it is important to estimate the minimum time after which there is a possibility for the biomolecules to be released from the material surfaces as a weak binding between the biomolecules and the nanomaterials may lead to premature detachment, while a very strong binding reduces the opportunity of the biomolecule to independently interact with a third substance such as lipid membranes. Although experimental techniques can reveal such release times of biomolecules from material surfaces, MD simulations can provide an alternative route to evaluate the same. From the magnitudes of the calculated PMF's, it is evident that the cyclotides have to overcome free energy barriers much larger than the energy available at biological temperatures. Therefore, the release of adsorbed cyclotides from the 2D material surfaces may be considered as a rare event and, consequently, the release of the molecules is expected to occur at time scales far beyond the accessibility of current tractability in MD simulations. The minimum time required for the release of cyclotides (τrelease) can be modelled in the framework of Smoluchowski theory and can be calculated in terms of the mean first-passage time using the following equation
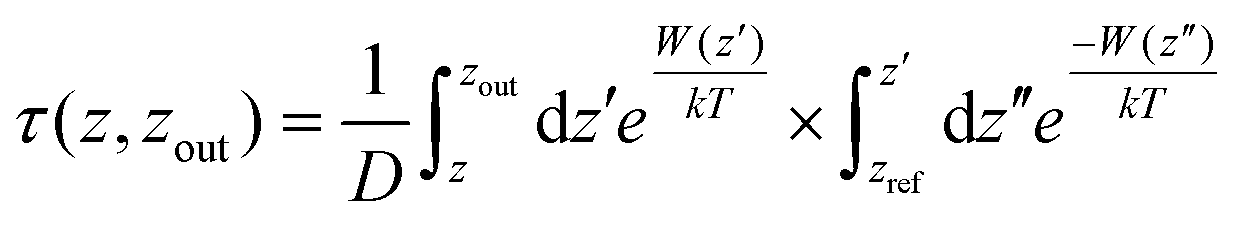 | (1) |
| Cyclotide | 2D material |
|
ΔGsinglead (kcal mol−1) | ΔGloss (kcal mol−1) | τsinglerelease (ns) | τaggrelease (ns) |
|---|---|---|---|---|---|---|
| Cycloviolacin O1 | Graphene | −12.8 | −30.4 | 17.6 | 1.30 × 1023 | 1.46 × 1010 |
| h-BN | −15.0 | −34.2 | 19.2 | 1.63 × 1026 | 2.60 × 1012 | |
| Katala B1 | Graphene | −11.8 | −25.3 | 13.5 | 9.03 × 1019 | 1.55 × 1010 |
| h-BN | −13.8 | −31.9 | 18.1 | 1.42 × 1024 | 1.25 × 1011 | |
| MCoTI-II | Graphene | −15.9 | −34.1 | 18.2 | 2.99 × 1026 | 2.32 × 1013 |
| h-BN | −18.3 | −43.2 | 24.9 | 2.27 × 1032 | 2.65 × 1014 | |
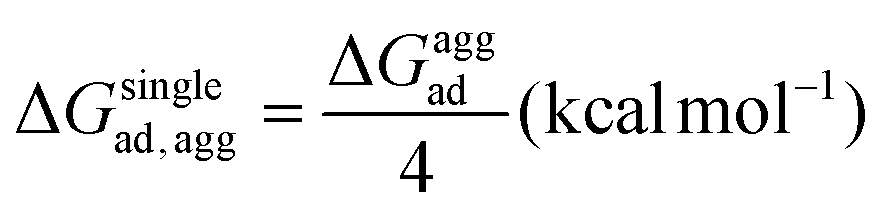
As observed from Table 1, the release times follow the same trend as observed for the adsorption free energies on any of the 2D materials, i.e. Katala B1 < cycloviolacin O1 < MCoTI-II and the release time from h-BN is 103–106 times higher than that observed for graphene. The minimum release times from graphene and h-BN for any of the cyclotides are calculated to be in the order of 1019 ns and 1024 ns, respectively, which are much higher than the usual interaction times between common biomolecules, as well as between nanomaterials and biomolecules. Therefore, the single cyclotide molecules are not expected to be easily desorbed or released from the surfaces of the 2D materials during the course of interaction with other biomolecules. Consequently, we can safely conclude that the adsorption of the cyclotides on 2D materials is mild and the molecular flexibility and structural properties remain nearly intact upon adsorption. However, the adsorption strengths are sufficient enough to form stable heterostructures.
As mentioned previously, in most plant organs, cyclotides remain in their aggregated states. Therefore, we studied the adsorption of aggregates of four cyclotides on both graphene and h-BN and observed neither dissociation of the monomers from the aggregated state nor their structural disruption. Next, we calculated the adsorption free energies (ΔGaggad) of such aggregates (Fig. S4(a–c)†) and compared them with the same calculated for single peptide molecules according to the equation 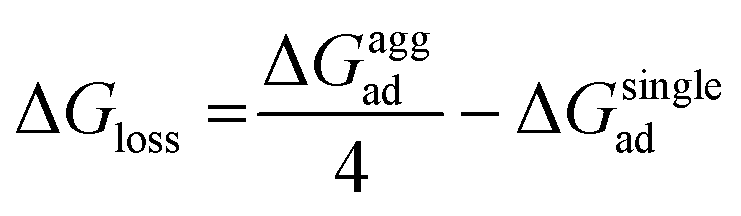 , where ΔGloss is the free energy loss per peptide molecule in an aggregated state upon adsorption on the 2D materials, the quantity
, where ΔGloss is the free energy loss per peptide molecule in an aggregated state upon adsorption on the 2D materials, the quantity 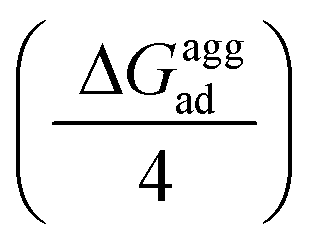 is the average adsorption free energy per peptide molecule in their aggregated state
is the average adsorption free energy per peptide molecule in their aggregated state 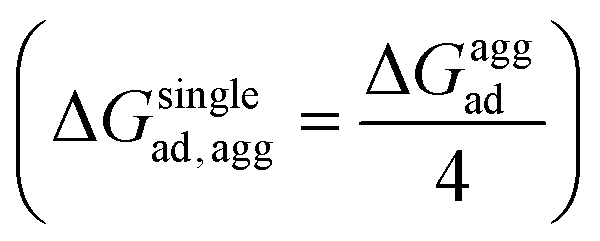 and ΔGsinglead is the free energy of adsorption for a single peptide molecule. A positive value of ΔGloss suggests lesser average adsorption free energy of a single peptide molecule in an aggregate and therefore quantifies the ease to get released from the 2D materials. Table 1 provides the magnitudes of the ΔGloss for the three cyclotide molecules on both graphene and h-BN. It is observed that for all three cyclotides and both 2D materials ΔGloss is positive, which in turn suggests that when the cyclotides are adsorbed as aggregates, the individual adsorption affinity is weaker. The trend of the magnitudes of ΔGloss is the same as that observed for the adsorption free energies of single cyclotides, clearly suggesting that the greater the adsorption strength of the single cyclotide molecules, the greater the loss in free energy upon adsorption as an aggregate. Interestingly, the release times calculated for single cyclotides from the adsorbed aggregates (τaggrelease) show an enormous decrease compared to those observed for the single cyclotides (τsinglerelease), evidently due to the decrease in the average adsorption free energies. For instance, MCoTI-II, the cyclotide having the highest adsorption free energy, shows a release time of 2.99 × 1026 ns and 2.27 × 1032 ns from graphene and h-BN, respectively, when adsorbed as single molecules, while the release times are reduced to only 2.32 × 1013 ns and 2.65 × 1014 ns respectively, when released from the adsorbed state of aggregates. In line with the above results, it may be envisioned that the larger the size of the aggregates of cyclotides, the more lower the adsorption free energies of the individual peptides, which would increase the releasing tendency of the individual peptides from the adsorbed aggregates, thereby providing a better opportunity to interact with other biomolecules of therapeutic concern. In fact, controlling the size of the aggregates may unfold an opportunity to fine-tune the extent of interaction between the 2D materials and cyclotides and develop precise heterostructures for their biomedical targeting and controlled release. Nonetheless, the affinity of the adsorption of cyclotides can, in principle, be further tuned by introducing various functionalization groups into the 2D materials or by controlling the extent of material oxidation, in accordance with the requirements for a specific therapeutic or biomedical purpose. It is worthwhile to mention that the adsorption free energy of large cyclotide aggregates on the 2D materials would certainly be higher than the interaction of single cyclotide molecules with the rest of the aggregate, and therefore, the cyclotides are expected to be released from the material surface as single peptides or smaller aggregates. However, as the cyclotides have a spontaneous tendency for aggregation, as observed in our simulations, they would tend to aggregate immediately after release, restoring their biological activities related to the formation and stability of aggregates.
and ΔGsinglead is the free energy of adsorption for a single peptide molecule. A positive value of ΔGloss suggests lesser average adsorption free energy of a single peptide molecule in an aggregate and therefore quantifies the ease to get released from the 2D materials. Table 1 provides the magnitudes of the ΔGloss for the three cyclotide molecules on both graphene and h-BN. It is observed that for all three cyclotides and both 2D materials ΔGloss is positive, which in turn suggests that when the cyclotides are adsorbed as aggregates, the individual adsorption affinity is weaker. The trend of the magnitudes of ΔGloss is the same as that observed for the adsorption free energies of single cyclotides, clearly suggesting that the greater the adsorption strength of the single cyclotide molecules, the greater the loss in free energy upon adsorption as an aggregate. Interestingly, the release times calculated for single cyclotides from the adsorbed aggregates (τaggrelease) show an enormous decrease compared to those observed for the single cyclotides (τsinglerelease), evidently due to the decrease in the average adsorption free energies. For instance, MCoTI-II, the cyclotide having the highest adsorption free energy, shows a release time of 2.99 × 1026 ns and 2.27 × 1032 ns from graphene and h-BN, respectively, when adsorbed as single molecules, while the release times are reduced to only 2.32 × 1013 ns and 2.65 × 1014 ns respectively, when released from the adsorbed state of aggregates. In line with the above results, it may be envisioned that the larger the size of the aggregates of cyclotides, the more lower the adsorption free energies of the individual peptides, which would increase the releasing tendency of the individual peptides from the adsorbed aggregates, thereby providing a better opportunity to interact with other biomolecules of therapeutic concern. In fact, controlling the size of the aggregates may unfold an opportunity to fine-tune the extent of interaction between the 2D materials and cyclotides and develop precise heterostructures for their biomedical targeting and controlled release. Nonetheless, the affinity of the adsorption of cyclotides can, in principle, be further tuned by introducing various functionalization groups into the 2D materials or by controlling the extent of material oxidation, in accordance with the requirements for a specific therapeutic or biomedical purpose. It is worthwhile to mention that the adsorption free energy of large cyclotide aggregates on the 2D materials would certainly be higher than the interaction of single cyclotide molecules with the rest of the aggregate, and therefore, the cyclotides are expected to be released from the material surface as single peptides or smaller aggregates. However, as the cyclotides have a spontaneous tendency for aggregation, as observed in our simulations, they would tend to aggregate immediately after release, restoring their biological activities related to the formation and stability of aggregates.
4. Conclusion
In summary, in the present article, we have ventured into the interactions between cyclotides, a structurally unique class of plant-derived peptides, with state-of-the-art 2D materials to evaluate the prospects of fabrication of their nanocomposites for impending bio-nano applications. Three representative cyclotides were taken, viz. Katala B1, cycloviolacin O1, and MCoTI-II, one from each of the three structurally distinct subclasses of peptides and classical molecular dynamics simulations were used to study the adsorption of cyclotides on graphene and h-BN. The evolution of the secondary structures shows that the cyclotides adsorb without significant structural perturbation and the exceptional stability is attributed to the presence of the weakly adsorbing cystine residues forming the cyclic “knots” and the absence of higher populations of aromatic amino acid residues. Similar to single peptides, the aggregates of these macrocyclic molecules are also adsorbed without inflicting any perturbation towards the inter-peptide non-covalent bonding. Free energy analyses show that the strength of adsorption of cyclotides is moderate as compared to other similar-sized biomolecules, and consequently, the release times of cyclotides from these 2D materials are orders of magnitude less than those observed for various categories of biomolecules such as nucleic acids. Furthermore, it is demonstrated that the free energy of adsorption per peptide is less for an adsorbed aggregate of cyclotides when compared with single peptide molecules. It is realized that with the increase in the size of the aggregates, the adsorption strength of individual peptides decreases, thereby increasing the efficiency of the interaction of the cyclotide molecules with other biomolecules and release from the adsorbed state. It is envisioned that the aggregation-dependent adsorption affinity of cyclotides may be exploited to prepare precise heterostructures with 2D materials for their biomedical targeting and sustained release. As a whole, it may be concluded that cyclotides and their aggregates are able to form stable heterostructures with common 2D materials maintaining their native structural properties unlike usual protein molecules, and the hybrid systems might be able to release individual peptides when the aggregate size is significantly large and the adsorption affinity is properly tuned. We believe that the so-formed hybrid bio-nano architectures can be used for targeting specific biomolecular domains, such as the lipid membrane, thereby leading to the plausible development of next-generation antifungal, antibacterial, and anti-tumour agents of bio-nano origin.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
AG acknowledges CSIR for a Senior Research Fellowship (SRF). AD thanks SERB (grant CRG/2020/000301) for partial funding. We thank IACS for the use of TRC and CRAY supercomputers for computational purposes.References
- D. J. Craik, N. L. Daly, T. Bond and C. Waine, Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif, J. Mol. Biol., 1999, 294, 1327–1336 CrossRef CAS PubMed.
- D. J. Craik, Seamless Proteins Tie Up Their Loose Ends, Science, 2006, 311, 1563–1564 CrossRef PubMed.
- D. J. Craik, M. A. Anderson, D. G. Barry, R. J. Clark, N. L. Daly, C. V. Jennings and J. Mulvenna, Discovery and structures of the cyclotides: novel macrocyclic peptides from plants, Lett. Pept. Sci., 2001, 8, 119–128 CrossRef CAS.
- M. L. Colgrave and D. J. Craik, Thermal, Chemical, and Enzymatic Stability of the Cyclotide Kalata B1: The Importance of the Cyclic Cystine Knot, Biochemistry, 2004, 43, 5965–5975 CrossRef CAS PubMed.
- S. M. Simonsen, L. Sando, D. C. Ireland, M. L. Colgrave, R. Bharathi, U. Göransson and D. J. Craik, A Continent of Plant Defense Peptide Diversity: Cyclotides in Australian Hybanthus (Violaceae), Plant Cell, 2005, 17, 3176–3189 CrossRef CAS PubMed.
- U. Göransson and D. J. Craik, Disulfide Mapping of the Cyclotide Kalata B1: CHEMICAL PROOF OF THE CYCLIC CYSTINE KNOT MOTIF, J. Biol. Chem., 2003, 278, 48188–48196 CrossRef PubMed.
- G. K. T. Nguyen, S. Zhang, N. T. K. Nguyen, P. Q. T. Nguyen, M. S. Chiu, A. Hardjojo and J. P. Tam, Discovery and Characterization of Novel Cyclotides Originated from Chimeric Precursors Consisting of Albumin-1 Chain a and Cyclotide Domains in the Fabaceae Family, J. Biol. Chem., 2011, 286, 24275–24287 CrossRef CAS PubMed.
- J. Gracy, D. Le-Nguyen, J.-C. Gelly, Q. Kaas, A. Heitz and L. Chiche, KNOTTIN: the knottin or inhibitor cystine knot scaffold in 2007, Nucleic Acids Res., 2007, 36, 314–319 CrossRef PubMed.
- M. F. S. Pinto, I. C. M. Fensterseifer, L. Migliolo, D. A. Sousa, G. de Capdville, J. W. Arboleda-Valencia, M. L. Colgrave, D. J. Craik, B. S. Magalhães, S. C. Dias and O. L. Franco, Identification and Structural Characterization of Novel Cyclotide with Activity against an Insect Pest of Sugar Cane, J. Biol. Chem., 2012, 287, 134–147 CrossRef CAS PubMed.
- B. L. Barbeta, A. T. Marshall, A. D. Gillon, D. J. Craik and M. A. Anderson, Plant cyclotides disrupt epithelial cells in the midgut of lepidopteran larvae, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1221–1225 CrossRef CAS PubMed.
- M. A.-O. Pinto, J. Z. G. Najas, L. G. Magalhães, A. F. Bobey, J. N. Mendonça, N. A.-O. Lopes, F. M. Leme, S. P. Teixeira, M. Trovó, A. D. Andricopulo, J. Koehbach, C. W. Gruber, E. A.-O. Cilli and V. A.-O. Bolzani, Inhibition of Breast Cancer Cell Migration by Cyclotides Isolated from Pombalia calceolaria, J. Nat. Prod., 2018, 81, 1203–1208 CrossRef CAS PubMed.
- Q. Du, L. A.-O. Chan, E. K. Gilding, S. T. Henriques, N. D. Condon, A. S. Ravipati, Q. Kaas, Y. A.-O. Huang and D. A.-O. Craik, Discovery and mechanistic studies of cytotoxic cyclotides from the medicinal herb Hybanthus enneaspermus, J. Biol. Chem., 2020, 295, 10911–10925 CrossRef CAS PubMed.
- C. Jennings, J. West, C. Waine, D. Craik and M. Anderson, Biosynthesis and insecticidal properties of plant cyclotides: The cyclic knotted proteins from Oldenlandia affinis, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10614–10619 CrossRef CAS PubMed.
- D. C. Ireland, C. K. Wang, J. A. Wilson, K. R. Gustafson and D. J. Craik, Cyclotides as natural anti-HIV agents, Pept. Sci., 2007, 50, 51–60 Search PubMed.
- E. K. Gilding, M. A. Jackson, A. G. Poth, S. T. Henriques, P. J. Prentis, T. Mahatmanto and D. J. Craik, Gene coevolution and regulation lock cyclic plant defence peptides to their targets, New Phytol., 2016, 210, 717–730 CrossRef CAS PubMed.
- M. Pränting, C. Lööv, R. Burman, U. Göransson and D. I. Andersson, The cyclotide cycloviolacin O2 from Viola odorata has potent bactericidal activity against Gram-negative bacteria, J. Antimicrob. Chemother., 2010, 65, 1964–1971 CrossRef PubMed.
- K. Jagadish and J. A. Camarero, Cyclotides, a promising molecular scaffold for peptide-based therapeutics, Pept. Sci., 2010, 94, 611–616 CrossRef CAS PubMed.
- S. T. Henriques and D. J. Craik, Cyclotides as templates in drug design, Drug Discovery Today, 2010, 15, 57–64 CrossRef CAS PubMed.
- P. G. Ojeda, M. H. Cardoso and O. L. Franco, Pharmaceutical applications of cyclotides, Drug Discovery Today, 2019, 24, 2152–2161 CrossRef CAS PubMed.
- A. Jayakumar, A. Surendranath and M. Pv, 2D materials for next generation healthcare applications, Int. J. Pharm., 2018, 551, 309–321 CrossRef CAS PubMed.
- K. M. L. Taylor-Pashow, J. Della Rocca, R. C. Huxford and W. Lin, Hybrid nanomaterials for biomedical applications, Chem. Commun., 2010, 46, 5832–5849 RSC.
- R. Mas-Ballesté, C. Navarro, J. Gómez-Herrero and F. Zamora, 2D materials: To graphene and beyond, Nanoscale, 2011, 3, 20–30 RSC.
- F.-M. Chen and X. Liu, Advancing biomaterials of human origin for tissue engineering, Prog. Polym. Sci., 2016, 53, 86–168 CrossRef CAS PubMed.
- R. Kurapati, K. Kostarelos, M. Prato and A. Bianco, Biomedical Uses for 2D Materials Beyond Graphene: Current Advances and Challenges Ahead, Adv. Mater., 2016, 28, 6052–6074 CrossRef CAS PubMed.
- C. A. Jimenez-Cruz, S.-g. Kang and R. Zhou, Large scale molecular simulations of nanotoxicity, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2014, 6, 329–343 CAS.
- Z. Ruhong, Modeling of nanotoxicity: Molecular interactions of nanomaterials with bionanomachines, 2015, pp. 1–189 Search PubMed.
- C. T. Lim and K. Kenry, Biocompatibility and Nanotoxicity of Layered Two-Dimensional Nanomaterials, ChemNanoMat, 2016, 3, 5–16 Search PubMed.
- S. Friedrichs and J. Schulte, Environmental, health and safety aspects of nanotechnology—implications for the R&D in (small) companies, Sci. Technol. Adv. Mater., 2007, 8, 12–18 CrossRef CAS.
- A. Nel, Y. Zhao and L. Mädler, Environmental Health and Safety Considerations for Nanotechnology, Acc. Chem. Res., 2013, 46, 605–606 CrossRef CAS PubMed.
- M. Fojtů, W. Z. Teo and M. Pumera, Environmental impact and potential health risks of 2D nanomaterials, Environ. Sci.: Nano, 2017, 4, 1617–1633 RSC.
- W. Hu, C. Peng, W. Luo, M. Lv, X. Li, D. Li, Q. Huang and C. Fan, Graphene-Based Antibacterial Paper, ACS Nano, 2010, 4, 4317–4323 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, Toxicity of Graphene and Graphene Oxide Nanowalls Against Bacteria, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS PubMed.
- M. Feng, H. Kang, Z. Yang, B. Luan and R. Zhou, Potential disruption of protein-protein interactions by graphene oxide, J. Chem. Phys., 2016, 144, 225102 CrossRef PubMed.
- B. Luan, T. Huynh, L. Zhao and R. Zhou, Potential Toxicity of Graphene to Cell Functions via Disrupting Protein–Protein Interactions, ACS Nano, 2015, 9, 663–669 CrossRef CAS PubMed.
- L. Horváth, A. Magrez, D. Golberg, C. Zhi, Y. Bando, R. Smajda, E. Horváth, L. Forró and B. Schwaller, In Vitro Investigation of the Cellular Toxicity of Boron Nitride Nanotubes, ACS Nano, 2011, 5, 3800–3810 CrossRef PubMed.
- S. Mateti, C. S. Wong, Z. Liu, W. Yang, Y. Li, L. H. Li and Y. Chen, Biocompatibility of boron nitride nanosheets, Nano Res., 2018, 11, 334–342 CrossRef CAS.
- T. K. Mukhopadhyay, K. Bhattacharyya and A. Datta, Gauging the Nanotoxicity of h2D-C2N toward Single-Stranded DNA: An in Silico Molecular Simulation Approach, ACS Appl. Mater. Interfaces, 2018, 10, 13805–13818 CrossRef CAS PubMed.
- T. K. Mukhopadhyay and A. Datta, Screening two dimensional materials for the transportation and delivery of diverse genetic materials, Nanoscale, 2020, 12, 703–719 RSC.
- T. K. Mukhopadhyay, A. Ghosh and A. Datta, Molecular Dynamics Simulations Reveal Orientation-Dependent Nanotoxicity of Black Phosphorene toward Dimeric Proteins, ACS Appl. Nano Mater., 2021, 4, 3095–3107 CrossRef CAS.
- B. Li, D. R. Bell, Z. Gu, W. Li and R. Zhou, Protein WW domain denaturation on defective graphene reveals the significance of nanomaterial defects in nanotoxicity, Carbon, 2019, 146, 257–264 CrossRef CAS.
- X. Lei, S. Liu, R. Zhou and X.-Y. Meng, Molecular Dynamics Simulation Study on Interactions of Cycloviolacin with Different Phospholipids, J. Phys. Chem. B, 2021, 125, 3476–3485 CrossRef CAS PubMed.
- M. Trabi and D. J. Craik, Tissue-Specific Expression of Head-to-Tail Cyclized Miniproteins in Violaceae and Structure Determination of the Root Cyclotide Viola hederacea root cyclotide1[W], Plant Cell, 2004, 16, 2204–2216 CrossRef CAS PubMed.
- K. J. Rosengren, N. L. Daly, M. R. Plan, C. Waine and D. J. Craik, Twists, Knots, and Rings in Proteins: STRUCTURAL DEFINITION OF THE CYCLOTIDE FRAMEWORK, J. Biol. Chem., 2003, 278, 8606–8616 CrossRef CAS PubMed.
- T. K. Mukhopadhyay and A. Datta, Delicate Balance of Non-Covalent Forces Govern the Biocompatibility of Graphitic Carbon Nitride towards Genetic Materials, ChemPhysChem, 2020, 21, 1836–1846 CrossRef CAS PubMed.
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, Constant pressure molecular dynamics simulation: The Langevin piston method, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS.
- R. L. Davidchack, R. Handel and M. V. Tretyakov, Langevin thermostat for rigid body dynamics, J. Chem. Phys., 2009, 130, 234101 CrossRef PubMed.
- O. Farago, Langevin thermostat for robust configurational and kinetic sampling, Phys. A, 2019, 534, 122210 CrossRef.
- H. C. Andersen, Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations, J. Comput. Phys., 1983, 52, 24–34 CrossRef CAS.
- L. Kalé, R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan and K. Schulten, NAMD2: Greater Scalability for Parallel Molecular Dynamics, J. Comput. Phys., 1999, 151, 283–312 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. K. Mukhopadhyay and A. Datta, Deciphering the Role of Solvents in the Liquid Phase Exfoliation of Hexagonal Boron Nitride: A Molecular Dynamics Simulation Study, J. Phys. Chem. C, 2017, 121, 811–822 CrossRef CAS.
- T. K. Mukhopadhyay and A. Datta, Ordering and Dynamics for the Formation of Two-Dimensional Molecular Crystals on Black Phosphorene, J. Phys. Chem. C, 2017, 121, 10210–10223 CrossRef CAS.
- T. K. Mukhopadhyay and A. Datta, Exfoliation and dispersion of 2D materials in polar solvents: A molecular simulation approach, J. Indian Chem. Soc., 2019, 96, 753–766 CAS.
- T. K. Mukhopadhyay and A. Datta, Disentangling the liquid phase exfoliation of two-dimensional materials: an “in silico” perspective, Phys. Chem. Chem. Phys., 2020, 22, 22157–22179 RSC.
- E. Darve, D. Rodríguez-Gómez and A. Pohorille, Adaptive biasing force method for scalar and vector free energy calculations, J. Chem. Phys., 2008, 128, 144120 CrossRef PubMed.
- E. Darve and A. Pohorille, Calculating free energies using average force, J. Chem. Phys., 2001, 115, 9169–9183 CrossRef CAS.
- T. Mukhopadhyay and A. Datta, Design Rules for the Generation of Stable Quartet Phases of Nucleobases over 2D Materials, J. Phys. Chem. C, 2018, 122, 28918–28933 CrossRef CAS.
- Z. E. Hughes and T. R. Walsh, What makes a good graphene-binding peptide? Adsorption of amino acids and peptides at aqueous graphene interfaces, J. Mater. Chem. B, 2015, 3, 3211–3221 RSC.
- J.-H. Lee, Y.-K. Choi, H.-J. Kim, R. H. Scheicher and J.-H. Cho, Physisorption of DNA Nucleobases on h-BN and Graphene: vdW-Corrected DFT Calculations, J. Phys. Chem. C, 2013, 117, 13435–13441 CrossRef CAS.
- M. Shakourian-Fard, G. Kamath and Z. Jamshidi, Trends in Physisorption of Ionic Liquids on Boron-Nitride Sheets, J. Phys. Chem. C, 2014, 118, 26003–26016 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05096j |
| This journal is © The Royal Society of Chemistry 2023 |