
Synthetic and computational investigation of neighboring group participation by a nucleophilic disulfide bond†
Takuhei
Yamamoto
 *ab,
Koki
Fukuta
a,
Yuki
Kariya
a,
Taiki
Matsuura
a,
Hiroaki
Hagiwara
c,
Bunji
Uno
d and
Yukihiro
Esaka
ab
*ab,
Koki
Fukuta
a,
Yuki
Kariya
a,
Taiki
Matsuura
a,
Hiroaki
Hagiwara
c,
Bunji
Uno
d and
Yukihiro
Esaka
ab
aGifu Pharmaceutical University, 1-25-4 Daigaku-nishi, Gifu 501-1196, Japan
bUnited Graduate School of Drug Discovery and Medical Information Science, Gifu University, 1-1 Yanaido, Gifu 501-1194, Japan
cDepartment of Chemistry, Faculty of Education, Gifu University, 1-1 Yanaido, Gifu 501-1193, Japan
dFaculty of Pharmacy, Gifu University of Medical Science, 4-3-3 Nijigaoka, Kani, Gifu 509-0923, Japan
First published on 18th November 2022
Abstract
Disulfide bonds of 2-isocyanatophenyl methyl disulfide and 2-endo-isocyanato-6-endo-(methyldisulfanyl)bicyclo[2.2.1]heptane showed neighboring group participation in the formation of thiocarbamates. Natural Bond Orbital (NBO) analyses revealed that the unusual nucleophilicity requires a rigid through-space interaction between a lone pair of the disulfide bond and an antibonding orbital of isocyanate.
The disulfide bond is known to be cleaved by nucleophiles such as hydrides,1 thiolates,2 and organometals.3 Concurrently, it indicates that lone pairs of the disulfide bond are relatively low in energy to be nucleophilic. It is partly because the lone pairs are stabilized by hyperconjugation with neighboring antibonding orbitals of the sulfur–carbon bond.4 Although nucleophilic reactions of the disulfide bond can occur with inorganic electrophiles such as Cl2,5 NaOCl,6 and SO2Cl2,7 little is known about the nucleophilicity of the disulfide bond toward organic electrophiles.
To overcome the low orbital energy and to gain good nucleophilicity, we hypothesized that it is necessary for a disulfide bond to have a significant orbital overlap with an antibonding orbital of an electrophile. According to the Klopman–Salem equation8 which describes chemical reactivity, the stabilizing energy, ΔE, for reactions between a nucleophile and an electrophile can be approximated using eqn (1).
The Klopman–Salem equation.
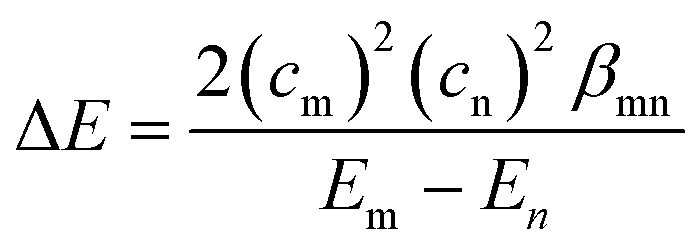 | (1) |
In this study, we performed the synthesis of thiocarbamates directly from disulfide bonds and isocyanates. A variety of synthetic methods9 of thiocarbamates have been extensively studied because they have attracted much attention as biologically active molecules.10 However, to the best of our knowledge, there is no synthetic method of thiocarbamates directly from disulfide bonds. Thus, insights into the nucleophilicity of disulfide bonds would provide a new synthetic route toward sulfur-containing compounds. Herein, we report neighboring group participation by disulfide bonds to produce cyclic thiocarbamates and the computational study.
For the purpose of changing the degree of molecular orbital overlap between a lone pair of the disulfide bond and an antibonding orbital of isocyanate, four model compounds were designed as shown in Fig. 1. The reaction of dimethyl disulfide 1 with 2-endo-isocyanatobicyclo[2.2.1]heptane is an intermolecular reaction with the least opportunity for the molecular orbital overlap (Fig. 1(a)). The intramolecular reaction of 3-isocyanatopropyl methyl disulfide 2 should provide a good chance for the molecular orbital overlap (Fig. 1(b)). The phenyl-enforced intramolecular reaction of 2-isocyanatophenyl methyl disulfide 3 and the norbornyl-enforced intramolecular reaction of 2-endo-isocyanato-6-endo-(methyldisulfanyl)bicyclo[2.2.1]heptane 4 should have excellent intramolecular orbital overlaps which are stable due to their rigid conformations (Fig. 1(c) and (d)).
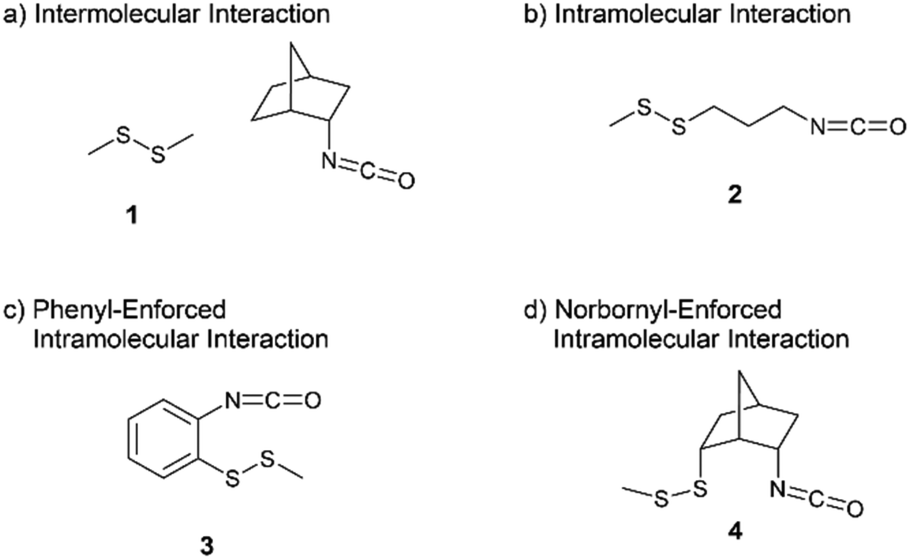 | ||
| Fig. 1 Model compounds to study the nucleophilicity of the disulfide bond. | ||
Nucleophilic reactions of disulfides 1–4 with isocyanates are shown in Scheme 1. Since isocyanates are not very stable, we started with the corresponding carboxylic acids and then converted them into the isocyanates by a Curtius rearrangement in the first step. After evaporation of the solvent, the nucleophilic reactions were carried out in a mixture of 5 M HCl and dioxane overnight. For the intermolecular reaction of disulfide 1 (Scheme 1(a)), TLC analysis showed a long streak without distinct spots. Although a small amount of the corresponding amine might be produced based on the ninhydrin stain, the methyl peak bonded to the sulfur was not found in the 1H NMR spectrum of the crude product. Therefore, it was unlikely that the intermolecular reaction occurred. For the flexible intramolecular reaction (Scheme 1(b)), 4-(methyldisulfanyl)butanoic acid 2-COOH was prepared using procedures reported by Watson et al.11 After the Curtius rearrangement and treatment with the 5 M HCl/dioxane solution, a major product (16 w/w% based on the starting material) was isolated by using a preparative TLC plate. It showed an isocyanate peak in the IR spectrum. However, we could not confirm whether or not the compound corresponded to the model isocyanate 2 because the compound slowly decomposed. Up to this point, we could not find any evidence of the nucleophilic character of the disulfide bond.
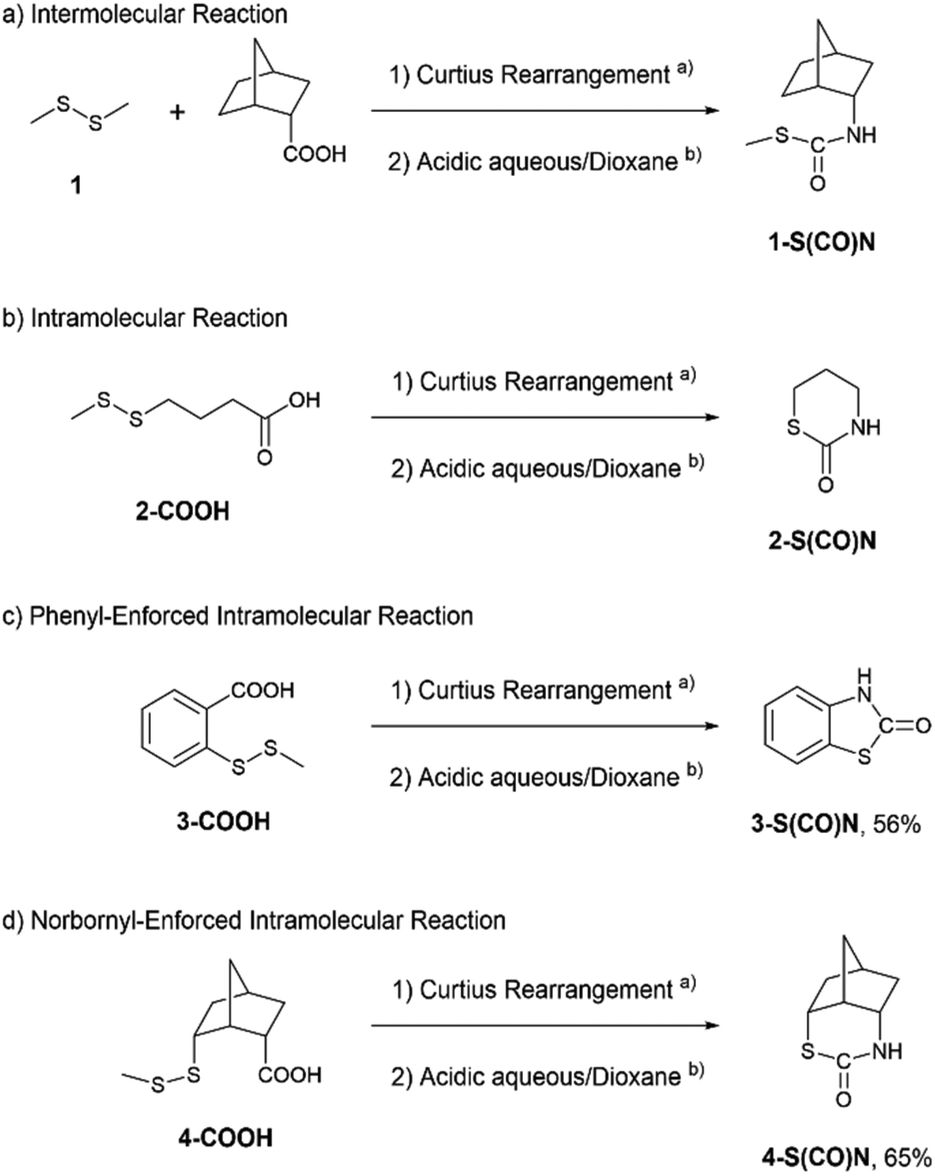 | ||
Scheme 1 Thiocarbamate synthesis of disulfide 1–4. (a) Diphenylphosphoryl azide (1.1 equiv.), triethylamine (2.2 equiv.), toluene, N2, 65 °C, 4 h. (b) 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture of 5 M HCl and dioxane, rt, overnight. :4 mixture of 5 M HCl and dioxane, rt, overnight. | ||
For the phenyl-enforced intramolecular reaction, 2-(methyldisulfanyl)benzoic acid 3-COOH was prepared using a procedure reported by Schirmeister et al.12 A certain amount of disulfide 3 was already converted into 2-benzothiazolinone, 3-S(CO)N, after the Curtius rearrangement based on the observation by TLC. Eventually, the corresponding thiocarbamate, 3-S(CO)N, was obtained in 56% yield (Scheme 1(c)). Finally, the norbornyl-enforced intramolecular reaction of disulfide 4 produced an isocyanate intermediate (see page S12 in the ESI†) and then gave the expected thiocarbamate 4-S(CO)N in 65% yield (Scheme 1(d)). The structure of thiocarbamate 4-S(CO)N was unequivocally established by an X-ray crystallographic structure study, which is shown in Fig. 2. Therefore, for the nucleophilic reaction of a disulfide bond with an isocyanate, rigid conformations that can juxtapose a disulfide bond with an isocyanate could be necessary.
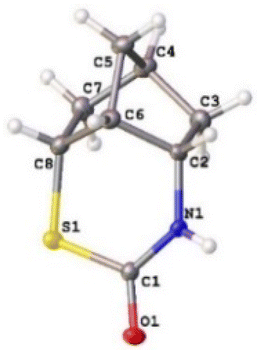 | ||
| Fig. 2 The molecular structure of thiocarbamate 4-S(CO)N. | ||
In order to clarify the nucleophilicity of the disulfide bonds at a quantum level, natural bond orbital (NBO) analyses were carried out at the M06-2X/6-311+G(d,p) level.13 NBO analysis is an outstanding method to examine through-space donor–acceptor interactions within a molecule.14 In addition, the M062X/6-311+G(d,p) level of theory has been used to rationalize through-space interactions between disulfide bonds and neighboring carbonyls by Raines and co-workers.15 Therefore, this method with the inclusion of solvent effects was employed to calculate isocyanate 3 and 4 in toluene or water and protonated isocyanate 3 and 4 in water based on the reaction conditions. The result of NBO analysis of isocyanate 3 is shown in Scheme 2. For isocyanate 3 produced by the Curtius rearrangement in toluene, which is 3-toluene, there is an interaction of 1.96 kcal mol−1 between a lone pair of the phenyl-bonded sulfur (Sph) and an antibonding orbital of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the isocyanate (see Fig. S7 in the ESI†). This interaction can be the driving force for 3-S(CO)N as observed by TLC after the Curtius rearrangement. 3-Water is an unprotonated form of the isocyanate in the acidic aqueous solution and has a significant interaction of 5.18 kcal mol−1 between a lone pair of Sph (HOMO−4) and an antibonding orbital of a lone pair of carbon of the isocyanate (LUMO). This interaction would lead to S–C bond formation of the thiocarbamate moiety. Interestingly, the protonated form of the isocyanate in the acidic aqueous solution, 3-NCOH+–water, does not exhibit any interaction between the lone pair of Sph and the antibonding orbital of the isocyanate. However, the methyl-bonded sulfur (SMe) exerts interactions with the protonated isocyanate (see Fig. S8 in the ESI†) as follows: 3.77 kcal mol−1 interaction between a lone pair of SMe (HOMO−15) and an antibonding orbital of NC (LUMO+24). 0.58 kcal mol−1 interaction between a lone pair of SMe (HOMO−15) and an antibonding orbital of NC (LUMO+2). Although the acidic conditions were intended to facilitate the intramolecular cyclization by protonation, the interactions in the protonated isocyanate would not lead to the thiocarbamate compound.
O of the isocyanate (see Fig. S7 in the ESI†). This interaction can be the driving force for 3-S(CO)N as observed by TLC after the Curtius rearrangement. 3-Water is an unprotonated form of the isocyanate in the acidic aqueous solution and has a significant interaction of 5.18 kcal mol−1 between a lone pair of Sph (HOMO−4) and an antibonding orbital of a lone pair of carbon of the isocyanate (LUMO). This interaction would lead to S–C bond formation of the thiocarbamate moiety. Interestingly, the protonated form of the isocyanate in the acidic aqueous solution, 3-NCOH+–water, does not exhibit any interaction between the lone pair of Sph and the antibonding orbital of the isocyanate. However, the methyl-bonded sulfur (SMe) exerts interactions with the protonated isocyanate (see Fig. S8 in the ESI†) as follows: 3.77 kcal mol−1 interaction between a lone pair of SMe (HOMO−15) and an antibonding orbital of NC (LUMO+24). 0.58 kcal mol−1 interaction between a lone pair of SMe (HOMO−15) and an antibonding orbital of NC (LUMO+2). Although the acidic conditions were intended to facilitate the intramolecular cyclization by protonation, the interactions in the protonated isocyanate would not lead to the thiocarbamate compound.
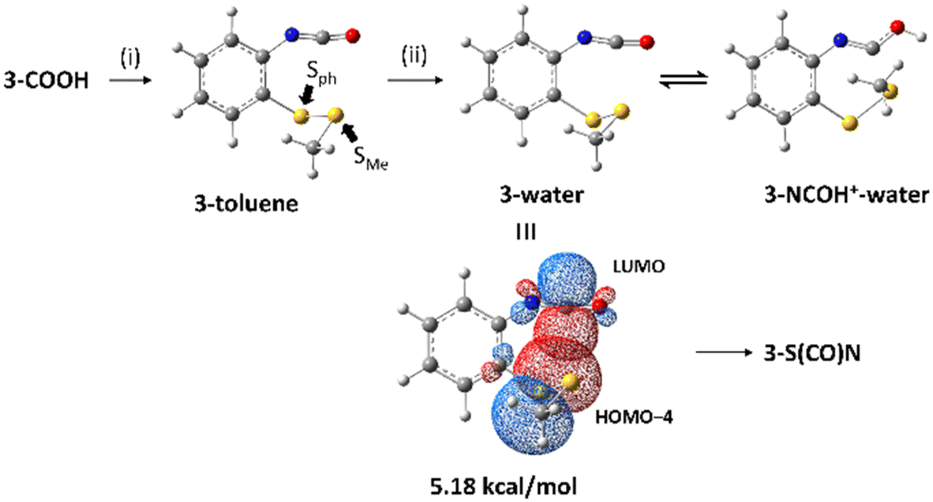 | ||
| Scheme 2 NBO analysis of isocyanate 3 in toluene and acidic aqueous solution. (i) Diphenylphosphoryl azide (1.1 equiv.), triethylamine (2.2 equiv.), toluene, N2, 65 °C, 4 h. (ii) 5:4 mixture of 5 M HCl and dioxane, rt, overnight. | ||
The result of NBO analysis of isocyanate 4 is shown in Scheme 3. Unlike 3-toluene, 4-toluene did not show any interaction between the lone pair of norbornyl-bonded sulfur (Snor) and the antibonding orbital of the isocyanate, which is consistent with the experimental result that the thiocarbamate 4-S(CO)N was not observed by TLC before the treatment with the 5 M HCl/dioxane solution. Unprotonated and protonated isocyanate 4, which are 4-water and 4-SSH+–water, are the equilibrium forms in the acidic aqueous solution. Interestingly, there is again no interaction between the lone pairs of Snor and the antibonding orbitals of the isocyanate.
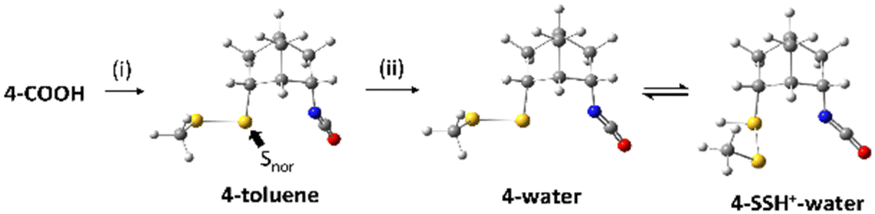 | ||
| Scheme 3 NBO analysis of isocyanate 4 in toluene and acidic aqueous solution. (i) Diphenylphosphoryl azide (1.1 equiv.), triethylamine (2.2 equiv.), toluene, N2, 65 °C, 4 h. (ii) 5:4 mixture of 5 M HCl and dioxane, rt, overnight. | ||
Although the lowest-energy optimized structures in Scheme 3 did not show any interactions that could lead to 4-S(CO)N, it was found that there is an oxygen-protonated isocyanate 4-NCOH+–water which is 11.0 kcal mol−1 higher in energy than 4-SSH+–water (Scheme 4) and it has significant interactions between the lone pair of Snor and the antibonding orbitals of NC (Fig. 3) as follows: 3.22 kcal mol−1 for an interaction of HOMO−26 and LUMO+29 and 1.69 kcal mol−1 for an interaction of HOMO−26 and LUMO+1. 11.0 kcal mol−1 energy barrier can be overcome at room temperature. Therefore, this structure is believed to lead to the thiocarbamate 4-S(CO)N.
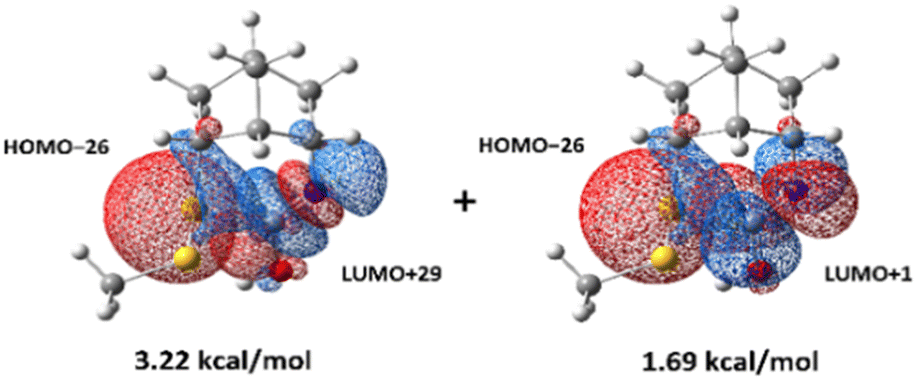 | ||
| Fig. 3 n → π* interactions in 4-NCOH+–water. | ||
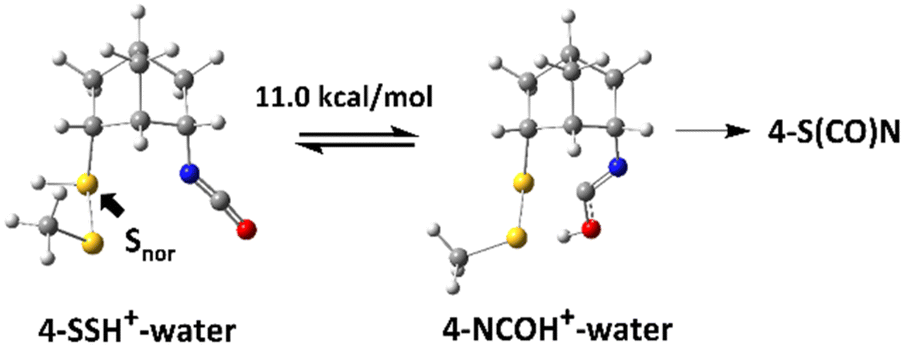 | ||
| Scheme 4 Equilibrium of isocyanate 4 in acidic aqueous solution. | ||
Based on the experimental and computational results, a plausible mechanism of formation of the thiocarbamate is proposed as shown in Scheme 5. Initially, the disulfide bond of 4-NCOH+–water attacks the isocyanate carbon to produce the intermediate I. This step requires a neighboring isocyanate in the rigid conformation. After proton translocation, the H2O molecule could remove the SMe moiety from the intermediate II to yield 4-S(CO)N.
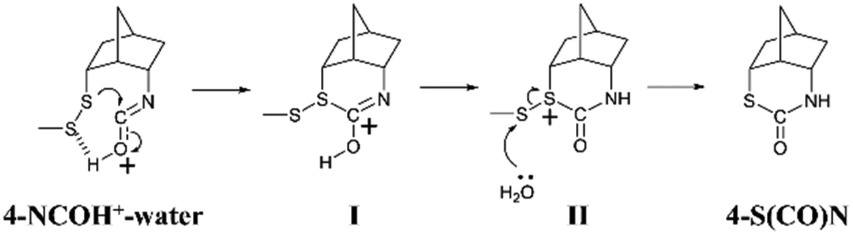 | ||
| Scheme 5 A plausible mechanism of 4-S(CO)N formation. | ||
In summary, we have reported first neighboring group participation by disulfide bonds. When a disulfide bond and an isocyanate moiety were juxtaposed by a rigid conformation, such as in 2-isocyanatophenyl methyl disulfide and 2-endo-isocyanato-6-endo-(methyldisulfanyl)bicyclo[2.2.1]heptane, the disulfide bond showed moderate nucleophilicity toward the isocyanates as expected from the Klopman–Salem equation. NBO analyses showed that the rigid conformations would render significant interactions between the lone pairs of disulfide bonds and the antibonding orbitals of isocyanates. This finding will open up a new synthetic route toward sulfur-containing compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded by JSPS KAKENHI Grant Number JP19K15545.References
- (a) R. C. Arnold, A. P. Lien and R. M. Alm, J. Am. Chem. Soc., 1950, 72, 731–733 CrossRef CAS; (b) R. Mozingo, D. E. Wolf, S. A. Harris and K. Folkers, J. Am. Chem. Soc., 1943, 65, 1013–1016 CrossRef CAS; (c) J. A. Pappas, J. Am. Chem. Soc., 1977, 99, 2926–2930 CrossRef CAS.
- (a) J. P. Danehy and K. N. Parameswaran, J. Org. Chem., 1968, 33, 568–572 CrossRef CAS; (b) P. W. Preisler and L. Berger, J. Am. Chem. Soc., 1947, 69, 322–325 CrossRef CAS.
- H. Gilman and F. J. Webb, J. Am. Chem. Soc., 1949, 71, 4062–4066 CrossRef CAS.
- (a) M. Aida and C. Nagata, Theor. Chim. Acta, 1986, 70, 73–80 CrossRef CAS; (b) A. D. Clauss, S. F. Nelsen, M. Ayoub, J. W. Moore, C. R. Landis and F. Weinhold, Chem. Educ. Res. Pract., 2014, 15, 417 RSC.
- (a) I. B. Douglass and B. S. Farah, J. Org. Chem., 1958, 23, 330 CrossRef CAS; (b) I. B. Douglass, B. S. Farah and E. G. Thomas, J. Org. Chem., 1961, 26, 1996–1999 CrossRef CAS.
- M. Kirihara, T. Okada, Y. Sugiyama, M. Akiyoshi, T. Matsunaga and Y. Kimura, Org. Process Res. Dev., 2017, 21, 1925–1937 CrossRef CAS.
- J. D. Buckman, M. Bellas, H. K. Kim and L. Field, J. Org. Chem., 1967, 32, 1626–1627 CrossRef CAS.
- (a) G. Klopman, J. Am. Chem. Soc., 1968, 90, 223–234 CrossRef CAS; (b) L. Salem, J. Am. Chem. Soc., 1968, 90, 543–552 CrossRef CAS.
- (a) H. Chabane, Y. Adjeroud and M. Liacha, Org. Commun., 2017, 10(1), 24–31 CrossRef CAS; (b) P. Bao, L. Wang, H. Yue, Y. Shao, J. Wen, D. Yang, X. Zhao, H. Wang and W. Wei, J. Org. Chem., 2019, 84, 2976–2983 CrossRef CAS PubMed; (c) W.-H. Bao, M. He, J.-T. Wang, X. Peng, M. Sung, Z. Tang, S. Jiang, Z. Cao and W.-M. He, J. Org. Chem., 2019, 84, 6065–6071 CrossRef CAS PubMed; (d) P. Mampuys, Y. Zhu, S. Sergeyev, E. Ruijter, R. V. A. Orru, S. V. Doorslaer and B. U. W. Maes, Org. Lett., 2016, 18, 2808–2811 CrossRef CAS PubMed; (e) G. Rescourio and H. Alper, J. Org. Chem., 2008, 73, 1612–1615 CrossRef CAS PubMed.
- (a) M. Erdogan, B. Kilic, R. I. Sagkan, F. Aksakal, T. Ercetin, H. O. Gulcan and D. S. Dogruer, Eur. J. Med. Chem., 2021, 212, 113124 CrossRef CAS PubMed; (b) S. M. Bronner, J. Murray, F. A. Romero, K. W. Lai, V. Tsui, P. Cyr, M. H. Beresini, G. D. L. Boenig, Z. Chen, E. F. Choo, K. R. Clark, T. D. Crawford, H. Jayaram, S. Kaufman, R. Li, Y. Li, J. Liao, X. Liang, W. Liu, J. Ly, J. Maher, J. Wai, F. Wang, A. Zheng, X. Zhu and S. Magnuson, J. Med. Chem., 2017, 60, 10151–10171 CrossRef CAS; (c) K. M. Gardinier, D. L. Gernert, W. J. Porter, J. K. Reel, P. L. Ornstein, P. Spinazze, F. C. Stevens, P. Hahn, S. P. Hollinshead, D. Mayhugh, J. Schkeryantz, A. Khilevich, O. D. Frutos, S. D. Gleason, A. S. Kato, D. Luffer-Atlas, P. V. Desai, S. Swanson, K. D. Burris, C. Ding, B. A. Heinz, A. B. Need, V. N. Barth, G. A. Stephenson, B. A. Diseroad, T. A. Woods, H. Yu, D. Bredt and J. M. Witkin, J. Med. Chem., 2016, 59, 4753–4768 CrossRef CAS.
- L. M. Miller, W.-J. Keune, D. Castagna, L. C. Young, E. L. Duffy, F. Potjewyd, F. Salgado-Polo, P. E. García, D. Semaan, J. M. Pritchard, A. Perrakis, S. J. F. Macdonald, C. Jamieson and A. J. B. Watson, J. Med. Chem., 2017, 60, 722–748 CrossRef CAS PubMed.
- F. Barthels, G. Marincola, T. Marciniak, M. Konhäuser, S. Hammerschmidt, J. Bierlmeier, U. Distler, P. R. Wich, S. Tenzer, D. Schwarzer, W. Ziebuhr and T. Schirmeister, ChemMedChem, 2020, 15, 839–850 CrossRef CAS PubMed.
- >M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, version 3.1, 1992 Search PubMed.
- H. R. Kilgore, C. R. Olsson, K. A. D'Angelo, M. Movassaghi and R. T. Raines, J. Am. Chem. Soc., 2020, 142, 15107–15115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2194462. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01574a |
| This journal is © The Royal Society of Chemistry 2023 |