
Enantio- and diastereoselective boron conjugate addition to α-alkyl α,β-unsaturated esters†
Meng
Li
,
Guang-Rui
Peng
,
Xuan
Yang
,
Zhen-Ning
Ma
and
Jian-Bo
Xie
 *
*
Shaanxi Key Laboratory of Natural Products & Chemical Biology, College of Chemistry & Pharmacy, Northwest A&F University, Yangling 712100, China. E-mail: jianbo xie@nwafu.edu.cn
First published on 25th November 2022
Abstract
We developed a copper-catalyzed enantio- and diastereoselective boron conjugate addition to α-alkyl α,β-unsaturated esters under base-free conditions. The approach showed excellent enantioselectivities (87–99% ee) and moderate to good conversions (51–99%), albeit with moderate diastereoselectivities (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–17:1 dr). The synthetic utility of this protocol was demonstrated.
:1–17:1 dr). The synthetic utility of this protocol was demonstrated.
Organic boron is one of the most important synthetic blocks in modern organic syntheses.1 Complex compounds could be assembled and synthesized by modular and general strategies, attributed to the potential of the boron functional group in various transformations.2 Introducing the boron group to readily available starting materials is thus of great significance.3 The copper-catalyzed asymmetric boron conjugate addition to α,β-unsaturated compounds has been studied intensively for the last two decades,4 providing chiral β-substituted boron products that are versatile building blocks for chemical synthesis and drug development. A typical reaction process4b is that under the promotion of alkali additives, the copper salt complex reacts with a diboron reagent to form a copper boron intermediate, which adds to an unsaturated substrate via conjugate addition, resulting a β-boron substituted enol copper intermediate. Alcohol protonates the enol copper intermediate to release the final product and alkoxy copper. It is worth noting that the addition of alkali was crucial to the catalytic activity. Due to this issue, α-substituted α,β-unsaturated compounds were not suitable substrates as the epimerization of the product under alkaline conditions would lead to a mixture of diastereoisomers, which limits the practical applicability of these methods.5
We previously reported the highly enantio- and diastereoselective boron conjugate addition to α-functionalized α,β-unsaturated compounds, using a copper catalyst (CuCl/(S,R)-ppfa/AgNTf2) under base-free conditions.6 For α-amino unsaturated esters and α-alkyl unsaturated ketones, excellent enantioselectivities and moderate to good diastereoselectivities were obtained, accompanied by good conversions. However, α-alkyl α,β-unsaturated esters gave only 10% conversion under similar conditions due to their comparatively low reactivities (Fig. 1A). Thus, development of a better catalytic method for α-alkyl α,β-unsaturated esters is desirable. Herein, we report an optimized method to achieve the highly enantioselective boron conjugate addition to α-alkyl α,β-unsaturated esters, albeit with moderate diastereoselectivity (Fig. 1B).
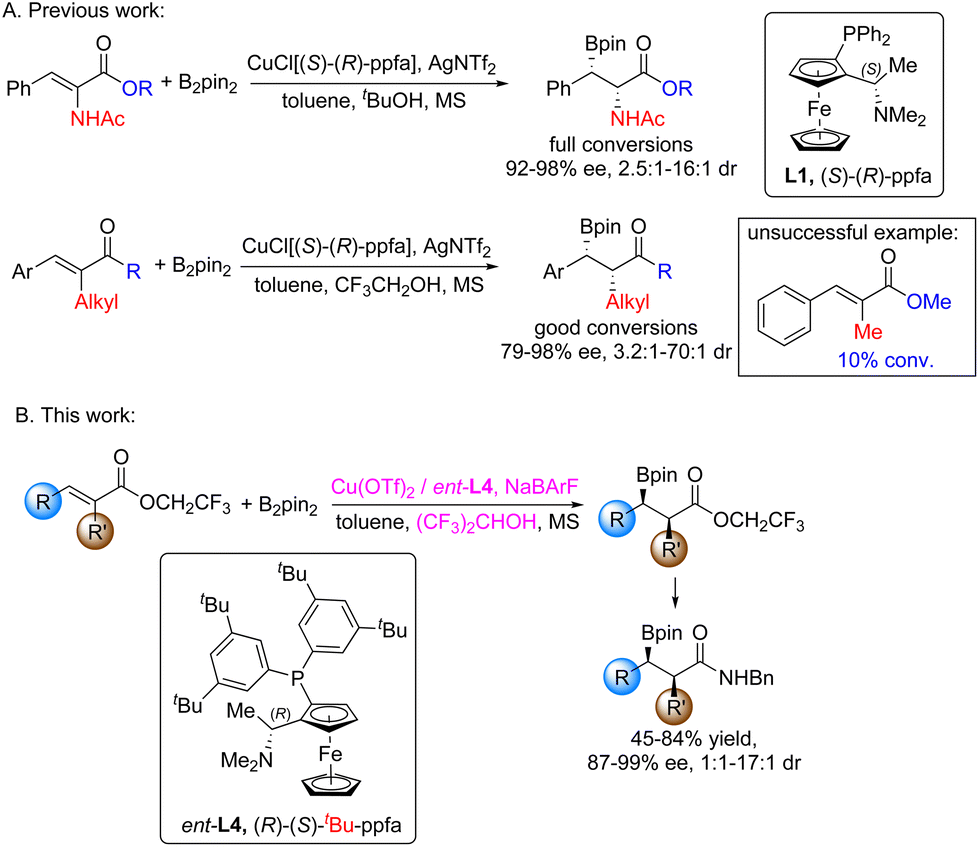 | ||
| Fig. 1 (A) Boron conjugate addition to α-amino α,β-unsaturated esters and α-alkyl α,β-unsaturated ketones (previous work). (B) Boron conjugate addition to α-alkyl α,β-unsaturated esters (this work). | ||
Compared with α,β-unsaturated ketones, the antibonding orbital (π*) energy of α,β-unsaturated esters is higher, while the electrons of the olefin bond are more active. In the copper-catalyzed boron conjugate addition reaction, the reactivity of α,β-unsaturated esters is lower. This shows that the π* orbital of the substrate plays a decisive role in the reaction rate: the substrate acts as the electron acceptor and the other key species (boryl copper compound) acts as the electron donor. In order to increase the reactivity to α,β-unsaturated esters, it is logical to increase the energy of electron donors and decrease the energy of the π* orbital of the substrate. Feringa et al. studied the relationship between the oxidative potential of copper(I) catalysts and the reactivity towards nonactivated substrates in the copper-catalyzed 1,4-addition of Grignard reagents, and they found that a more electron rich catalyst (easier to oxidize) would be beneficial for nonactivated substrates.7 They concluded that the difference in reactivity might be related to the energy match of the substrate and catalyst. This could explain that the amino phosphine ligand, (S,R)-ppfa, was proven to be superior to the biphosphine ligands in the copper-catalyzed boron conjugate addition to α-functionalized α,β-unsaturated compounds, as the amino ligand is purely electron donating, while the phosphine ligand could form π-backbonding.6 Moreover, Feringa et al. found that the presence of Mg2+ and Br− was important to the reactivity and selectivity.7 Similarly, we found that additives played critical roles in the reactivity and selectivity, and acidic alcohol additives [CF3CH2OH, (CF3)2CHOH] were proven to be beneficial for the reactivity.6
Inspired by the above analyses, a systematic optimization of the copper precursor, ligand, additive, etc., was performed on the basis of our previous report. We initiated our study with 2,2,2-trifluoroethyl (E)-2-methyl-3-phenylacrylate (1a) as the template substrate which has a lower π* energy than the normal alkyl esters. The effect of the anion exchange reagent as an additive was studied first, with CuCl as the catalyst precursor and tert-butyl substituted amino phosphine (L4) as the ligand (Table 1, entries 1–4). The bulky noncoordinating counterions, such as NTf2− and BArF−, could initiate the reaction with moderate conversion. The relatively low diastereoselectivity (1.6:1 dr) obtained by using AgNTf2 might be attributed to its strong Lewis acidity. For comparison, the weakly coordinating counterions, such as BF4− and PF6−, gave nearly no conversion.8 While NaBArF proved to be superior to other anion exchange reagents, we then studied the effect of alcohol additives (Table 1, entries 5 and 6). The more acidic alcohol, (CF3)2CHOH, gave a higher conversion, albeit with lower diastereoselectivity (52% conv., 6:1 dr). In the meantime, when ethanol was applied to the system, nearly no conversion was observed. This indicated that the proton on the alcohol was involved in the transition state of the decisive step, probably similar to Mg2+ in Feringa's system.7 Then we turned to examine copper precursors, including univalent and divalent copper salts with different counterions (Table 1, entries 7–11). Copper salts with different valence states, Cu(OTf)2 and CuOTf, gave close results with good conversion and diastereoselectivity and excellent enantioselectivity (entries 7 and 8). This might be attributed to the oxidizability of Cu(OTf)2 and the reducibility of diboron compounds, which led to the in situ formation of the Cu(I) catalyst. The copper carboxylates were less reactive (entries 9 and 10), probably due to the stronger coordination ability of carboxylates. The enolate type copper precursor, Cu(acac)2, was also reactive and gave similar results to CuOTf (entry 11). With Cu(OTf)2 as the optimal precursor, we tested anion exchange reagents as additives again (Table 1, entries 12–16). The coordinating anions, such as F− (entries 12 and 13) and tBuCO2− (entry 15), were harmful to the reactivity. When a common activator, NaOtBu (entry 14), was used in copper-catalyzed boron conjugate addition, only 20% conversion was obtained as well as non-diastereoselectivity (1.2:1 dr). The lower conversion with NaOtBu disclosed that the activation mode with bulky noncoordinating counterions might be different from that of the base additives: while the NaOtBu additive proved to produce the intermediate CuOtBu which led to CuBpin,9 the NaBArF additive might provide the CuBpin(HFIP) intermediate instead which involves the function of acidic HFIP. The precursor itself, Cu(OTf)2, could also initiate the reaction, albeit with lower conversion than that with NaBArF (entry 16).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R | Cu precursor | L* | Additive | Alcohol | Conv. | dr (syn/anti) | ee (%) |
|
a Conditions: 1a (0.122 mmol), B2pin2 (2 equiv.), Cu precursor (0.012 mmol, 10 mol%), L* (0.018 mmol, 15 mol%), alcohol (1.22 mmol), additive (0.024 mmol), 4 Å MS (30 mg), toluene (1 mL), rt, 48 h. Conversions and diastereoselectivities were determined by 1H NMR analyses of the crude mixtures. The enantiomeric excess was determined by transforming 2 into the corresponding alcohol using NaBO3·4H2O as the oxidant4e and then analyzed by chiral HPLC.
b A E/Z = 3:1 mixture of the substrate was used.
|
||||||||
| 1 | OCH2CF3 (1a) | CuCl | L4 | AgNTf2 | CF3CH2OH | 46% | 1.6:1 |
ND |
| 2 | OCH2CF3 | CuCl | L4 | NaBArF | CF3CH2OH | 42% | 10:1 |
ND |
| 3 | OCH2CF3 | CuCl | L4 | AgBF4 | CF3CH2OH | <5% | — | — |
| 4 | OCH2CF3 | CuCl | L4 | AgPF6 | CF3CH2OH | Trace | — | — |
| 5 | OCH2CF3 | CuCl | L4 | NaBArF | (CF3)2CHOH | 52% | 6:1 |
ND |
| 6 | OCH2CF3 | CuCl | L4 | NaBArF | CH3CH2OH | Trace | — | — |
| 7 | OCH 2 CF 3 | Cu(OTf) 2 | L4 | NaBArF | (CF 3 ) 2 CHOH | 84% |
7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
97 |
| 8 | OCH2CF3 | (CuOTf)·1/2 toluene | L4 | NaBArF | (CF3)2CHOH | 80% | 5:1 |
98 |
| 9 | OCH2CF3 | CuTc | L4 | NaBArF | (CF3)2CHOH | 45% | 6:1 |
98 |
| 10 | OCH2CF3 | CuOAc | L4 | NaBArF | (CF3)2CHOH | 47% | 3:1 |
91 |
| 11 | OCH2CF3 | Cu(acac)2 | L4 | NaBArF | (CF3)2CHOH | 79% | 5:1 |
>99 |
| 12 | OCH2CF3 | Cu(OTf)2 | L4 | CsF | (CF3)2CHOH | 22% | 3:1 |
ND |
| 13 | OCH2CF3 | Cu(OTf)2 | L4 | KF | (CF3)2CHOH | 25% | 3:1 |
ND |
| 14 | OCH2CF3 | Cu(OTf)2 | L4 | NaOtBu | (CF3)2CHOH | 20% | 1.2:1 |
ND |
| 15 | OCH2CF3 | Cu(OTf)2 | L4 | CsOPiv | (CF3)2CHOH | Trace | — | — |
| 16 | OCH2CF3 | Cu(OTf)2 | L4 | — | (CF3)2CHOH | 44% | 3:1 |
ND |
| 17 | OCH2CF3 | Cu(OTf)2 | L1 | NaBArF | (CF3)2CHOH | 67% | 4:1 |
90 |
| 18 | OCH2CF3 | Cu(OTf)2 | L2 | NaBArF | (CF3)2CHOH | 36% | 5:1 |
89 |
| 19 | OCH2CF3 | Cu(OTf)2 | L3 | NaBArF | (CF3)2CHOH | 63% | 6:1 |
90 |
| 20 | OCH2CF3 | Cu(OTf)2 | L5 | NaBArF | (CF3)2CHOH | 62% | 10:1 |
97 |
| 21b | OCH2CF3 | Cu(OTf)2 | L4 | NaBArF | (CF3)2CHOH | 89% | 5:1 |
ND |
| 22 | OEt (1b) | Cu(OTf)2 | L4 | NaBArF | (CF3)2CHOH | 42% | 10:1 |
91 |
| 23 | OPh (1c) | Cu(OTf)2 | L4 | NaBArF | (CF3)2CHOH | >99% | 1.5:1 |
94/94 |
| 24 | NHC6H4(p-OMe) (1d) | Cu(OTf)2 | L4 | NaBArF | (CF3)2CHOH | 4% | — | — |
We then studied the substitution effect on chiral ligands (Table 1, entries 17–20). When increasing the volume of groups on phosphine atoms, the enantioselectivity and diastereoselectivity could be regularly enhanced. However, there was no obvious law on the effect on conversion. This may be because the copper catalyst was not stable, and the conversion was determined by both the lifetime of the catalyst and the reaction rate. A less bulky catalyst might favour the reaction rate; however, it might be less stable at the same time. A piece of evidence about the relationship between the reaction rate and the catalyst's lifetime was that the conversion could be enhanced at lower temperature (94% conv. at 0 °C, see the ESI† for details), and this was probably due to the higher lifetime of the catalyst at lower temperature, albeit with a slower reaction rate. The effect of the configuration of the substrate on the diastereoselectivity was investigated, and a slightly lower diastereoselectivity was obtained with the E/Z = 3:1 mixture (Table 1, entry 21). With the optimal conditions (Table 1, entry 7), we finally compared the types of substrates (Table 1, entries 22–24). The ethyl ester (1b) showed lower reactivity and enantioselectivity, but higher diastereoselectivity. The phenolic ester (1c) gave full conversion, albeit with low diastereoselectivity (1.5:1 dr). The amide type substrate was also tested (1d) and only 4% conversion was found. These results were consistent with Feringa's conclusion that the reactivity could be related to the energy match of the substrate and catalyst.7
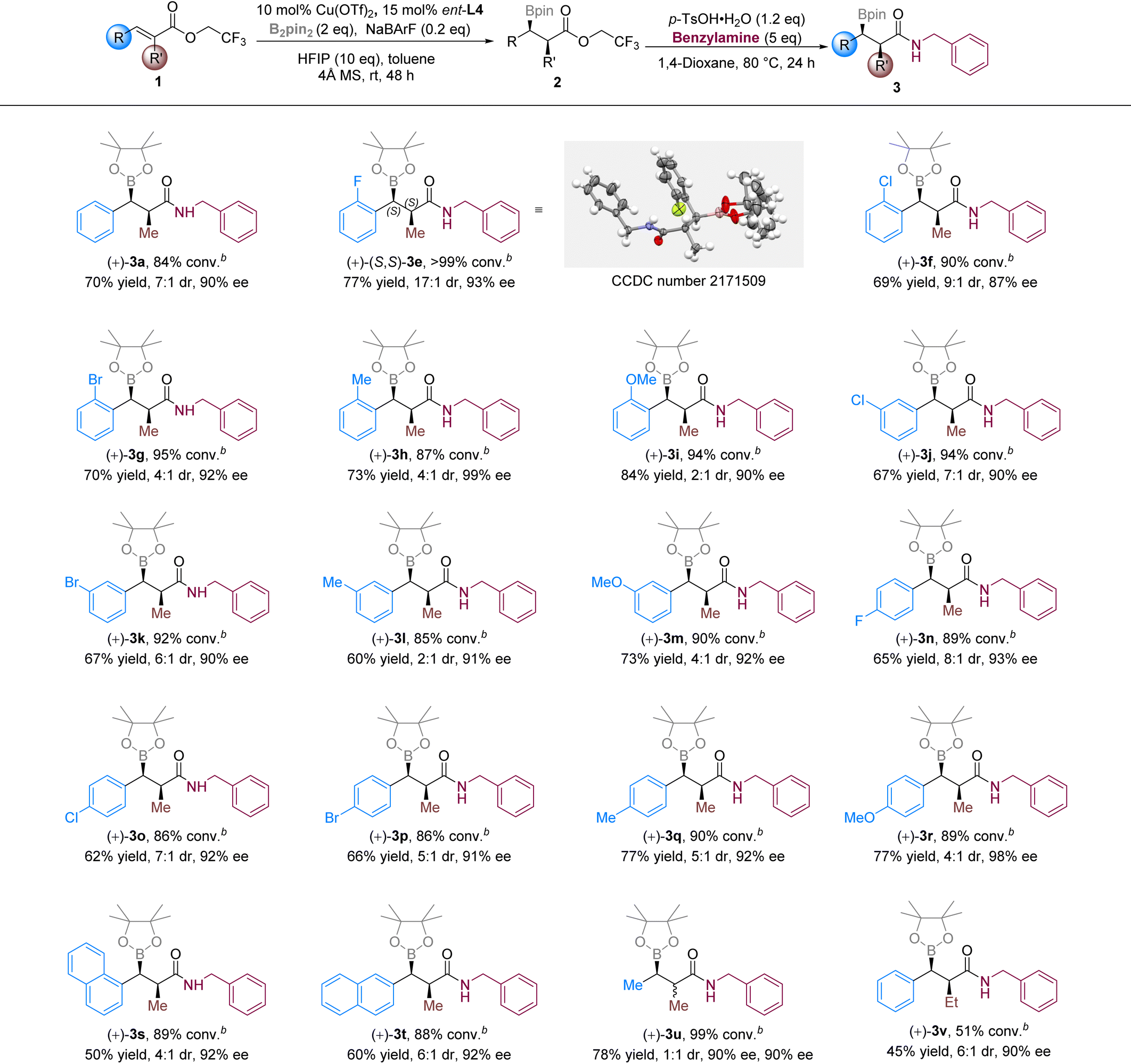
With the optimized conditions in hand, we sought to explore the scope of substrates with α-methyl α,β-unsaturated trifluoroethyl esters (Table 2). As the polarity of 2 was very small, we converted it into the corresponding amide (3a, 3e–v) for separation and identification. Good conversions (84–99%) and enantioselectivities (87–99% ee) were obtained for all the α-methyl substrates, while the α-ethyl substrate (1v) gave only moderate conversion (51%). Changing the substitution group and its position on the phenyl ring did not affect the reactivity significantly (3e–t). However, poor diastereoselectivities were observed in some cases (3i and 3l) without obvious regularity. For the β-methyl substrate, no diastereoselectivity was found (3u, 1:1 dr). It disclosed that β-substitution affected the face selectivity in the protonation process that determined the diastereoselectivity. The absolute configuration was determined by an X-ray study of the crystal of 3e, and the kinetically selective syn-product was dominant in the reaction.
| a Conditions: 1 (0.122 mmol), B2pin2 (0.244 mmol, 2 equiv.), Cu(OTf)2 (0.012mmol, 10 mol%), ent-L4 (0.018 mmol, 15 mol%), HFIP (0.13 mL, 1.22 mmol), NaBArF (0.024 mmol), 4 Å MS (30 mg), toluene (1 mL), rt, 48 h. Isolated yields of 3 are given. Diastereoselectivities were determined by 1H NMR analysis of 3. The enantiomeric excess was determined by chiral HPLC. b Conversions were determined by 1H NMR analyses of the crude mixtures after the boron conjugate addition step (those of 1). |
|---|
|
To demonstrate the utility of our method in organic synthesis, the template substrate 1a was transformed into 2a on a 500 mg scale under the optimized conditions (Fig. 2). The trifluoroethyl ester product could be converted directly into the benzyl amide (3a) under the promotion of p-toluenesulfonic acid, or be hydrolyzed into the corresponding acid and then transformed into the amide (5) with the promotion of the condensation agent. The trifluoroborate 4 could be obtained with KHF2 with an enhanced diastereomeric ratio (8:1 dr from 5:1 dr) from 3a. A typical Zweifel alkenylation reaction10 was performed with 5, and the enantioenriched alkene product 6 was obtained after the well-known stereospecific reaction.
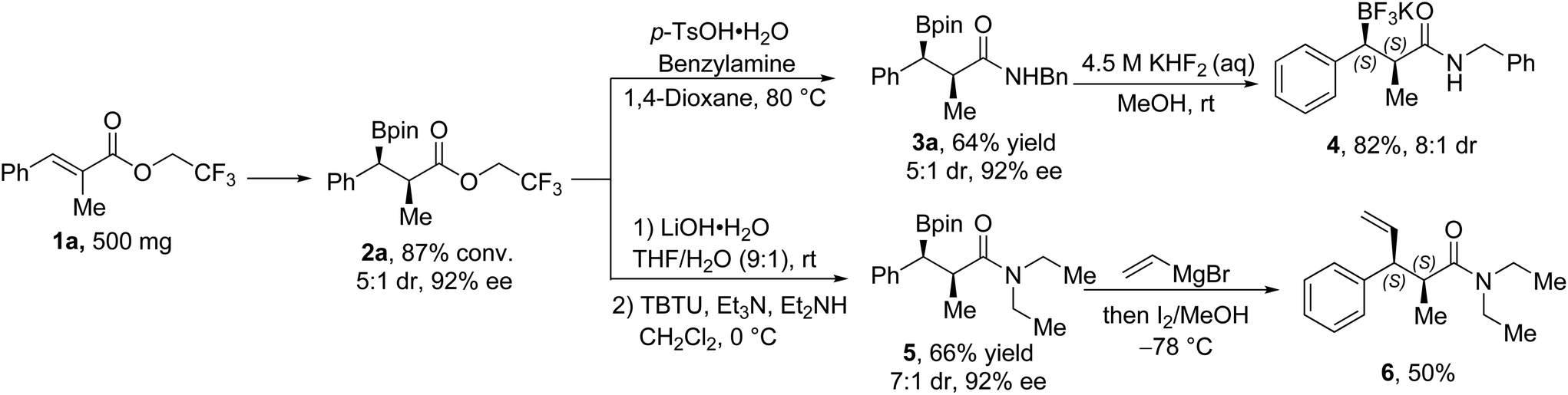 | ||
| Fig. 2 Transformation applications. | ||
As the catalytic mode in this report was probably different from the previous copper-catalyzed boron conjugate addition promoted by a base additive, we proposed a synergistic catalytic mode, involving the copper catalyst and the promotion with an acidic alcohol additive, on the basis of our findings and Feringa's report7 (Fig. 3). The active copper species, complex A, is produced after reduction and anion exchange. It interacts with the substrate on two sites: the proton activates the carbonyl groups; in the meantime, the copper catalyst contributes its d electron pair to the π* orbital of the substrate. These interactions are probably synergistic and the transition state in this step is described as a π-complex. The σ-complex (Cu(III) species) is then formed. The product enolate is quickly released as the σ-complex is highly unstable. The diastereomeric ratio is determined via the controlled protonation, in which the volume of the substitution at the β-position affects the efficiency of face selection.6 Complex A is regenerated after the reaction between alkoxy copper and diboron.
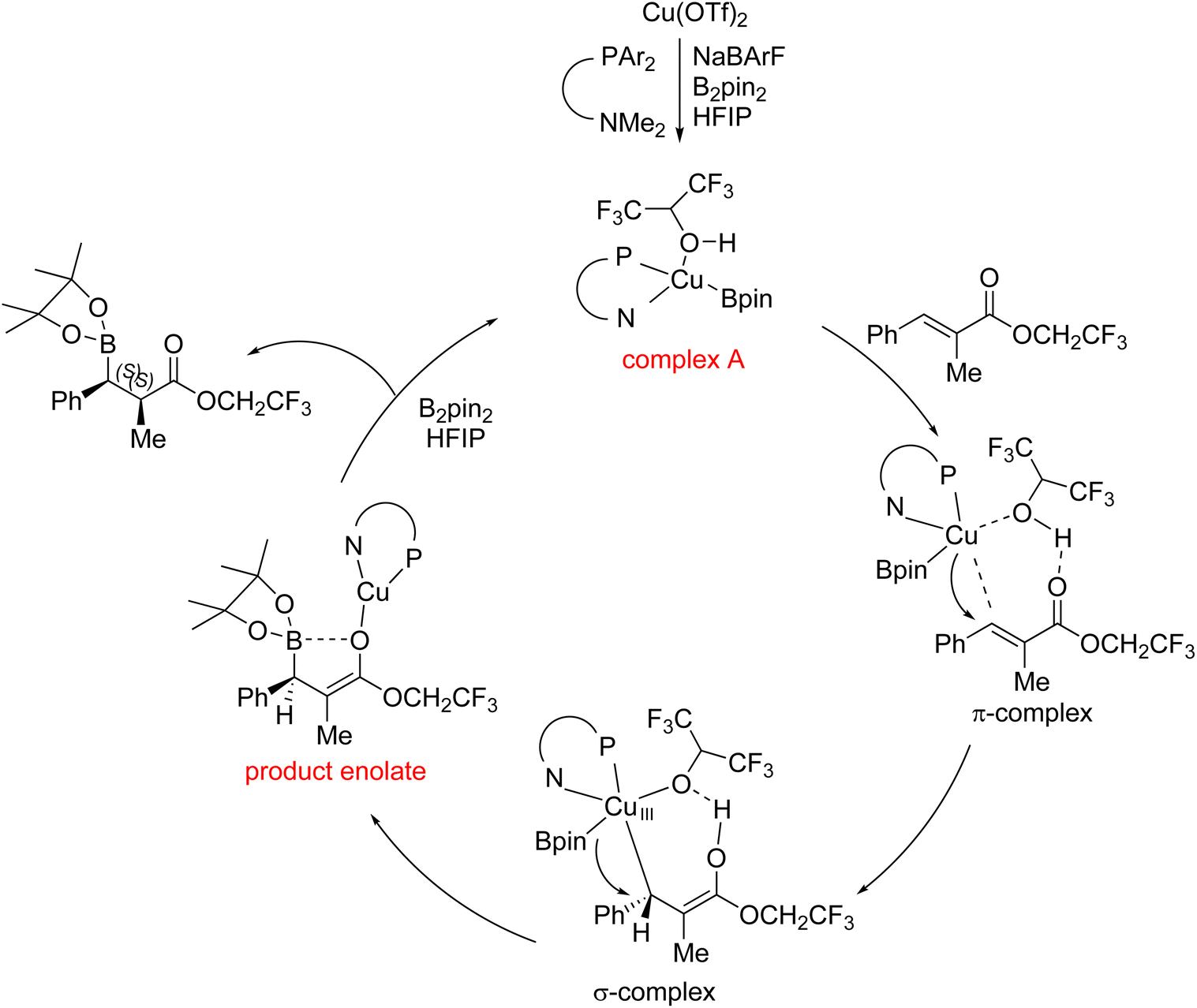 | ||
| Fig. 3 Proposed catalytic mode. | ||
Conclusions
In conclusion, a copper-catalyzed enantio- and diastereoselective boron conjugate addition to α-alkyl α,β-unsaturated esters under base-free conditions was developed. A systematic optimization of the copper precursor, ligand, additive, etc., was performed. Unlike the common base-promoted copper-catalyzed boron conjugate addition, the bulky noncoordinating counterions and acidic alcohols proved to be crucial to initiate the reaction. Excellent enantioselectivities and good conversions were obtained, albeit with moderate diastereoselectivities. The synthetic utility of this protocol was also demonstrated. A synergistic catalytic mode, including the copper catalyst and the promotion with an acidic alcohol additive, was proposed on the basis of our results and previous reports.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NSFC (21702164) for financial support. The project was also supported by the Guangdong Provincial Key Laboratory of Catalysis (Grant No. 2020B121201002).References
- For selected examples of chiral organic boron compounds, see: (a) T. Awano, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2011, 133, 20738–20741 CrossRef CAS; (b) M. Tortosa, Angew. Chem., Int. Ed., 2011, 50, 3950–3953 CrossRef CAS; (c) T. Ohmura, T. Awano and M. Suginome, J. Am. Chem. Soc., 2010, 132, 13191–13193 CrossRef CAS PubMed; (d) F. Meng, K. P. McGrath and A. H. Hoveyda, Nature, 2014, 513, 367–374 CrossRef CAS; (e) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584–589 CrossRef CAS PubMed.
- For selected examples of syntheses with chiral organic boron compounds, see: (a) S. L. Poe and J. P. Morken, Angew. Chem., Int. Ed., 2011, 50, 4189–4192 CrossRef CAS; (b) R. P. Sonawane, V. Jheengut, C. Rabalakos, R. Larouche-Gauthier, H. K. Scott and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 3760–3763 CrossRef CAS PubMed; (c) S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 17096–17098 CrossRef CAS; (d) J. Chang, H. Lee and D. G. Hall, J. Am. Chem. Soc., 2010, 132, 5544–5545 CrossRef; (e) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207–2293 CrossRef CAS.
- For selected examples of the syntheses of chiral organic boron compounds, see: (a) J. K. Park, H. H. Lackey, B. A. Ondrusek and D. T. McQuade, J. Am. Chem. Soc., 2011, 133, 2410–2413 CrossRef CAS PubMed; (b) D. Imao, B. W. Glasspoole, V. S. Laberge and C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024–5025 CrossRef CAS; (c) E. Hupe, P. Marek and I. Knochel, Org. Lett., 2002, 4, 2861–2863 CrossRef CAS PubMed.
- For selected reviews, see: (a) A. Schiffner, K. Müther and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 1194–1196 CrossRef; (b) J. Cid, H. Gulyas, J. J. Carbo and E. Fernandez, Chem. Soc. Rev., 2012, 41, 3558–3570 RSC. For selected examples, see: (c) J.-E. Lee and J. Yun, Angew. Chem., Int. Ed., 2008, 47, 145–147 CrossRef CAS; (d) I.-H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664–11665 CrossRef CAS PubMed; (e) K.-s. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253–7255 CrossRef CAS; (f) G. Palau-Lluch and E. Fernandez, Adv. Synth. Catal., 2013, 355, 1464–1470 CrossRef CAS; (g) Y. Luo, I. D. Roy, A. G. E. Madec and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 4186–4190 CrossRef CAS PubMed; (h) S. Kobayashi, P. Xu, T. Endo, M. Ueno and T. Kitanosono, Angew. Chem., Int. Ed., 2012, 51, 12763–12766 CrossRef CAS; (i) G. Stavber and Z. Časar, Appl. Organomet. Chem., 2013, 27, 159–165 CrossRef CAS.
- For acyclic substrates, non-diastereoselectivity could be achieved, see: (a) V. Lillo, A. Prieto, A. Bonet, M. M. Diaz-Requejo, J. Ramirez, P. J. Perez and E. Fernandez, Organometallics, 2009, 28, 659–662 CrossRef CAS; (b) Z.-T. He, Y.-S. Zhao, P. Tian, C.-C. Wang, H.-Q. Dong and G.-Q. Lin, Org. Lett., 2014, 16, 1426–1429 CrossRef CAS. For cyclic substrates, good diastereoselectivity could be achieved, see: (c) K. Kubota, K. Hayama, H. Iwamoto and H. Ito, Angew. Chem., Int. Ed., 2015, 54, 8809–8813 CrossRef CAS; (d) L. Chen, J.-J. Shen, Q. Gao and S. Xu, Chem. Sci., 2018, 9, 5855–5859 RSC; (e) H. A. Clement, M. Boghi, R. M. McDonald, L. Bernier, J. W. Coe, W. Farrell, C. J. Helal, M. R. Reese, N. W. Sach, J. C. Lee and D. G. Hall, Angew. Chem., Int. Ed., 2019, 58, 18405–18409 CrossRef CAS; (f) K. Hayama, R. Kojima, K. Kubota and H. Ito, Org. Lett., 2020, 22, 739–744 CrossRef CAS PubMed.
- J.-B. Xie, S. Lin, S. Qiao and G. Li, Org. Lett., 2016, 18, 3926–3929 CrossRef CAS.
- S. R. Harutyunyan, F. López, W. R. Browne, A. Correa, D. Peña, R. Badorrey, A. Meetsma, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 9103–9118 CrossRef CAS PubMed.
- M. Brookhart, B. Grant and A. F. Volpe, Organometallics, 1992, 11, 3920–3922 CrossRef CAS.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036–11037 CrossRef CAS.
- (a) G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652–3653 CrossRef CAS; (b) Y. Wang, A. Noble, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 4270–4274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2171509. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01928k |
| This journal is © The Royal Society of Chemistry 2023 |