
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanostructured graphene oxide enriched with metallic nanoparticles as a biointerface to enhance cell adhesion through mechanosensory modifications
Michał
Pruchniewski
 a,
Ewa
Sawosz
a,
Malwina
Sosnowska-Ławnicka
a,
Agnieszka
Ostrowska
a,
Maciej
Łojkowski
b,
Piotr
Koczoń
c,
Paweł
Nakielski
d,
Marta
Kutwin
a,
Sławomir
Jaworski
a and
Barbara
Strojny-Cieślak
*a
a,
Ewa
Sawosz
a,
Malwina
Sosnowska-Ławnicka
a,
Agnieszka
Ostrowska
a,
Maciej
Łojkowski
b,
Piotr
Koczoń
c,
Paweł
Nakielski
d,
Marta
Kutwin
a,
Sławomir
Jaworski
a and
Barbara
Strojny-Cieślak
*a
aDepartment of Nanobiotechnology, Institute of Biology, Warsaw University of Life Sciences, Warsaw, Poland. E-mail: barbara_strojny-cieslak@sggw.edu.pl
bFaculty of Material Sciences and Engineering, Warsaw University of Technology, Warsaw, Poland
cDepartment of Chemistry, Institute of Food Sciences, Warsaw University of Life Sciences, Warsaw, Poland
dDepartment of Biosystems and Soft Matter, Institute of Fundamental Technological Research, Polish Academy of Sciences, Warsaw, Poland
First published on 17th November 2023
Abstract
Nanostructuring is a process involving surface manipulation at the nanometric level, which improves the mechanical and biological properties of biomaterials. Specifically, it affects the mechanotransductive perception of the microenvironment of cells. Mechanical force conversion into an electrical or chemical signal contributes to the induction of a specific cellular response. The relationship between the cells and growth surface induces a biointerface-modifying cytophysiology and consequently a therapeutic effect. In this study, we present the fabrication of graphene oxide (GO)-based nanofilms decorated with metallic nanoparticles (NPs) as potential coatings for biomaterials. Our investigation showed the effect of decorating GO with metallic NPs for the modification of the physicochemical properties of nanostructures in the form of nanoflakes and nanofilms. A comprehensive biocompatibility screening panel revealed no disturbance in the metabolic activity of human fibroblasts (HFFF2) and bone marrow stroma cells (HS-5) cultivated on the GO nanofilms decorated with gold and copper NPs, whereas a significant cytotoxic effect of the GO nanocomplex decorated with silver NPs was demonstrated. The GO nanofilm decorated with gold NPs beneficially managed early cell adhesion as a result of the transient upregulation of α1β5 integrin expression, acceleration of cellspreading, and formation of elongated filopodia. Additionally, the cells, sensing the substrate derived from the nanocomplex enriched with gold NPs, showed reduced elasticity and altered levels of vimentin expression. In the future, GO nanocomplexes decorated with gold NPs can be incorporated in the structure of architecturally designed biomimetic biomaterials as biocompatible nanostructuring agents with proadhesive properties.
1. Introduction
Precisely designed cell growth surfaces can trigger a cascade of molecular events, ultimately achieving the desired therapeutic effect. In this case, a comprehensive understanding of the tissue environment, both physiological and pathological, for a specific disease state is considered the starting point in the development of therapies using the processing of mechanical and chemical stimuli from a substrate into an intracellular signal,1 and consequently modifying the polarization of the structure and functions of cells.2 Cells, which sense the topography and chemistry of the nanostructured surface, receive specific mechanical stimuli and convert them into a set of biochemical reactions, influencing the cellular response.3–5 The mechanotransduction process initiated by the binding of the cell membrane to the growth surface by cell-adhesion molecules (CAMs), including integrins, leads to significant changes in the cellular machinery, and thus the manipulation of the adhesion phenomenon,6 cytoskeleton architecture,7,8 and secretory profile of cells.9,10A peculiar example of the growth surface for cells is graphene oxide (GO) in the form of a nanofilm11,12 or incorporated in the structure of more sophisticated biomaterials as a coating13,14 or filling,15,16 enhancing the biological properties of the constructs. At the molecular level, GO is a one-atom-thick highly oxidized form of graphene, which is composed of two-dimensional conjugated sp2 carbon atoms arranged in six-membered rings, forming a honeycomb-like structure. The nanobands of the edges surrounding the carbonaceous GO ground plane are equipped with various dangling bonds and oxygen-containing molecular species, such as hydroxyl, carboxyl, carbonyl, and epoxide groups.17 The reactive nature and molecular dynamics of GO determine the ease of functionalization and modifiability of the oxidized side edges, which translates into abundant possibilities for introducing bioactive substances in the structure of GO nanosheets and enabling them to be used as nanocarriers of therapeutic agents. Interestingly, the introduced substances affect not only the chemical properties of the composites but also the physical properties of GO, which then has a significant impact on the biological activity of the construct by modifying the mechanotransduction signaling pathways.18–20 GO-enriched platforms composed of growth substrates for cells act as a type of mechanotransmitter, propagating a mechanical signal from the substrate–cell binding site to the intracellular space. Therefore, any subtle modification, even at the nanometric scale, may induce distinct differences in the topography, stiffness/elasticity, and adhesive properties of the substrate, and thus cellular mechanosensing.
Synthetic biomaterials dedicated to regenerative medicine applications are often associated with the problem of poor adhesive properties, which can be solved by decorating their surface with GO nanosheets to enhance their bioactivity and enrich their nanostructure.21–23 Furthermore, GO nanosheets can also be decorated by incorporating secondary phases with precious and semi-precious metals. In this case, silver (Ag), gold (Au), and copper (Cu) nanoparticles (NPs), considering their antimicrobial properties and multifaceted participation in tissue remodeling and regeneration processes, provide the desired enrichment of GO nanosheets. Derived GO–metallic NP nanocomposites differ from their components in terms of their physicochemical and biological properties. The mutual interactions of the nanocomposite components lead to the modification of the properties of the GO nanosheets composed of nanocarriers of metallic NPs, determining their release profile,24,25 and consequently access to biological systems. Therefore, in the design and manufacture of hybrid GO–metallic NP coatings, the molecular interactions in the nanocomposite and the possibility of releasing both metallic NPs from the GO nanocarrier and exfoliated GO nanoflakes entirely from the surface of the biomaterial should be considered. The possibility of the biodegradation of the nanocomplex in the tissue environment makes it necessary to investigate the biological impact of the tested materials in colloidal form, representing released/exfoliated nanostructures, and as a nanofilm ground on the solid surface of biomaterials.
This study aimed at deriving and evaluating the biological effect of GO nanofilms decorated with Ag, Au, and Cu NPs as enhancers of the bioactivity of GO coatings. The biocompatibility of the prepared nanocomplexes, both in the colloidal form and as nanofilms, was verified, together with the proadhesive properties of the nanofilms. Moreover, preliminary insights into the nanofilm–cell mechanistic relationship are presented. We further report that biocompatible GO nanofilms decorated with Au and Cu NPs accelerated the processes of early cell adhesion by initiating a molecular cascade, leading to the stable attachment of the cell to the GO–metallic NP nanofilm constituting an artificial extracellular matrix.
2. Results and discussion
A frequent impediment in the use of biomaterials as effective tools in regenerative medicine is the impaired integration of the biomaterial surface with the surrounding tissues because of its low adhesive properties and the disturbance of the mechanical interactions between cells and the surface, thus hindering the anchoring of cells on the surface of biomaterials. Accordingly, these limitations can be addressed by incorporating bioactive coatings with proadhesive and proregenerative properties in the surface structures of biomaterials. The main goal of this study was to develop GO decorated with metallic NP nanofilms as a candidate nanocomposite coating to supplement biomaterials. This nanocomposite was characterized by high cytocompatibility and excellent adhesive properties, stimulating the formation of a native cellular scaffold (extracellular matrix) predisposed to supplement biomaterials. The physicochemical properties and biological activity of pure GO and GO enriched with metallic NPs in colloidal form and as a nanofilm were characterized in the context of proadhesive and proregenerative properties. The assessment of the impact of the GO–metallic NP nanocomplexes on the cell response, including mechanotransduction, was performed using biological models of fetal foreskin fibroblast HFFF2 and bone marrow stromal HS-5 cells with a fibroblast-like morphology as cellular representations of the skin and bone tissue environment, respectively.The analysis of the hydrodynamic diameter determined by the DLS method (Fig. 1a–d) showed homogeneous populations of all nanostructures dispersed in water at time point 0 h, without the presence of agglomerates in the samples. The hydrodynamic diameter was 295 nm, 197 nm, 267 nm, and 384 nm for GO, GO–Ag NPs, GO–Au NPs, and GO–Cu NPs, respectively. After 24 h, an increase in the hydrodynamic diameter and the presence of agglomerates were observed for all the nanocomplexes dispersed in water. The hydrodynamic diameter increased by 16%, 20%, 55%, and 112% at the 0 h time point for GO, GO–Ag NPs, GO–Au NPs, and GO–Cu NPs, respectively. The nanocomplexes dispersed in the culture medium (DMEM) supplemented with 10% FBS showed significant heterogeneity at 0 h and 24 h. However, agglomerates were observed at 0 h for GO and GO–Ag NPs and after 24 h for GO–Au NPs and GO–Cu NPs. The hydrodynamic diameter of GO without metallic NPs in the culture medium was 413 nm immediately after their addition to the medium, which subtly increased after 24 h. In general, the nanocomplexes increased the hydrodynamic diameter to >1000 nm when introduced in the culture medium, and after 24 h, the hydrodynamic diameter was reduced to 888 nm, 436 nm, and 604 nm for GO–Ag NPs, GO–Au NPs, and GO–Cu NPs, respectively. However, DLS is not a recommended method for the analysis of non-spherical materials such as GO. Here, the results of the analysis were only used to estimate the behavior of the nanostructures depending on the dispersion medium and time; therefore, the obtained values may differ from reality to some extent. The size of the nanoflakes determined by microscopic analysis was 3767 ± 620 nm, 4732 ± 1073 nm, 3849 ± 988 nm and 4501 ± 928 nm for GO, GO–Ag NPs, GO–Au NPs and GO–Cu NPs, respectively.
 | ||
| Fig. 1 The influence of choice of dispersion medium and time on the physicochemical properties of GO nanocomplexes. Graphs representing the hydrodynamic diameter measured by dynamic light scattering (DLS) for GO (a), GO–Ag NPs (b), GO–Au NPs (c), and GO–Cu NPs (d) expressed as intensity (%) as a function of size (hydrodynamic diameter, nm; logarithmic scale). Polydispersity index measurements (e), where the dots represent a single measurement and the bar indicates the median values (n = 3). Changes in the zeta potential (f), where the results are presented as mean (n = 4) with standard deviation. Each measurement was performed in ultrapure water at 0 hour (blue) and after 24 hours (navy blue) and the culture medium (DMEM) at 0 hour (pink) and after 24 hours (red). The size of nanoflakes based on high-resolution scanning electron microscopy visualization (g), where the dots represent a single measurement and bar indicates the median (n = 50). | ||
Based on the polydispersity index (PDI), the size distribution of the nanostructures was determined (Fig. 1f). In the case of the nanostructures dispersed in water at the time point 0 h, the lowest PDI of approximately 0.1, was demonstrated. After 24 h, the PDI increased to 0.23, 0.23, 0.35, and 0.27 for GO, GO–Ag NPs, GO–Au NPs, and GO–Cu NPs, respectively, thus proving an increase in colloid heterogeneity. The nanocomplexes introduced in the culture medium also formed a heterogeneous dispersion within 0 h. However, after 24 h, the PDI decreased to a value generally considered acceptable for nanomaterials (0.3), but the colloids still maintained a relatively high particle size distribution.
Electrophoretic light scattering was performed to determine the stability of the nanocomplexes. For all the nanomaterials, a negative zeta potential was demonstrated (Fig. 1e). The zeta potential value for the water-dispersed nanostructures was <−50 mV, excluding GO–Cu NPs, indicating their good colloidal stability. After 24 h, the zeta potential remained at the original level or decreased slightly. The zeta potential for GO–Cu NPs in water was −33 mV and −35 mV for 0 hours and 24 h, respectively. After introducing the nanostructures in the culture medium, a significant decrease in the zeta potential value was observed, hence destabilization of the colloid. The zeta potential value for the nanostructures dispersed in the medium was about −10 mV at 0 h and remained at a similar level after 24 h.
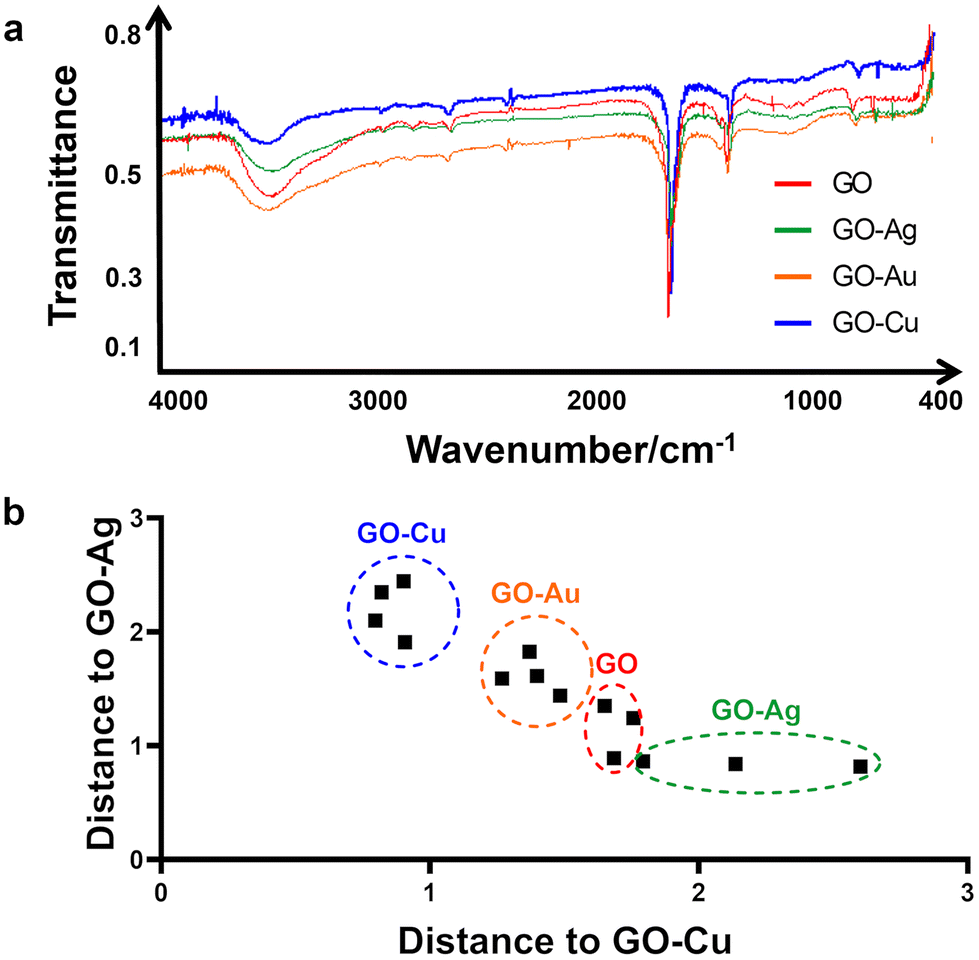
The raw infrared spectra recorded in the middle range, i.e., 400–4000 cm−1 are presented in Fig. 2a, with transmittance/% against wavenumber/cm−1 on the y- and x-axis, respectively. All samples produced very similar spectra. Thus, it was impossible to distinguish the samples solely by visual inspection without a legend. In the high spectral region at 3600–3300 cm−1, an intense broad band generated by the O–H stretches or hydrogen bonding between O–H groups is present in the spectrum of all the samples. This band is asymmetrical, with a characteristic broad shoulder on the right-hand side, which is due to the presence of C–H stretching of carbon and hydrogen from the benzene ring. Next, in the range of 3000–2600 cm−1, several weak bands assigned to C–H stretches were observed. Given that all of them are located at wavenumbers lower than 3000 cm−1, they evidence the presence of aliphatic carbon chains. The noise present at around 2300 cm−1 is due to carbon dioxide. At 1619 cm−1, a strong, sharp band generated by very characteristic >C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C< stretches is present. Next, a middle-intense band is present at 1349 cm−1, corresponding to C–H deformation, and a weak band at 758 cm−1, which was not assigned. Although the shape of all the spectra looks very similar, a closer look at the band assigned to the carbon–carbon double bond stretching vibrations allowed us to conclude that the spectra are not identical. Therefore, this spectral region was used in the discriminant analysis to distinguish among the GO–Ag NP, GO–Au NP, GO–Cu NP and GO samples.
C< stretches is present. Next, a middle-intense band is present at 1349 cm−1, corresponding to C–H deformation, and a weak band at 758 cm−1, which was not assigned. Although the shape of all the spectra looks very similar, a closer look at the band assigned to the carbon–carbon double bond stretching vibrations allowed us to conclude that the spectra are not identical. Therefore, this spectral region was used in the discriminant analysis to distinguish among the GO–Ag NP, GO–Au NP, GO–Cu NP and GO samples.
 | ||
| Fig. 2 Fourier-transform infrared spectra of (a) GO and GO–metallic nanocomplexes in the middle region (4000–500 cm−1). Discrimination of GO sample and GO–Ag NP, GO–Au NP and GO–Cu NP samples into homologous groups based on the spectral data (b). Color markings: red, GO; green, GO–Ag NPs; yellow, GO–Au NPs; and blue, GO–Cu NPs. | ||
Fig. 2b presents the result of the discriminant analysis conducted with three samples of every type of sample, i.e., GO–Ag NPs, GO–Au NPs, GO–Cu NPs and GO. Separate homologous groups were calculated with the use of Mahalanobis distances. This separation evidenced that the samples in the same group are different from that in the other groups. The data in the 1920–1218 cm−1 spectral region was used for calibrating the discriminant model. The above-mentioned statement that the region where the carbon–carbon double bonds appeared is very important given that if the spectral data from the other regions, including the total spectral region or O–H band region alone, were used for calibrating the statistical model, the discrimination did not occur. This indicates that the specific difference among the samples is in the CC bonds but not alone. If considered together with the C–H deformation region, then discrimination at a 100% level occurs, specifically, in the energy value of the carbon–carbon double bonds. Given that one of the carbons from the CC bond is bonded to a specific metal, e.g. Ag, Cu, and Au, in all the samples, the strength of the CC bond is different. Hence, the energy for the CC stretching is different. Consequently, this presents itself as the above-mentioned small difference in the spectrum. Therefore, this difference can serve to distinguish samples of GO combined with different metals.
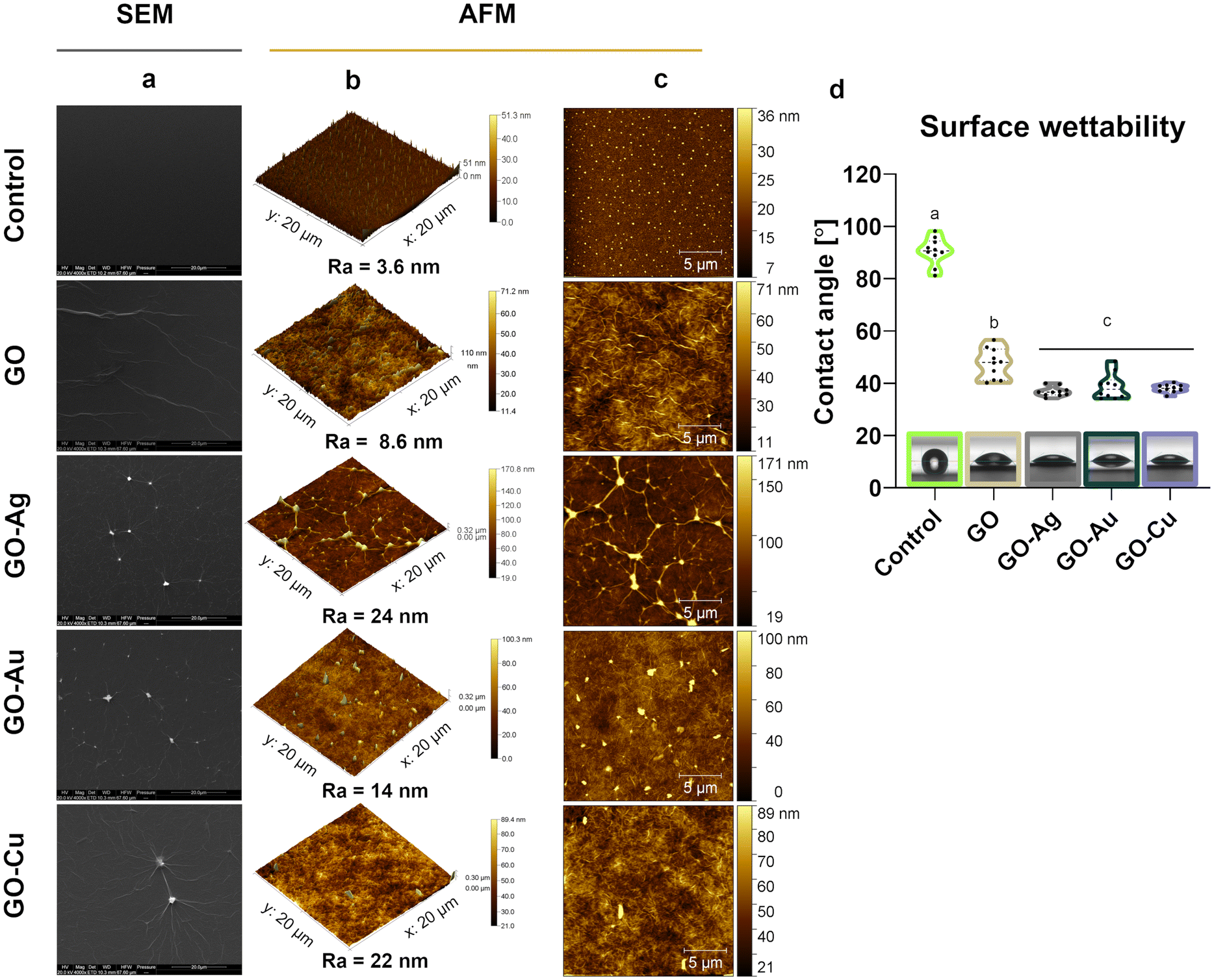
The topographic assessment of the nanofilms was performed using SEM (Fig. 3a) and AFM visualization (Fig. 3b and c). In general, the GO and GO–metallic NPs formed irregular, rough, and subtly wrinkled surfaces. Decorating GO with metallic NPs enhanced the draping, and thus the roughness and heterogeneity in terms of surface height. The average roughness value (Ra) was 3.6 nm, 8.6 nm, 24 nm, 14 nm, and 22 nm for the uncoated surface (control), GO, GO–Ag NPs, GO–Au NPs, and GO–Cu NPs, respectively. Evenly distributed Ag NPs were observed on the surface of the GO–Ag nanofilm, which increased the height to a maximum of 171 nm. In the case of the GO–Au and GO–Cu nanofilms, the presence of uniformly distributed metallic NPs of various sizes and a maximum nanofilm height of 100 nm (GO–Au NPs) and 89 nm (GO–Cu NPs) were observed.
 | ||
| Fig. 3 The surface topography of GO and GO–metallic NP nanofilms. Scanning electron microscopy (SEM) (a) and atomic force microscopy (AFM) (b, c) visualization. 3D images (b) and 2D images (c) with results of average roughness (Ra) of the control (uncoated surface), GO, GO–Ag NP, GO–Au NP, and GO–Cu NP nanofilm topography. Scale bars: (a) 20 μm, (b) 20 μm × 20 μm × sample height [μm] (AFM), and (c) 5 μm. Sessile drop contact angle (CA) measurement of nanofilms (d), where the dots represent a single measurement and the black crossbar the median (n = 10), while the different letters (a, b, and c) indicate statistically significant differences among the groups (p ≤ 0.05). | ||
Goniometric analysis showed increased wettability of the control surface after coating with GO and GO–metallic NP nanocomplexes (Fig. 3d). Moreover, the incorporation of metallic NPs in the GO structure significantly decreased the CA of the nanofilm surface compared to the pure GO nanofilm. The average CA value was 38.7°, 36.8°, and 38.0° for the GO–Ag NPs, GO–Au NPs, and GO–Cu NPs, respectively, while that for GO was approximately 7.3° lower.
Given that biocompatibility is an inherent feature of biomaterials in direct contact with patient tissue, a comprehensive biocompatibility analysis was performed, capturing the cellular response at various levels, as presented in Fig. 4–6. In this study, two elements differentiated the tested factor, and thus the impact on in vitro cytobiocompatibility was distinguished, i.e., the treatment approach (colloid or nanofilm) and decorating GO nanosheets with metallic NPs (Ag NPs, Au NPs, and Cu NPs). The importance of considering the colloidal form of GO and the nanocomplexes in the assessment of biocompatibility is justified given the possibility of the exfoliation of nanoflakes from the surface of the nanofilm.
 | ||
| Fig. 4 The viability of HFFF2 (a and b) and HS-5 (c and d) cells after 24-hour treatment with GO and GO–metallic NPs in colloidal (a and c) or nanofilm (b and d) form determined by the neutral red assay. The results are expressed as percentage of the control (mean with standard deviation; n = 4). Different letters above the bars indicate statistically significant differences among the groups (p ≤ 0.05). Bar color markings: green, control; beige, GO; gray, GO–Ag NPs; dark green, GO–Au NPs; and purple, GO–Cu NPs. | ||
 | ||
| Fig. 5 The effect of GO and GO–metallic NPs in colloid (a and c) and nanofilm (b and d) form on HFFF2 (a and b) and HS-5 (c and d) cell viability after 24-hour treatment determined by the MTT assay. The results are expressed as percentage of the control (mean with standard deviation; n = 4). Different letters above the bars indicate statistically significant differences among the groups (p ≤ 0.05). Bar color markings: green, control; beige, GO; gray, GO–Ag NPs; dark green, GO–Au NPs; and purple, GO–Cu NPs. | ||
 | ||
| Fig. 6 The evaluation of the membrane integrity of HFFF2 (a and b) and HS-5 (c and d) cells after 24 hours of treatment with GO and GO–metallic NPs in the form of colloid (a and c) and nanofilm (b and d) determined by the LDH leakage assay. The results are presented as the percentage of LDH max (mean with standard deviation; n = 4). Different letters above the bars indicate statistically significant differences among the groups (p ≤ 0.05). Bar color markings: green, control; red, LDH max; beige, GO; gray, GO–Ag NPs; dark green, GO–Au NPs; and purple, GO–Cu NP. | ||
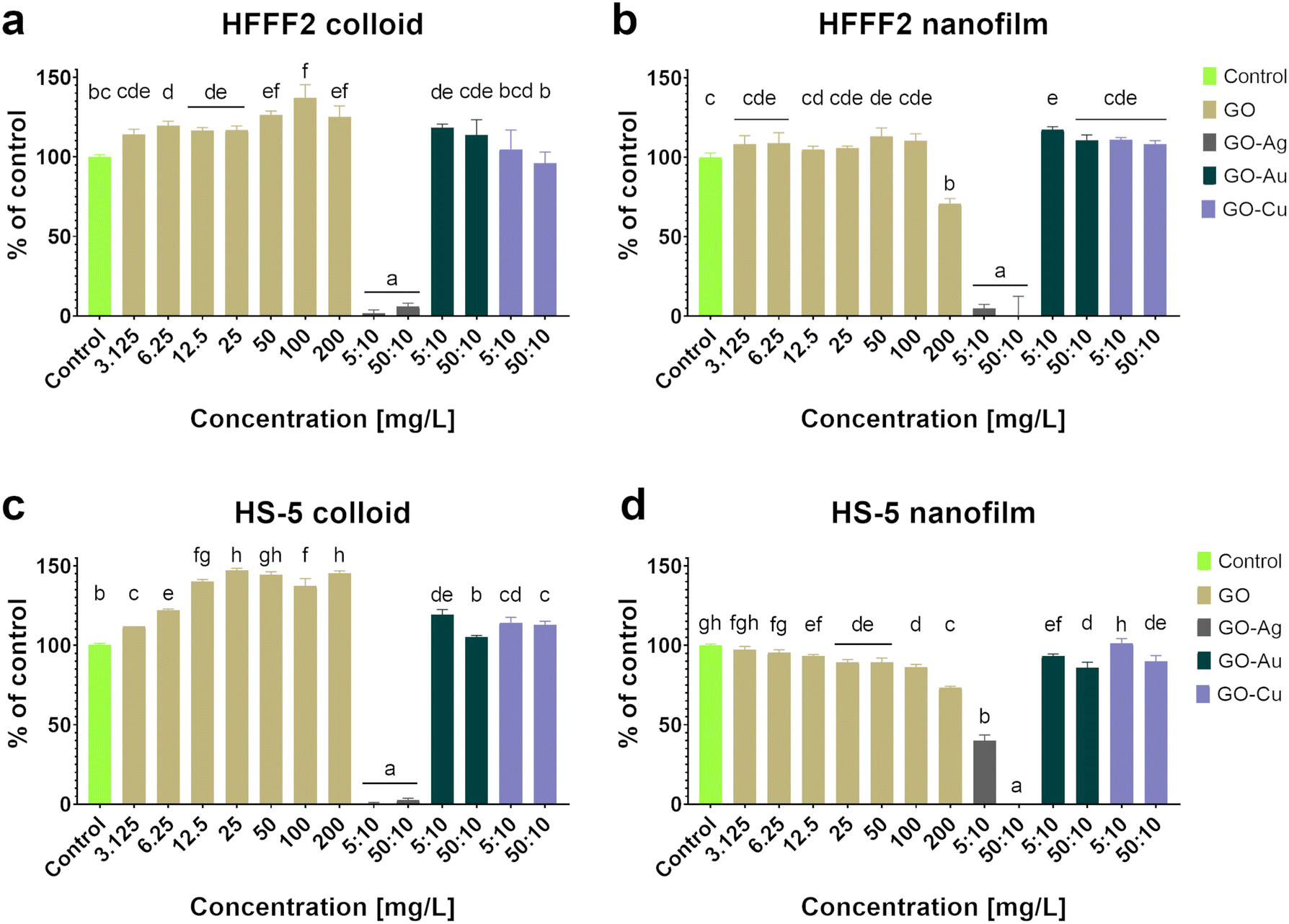
Based on the neutral red test (Fig. 4), which assesses the cell viability through the uptake and accumulation of the dye in lysosomes, no cytotoxic effect of GO in the colloidal form was demonstrated on HFFF2 cells, while in the form of a nanofilm, a reduction in the viability of HFFF2 cells was observed only for the highest concentration of GO (200 μg mL−1). Alternatively, a considerable decrease in HFFF2 cell viability treated with GO–Ag NPs was observed regardless of the treatment approach. The other nanocomplexes did not reduce the viability of the HFFF2 cells. Colloidal GO without metallic NP decoration did not reduce the HS-5 cell viability. However, the GO nanofilm caused a statistically significant dose-dependent decrease in HS-5 cell viability starting at 12.5 μg mL−1 concentration, which is an approximately 27% decrease compared to the control cells for the highest GO concentration (200 μg mL−1). In the nanocomplexes, a significant reduction in the viability of HS-5 cells cultivated in the presence of GO–Ag NPs was demonstrated, regardless of the concentration and treatment approach. A decrease in the viability of HS-5 cells was also observed for the GO–Au NP (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 μg mL−1 and 50:10 μg mL−1) and GO–Cu NP (50:10 μg mL−1) nanofilms.
:10 μg mL−1 and 50:10 μg mL−1) and GO–Cu NP (50:10 μg mL−1) nanofilms.
Colloidal GO reduced the activity of mitochondrial dehydrogenase, as determined by the MTT assay (Fig. 5), and consequently, the HFFF2 cell viability. A dose-dependent decrease in viability was observed for the HFFF2 cells treated with colloidal GO starting at a concentration of 25 μg mL−1. A significant decrease in viability was noted for the HFFF2 cells treated with the colloidal GO–Ag NP nanocomplex. A dose-dependent decrease in viability was also demonstrated for the HFFF2 cells cultivated in the presence of colloidal GO–Au NPs and GO–Cu NPs. A subtle decrease in HFFF2 cell viability on GO nanofilms was noted from 12.5 μg mL−1, while a significant decrease was observed only at GO concentrations of 100 μg mL−1 and 200 μg mL−1. The GO–Ag NP nanofilms contributed to a complete reduction in HFFF2 cell viability to 0%, while the GO–Cu NP nanofilms significantly decreased the HFFF2 cell viability to approximately 75%. Slightly reduced HS-5 cell viability was demonstrated from the lowest concentration of colloidal GO without a clear dose-dependent effect and for the cells in the presence of GO–Ag NPs regardless of the treatment approach. In the remaining cases, the tested factors did not reduce the activity of mitochondrial dehydrogenase.
Increased LDH leakage (Fig. 6), and thus impaired cell membrane integrity, was observed in the HFFF2 cells treated with the GO–Ag NP nanocomplex in the nanofilm form at a concentration of 5:10 μg mL−1. In the case of HS-5 cells, LDH leakage was observed for the cells treated with colloidal GO at 100 μg mL−1 and 200 μg mL−1 and cultivated on the nanofilm at a concentration of 200 μg mL−1. In the remaining cases, no cytotoxic effect was found, which is understood as a disruption of the cell membrane integrity, and consequently the leakage of LDH above the level of the untreated cells.
The main premise for the cytotoxic effect of GO is its high affinity for the cell membrane, contributing to the formation of pores on the cell as a result of the cooperative extraction of lipids driven by the nanostructure,26 wrapping the cells, and thus isolating them from the environment27 and cutting the cell membrane through the sharpened edges of GO nanoflakes.28 As a result of the destruction of the lipid bilayer of the cell, small fragments of GO are internalized, followed by the induction of oxidative stress,29 mitochondrial dysfunctions,30 epigenetic changes,31 and finally cell death. However, a thorough understanding of the nature of GO and its biointeractions with biological systems lays the foundation for designing a precise therapeutic approach by adjusting the dose, physicochemical properties, mechanotransduction relationship, and mode of application that will exclude the cytotoxic effects of GO and reveal its therapeutic properties.
Cells applied to nanofilms initially sediment toward the surface, contacting the ventral side of the cell body with the substrate. After adhering to the substrate, the cells can carry out chemical surface modifications and migration to generate the most favorable growth conditions. Therefore, GO nanoplatforms can be a stimulating basis for building a native protein scaffold, forming a cellular niche, and thus supporting and directing cell growth. Given that pure GO nanofilms are considered biocompatible nanostructures, the aforementioned reduction in cell viability for high GO concentrations may result from increasing the nanofilm roughness to a critical value for cells. Cells, depending on their type and origin, have certain preferences related to their surface roughness because of the differences in the mechanical properties in the niche they inhabit. For example, less rough surfaces are more favorable for the growth and development of fibroblasts compared to bone-derived cells.32,33 Moreover, cells show varying degrees of tolerance to changes in surface roughness. The high surface roughness value of micro-structured surfaces may result in the downregulation of adhesion-related genes such as integrins or laminins by epithelial cells compared to cells cultivated on nanostructured surfaces with lower roughness, while fibroblasts growing on both nano- and micro-structured surfaces express genes on a similar level, but higher than the control cells.34
As the surface roughness increases, cell adhesion decreases, while the nuclear tension and formation of stress fibers increase.35 Increasing the concentration of GO for the derivation of nanofilms is directly proportional to the number of overlapping layers of carbon nanostructures, resulting in an increase in thickness and decrease in the surface roughness.36 Moreover, the increased roughness of GO nanolayers was associated with the enhanced hydrophilicity of the surface, facilitating the initiation of contact between the cell and the surface.37 Increased hydrophilicity may increase the amount of adsorbed proteins,38 including cell receptor proteins, and change their conformation,39 thereby enhancing the substrate–cell interaction.
In contrast to the nanofilm treatment approach, the cells treated with a colloid are doomed to nanoflakes freely sedimenting on their dorsal surface. Given the lack of cellular mechanisms enabling the shedding of the GO coat or GO–metallic NPs, cells are arbitrarily isolated from the extracellular environment by nanoflakes in a size- and dose-dependent manner, which excludes the intercellular communication essential for cell survival and hinders the exchange of molecules between the cell and the environment and receiving extracellular stimuli. Therefore, the treatment of cells with the colloidal form of GO considers any type of physical damage to the cells, i.e., pore formation, isolation by insulating, and cutting with sharp edges, while in the case of using GO as a nanofilm, the cytotoxic effect of GO by isolation and wrapping was minimized or completely excluded. As demonstrated by DLS analysis (Fig. 1a), GO, when introduced in DMEM enriched with 10% FBS, aggregated, which may result in the accelerated sedimentation of large GO aggregates in the cells. Subsequently, GO aggregated, constituting a mechanical stressor for the cells by covering, burdening, and isolating them from the extracellular environment. The application of a mechanical stressor, even assuming its biocompatibility, can lead to changes in cytophysiology through modifications in mechanical stresses in the maturation of focal adhesion,40 an increase in interstitial fluid pressure, and an increase in cell–cell and cell–ECM tension.41
Here, Ag NPs with the concentration of 10 μg mL−1 deposited in the GO nanoflakes as a GO–Ag NP nanocomposite was observed to be extremely cytotoxic to the cells, and decorating GO with Ag NPs did not cancel the deleterious bioeffect of the Ag NPs. The inclusion of Ag NPs in the structure of GO nanoflakes resulted in a complete reduction in cell viability (Fig. 4 and 5) regardless of the treatment approach and the concentration of GO as a carrier. The cells treated with GO–Ag NPs possessed an apoptotic cell morphology (Fig. 9), while both mitochondrial activity and neutral red uptake were inhibited (Fig. 4 and 5). Before the Ag NPs gain access to the cell membrane, there is an earlier interaction with the serum-containing culture medium, which increases the release of Ag ions and contributes to the formation of a protein crown on the surface of the Ag NPs composed mainly of albumin.42 The NPs receive their biological identity because of proteins included in the protein corona, which strengthens the affinity of the NPs to the membrane receptors.43 Ag NPs are internalized by endocytosis and clathrin-dependent macropinocytosis.44 Subsequently, Ag NPs cause many abnormalities, and the main causes of their cytotoxicity are thought to be membrane pore formation, oxidative stress, lipid peroxidation,45 and the induction of mitochondrial dysfunction.46 Ag NPs cannot be completely rejected for biomedical applications given their high antibacterial potential; however, the dose and method of their application should be adjusted so that they annihilate bacteria, and simultaneously do not exhibit any deleterious effect on the cells and tissues of patients. GO abolishes the cytotoxic effects of Ag NPs against human keratinocytes, while enhancing the antifungal effect (Candida albicans) for the nanocomplex at concentrations of 50 μg mL−1 and 4.8 μg mL−1 for GO and Ag NPs, respectively.47 The GO–Ag NP nanocomposite (200 mg L−1) is also an excellent complement to polyurethane foils because of the increased biocompatibility compared to solitary Ag NPs evaluated on fibroblasts and umbilical vein endothelial cells, both of human origin, and the chorioallantoic membrane model in ovo as well as antibacterial properties assessed with Salmonella enteritidis.48 The incorporation of GO–Ag NPs in hydroxyapatite coatings improves the mechanical properties of the implant, maintains the antibacterial effect of Ag NPs, and is biocompatible for osteoblast-like cells, even at concentrations of 3% for GO–Ag NPs.49 Moreover, the GO–Ag NP nanocomplex at a concentration of 5:25 μg mL−1 (GO–Ag NPs) is highly biocompatible with skin tissue based on a 3D EpiDerm model, and simultaneously affect tissue reconstruction by modulating the secretory profile of cytokines.50
GO–Au NP nanocomposites in the colloidal form and GO–Cu NPs in both forms reduced the activity of mitochondrial dehydrogenases in HFFF2 cells (Fig. 5). However, the decrease in the mitochondrial metabolic activity of the HFFF2 cells treated with GO–Au NPs and GO–Cu NPs in colloidal form corresponded to a decrease in the viability of the cells treated with pure GO. The HFFF2 cell viability was 54.25%, 59%, and 51% for 50 μg mL−1 GO, 50:10 μg mL−1 GO–Au NPs, and 50:10 μg mL−1 GO–Cu NPs, respectively, suggesting that not only a high concentration of colloidal GO with metallic NPs contributed to the reduction in metabolic activity. However, the decrease in the viability of the HFFF2 cells cultivated on the GO–Cu NP nanofilm to 73% for the concentration of 50:10 μg mL−1 did not correspond to pure GO at the concentration 50 μg mL−1 (94.5%). Therefore, it can be argued that the adverse effect of surface topography on cell viability occurs through the activity of mitochondria. The GO–Au NP and GO–Cu NP nanofilms slightly reduced the viability of the HS-5 cells (86% and 89.75% for GO–Au NPs and GO–Cu NPs, respectively), as assessed by the neutral red assay (Fig. 4), also to the extent corresponding to pure GO (89.25%). Thus, the most biocompatible nanocomposite turned out to be GO–Au NPs, especially in the form of a nanofilm, which makes it suitable to be used as a coating for biomaterials for various purposes.
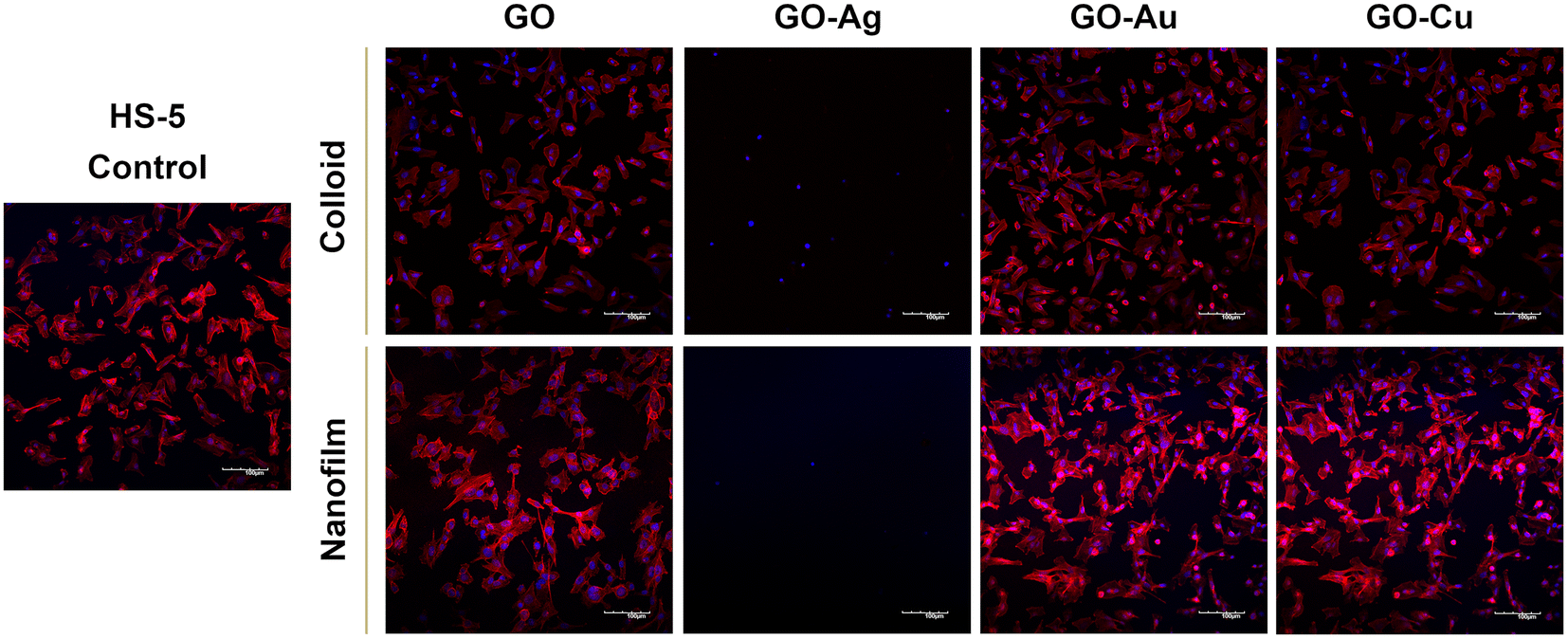
To assess the morphology of the actin cytoskeleton and cell nuclei, visualization using a confocal microscope was performed. Despite maintaining the relatively normal morphology of HFFF2 cells (Fig. 7) treated with GO, GO–Au NPs, and GO–Cu NPs, subtle differences in the organization of the actin cytoskeleton depending on the treatment approach can be observed. The HFFF2 cells cultivated on the nanofilms showed a very comparable actin cytoskeleton architecture compared to the control cells, while the colloid-treated cells showed slightly increased dynamics of the actin cytoskeleton system. Microscopic analysis confirmed the cytotoxic effect of the GO–Ag NPs on the HFFF2 cells regardless of the treatment method, as manifested by the significant disorganization of the actin cytoskeleton and shrinkage and reduction of the surface area of the GO–Ag NP-treated cells. Neither the colloids nor nanofilms of GO, GO–Au NPs, and GO–Cu NPs showed a cytotoxic effect on the HS-5 cells, as evidenced by the correct organization of the actin cytoskeleton and the morphology of the cell nuclei (Fig. 8). However, in the case of the HS-5 cells maintained in the presence of both the colloidal and nanofilm GO–Ag NPs, significantly shrunken spherical cells and small fragments of genetic material, possibly apoptotic bodies, were observed.
 | ||
| Fig. 7 Confocal microscopy visualization of the actin cytoskeleton architecture of HFFF2 cells after 24-hour exposure to GO and GO–metallic NPs in colloid and nanofilm form. Blue, cell nuclei (DAPI) and red, actin cytoskeleton (Alexa Fluor 633 phalloidin). Scale: 80 μm. | ||
 | ||
| Fig. 8 Organization of the actin cytoskeleton of HS-5 cells visualized by confocal microscopy after 24-hour treatment with GO and GO–metallic NPs in colloidal and nanofilm form. Blue, cell nuclei (DAPI) and red, actin cytoskeleton (Alexa Fluor 633 phalloidin). Scale: 100 μm. | ||
In this experiment, we demonstrated the effect of nanostructures on the cytoskeleton, which plays an important role in cellular adhesion, and simultaneously disturbances in its architecture may indicate the cytotoxicity of nanocomplexes. Actin filaments act as a mechanosensor of the mechanical forces of cells, including tensile forces;51 therefore, the composition of the actin cytoskeleton was also investigated in this study. Microscopic analysis of the actin cytoskeleton showed differences in the actin filament architecture depending on the treatment approach (Fig. 7 and 8). GO nanoflakes can bind directly to actin filaments, increase the distance between actin–actin subunits, and consequently increase the cell stiffness, and thus the possibility of cell migration.52 GO nanosheets can be internalized by cells by localizing to F-actin filaments, affecting the cytoskeletal fluctuations and rheology, resulting in the induction of cell cycle changes, the induction of oxidative stress, and ultimately cell death.53 Moreover, colloidal GO may alter the dynamics of the cytoskeleton and cell membrane by attenuating the transduction of integrin-FAK-Rho-ROCK signaling.54 However, GO modulates the assembly kinetics of actin filaments in a concentration- and application-dependent manner, making cells grown on the GO surface more elongated and spread out compared to the cells treated with GO nanoflakes accompanied by enhanced actin polymerization.55
The visualization of the cells using SEM enabled the assessment of the morphology of the organelles actively involved in the process of cell adhesion and attachment to the substrate, i.e., filopodia and lamellipodia, as well as the general morphology of the cell and the structure of the cell membrane (Fig. 9). The HFFF2 cells in contact with GO, GO–Au NP, and GO–Cu NP nanofilms achieved a slightly more three-dimensional and convex structure of the cell soma compared to the control cells, while being firmly attached to the substrate. The cells developed a more extensive network of elongated and branched filopodia, growing directly in front of the lamellipodium compared to the cells on the uncoated surfaces as well as short microspikes protruding from the side edges of the cell, independent of the lamellipodium. In addition, in the case of the cells treated with the GO–Au NP nanofilm, few long protrusions in direct contact with the substrate were visible. The HFFF2 cells on the GO–Ag NP nanofilms showed significant membrane permeabilization, cell soma shrinkage, apoptotic bodies, and vesicles typical of apoptotic cells. The HS-5 cells cultivated on the GO, GO–Au NP, and GO–Cu NP nanofilms showed a similar morphology to the control cells. However, only the GO–Au NP nanofilms contributed to the formation of a dense network of chaotically arranged filopodia by the cells. The HS-5 cells on the GO–Ag NP nanofilm exhibited a change in morphology from the characteristic spindle-shaped to spherical and were additionally surrounded by apoptotic bodies and blebs in their vicinity.
 | ||
| Fig. 9 Evaluation of the adhesion of HFFF2 and HS-5 cells to GO and GO–metallic NP nanofilms after 24 h incubation. The arrows indicate filopodia (white), lamellipodia (green), and apoptotic bodies (red). Scale: 20 μm. | ||
Microscopic analysis of the cell coverage of the nanofilms demonstrated increased coverage areas for the GO–Au NP and GO–Cu NP nanofilms by HFFF2 cells at the 30 min time point compared to the control (Fig. 10a). At 60 min, a greater coverage area of the HFFF2 cells on the GO–Cu NP nanofilm was observed, while a reduction in the coverage area was observed for the cells cultivated on the GO–Au NP nanofilm at 120 and 240 min. A significantly increased coverage area by the HFFF2 cells was also noted for the GO–Au NP nanofilm after 240 min. In the case of the HS-5 cells, at 30 min, a reduction in the area covered by cells on the GO–Ag NP nanofilm was demonstrated (Fig. 10b). Consequently, an increase in the coverage area was demonstrated for the cells cultivated on the GO (60 min), GO–Au NP (120 min), and GO–Cu NP (60 min, 120 min, and 240 min) nanofilms.
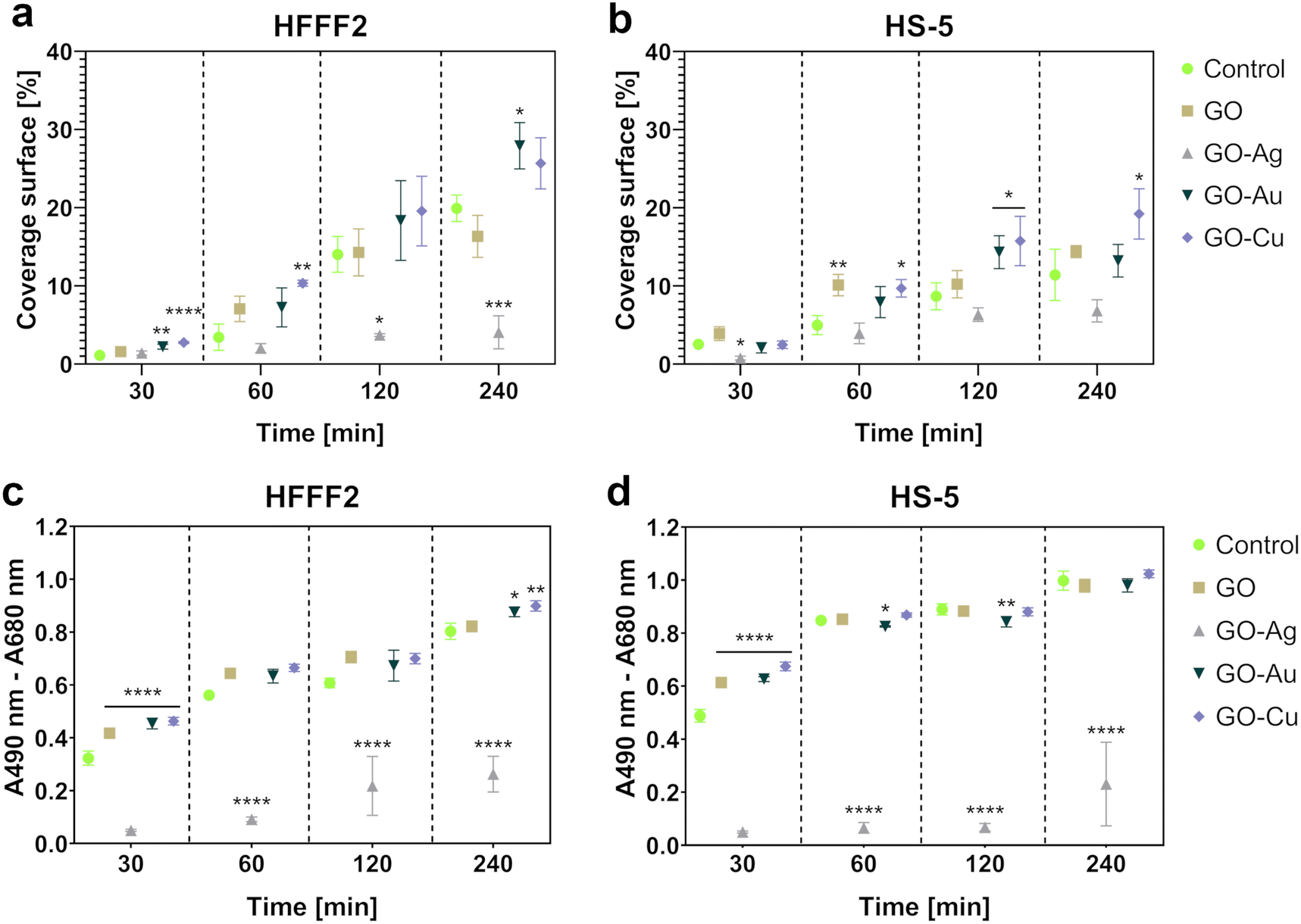 | ||
| Fig. 10 The adhesion assessment of HFFF2 (a and c) and HS-5 (b and d) cells to GO and GO–metallic NP nanofilms determined based on microscopic analysis (a and b) and total amount of intracellular LDH (c and d) at 30, 60, 120, and 240 minutes. The microscopic analysis results are expressed as the mean % coverage (n = 3, ±SD) of the total visual area (100%), while the total LDH assay results are expressed as the mean (n = 5, ±SD) of the spectrophotometric reading (A490 nm, ref A680 nm). Statistically significant differences between the control and study groups within a specific time point are marked as *p < 0.0332, **p < 0.0021, ***p < 0.0002, and ****p < 0.0001. Bar color markings: green, control; beige, GO; gray, GO–Ag NPs; dark green, GO–Au NPs; and purple, GO–Cu NPs. | ||
The LDH assay (Fig. 10c and d) complemented the microscopic analysis and expressed the adhesive properties of the nanofilms by the amount of intracellular LDH of the cells attached to the nanofilms after a series of washes. In contrast to the previously performed LDH assay, this variant assessed the intracellular LDH level of the cells attached to the growth surface, and subsequently lysed instead of the amount of LDH released extracellularly. Nanofilms, excluding the GO–Ag NP nanofilm, enhanced early HFFF2 cell adhesion within 30 min. Subsequently, the GO–Ag NP nanofilms contributed to a marked reduction in HFFF2 cell adhesion at all time points compared to the controls. HFFF2 cell adhesion was supported by the GO–Au NP and GO–Cu NP nanofilms at 240 min. A similar trend was observed for the HS-5 cells. Early adhesion of HS-5 cells at the 30 min time point was supported on the nanofilms, excluding the GO–Ag NP nanofilm. A significant decrease in adhesion was observed for the HS-5 cells on the GO–Ag NP nanofilms at 60, 120, and 240 min, and a slight reduction compared to the control was noted on the GO–Au NP nanofilm at 60 and 120 min.
May-Grünwald Giemsa cell staining at a specific time point demonstrated cell morphological differences depending on the adhesion time as well as the nanofilm constituting the growth surface. After 30 min, increased surface area and flattening within a single cell were observed for the HFFF2 (Fig. 11) cell line grown on nanofilms compared to the control cells. Moreover, sprouting filopodia were observed on the GO–Au NP and GO–Cu NP nanofilms. At 60 min, a high similarity of cells in all the study groups was observed in terms of flattening and general morphology. However, relatively more cells remained on the nanofilms after washing. After 120 min, the cells cultivated on the GO, GO–Au NP, and GO–Cu NP nanofilms developed longer and more robust networks of filopodia compared to the control cells, while after 240 min, the cells acquired an elongated and spindle-shaped morphology characteristic for the HFFF2 cell line. In the cells on the GO–Ag NP nanofilms, initiation of the vacuolization process was observed after 60 min, progressing at successive time points, accompanied by a significant change in morphology, and finally the shrinkage of the cells to achieve a spherical or irregular shape with sprouting apoptopodia, regular or beaded, at 120 and 240 min.
 | ||
| Fig. 11 The evaluation of the overall morphology and adhesion of HFFF2 cells to GO and GO–metallic NP nanofilms at 30, 60, 120, and 240 minutes. May-Grünwald Giemsa staining. Scale: 50 μm. | ||
The GO and GO–Cu NP nanofilms elicited a cellular response, and subsequently managed HS-5 cell adhesion (Fig. 12) in a similar manner to the HFFF2 cells. In the early phase of adhesion, the HS-5 cells on the GO, GO–Au NP, and GO–Cu NP nanofilms were more flattened (30 min and 60 min) and equipped with sprouting filopodia (60 min) in contrast to the control cells. Shrunken and unexpanded HS-5 cells were observed on the GO–Ag NP nanofilm at the 30 min and 60 min time points. After 120 min, the cells on the GO, GO–Au NP, and GO–Cu NP nanofilms assumed the spindle-shaped morphology typical of the HS-5 cell line, while the cells on the uncoated surface finally reached the correct morphology after only 240 min. At 120 min, the HS-5 cells were subtly flattened, thus revealing progressive vacuolization. Moreover, the morphology of the cells was significantly disturbed and different compared to the control cells.
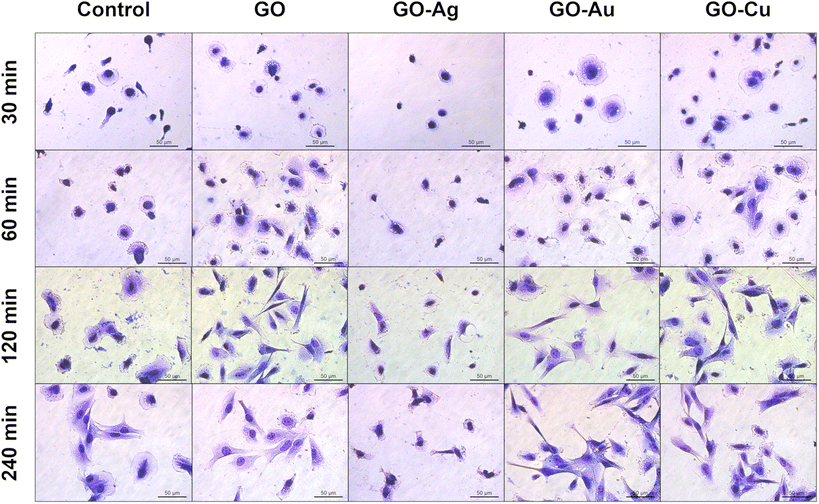 | ||
| Fig. 12 The general morphology and adhesion of HS-5 cells to GO and GO–metallic NP nanofilms at 30, 60, 120, and 240 minutes. May-Grünwald Giemsa staining. Scale: 50 μm. | ||
In this study, we proved the proadhesive effect of the surfaces formed from GO nanocomposites. Given that the main function of the coatings is to manage and enhance cell adhesion and develop an effective cell–surface biointerface, several tests were performed to assess the cell adhesion to GO and GO–metallic NPs. The in vitro analyses demonstrated the proadhesive properties of the GO, GO–Au NP, and GO–Cu NP nanofilms. As presented in Fig. 10 and 11, the GO–Au NP and GO–Cu NP nanofilms enhanced the cell adhesion to a greater extent compared to the pure GO nanofilm, especially within the first hour after cell seeding. SEM visualization of the cells cultivated on the GO–Au NP and GO–Cu NP nanofilms revealed the formation of numerous branched filopodia (Fig. 9), which are responsible for, among others, cell migration and binding to the substrate. Moreover, on GO nanofilms enriched with Au NPs and Cu NPs, a greater cell coverage area and increased number of cells were observed (Fig. 10a and b). Decorating GO with metallic NPs led to an increase in surface hydrophilicity (Fig. 3d), suggesting enhanced cell adhesion driven by increased wettability. Many studies prove the proadhesive properties of GO by, for example, increasing the degree of cell flattening56 and the development of cell cytoplasmic protrusions, including filopodia.57
The expression of genes related to the process of cell adhesion was determined using the real-time PCR technique (Fig. 13). The research group treated with the GO–Ag NP nanofilm was excluded from the analysis given its high cytotoxicity, and thus lack of proadhesion properties.
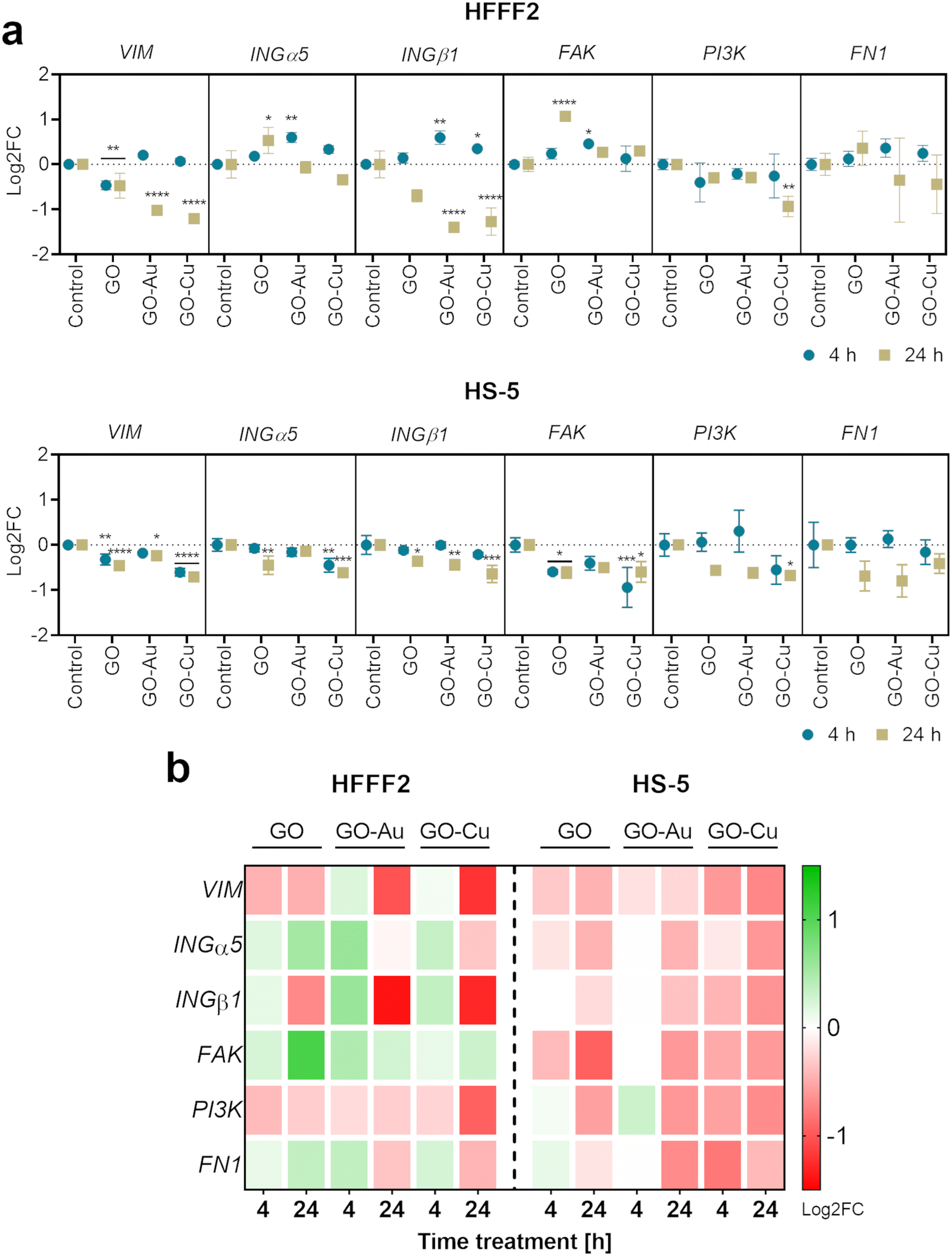 | ||
| Fig. 13 The analysis of gene expression at the mRNA level in HFFF2 and HS-5 cells 4 and 24 h after treatment with GO, GO–Au NP, and GO–Cu NP surfaces. The real-time PCR technique was used to determine the expression of the following genes: VIM (vimentin), INGα5 (alpha-5 integrin), INGβ1 (beta-1 integrin), FAK, PI3K (phosphoinositide 3-kinase), and FN1 (fibronectin). The points in the graphs (a) represent the mean with deviation (n = 3), and the treatment time is indicated as follows: blue dot = 4 hours; beige square = 24 hours. The results are presented as log2FC, where log2FC > 0 indicates upregulation and log2FC < 0 downregulation relative to the controls (log2FC = 0) within a single gene. Statistically significant differences between the control and study groups are marked as *p < 0.0332, **p < 0.0021, ***p < 0.0002, and ****p < 0.0001. The columns and rows of the heat map (b) represent the samples and genes, respectively, while the colors indicate the fold changes in gene expression (green = upregulation; red = downregulation). | ||
The analysis demonstrated vimentin (VIM) downregulation for HFFF2 cells on the nanofilms after 24 h, and additionally for cells on the GO nanofilm after 4 h. In the HS-5 cells on the nanofilms, VIM downregulation was also observed at both time points except for the cells on the GO–Au NP nanofilm at 4 h.
Vimentin maintains the proper shape of the cell, anchors the organelles in the cytosol, stabilizes the cytoskeleton, and is responsible for maintaining appropriate cell elasticity. Moreover, vimentin modulates cell mechanosensing and influences the cell shape and intracellular tension. The rearrangement of vimentin fibers can occur in response to surface topography.58 Vimentin translocation to the plasma membrane area results in enhanced integrin–ligand interactions by inducing changes in the clustering of integrins mediated by vimentin heads.59 However, the overexpression of vimentin heads is not a favorable phenomenon given the possibility of integrin cleavage from the vimentin fibers, and consequently impaired cell adhesion. Thus, vimentin is an important molecular link in mechanotransduction given that it is responsible for maintaining the mechanical homeostasis of the cell in response to the physicochemical properties of the growth surface. In this study (Fig. 13), after 4 h cell cultivation on nanofilms, no differences in VIM expression were observed except for a slight downregulation for the cells seeded on the pure GO nanofilm. In contrast, after 24 h treatment, significant downregulation of VIM was observed by the cells, especially those grown on the nanofilms decorated with metallic NPs. It is assumed that the cells, in response to the increased roughness of the substrate, changed their elasticity as a result of changes in the VIM levels. Generally, it is believed that the downregulation or mutations of VIM reduce the migratory capacity of cells.60,61 However, considerations apply to cells cultivated in two-dimensional planar surfaces. Surfaces made of GO nanoflakes and their nanocomposites gain not only roughness but also three-dimensionality. Actively migrating cells cultivated on two-dimensional surfaces upregulate VIM, whereas when cells are maintained on three-dimensional surfaces and spatial layouts, the downregulation of VIM is accelerated and cell migration processes promoted,60 and consequently may promote wound healing processes.
The expression of INGα5 and INGβ1 was shown to be upregulated within 4 h in the HFFF2 cells on the GO–Au NP and GO–Cu NP nanofilms. However, after 24 h, the expression returned to the control level (INGα5) or was downregulated (INGβ1). In the case of the HS-5 cells, INGα5 was downregulated for cells on the GO (4 h) and GO–Cu NP (4 h and 24 h) nanofilms, and INGβ1 was downregulated for the cells on all the nanofilms after 24 h.
The α5β1 integrin (ING α5β1) is the primary fibronectin (FN) transmembrane receptor facilitating the formation of the cell–surface junction, and thus participating in cell–surface-mediated signaling. In our study (Fig. 13), in the case of HFFF2 cells cultivated on nanofilms enriched with metallic NPs, a trend was observed consisting of the increased expression of integrins (INGs) at the early adhesion stage (4 h), while after 24 h, the expression was normalized (ING α5) or downregulated (ING β1). ING β1 interacts directly with vimentin, which enables the binding of heterodimer α5β1 to FN.62 Vimentin regulates the assembly of INGs and their depletion reduces the clustering of the ING α5β1 heterodimer.63 However, INGs do not lose their activity because of other molecular sources of activation stimulus, such as talin, kindlin,64 and protease-activated receptors.65 Moreover, the downregulation or knockout of vimentin results in an enhancement in the integrin-driven adhesion machinery,66,67 which is consistent with our results by the downregulation of vimentin with the simultaneous upregulation of INGs in the HFFF2 cells cultivated on GO–Au NP and GO–Cu NP surfaces in the early phase of adhesion (after 4 h).
The upregulated expression of FAK was observed in the HFFF2 cells cultured on the GO and GO–Au NP nanofilms for 24 h and 4 h, respectively. However, FAK downregulation was demonstrated in the HS-5 cells on the GO and GO–Cu NP nanofilms at both time points. Downregulation of PI3K expression was observed for both cell lines on the GO–Cu NP nanofilm over 24 h. No changes in FN1 expression were observed for both cell lines.
FAK is a protein involved in the adhesion, migration, cell cycle regulation, and phosphorylation of cytoskeletal proteins. The primary function of FAK is to launch an intracellular cascade of events, leading to the formation of focal adhesions via ING β1, which is a direct recruiter of the FAK protein.68 The FAK–Src–PI3K (phosphoinositide 3-kinase) pathway regulates Hippo signaling, which integrates extracellular signals with intracellular control, thereby regulating cell proliferation and adhesion during organ development and wound healing.69 Maintaining the balanced expression of FAK and PI3K seems to be crucial in the early adhesion stages given the fact that they link INGs with FN,70–72 allowing for the appropriate allocation and modeling of the FN matrix, which is a native cell scaffold. The equilibrium of cell signaling consists of promoting the adhesion of cells to the growth surface in the early stages of adhesion, and then normalizing the process as further promotion contributes to the formation of physiologically non-functional tissues such as keloids or fibrosis, which may be the result of FN overexpression.73,74 This trend was maintained only in the case of HFFF2 cells cultivated on the surface of GO–Au NPs (Fig. 13), which turned out to be a platform that manages biological processes in the most appropriate way.
According to the previous analyses, the GO–Au NP nanofilm was found to be the most biocompatible and proadhesive nanofilm derived from the GO–metallic nanocomplex; therefore, it was selected for further research and compared to the control (uncoated surface) and the GO nanofilm not decorated with metallic NPs. To assess the cell stiffness expressed as the modulus of elasticity depending on the adhesion surface, AFM analysis was performed (Fig. 14). Overall, an increase in cell stiffness was observed for both cell lines, which followed the order of control < GO < GO–Au NPs. The average modulus of elasticity of the HFFF2 cells was 5.1 kPa, 6.6 kPa, and 10.4 kPa for the cells on the uncoated surface (control), GO nanofilms, and GO–Au NPs, while for the HS-5 cells, the average modulus of elasticity was 3.1 kPa, 3.2 kPa, and 4.9 kPa for the uncoated surface nanofilms, GO, and GO–Au NPs, respectively. Characteristic higher values of the modulus of elasticity were observed, moving the average upward for the HFFF2 cells on the GO nanofilm, with an even greater increase for the GO–Au NP nanofilm. A highly heterogeneous structure in terms of stiffness was observed in the HFFF2 cells, especially in the case of the cells grown on the GO–Au NP nanofilm.
 | ||
| Fig. 14 Atomic force microscopy three-dimensional map of elastic modulus (a) and distribution of elastic modulus (b) for HFFF2 and HS-5 cells on the uncoated surface (control), GO, and GO–Au NP nanofilms after 24 hours. The dots represent a single cell puncture point by the microlever and the black dashed lines indicate the median. Cells on the following growth surfaces are color-coded: green – uncoated surface; beige = GO nanofilm; and dark green = GO–Au NP nanofilm. | ||
3. Experimental
3.1. Nanomaterials
:9 ratio (v/v) with culture medium to final concentrations of 3.125, 6.25, 12.5, 25, 50, 100, and 200 μg mL−1. To produce the nanofilms, solutions of 10× concentrated GO were combined with ultrapure water in a ratio of 1:4 (v/v) in such a way that the same mass of GO was found in the corresponding samples regardless of the cell treatment approach used. Drying in sterile conditions of the nanofilms was carried out under a laminar chamber until complete evaporation of the solvent and completely dry nanolayers were obtained. The water-suspended nanostructure concentrates were sonicated prior to every experiment.
:1 ratio (v/v) to obtain final concentrations of 5:10 μg mL−1 and 50:10 μg mL−1 (5 μg mL−1 and 50 μg mL−1 for GO and 10 μg for the metallic NPs). The nanofilms composed of nanocomplexes were formed by diluting concentrated solutions of nanocomplexes in a 1:1 ratio (v/v) with ultrapure water and drying as for pure GO.
3.1.3.1. Nanoflakes. An evaluation of the size distribution and stability of the hydrocolloid was performed for selected concentrations of nanostructures, i.e., 50 μg mL−1 for GO and 50
:10 μg mL−1 for GO–metallic NP nanocomplexes. Zeta potential measurements were performed by microelectrophoresis with Smoluchowski approximation and size distribution measurements by dynamic light scattering (DLS) using a Zetasizer Nano-ZS90 analyzer (Malvern Instruments, Worcestershire, UK) after stabilization of the colloids for 120 s at 25 °C in triplicate. The measurements were performed in two media to determine the physicochemical behaviour of the nanostructures during in vitro assays, i.e., ultrapure water and a culture medium supplemented with 10% fetal bovine serum (FBS) at two time points (0 and 24 h). The size of the nanoflakes was determined using a high-resolution scanning electron microscope (SEM) (Quanta FEG 250). Briefly, 2.5 μL of GO–metallic NP solution at a concentration of 50:10 μg mL−1 was applied to a cover slip and dried. Then, visualization with a beam voltage of 5 kV was performed. Measurements were performed for 50 nanoflakes from at least 3 images using the ImageJ software (Eliceiri/LOCI lab, Madison, USA).
To detect the chemical bonds present in the studied nanostructures, infrared spectra were recorded with the use of the Fourier transform technique (FT-IR). A PerkinElmer System 2000 spectrometer (PerkinElmer Inc., Waltham, MA, USA) was used as the working instrument. The samples were dried and thoroughly mixed with KBr crystals (ratio 1:300 m/m) for 5 min in a laboratory mill to obtain a fine powder. Then, using a pellet-maker and a 5-ton hydraulic press, transparent pellets with a diameter of 13 mm were prepared. The pellets were placed into a dedicated holder that in turn was placed in the spectrometer measuring chamber and radiated with infrared radiation in the spectral range of 4000–400 cm−1 to collect spectral data. The measurements were preceded by the collection of the background spectrum containing the bands generated by some vibrations, translations and rotations of gaseous present, e.g. CO2 and H2O. Every spectrum was recorded with 4 cm−1 resolution and 25 scans. Raw spectra were used for statistical analysis.
3.1.3.2. Nanofilms. The assessment of the morphology of the GO–metallic NP surfaces was performed using a scanning electron microscope (SEM) (Quanta 200, FEI, Hillsboro, OR, USA). A 50
:10 μg mL−1 solution of GO–metallic NPs was applied to a 12-well plate (500 μL). After complete drying, the nanostructures were visualized with a beam voltage of 20 kV.
The assessment of the topography and surface formation of the nanofilms was analyzed via atomic force microscopy (AFM) (Asylum Research, MFP3D Bio, Santa Barbara, CA, USA). AFM imaging was performed in air and AC mode using a commercial triangular cantilever (MLCT Bruker, Camarillo, CA, USA), with a spring constant of k = 0.10 N m−1. The area of the mapped surfaces was 5 μm × 5 μm.
The contact angle (CA) measurement and determination of the hydrophilic nature of the nanofilms, derived as in the case of SEM analysis, were performed using an OCA15 goniometer (Data Physics Instruments GmbH, Filderstadt, Germany). Measurements were carried out using the sessile drop method directly after a drop of deionized water (1 μL) came into contact with the nanofilm. For each sample, 10 measurements were performed at room temperature.
3.2. In vitro cell cultures
:10 μg mL−1 and 50:10 μg mL−1) of GO–metallic NP nanocomplexes were selected to determine their effect on the cellular response. After cytotoxicity screening, the concentrations of 50 μg mL−1 for GO and 50:10 μg mL−1 for GO–metallic NPs were selected for the remaining tests.
3.3. Cytotoxicity screening panel
Given that cytotoxicity assays often rely on different cellular mechanisms at the molecular level or within individual organelles, cytotoxicity screening was performed with three different assays to provide a complete description of the cytotoxicity of the tested nanomaterials including neutral red uptake, MTT mitochondrial activity, and lactate dehydrogenase (LDH) leakage assays.:1 (v/v) per well. Previously, the prepared neutral red stock solution was centrifuged (600g, 10 min), and the obtained supernatant was additionally filtered and heated to 37 °C before being added to the cells. Subsequently, after 3 h of incubation at 37 °C, the medium was discarded, and the cells were rinsed with phosphate buffered saline (PBS), and finally dissolved in 100 μL of a solubilization buffer (ethanol, acetic acid, and ultrapure water at a ratio of 50:1:49 [v/v]) per well for 20 min at room temperature on an orbital shaker. Prior to spectrophotometric reading, the plates were centrifuged (250g, 10 min), and then the supernatant was transferred to clean multi-well plates to avoid the falsification of the results by nanofilms. Spectrophotometric measurement was performed using an Infinite®200 PRO microplate reader with i-control™ software (Tecan Group Ltd, Germany) at a wavelength of 540 nm. Four repetitions were performed for each group. The results were expressed as the mean percentage of the test sample relative to the mean control (100%).
| Cytotoxicity [%] = (exp. value)/(high control) × 100 |
3.4. Visualization of cells
3.5. Adhesion assessment
The effect of the GO–metallic NP nanofilms on cell adhesion was evaluated by measuring the surface coverage of the nanofilms by the cells and the total LDH leakage assay. The cells were plated in 12-well (coverage area measurements) and 96-well (LDH assay) plates coated with nanofilms and incubated at 4 time points of 30, 60, 120, and 240 min. The last time point was chosen as the time needed to achieve correct morphology and flattening of the control cells. In addition, real-time PCR analysis was performed to assess the expression of genes related to the adhesion and cellular cytoskeleton.3.5.3.1. RNA isolation. Total RNA was isolated using the PureLink® RNa Mini Kit (ThermoFisher Scientific, Wilmington, DE, USA). The cells were seeded on a nanofilm-coated 6-well plate incubated for 4 h or 24 h. Next, the cells were washed with warm PBS, and then trypsinized and neutralized with DMEM supplemented with 10% FBS. The detached cells were centrifuged (5 min, 300g), and the resulting pellet was rinsed twice with PBS. Afterward, 300 μL of lysis buffer containing 1% β-mercaptoethanol was added to the pellet and lysed for 15 min at room temperature with occasional vortexing. The lysate was centrifuged (10 min, 12
000g, 4 °C) and the supernatant was transferred to a clean tube with one volume of 70% ethanol. The remaining stages of isolation were carried out in accordance with the protocol provided by the manufacturer. Finally, the elution of total RNA was performed in 30 μL of RNase-free water. The concentration of the obtained RNA was measured using a NanoDrop 2000 (Thermo Fisher Scientific, Wilmington, DE, USA), equalized, and stored at −80 °C. At the stage of RNA isolation, the groups treated with GO–Ag NP nanofilms were excluded because of the inability to isolate a sufficient amount of genetic material.
3.5.3.2. RT-PCR. The cDNA High Capacity Reverse Transcription Kit (AppliedBiosystem, Foster City, Ca, USA) was used for the reverse transcription of RNA to derive cDNA via reverse transcriptase polymerase chain reaction (RT-PCR) according to the manufacturer's protocol. A total of 500 ng of RNA was used for each reaction. The samples were placed in a 2720 Thermal Cycler (Thermo Fisher Scientific, USA), and the temperature profile was set as 25 °C (10 min), 37 °C (120 min), and 4 °C (5 min). The cDNA concentration was measured on the NanoDrop instrument and adjusted to 10 ng μL−1 in RNase-free water. The samples were stored at −20 °C.
3.5.3.3. Real-time PCR. Relative gene expression at the mRNA level was performed using real-time quantitative PCR and the ΔΔCT method for the following human genes: vimentin (VIM), α5 integrin (ITGA5), β1 integrin (ITGB1), focal adhesion kinase (FAK), phosphoinositide 3-kinase (PI3K), and fibronectin (FN1) (Table 1). Glyceraldehyde-3-phosphate dehydrogenase (GADPH) was used as the housekeeping gene (endogenous control). The reaction mix consisted of 5 μL of PowerUp SYBR Green Master Mix (Applied Biosystem, Waltham, MA, USA), 5 μL 50 ng cDNA template, 3.5 μL of RNase-free water, and 0.75 μL 10 μM forward and reverse primer (Genomed, Warsaw, Poland). Finally, 15 μL of the reaction mix was dropped per well on a MicroAmp™ Fast Optical 48-Well Reaction Plate (Applied Biosystems, Waltham, MA, USA). The reactions for each treatment group were performed in triplicate. The reaction was performed on the StepOnePlus™ Real-Time PCR System with the following temperature profile: 95 °C (10 min), followed by 40 cycles of 95 °C (15 s) and 60 °C (60 s). Relative gene expression was calculated according to the 2−ΔΔCT method, where ΔΔCT = ΔCT of the target gene −ΔCT of calibrator (untreated sample) and ΔCT = mean CT of the target gene – mean CT of endogenous control (GADPH). The results were normalized to the calibrator.
| Gene | Sequence of primer 5′→3′ | Amplicon size [bp] | GenBank accession number |
|---|---|---|---|
| VIM | F: CCTCACCTGTGAAGTGGATGC | 112 | NC_000010.11 |
| R: CAACGGCAAAGTTCTCTTCCA | |||
| INGα5 | F: GCCGATTCACATCGCTCTCAAC | 139 | NM_002205.5 |
| R: GTCTTCTCCACAGTCCAGCAAG | |||
| INGβ1 | F: GGATTCTCCAGAAGGTGGTTTCG | 143 | NM_133376.3 |
| R: TGCCACCAAGTTTCCCATCTCC | |||
| FAK | F: CCCACCAGAGGAGTATGTCC | 150 | XM_017013688 |
| R: CCCAGGTCAGAGTTCAATAG | |||
| PI3K | F: GGTTGTCTGTCAATCGGTGACTGT | 108 | NG_012113.2 |
| R: GAACTGCAGTGCACCTTTCAAGC | |||
| FN1 | F: ACAACACCGAGGTGACTGAGAC | 143 | NG_012196.2 |
| R: GGACACAACGATGCTTCCTGAG | |||
| GADPH | F: GAGAAGGCTGGGGCTCATTTG | 97 | NM_002046 |
| R: CATGGTTCACACCCATG |
3.6. Mechanical measurements of cells
Cell stiffness expressed as elastic modulus (kPa) was determined using AFM. The tests were performed for cells grown on GO and GO–Au NP nanofilms. Conical cantilevers (Applied NanoStructures; height of 4–6 μm) were used for the measurements. The angle of inclination of the cantilever sidewalls was α ∼ 20°, the frequency was 17 kHz, and the nominal spring constant was 35 ph nm−1. To determine the spring constant and the sensitivity value of the optical lever, the probes were calibrated before the measurement using the thermal vibration method, in accordance with the algorithm provided by the microscope manufacturer. The probe deflection threshold was set at 0.25 V. AFM was set to operate in contact mode. The measurement was performed by indenting the cell at a fixed threshold. The indentation speed was fixed at 1 μm s−1. The assumed cut-off point guaranteed that the indentation depth was between 1 μm and 2 μm. The recoil distance of the probe was set to 2 μm. Cell maps were made of 45 rows × 44 columns of punctures (total 1980 F–D curves per cell). The Hertz model for conical indenters was used to analyze the curves.3.7. Statistical analysis
The significance of differences among the groups was verified through one-way analysis of variance (ANOVA) using Tukey's post-hoc test in GraphPad Prism 9.1.2 (GraphPad Software, San Diego, California USA) and Statistica 13.3 (StatSoft, Poland) software. Results with p < 0.05 were considered statistically significantly different.4. Conclusions
Our results demonstrated the modulation of cell mechanosensing in a nanotopographically dependent manner, thereby altering the biocompatibility and adhesive properties of nanofilms. GO decorated with gold NPs was revealed to be the most biocompatible surface, not disturbing the cell metabolism and morphology, and a proadhesive nanoplatform by accelerating the formation of a cell–surface junction guided by the temporary upregulation of integrin expression. In this study, we demonstrated the acceleration of cell flattening and the formation of an advanced network of filopodia by cells cultivated on the surface of GO with gold NPs as well as reduced cell elasticity and downregulation of vimentins. Overall, our results support the use of gold NP-enriched GO nanocomposites as surface nano-structuring agent. Subsequent advanced studies will focus on incorporating this nanocomplex into more sophisticated biomaterials and determining their molecular and physiological mechanisms as well as biointeraction with cellular and tissue models.Author contributions
Conceptualization – MP, BSC, ES, MK; data curation – MP; formal analysis – MP, BSC, PK; funding acquisition – BSC, SJ; investigation – MP, BSC, MSŁ, AO, MŁ, PN, PK; methodology – MP, BSC, MSŁ, MK; project administration – MP; resources – MP; software – MP, BSC; supervision – BSC, ES; validation – MP, BSC, ES; visualization – MP; writing – original draft – MP, MŁ (AFM), PK (FT-IR); writing – review & editing – BSC, MP, MSŁ. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The manuscript is part of the PhD thesis of Michał Pruchniewski.References
- H. Du, J. M. Bartleson, S. Butenko, V. Alonso, W. F. Liu, D. A. Winer and M. J. Butte, Nat. Rev. Immunol., 2023, 23, 174–188 CrossRef CAS.
- P. Romani, L. Valcarcel-Jimenez, C. Frezza and S. Dupont, Nat. Rev. Mol. Cell Biol., 2021, 22, 22–38 CrossRef CAS.
- P. Han, T. Guo, A. Jayasree, G. A. Gomez, K. Gulati and S. Ivanovski, Nano Res., 2023, 1–11 Search PubMed.
- A. Zhang, J. Fang, X. Li, J. Wang, M. Chen, H. J. Chen, G. He and X. Xie, Nanoscale Adv., 2022, 4, 1844–1867 RSC.
- K. Modaresifar, M. Ganjian, P. J. Díaz-Payno, M. Klimopoulou, M. Koedam, B. C. J. van der Eerden, L. E. Fratila-Apachitei and A. A. Zadpoor, Mater. Today Bio, 2022, 16, 100448 CrossRef CAS PubMed.
- M. A. Schwartz and D. W. DeSimone, Curr. Opin. Cell Biol., 2008, 20, 551–556 CrossRef CAS PubMed.
- F. Martino, A. R. Perestrelo, V. Vinarský, S. Pagliari and G. Forte, Front. Physiol., 2018, 9, 824 CrossRef PubMed.
- C. Zhou, M. Duan, D. Guo, X. Du, D. Zhang and J. Xie, Int. J. Oral Sci., 2022, 14, 1–10 CrossRef CAS PubMed.
- E. Maffioli, A. Galli, S. Nonnis, A. Marku, A. Negri, C. Piazzoni, P. Milani, C. Lenardi, C. Perego and G. Tedeschi, Front. Cell Dev. Biol., 2020, 8, 508 CrossRef PubMed.
- M. Yuan, P. J. Pai, X. Liu, H. Lam and B. P. Chan, Sci. Rep., 2018, 8, 1–15 Search PubMed.
- M. Wierzbicki, A. Hotowy, M. Kutwin, S. Jaworski, J. Bałaban, M. Sosnowska, B. Wójcik, A. Wędzińska, A. Chwalibog and E. Sawosz, Int. J. Mol. Sci., 2020, 21, 4173 CrossRef CAS PubMed.
- J. Bałaban, M. Wierzbicki, M. Zielińska, J. Szczepaniak, M. Sosnowska, K. Daniluk, D. Cysewski, P. Koczoń, A. Chwalibog and E. Sawosz, Molecules, 2020, 25, 1991 CrossRef PubMed.
- S. Bahrami, N. Baheiraei and M. Shahrezaee, Sci. Rep., 2021, 11, 1–10 CrossRef PubMed.
- J. M. Seok, G. Choe, S. J. Lee, M. A. Yoon, K. S. Kim, J. H. Lee, W. D. Kim, J. Y. Lee, K. Lee and S. A. Park, Mater. Des., 2021, 209, 109941 CrossRef CAS.
- V. Kheiri Mollaqasem, A. Asefnejad, M. R. Nourani, V. Goodarzi and M. R. Kalaee, J. Appl. Polym. Sci., 2021, 138, 49797 CrossRef CAS.
- F. Qi, C. Wang, S. Peng, C. Shuai, W. Yang and Z. Zhao, Mater. Chem. Front., 2021, 5, 2373–2386 RSC.
- W. Yuan, Y. Zhou, Y. Li, C. Li, H. Peng, J. Zhang, Z. Liu, L. Dai and G. Shi, Sci. Rep., 2013, 3, 1–7 Search PubMed.
- Z. Li, S. Xiang, Z. Lin, E. N. Li, H. Yagi, G. Cao, L. Yocum, L. Li, T. Hao, K. K. Bruce, M. R. Fritch, H. Hu, B. Wang, P. G. Alexander, K. A. Khor, R. S. Tuan and H. Lin, Biomaterials, 2021, 277, 121082 CrossRef CAS PubMed.
- H. Maleki-Ghaleh, M. Hossein Siadati, A. Fallah, A. Zarrabi, F. Afghah, B. Koc, E. Dalir Abdolahinia, Y. Omidi, J. Barar, A. Akbari-Fakhrabadi, Y. Beygi-Khosrowshahi and K. Adibkia, Chem. Eng. J., 2021, 426, 131321 CrossRef CAS.
- A. R. Unnithan, A. R. K. Sasikala, B. K. Shrestha, A. Lincoln, T. Thomson and A. J. El Haj, Adv. Funct. Mater., 2022, 32, 2201311 CrossRef CAS.
- A. O. Gee, B. M. Baker, A. M. Silverstein, G. Montero, J. L. Esterhai and R. L. Mauck, Cell Tissue Res., 2012, 347, 803–813 CrossRef CAS.
- H. Kihara, D. M. Kim, M. Nagai, T. Nojiri, S. Nagai, C. Y. Chen, C. Lee, W. Hatakeyama, H. Kondo and J. Da Silva, Int. J. Oral Sci., 2018, 10, 1–8 CrossRef CAS.
- H. Zhang and S. Hollister, J. Biomater. Sci., Polym. Ed., 2009, 20, 1975–1993 CrossRef CAS PubMed.
- S. Kang, J. Lee, S. Ryu, Y. Kwon, K. H. Kim, D. H. Jeong, S. R. Paik and B. S. Kim, Chem. Mater., 2017, 29, 3461–3476 CrossRef CAS.
- M. Kooti, A. N. Sedeh, H. Motamedi and S. E. Rezatofighi, Appl. Microbiol. Biotechnol., 2018, 102, 3607–3621 CrossRef CAS PubMed.
- G. Duan, Y. Zhang, B. Luan, J. K. Weber, R. W. Zhou, Z. Yang, L. Zhao, J. Xu, J. Luo and R. Zhou, Sci. Rep., 2017, 7, 1–12 CrossRef CAS PubMed.
- G. Duan, S. G. Kang, X. Tian, J. A. Garate, L. Zhao, C. Ge and R. Zhou, Nanoscale, 2015, 7, 15214–15224 RSC.
- Y. Li, H. Yuan, A. Von Dem Bussche, M. Creighton, R. H. Hurt, A. B. Kane and H. Gao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12295–12300 CrossRef CAS PubMed.
- S. Mittal, V. Kumar, N. Dhiman, L. K. S. Chauhan, R. Pasricha and A. K. Pandey, Sci. Rep., 2016, 6, 1–16 CrossRef PubMed.
- W. N. Zeng, Q. P. Yu, D. Wang, J. L. Liu, Q. J. Yang, Z. K. Zhou and Y. P. Zeng, J. Nanobiotechnol., 2021, 19, 1–19 CrossRef PubMed.
- X. Wang, W. Zhou, X. Li, J. Ren, G. Ji, J. Du, W. Tian, Q. Liu and A. Hao, J. Transl. Med., 2020, 18, 1–14 CrossRef PubMed.
- M. B. da Cruz, J. F. Marques, N. Silva, S. Madeira, Ó. Carvalho, F. S. Silva, J. Caramês and A. Mata, Materials, 2022, 15, 2481 CrossRef CAS PubMed.
- T. P. Kunzler, T. Drobek, M. Schuler and N. D. Spencer, Biomaterials, 2007, 28, 2175–2182 CrossRef CAS PubMed.
- X. Miao, D. Wang, L. Xu, J. Wang, D. Zeng, S. Lin, C. Huang, X. Liu and X. Jiang, Int. J. Nanomed., 2017, 12, 1415–1430 CrossRef CAS PubMed.
- Y. Hou, W. Xie, L. Yu, L. Cuellar Camacho, C. Nie, M. Zhang, R. Haag, Q. Wei, Y. Hou, L. Yu, L. C. Camacho, C. Nie, R. Haag, W. Xie, M. Zhang and Q. Wei, Small, 2020, 16, 1905422 CrossRef CAS PubMed.
- X. Lin, J. Jia, N. Yousefi, X. Shen and J. K. Kim, J. Mater. Chem. C, 2013, 1, 6869–6877 RSC.
- J. Meng, G. Yang, L. Liu, Y. Song, L. Jiang and S. Wang, Sci. China: Chem., 2017, 60, 614–620 CrossRef CAS.
- R. M. Visalakshan, M. N. Macgregor, S. Sasidharan, A. Ghazaryan, A. M. Mierczynska-Vasilev, S. Morsbach, V. Mailänder, K. Landfester, J. D. Hayball and K. Vasilev, ACS Appl. Mater. Interfaces, 2019, 11, 27615–27623 CrossRef CAS PubMed.
- C. Mücksch and H. M. Urbassek, Langmuir, 2016, 32, 9156–9162 CrossRef PubMed.
- F. Boccafoschi, M. Bosetti, P. M. Sandra, M. Leigheb and M. Cannas, Cell Adhes. Migr., 2010, 4, 19 CrossRef CAS PubMed.
- J. M. Northcott, I. S. Dean, J. K. Mouw and V. M. Weaver, Front. Cell Dev. Biol., 2018, 6, 17 CrossRef PubMed.
- U. Hansen and A. F. Thünemann, Langmuir, 2015, 31, 6842–6852 CrossRef CAS PubMed.
- S. Lara, F. Alnasser, E. Polo, D. Garry, M. C. Lo Giudice, D. R. Hristov, L. Rocks, A. Salvati, Y. Yan and K. A. Dawson, ACS Nano, 2017, 11, 1884–1893 CrossRef CAS PubMed.
- C. Greulich, J. Diendorf, T. Simon, G. Eggeler, M. Epple and M. Köller, Acta Biomater., 2011, 7, 347–354 CrossRef CAS PubMed.
- M. M. Rohde, C. M. Snyder, J. Sloop, S. R. Solst, G. L. Donati, D. R. Spitz, C. M. Furdui and R. Singh, Part. Fibre Toxicol., 2021, 18, 37 CrossRef CAS PubMed.
- J. S. Teodoro, A. M. Simões, F. V. Duarte, A. P. Rolo, R. C. Murdoch, S. M. Hussain and C. M. Palmeira, Toxicol. in Vitro, 2011, 25, 664–670 CrossRef CAS PubMed.
- J. Cui, Y. Yang, M. Zheng, Y. Liu, Y. Xiao, B. Lei and W. Chen, Mater. Res. Express, 2014, 1, 045007 CrossRef CAS.
- M. Wierzbicki, S. Jaworski, E. Sawosz, A. Jung, G. Gielerak, H. Jaremek, W. Łojkowski, B. Woźniak, L. Stobiński, A. Małolepszy and A. Chwalibog, Nanoscale Res. Lett., 2019, 14, 1–11 CrossRef CAS PubMed.
- R. Ahmadi and S. Izanloo, J. Mech. Behav. Biomed. Mater., 2022, 126, 105075 CrossRef CAS PubMed.
- M. Zielińska-Górska, E. Sawosz, M. Sosnowska, A. Hotowy, M. Grodzik, K. Górski, B. Strojny-Cieślak, M. Wierzbicki and A. Chwalibog, Pharmaceutics, 2022, 14, 1398 CrossRef PubMed.
- K. Ohashi, S. Fujiwara and K. Mizuno, J. Biochem., 2017, 161, 245–254 CAS.
- M. Ghorbani, H. Soleymani, H. Hashemzadeh, S. Mortezazadeh, M. Sedghi, S. Shojaeilangari, A. Allahverdi and H. Naderi-Manesh, Sci. Rep., 2021, 11, 1–13 CrossRef PubMed.
- M. C. Matesanz, M. Vila, M. J. Feito, J. Linares, G. Gonçalves, M. Vallet-Regi, P. A. A. P. Marques and M. T. Portolés, Biomaterials, 2013, 34, 1562–1569 CrossRef CAS PubMed.
- J. Zhu, M. Xu, M. Gao, Z. Zhang, Y. Xu, T. Xia and S. Liu, ACS Nano, 2017, 11, 2637–2651 CrossRef CAS PubMed.
- J. Park, P. Kravchuk, A. Krishnaprasad, T. Roy and E. H. Kang, Int. J. Mol. Sci., 2022, 23, 509 CrossRef CAS PubMed.
- J. T. Jeong, M. K. Choi, Y. Sim, J. T. Lim, G. S. Kim, M. J. Seong, J. H. Hyung, K. S. Kim, A. Umar and S. K. Lee, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- H. A. M. Ardoña, J. F. Zimmerman, K. Shani, S. H. Kim, F. Eweje, D. Bitounis, D. Parviz, E. Casalino, M. Strano, P. Demokritou and K. K. Parker, NanoImpact, 2022, 26, 100401 CrossRef PubMed.
- M. Swoger, S. Gupta, E. E. Charrier, M. Bates, H. Hehnly and A. E. Patteson, ACS Appl. Bio Mater., 2022, 5, 552–561 CrossRef CAS PubMed.
- J. Kim, C. Yang, E. J. Kim, J. Jang, S. J. Kim, S. M. Kang, M. G. Kim, H. Jung, D. Park and C. Kim, J. Cell Sci., 2016, 129, 2030–2042 CrossRef CAS PubMed.
- A. E. Patteson, K. Pogoda, F. J. Byfield, K. Mandal, Z. Ostrowska-Podhorodecka, E. E. Charrier, P. A. Galie, P. Deptuła, R. Bucki, C. A. McCulloch and P. A. Janmey, Small, 2019, 15, 1903180 CrossRef CAS PubMed.
- C. Velez-delValle, M. Marsch-Moreno, F. Castro-Muñozledo, I. J. Galván-Mendoza and W. Kuri-Harcuch, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- J. Kim, J. Jang, C. Yang, E. J. Kim, H. Jung and C. Kim, FEBS Lett., 2016, 590, 3517–3525 CrossRef CAS PubMed.
- Z. Ostrowska-Podhorodecka, I. Ding, W. Lee, J. Tanic, S. Abbasi, P. D. Arora, R. S. Liu, A. E. Patteson, P. A. Janmey and C. A. McCulloch, J. Cell Sci., 2021, 134, 1–16 CrossRef PubMed.
- Z. Sun, M. Costell and R. Fässler, Nat. Cell Biol., 2019, 21, 25–31 CrossRef CAS PubMed.
- P. M. Spoerri, N. Strohmeyer, Z. Sun, R. Fässler and D. J. Müller, Nat. Mater., 2020, 19, 218–226 CrossRef CAS PubMed.
- C. Dmello, S. Sawant, H. Alam, P. Gangadaran, R. Tiwari, H. Dongre, N. Rana, S. Barve, D. E. Costea, D. Chaukar, S. Kane, H. Pant and M. Vaidya, Int. J. Biochem. Cell Biol., 2016, 70, 161–172 CrossRef CAS PubMed.
- B. Langlois, E. Belozertseva, A. Parlakian, M. Bourhim, J. Gao-Li, J. Blanc, L. Tian, D. Coletti, C. Labat, Z. Ramdame-Cherif, P. Challande, V. Regnault, P. Lacolley and L. Zhenlin, Sci. Rep., 2017, 7, 1–15 CrossRef CAS PubMed.
- X. K. Zhao, Y. Cheng, M. Liang Cheng, L. Yu, M. Mu, H. Li, Y. Liu, B. Zhang, Y. Yao, H. Guo, R. Wang and Q. Zhang, Sci. Rep., 2016, 6, 1–12 CrossRef CAS PubMed.
- N. G. Kim and B. M. Gumbiner, J. Cell Biol., 2015, 210, 503 CrossRef CAS PubMed.
- M. Benito-Jardón, S. Klapproth, I. Gimeno-Lluch, T. Petzold, M. Bharadwaj, D. J. Müller, G. Zuchtriegel, C. A. Reichel and M. Costell, eLife, 2017, 6, e22264 CrossRef PubMed.
- D. Ilić, B. Kovačič, K. Johkura, D. D. Schlaepfer, N. Tomašević, Q. Han, J. B. Kim, K. Howerton, C. Baumbusch, N. Ogiwara, D. N. Streblow, J. A. Nelson, P. Dazin, Y. Shino, K. Sasaki and C. H. Damsky, J. Cell Sci., 2004, 117, 177–187 CrossRef PubMed.
- M. L. Matter and E. Ruoslahti, J. Biol. Chem., 2001, 276, 27757–27763 CrossRef CAS PubMed.
- R. M. Kelsh, P. J. McKeown-Longo and R. A. F. Clark, J. Invest. Dermatol., 2015, 135, 1714–1718 CrossRef CAS PubMed.
- I. Valiente-Alandi, S. J. Potter, A. M. Salvador, A. E. Schafer, T. Schips, F. Carrillo-Salinas, A. M. Gibson, M. L. Nieman, C. Perkins, M. A. Sargent, J. Huo, J. N. Lorenz, T. DeFalco, J. D. Molkentin, P. Alcaide and B. C. Blaxall, Circulation, 2018, 138, 1236–1252 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |