
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstitution pattern in ruthenium octa-n-butoxyphthalocyanine complexes influence their reactivity in N–H carbene insertions†
Andrey P.
Kroitor
ab,
Alexander A.
Dmitrienko
 c,
Alexander G.
Martynov
*b,
Yulia G.
Gorbunova
*bcd and
Alexander B.
Sorokin
*a
c,
Alexander G.
Martynov
*b,
Yulia G.
Gorbunova
*bcd and
Alexander B.
Sorokin
*a
aInstitut de recherches sur la catalyse et l'environnement de Lyon (IRCELYON), UMR 5256, CNRS – Université Lyon 1, 2 av. A. Einstein, 69626 Villeurbanne, France. E-mail: alexander.sorokin@ircelyon.univ-lyon1.fr
bFrumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninskii pr., 31, bldg. 4, 119071, Moscow, Russia. E-mail: martynov@phyche.ac.ru
cLomonosov Moscow State University, Leninskie Gory, 1-3, Moscow, 119991, Russia
dKurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Leninskii pr., 31, 119991 Moscow, Russia. E-mail: yulia@igic.ras.ru
First published on 23rd November 2022
Abstract
Ruthenium phthalocyanine complexes bearing n-OBu substituents in the peripheral or non-peripheral positions are efficient catalysts for the selective double or single carbene insertion to the amine N–H bonds. This complementary reactivity of two Ru complexes can be used for the synthesis of asymmetric tertiary amines and diamines bearing different substituents and has been demonstrated by two examples of readily available primary amines using different carbene precursors in successive reactions.
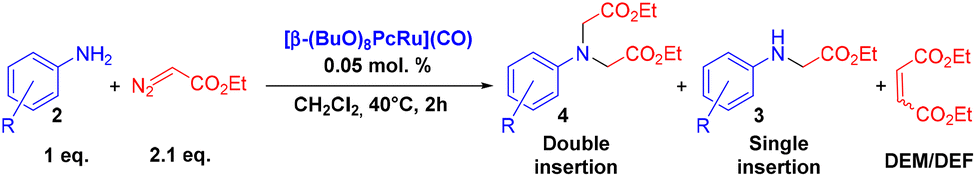
Catalytic X–H (X = N, C, O, S…) insertion of carbenes represents a powerful strategy for the construction of C–X bonds.1 In particular, porphyrin complexes2 and engineered hemoproteins3 are remarkably efficient in cyclopropanation and C–X carbene insertion reactions. However, it should be noted that carbene insertion into amine N–H bonds is more demanding compared to other C–X bonds because of the catalyst poisoning due to amine coordination to the metal sites. The selectivity to single N–H insertion products is also often limited. In recent years, a variety of transition metal complexes4 and engineered hemoproteins5 have been reported for the insertion of carbenes to the N–H bonds of amines. Along with Rh, Ir, Fe and Co complexes, considerable attention has been devoted to Ru compounds.6 Among them, Ru porphyrins have been extensively studied as efficient catalysts for carbene transfer reactions,7 principally in cyclopropanation of olefins.8 Noteworthily, other tetrapyrrolic complexes, e.g. Ru phthalocyanines, have been rarely studied in these reactions.9 In general, the nature of the substituents of the phthalocyanine ligand determines the electronic properties of the corresponding complexes and hence their catalytic activity.10 Our previous study showed that peripherally substituted Ru(II) octa-n-butoxyphthalocyaninate β-(BuO)8PcRu(CO) (1β) was an efficient catalyst for the carbene insertion into amine N–H bonds under practical reaction conditions.11 A large variety of structurally divergent amines were selectively converted into mono-substituted glycine derivatives using a 0.05 mol% catalyst loading, a high amine concentration (1 M) and 1.1 equiv. of ethyl diazoacetate (EDA) with up to quantitative yields and turnover numbers reaching 2000. In the course of our studies, we have found that the catalytic properties of the Ru(II) octa-n-butoxyphthalocyanine complex depend on the position of the substituents. Therein, we report that peripherally and non-peripherally substituted complexes β-(BuO)8PcRu(CO) and α-(BuO)8PcRu(CO) (1α) exhibit different properties in carbene insertion to amine N–H bonds, providing selectively either double or single N–H insertion products, respectively. We demonstrate how the complementary catalytic properties of 1α and 1β can in principle be used for the synthesis of elaborated amine derivatives with various substitution patterns.
The peripherally substituted complex 1β was prepared by refluxing the metal-free phthalocyanine H2[β-(BuO)8Pc] with Ru3(CO)12 in o-dichlorobenzene (o-DCB) according to an optimized published procedure9b with an 85% yield (Scheme 1). Our attempts to synthesize 1α from non-peripherally substituted phthalocyanine H2[α-(BuO)8Pc] and Ru3(CO)12 using the same protocol led to a mixture of unidentified products, as was indicated by MALDI-TOF MS analyses. The replacement of o-DCB with pure PhCN distilled in a vacuum over P2O5 and then over K2CO3 led to a high yield of the target 1α complex (Scheme 1). The PhCN purity plays a crucial role since even the traces of benzamide in the solvent result in a very low yield of 1α. Contrary to the preparation of peripherally substituted Ru phthalocyanines by the treatment with Ru3(CO)12 in o-DCB accompanied by the formation of μ-carbido dimers,9b no corresponding μ-carbido dimer was formed during the synthesis of 1α in PhCN. The 1H NMR spectrum of 1α confirmed its diamagnetic nature. The presence of the axial carbonyl ligand was evidenced by the strong CO stretching band at 1948 cm−1 in the IR spectrum. The comparison of the UV-vis spectra of the solutions of both Ru phthalocyanines shows the significant red-shift of the Q band from 655 nm for the peripheral 1β complex to 717 nm for the non-peripherally substituted species 1α (Fig. S4†). This means a difference in their electronic structures, which, in turn, can induce a difference in the catalytic properties.
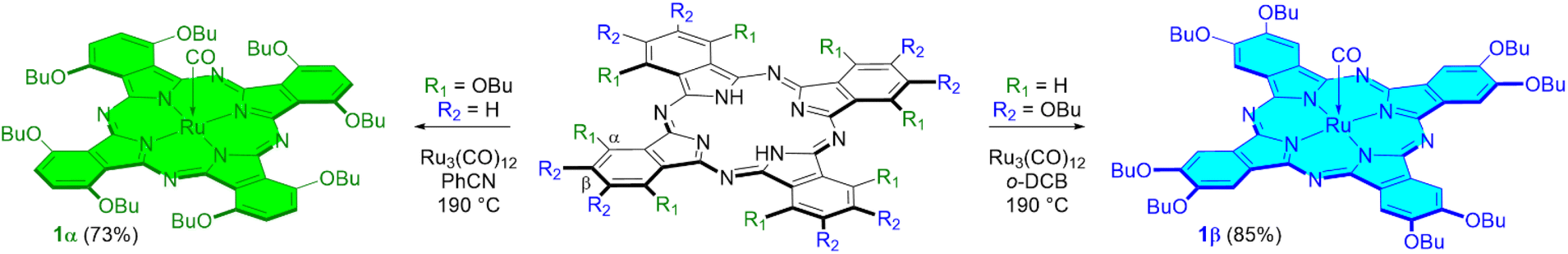 | ||
| Scheme 1 Synthesis of the peripherally and non-peripherally substituted ruthenium octa-n-butoxyphthalocyanines 1β and 1α. | ||
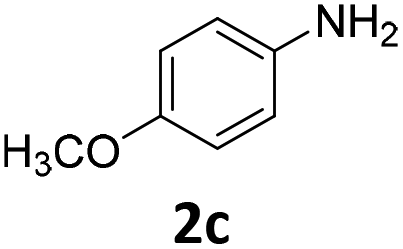
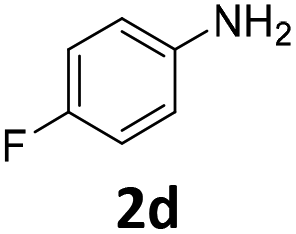
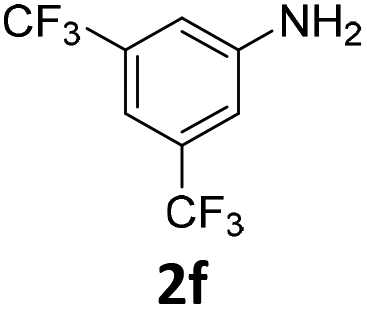
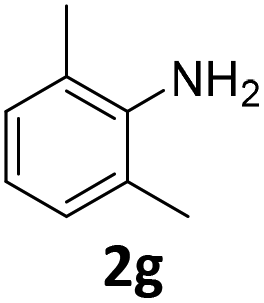
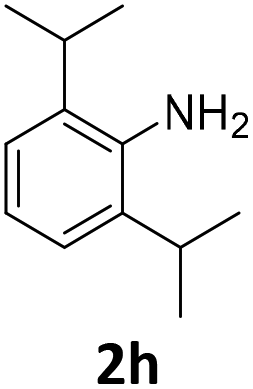
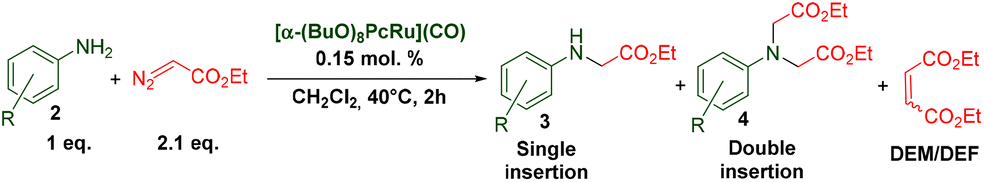
The catalytic properties of the complexes were evaluated in the reaction of 2.1 equiv. of EDA with aromatic amines 2a–2j (1 M) in CH2Cl2 at 40 °C using 0.05 mol% catalyst loading (Table 1). It is worth noting that carbene transfer reactions catalyzed by metal complexes are typically carried out under an inert atmosphere.1–3 Importantly, the carbene insertion to amines catalyzed by 1β and 1α can be performed under air without decreasing the product yields, which represents a significant practical advantage. Aniline derivatives bearing electron-donating (Table 1, entries 1–3 and 8) or electron-withdrawing groups (Table 1, entries 4 and 5) were converted into the N–H double insertion products 4 with almost quantitative yields. Two strong electron-withdrawing groups of 3,5-ditrifluoromethylaniline 2f decrease its nucleophilic properties, resulting in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the products of double and single N–H insertions – d- and s-products: 4f and 3f, respectively (Table 1, entry 6). The presence of methyl and isopropyl substituents in the ortho-positions is well tolerated and the corresponding d-products 4g and 4h were obtained in 89 and 97% yields, respectively (Table 1, entries 7 and 8). However, the bulky tert-butyl o-substituent prevents the second N–H insertion (Table 1, entry 9). The 1β – EDA system shows a clear preference for N–H insertion compared to the cyclopropanation of the double bond. The products of single and double carbene insertions to p-aminostyrenes 3j and 4j as well as the cyclopropanation product of 4j were obtained in 10, 62 and 18% yields, respectively (Table 1, entry 10).
:1 ratio of the products of double and single N–H insertions – d- and s-products: 4f and 3f, respectively (Table 1, entry 6). The presence of methyl and isopropyl substituents in the ortho-positions is well tolerated and the corresponding d-products 4g and 4h were obtained in 89 and 97% yields, respectively (Table 1, entries 7 and 8). However, the bulky tert-butyl o-substituent prevents the second N–H insertion (Table 1, entry 9). The 1β – EDA system shows a clear preference for N–H insertion compared to the cyclopropanation of the double bond. The products of single and double carbene insertions to p-aminostyrenes 3j and 4j as well as the cyclopropanation product of 4j were obtained in 10, 62 and 18% yields, respectively (Table 1, entry 10).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Conversion, % | Double N–H insertion, yields of 4,b % | Single N–H insertion, yields of 3, % | Yield of DEM (DEF),e % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Conditions: 0.5 mmol of amine, 1.05 mmol of EDA, 0.05 mol% of the catalyst, 0.5 mL of CH2Cl2, air, 40 °C. b Yields determined by 1H NMR (isolated yields). c The product of the single N–H insertion was isolated in a 51% yield. d The cyclopropanation product of the double N–H carbene insertion was obtained in an 18% isolated yield. e Yields of diethylmaleate (DEM) and diethylfumarate (DEF) determined by 1H NMR are based on the initial amount of EDA. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |

|
98 | 97 (92) | 1 | 9 (1) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |

|
99 | 98 (97) | 1 | 9 (<0.5) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
|
100 | 97 (97) | 3 | 10 (0.5) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 |
|
100 | 99 (96) | 1 | 8 (0.5) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |

|
100 | 94 (94) | 6 | 12 (1) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |
|
100 | 51 (43) | 49 | 28 (5) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 |
|
97 | 89 (89) | 8 | 10 (1) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 |
|
100 | 97 (97) | 3 | 25 (2) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 |

|
88 | 0 | 88c | 50 (5) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 |

|
90d | 62 (55) | 10 | 9 (0.5) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The 1α complex exhibited a lower catalytic activity in the reaction of aniline with EDA under conditions used for 1β, providing a 35% conversion into the s-product 3a. We optimized the protocol towards a quantitative substrate conversion (Table S1†). The replacement of CH2Cl2 with MeCN did not improve the aniline conversion. A slow addition of an EDA/aniline mixture to the solution of 1α in CH2Cl2 resulted in a sharp drop of the conversion. However, increasing of the catalyst amount to 0.1 and 0.15 mol% allowed for the improvement of aniline conversion into 71 and 96%, respectively (Table S1†). Importantly, only the s-product 3a was selectively formed. Thus, the substrate scope was explored using 0.15 mol% of the catalyst and 2.1 equiv. of EDA in CH2Cl2 at 40 °C (Table 2). Although the s-products can be obtained with 1.1 equiv. of EDA, we used excess EDA in order to compare the catalytic properties of the two complexes under the same conditions except the catalyst loading.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Conversion, % | Single N–H insertion, yields,b % | Double N–H insertion, yields, % | Yield of DEM (DEF),c % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Conditions: 0.5 mmol of amine, 1.05 mmol of EDA, 0.15 mol% of the catalyst, 0.5 mL of CH2Cl2, air, 40 °C. b Yields determined by 1H NMR (isolated yields). c Yields of diethylmaleate (DEM) and diethylfumarate (DEF) determined by 1H NMR are based on the initial amount of EDA. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |
|
96 | 96 (96) | 0 | 0.5 (tr.) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |

|
99 | 96 (93) | 3 | 2 (0) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
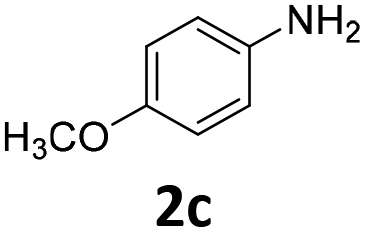
|
93 | 93 (93) | 0 | tr. (0) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 |
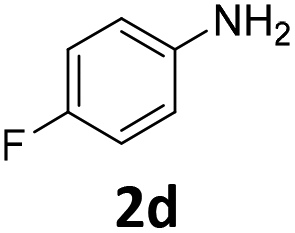
|
100 | 97 (97) | 3 | 2 (0) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |
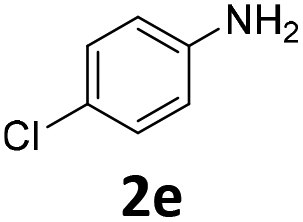
|
98 | 98 (85) | 0 | 2 (0) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |
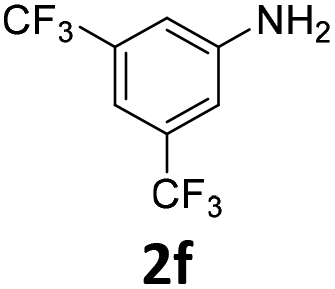
|
40 | 40 (n.d.) | 0 | 1 (0) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 |

|
100 | 80 (29) | 20 | 9 (tr.) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 |
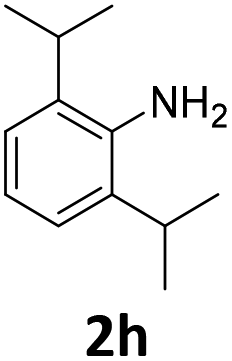
|
100 | 97 (97) | 3 | 9 (0) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 |
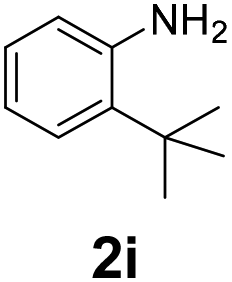
|
55 | 55 (40) | 0 | 3 (0) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 |
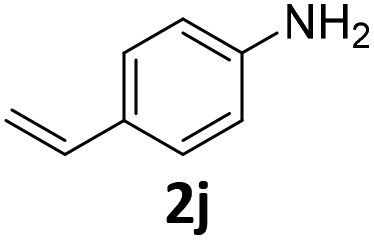
|
95 | 95 (95) | 0 | tr. (0) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In sharp contrast to 1β selectively providing d-products, 1α shows a very high selectivity to s-products even in the presence of 2.1 equiv. of EDA. Quasi-quantitative yields of mono-substituted glycine derivatives were obtained from aniline with electron-donating (Table 2, entries 1–3 and 8) and electron-withdrawing (Table 2, entries 4 and 5) substituents. While 2,6-diisopropylaniline 2h afforded a 97% yield of 3h, less hindered highly nucleophilic 2,6-dimethylaniline afforded an 80:20 ratio of the s- and d-products 3g and 4g, respectively (Table 2, entries 7 and 8). Noteworthily, a highly selective single N–H insertion was observed in the reaction with p-aminostyrene, and no double insertion and no cyclopropanation of the double bond were observed in this case (Table 2, entry 10).
The synthetic utility of Ru(II) octa-n-butoxyphthalocyaninates 1α and 1β due to their different catalytic properties was demonstrated in the derivatization of three aromatic diamines with different electronic properties and substitution patterns (Scheme 2). The carbene insertion to diamines leads to the formation of four products: single insertion (s), single–single insertion (ss), single–double insertion (sd) and double–double insertion (dd). Thus, a selective preparation of the desired product is more challenging compared to the synthesis of simple amines. Using 0.05 mol% of 1β and 4.1 equiv. of EDA, p-phenylenediamine 5 was converted into the dd-product 5dd in a quantitative yield (TON = 8000) (Scheme 2a).
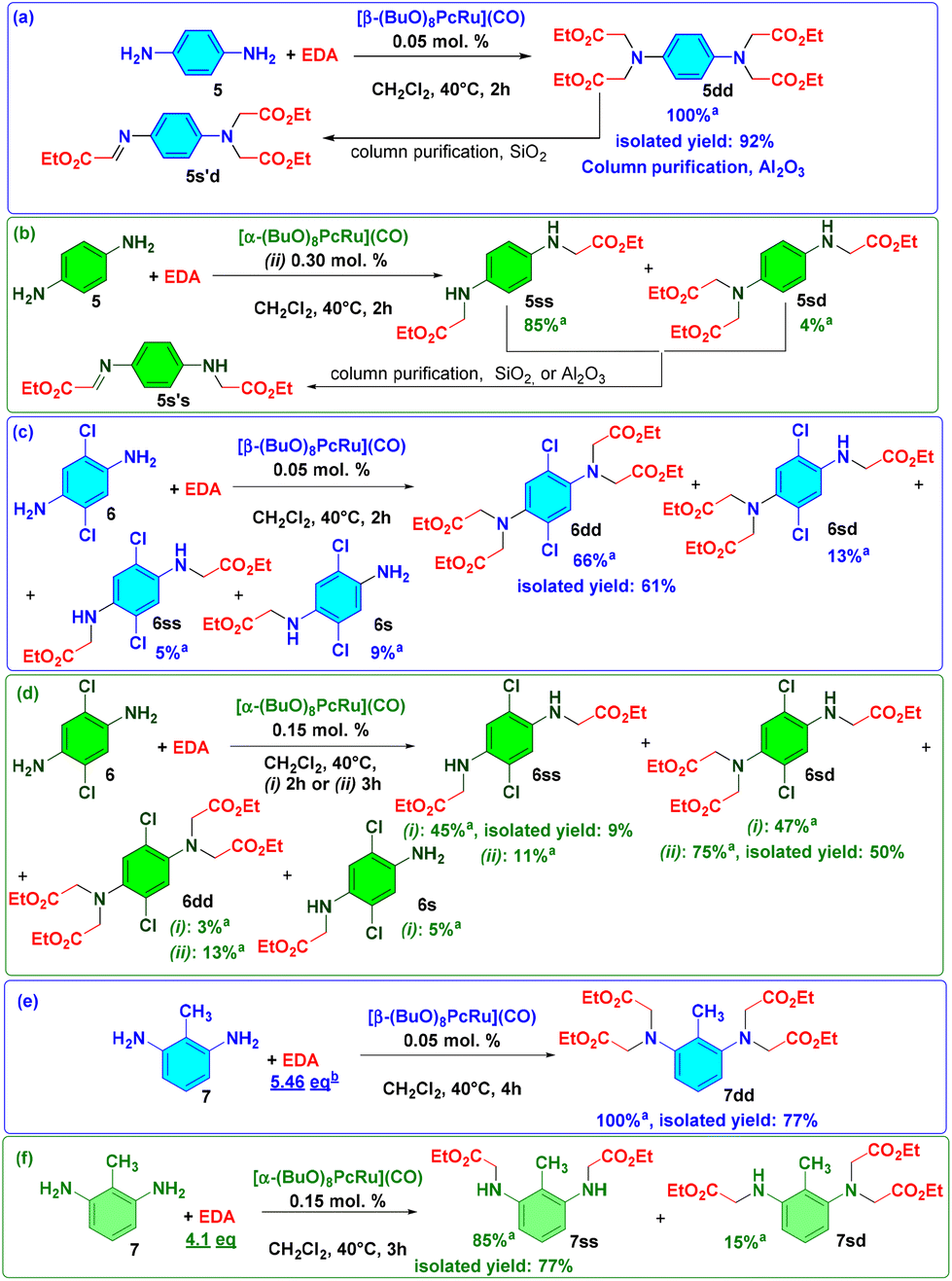 | ||
| Scheme 2 Functionalization of the aromatic diamines – (a, b) p-phenylenediamine, (c, d) 2,6-dichloro-1,4,-diaminobenzene and (e, f) 2,6-diaminotoluene with EDA in the presence of 1α and 1β. aYields were determined from the 1H NMR spectra. | ||
During its isolation by column chromatography with SiO2, 5dd underwent partial elimination of one CH2COOEt group, affording a double insertion-imine product 5s′d identified by GC-MS and 1H NMR. Purification using neutral Al2O3 prevented degradation and 5dd was obtained in a 92% isolated yield. As expected, the 1α complex was selective to the formation of the ss-product 5ss from p-phenylenediamine. In the presence of 0.15 mol% of 1α, 5ss was formed in 50% yield (Scheme 2b). Under the standard conditions with 0.3 mol% of 1α (0.15 mol% per NH2 group), 5ss and 5sd were obtained in 85% and 4% yields, respectively. Upon chromatographic purification either with SiO2 or Al2O3, these compounds underwent a partial transformation to imine derivatives, which could not be separated from the target product 5ss.
The 2 h reaction of 2,6-dichloro-1,4-diaminobenzene 6 with 4.1 equiv. of EDA mediated by 1α led to the formation of 6s, 6ss, 6sd and 6dd products in 5%, 45%, 47% and 3% yields, respectively (Scheme 2d). Owing to their similar polarity, this mixture was difficult to separate and 6ss could be isolated in a 9% yield. When the reaction was performed for 3 h, the product 6sd was formed in a 75% yield and was isolated in a 50% yield. In the presence of 1β, the products 6s, 6ss, 6sd and 6dd were formed in 9%, 5%, 13% and 66% yields, respectively (Scheme 2c). The target product 6dd was isolated in a 61% yield. This example indicates that the optimization of the reaction conditions depending on the properties of diamine can afford the desired product in a good yield.
Indeed, under standard conditions, 1β catalyzed the reaction of 2,6-diaminotoluene 7 with 4.1 equiv. of EDA, forming a mixture of 7dd (57%) and 7sd products (42%), which could not be completely separated. When 5.46 equiv. of EDA were added in 4 portions during 3 h, a quantitative formation of 7dd was observed and the target product was isolated in 82% yield (Scheme 2e). In a complementary manner, 1α provided 85% yield of 7ss, accompanied by 15% yield of 7sd (Scheme 2f). The desired 7ss was isolated in a 77% yield.
Such a different reactivity of two Ru phthalocyanines opens up new opportunities for the synthesis of elaborated amine derivatives, e.g., unsymmetrical tertiary amines bearing three different groups. In the first step, readily available primary amines and diamines can be converted into s- or ss-products using the 1α catalyst. Next, 1β can catalyze the N–H insertion of another carbene to an s-product to afford an unsymmetrical NR1R2R3 amine. The feasibility of this strategy was first evaluated using diazoacetonitrile as a carbene precursor and N-methylaniline 8 as a model substrate. N2CHCN is a particularly useful compound to introduce nitrile groups into organic molecules.8c,12
Compared to EDA, the use of N2CHCN as a carbene precursor has still been rather limited13 because of the explosion propensity of a neat compound.12 Recently, the safe generation of N2CHCN in situ or the use of diluted solutions has been proposed.12 The high yields of the tertiary amine 9 were obtained from N-methylaniline using both protocols (Fig. S131†). Among the organic solvents tested, the highest yield of 9 (83%) was obtained using 1,2-dichloroethane. The use of 0.57 M N2CHCN solution in DCE (1.1 equiv.) afforded the target unsymmetrical tertiary amine in 97% yield in the presence of 0.05 mol% of 1β instead of 0.1 mol% of the catalyst charge applied for the in situ protocol (Fig. S131†).
Upon applying the efficient protocols for the selective single carbene insertion from EDA to primary amines catalyzed by 1α and for the efficient functionalization of secondary amines using N2CHCN in combination with 1β, we have prepared unsymmetrical tertiary amines, starting from the simple aromatic amine and diamine (Scheme 3). First, ethyl p-tolylglycinate 3b was obtained from p-toluidine and EDA in the presence of 1α in 93% isolated yield (Scheme 3a). The subsequent treatment of 3b with N2CHCN and 1β afforded the target unsymmetrical tertiary amine 10 in 87% isolated yield. This approach was further extended to primary diamine exemplified by 2,6-diaminotoluene 7 converted into 7ss in 77% yield through the reaction with EDA and 1α (Scheme 3b). The second step was accomplished using N2CHCN and 0.2 mol% of 1β, providing 56% isolated yield of the asymmetric tertiary diamine 11. Asymmetric tertiary diamines can in principle be prepared in two steps using the same peripheral complex, but the product yields will be lower because of the formation of the side single–double insertion product and lower substrate conversion. Furthermore, the isolation of the target product is much more difficult in this case. Thus, the two-step strategy involving two catalysts is particularly useful to access the asymmetric tertiary amines.
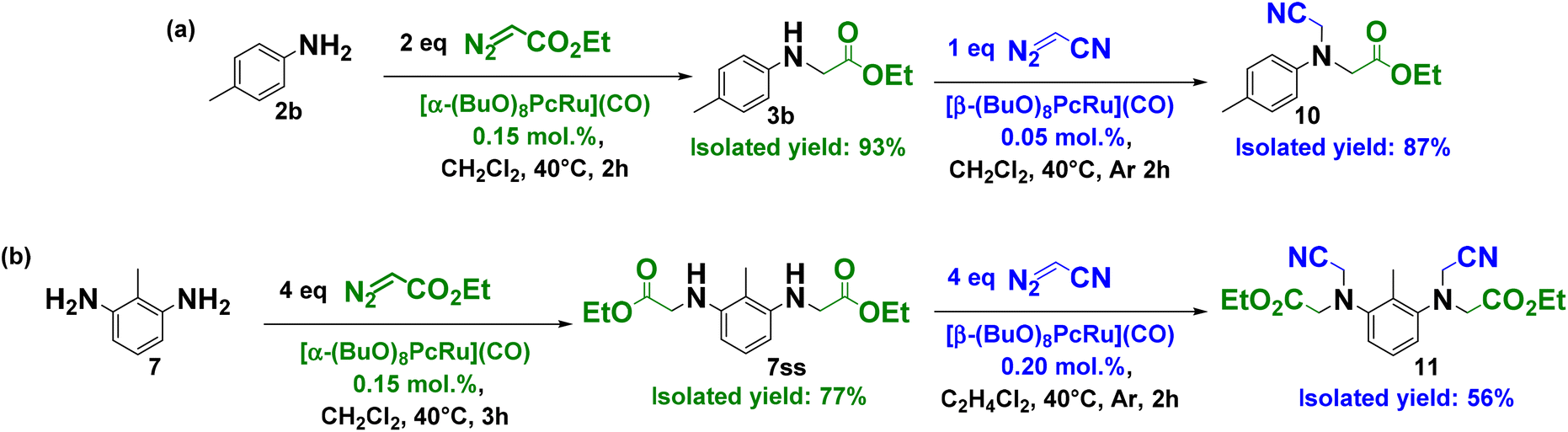 | ||
| Scheme 3 Preparation of unsymmetrical tertiary amines from primary amines – (a) p-toluidine and (b) 2,6-diaminotoluene by successive carbene transfer reactions catalyzed by 1α and 1β. | ||
In conclusion, the Ru(II) octa-n-butoxyphthalocyanine complexes are efficient catalysts for the carbene insertion to amine N–H bonds under practical reaction conditions. Using a low loading of these air-tolerant catalysts and a near-equivalent substrate/carbene precursor ratio, a wide range of simple amines and diamines were converted into different products in high yields. Importantly, the position of the substituents in the Ru(II) octa-n-butoxyphthalocyanine complexes controls their catalytic properties: the non-peripherally and peripherally substituted complexes 1α and 1β afford single and double N–H insertion products, respectively. Due to the complementary catalytic properties of the two Ru complexes, a novel synthetic strategy was proposed, which can provide access to asymmetric tertiary amines and diamines, starting from simple readily available compounds. This approach was validated using p-toluidine and 2,6-diaminotoluene as model substrates. Since N-aryl amines are important building blocks in natural products and synthetic compounds, this approach might be useful to access elaborated functionalized amines with interesting biological and pharmacological properties.14
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the CNRS and RFBR through the International Emerging Action 2021 and the research project 21-53-15004.References
- (a) C. Damiano, P. Sonzini and E. Gallo, Chem. Soc. Rev., 2020, 49, 4867–4905 RSC; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS; (c) M. J. Weissenborn and R. M. Koenigs, ChemCatChem, 2020, 12, 2171–2179 CrossRef CAS; (d) V. Carreras, N. Tanbouza and T. Ollevier, Synthesis, 2021, 53, 79–94 CrossRef CAS; (e) D. Intrieri, D. M. Carminati and E. Gallo, Dalton Trans., 2016, 45, 15746–15761 RSC; (f) I. Aviv and Z. Gross, Chem. – Eur. J., 2008, 14, 3995–4005 CrossRef CAS; (g) R. F. J. Epping, M. M. Hoeksma, E. O. Bobylev, S. Mathew and B. de Bruin, Nat. Chem., 2022, 14, 550–557 CrossRef CAS PubMed.
- D. Intrieri, D. M. Carminati and E. Gallo, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2016, vol. 38, pp. 1–99 Search PubMed.
- (a) Y. Yang and F. H. Arnold, Acc. Chem. Res., 2021, 54, 1209–1225 CrossRef CAS; (b) D. M. Carminati and R. Fasan, ACS Catal., 2019, 9, 9683–9697 CrossRef CAS; (c) B. J. Wittmann, A. M. Knight, J. L. Hofstra, S. E. Reisman, S. B. J. Kan and F. H. Arnold, ACS Catal., 2020, 10, 7112–7116 CrossRef CAS; (d) K. Chen and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 6891–6895 CrossRef CAS PubMed.
- (a) I. Aviv and Z. Gross, Chem. Commun., 2006, 4477–4479 RSC; (b) C. J. Moody, Angew. Chem., Int. Ed., 2007, 46, 9148–9150 CrossRef CAS; (c) B. Liu, S.-F. Zhu, W. Zhang, C. Chen and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834–5835 CrossRef CAS PubMed; (d) B. Xu, S.-F. Zhu, X.-L. Xie, J.-J. Shen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 11483–11486 CrossRef CAS.
- (a) Z. J. Wang, N. E. Peck, H. Renata and F. H. Arnold, Chem. Sci., 2014, 5, 598–601 RSC; (b) G. Sreenilayam and R. Fasan, Chem. Commun., 2015, 51, 1532–1534 RSC; (c) V. Steck, G. Sreenilayam and R. Fasan, Synlett, 2020, 31, 224–229 CrossRef CAS PubMed; (d) V. Steck, D. M. Carminati, N. R. Johnson and R. Fasan, ACS Catal., 2020, 10, 10967–10977 CrossRef CAS PubMed; (e) D. Nam, A. Tinoco, Z. Shen, R. D. Adukure, G. Sreenilayam, S. D. Khare and R. Fasan, J. Am. Chem. Soc., 2022, 144, 2590–2602 CrossRef CAS.
- G. Maas, Chem. Soc. Rev., 2004, 33, 183–190 RSC.
- (a) E. Galardon, P. Le Maux and G. Simonneaux, J. Chem. Soc., Perkin Trans. 1, 1997, 2455–2456 RSC; (b) C.-M. Ho, J.-L. Zhang, C.-Y. Zhou, O.-Y. Chan, J. J. Yan, F.-Y. Zhang, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2010, 132, 1886–1894 CrossRef CAS; (c) K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982–2987 CrossRef CAS; D. Carrié, T. Roisnel and G. Simonneaux, Polyhedron, 2021, 205, 115294 Search PubMed.
- (a) Y. Ferrand, P. Le Maux and G. Simonneaux, Org. Lett., 2004, 6, 3211–3214 CrossRef CAS PubMed; (b) C.-Y. Zhou, J.-S. Huang and C.-M. Che, Synlett, 2010, 2681–2700 CAS; (c) Y. Ferrand, P. Le Maux and G. Simonneaux, Tetrahedron: Asymmetry, 2005, 16, 3829–3836 CrossRef CAS; (d) E. Galardon, P. Le Maux and G. Simonneaux, Tetrahedron, 2000, 56, 615–621 CrossRef CAS; (e) H.-X. Wang, Q. Wan, K. Wu, K.-H. Low, C. Yang, C.-Y. Zhou, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2019, 141, 9027–9046 CrossRef CAS PubMed; (f) E. Galardon, P. Le Maux and G. Simonneaux, Chem. Commun., 1997, 927–928 RSC; (g) H. F. Srour, P. Le Maux, S. Chevance, D. Carrié, N. Le Yondre and G. Simonneaux, J. Mol. Catal. A: Chem., 2015, 407, 194–203 CrossRef CAS.
- (a) H.-H. Liu, Y. Wang, Y.-J. Shu, X.-G. Zhou, J. Wu and S.-Y. Yan, J. Mol. Catal. A: Chem., 2006, 246, 49–52 CrossRef CAS; (b) A. P. Kroitor, L. P. Cailler, A. G. Martynov, Yu. G. Gorbunova, A. Yu. Tsivadze and A. B. Sorokin, Dalton Trans., 2017, 46, 15651–15655 RSC.
- (a) A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS; (b) E. A. Lukyanets and V. N. Nemykin, J. Porphyrins Phthalocyanines, 2010, 14, 1–40 CrossRef CAS; (c) V. N. Nemykin and E. A. Lukyanets, ARKIVOC, 2010, 136–208 Search PubMed.
- L. P. Cailler, A. P. Kroitor, A. G. Martynov, Y. G. Gorbunova and A. B. Sorokin, Dalton Trans., 2021, 50, 2023–2031 RSC.
- P. K. Mykhailiuk and R. M. Koenigs, Chem. – Eur. J., 2020, 26, 89–101 CrossRef CAS PubMed.
- (a) K. Hock, R. Spitzner and R. M. Koenigs, Green Chem., 2017, 19, 2118–2122 RSC; (b) A. L. Chandgude and R. Fasan, Angew. Chem., Int. Ed., 2018, 57, 15852–15856 CrossRef CAS; (c) C. Empel, K. J. Hock and R. M. Koenigs, Org. Biomol. Chem., 2018, 16, 7129–7133 RSC; (d) C. Empel, K. J. Hock and R. M. Koenigs, Chem. Commun., 2019, 55, 338–341 RSC.
- (a) O. I. Afanasyev, E. A. Kuchuk, K. M. Muratov, G. L. Denisov and D. Chusov, Eur. J. Org. Chem., 2021, 543–586 CrossRef CAS; (b) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and compound characterization data. See DOI: https://doi.org/10.1039/d2ob01861f |
| This journal is © The Royal Society of Chemistry 2023 |