
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The arylvinylpyrimidine scaffold: a tunable platform for luminescent and optical materials
Sylvain
Achelle
 *a,
Julián
Rodríguez-López
*b and
Françoise
Robin-le Guen
a
*a,
Julián
Rodríguez-López
*b and
Françoise
Robin-le Guen
a
aUniv. Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: sylvain.achelle@univ-rennes1.fr
bUniversidad de Castilla-La Mancha, Área de Química Orgánica, Facultad de Ciencias y Technologías Químicas, 13071 Ciudad Real, Spain. E-mail: julian.rodriguez@uclm.es
First published on 10th November 2022
Abstract
The incorporation of electron-withdrawing pyrimidine rings within π-extended systems allows access to a wide variety of fluorescent push–pull molecules that display emission properties highly sensitive to external stimuli. A suitable design of these compounds leads to interesting materials for a variety of optoelectronic applications. In this context, a vast number of arylvinylpyrimidine-based chromophores have been extensively studied during the last two decades. Along with the main synthetic pathways, this review summarizes the photophysical features of these active compounds having great potential and their most important applications as sensors and luminescence materials.
 Sylvain Achelle | Sylvain Achelle received his PhD in organic chemistry in 2007 from INSA Rouen (France). After two post-doctoral stays at the Universidad de Castilla-La Mancha (Spain) and Institut Curie (France), he was recruited as associate professor at IUT Lannion, University of Rennes 1, in 2010. He obtained the habilitation to direct research in 2014. His main research topics include the luminescence and nonlinear optical properties of organic and organometallic heterocyclic derivatives. He has published 84 scientific articles (h-index: 31 according to the Web of Science) and he is currently associate editor of the journal Dyes & Pigments. |
 Julián Rodríguez-López | Julián Rodríguez López studied chemistry at the Universidad Complutense de Madrid (UCM) and obtained his PhD with honors (cum laude) in 1990. He was a Fulbright postdoctoral fellow at the University of Texas at Austin for one year, under the supervision of Professor Philip Magnus. After successive positions at the UCM, he joined the Universidad de Castilla-La Mancha at Ciudad Real in 1995, where he is currently full professor in Organic Chemistry. His research focuses on the design and synthesis and self-assembly of novel light-emitting π-conjugated systems in order to control their photophysical properties for various (opto)electronic applications. |
 Françoise Robin-le Guen | Françoise Robin-le Guen received her PhD in organometallic chemistry from the University of Bretagne Occidentale (Brest, France). She is currently full professor at IUT Lannion, University of Rennes 1 and head of the heterocyclic organometallic teams at the Chemical Sciences Institute of Rennes. Her main research topics are focused on the design of π-conjugated heterocyclic and organometallic systems for dye-sensitized solar cells, luminescence and nonlinear optics. |
1. Introduction
Intramolecular charge transfer (ICT) is one of the key elements that govern the properties of organic optical materials.1,2 ICT occurs when an electron-donating group (D) is linked to an electron-attracting fragment (A) via a π-conjugated linker (D–π-A, also called push–pull structures). Such structures are characterized by their color, with absorption bands in the visible spectral range that experience a significant red shift with ICT enhancement. Moreover, some push–pull chromophores exhibit intense photoluminescence and nonlinear optical properties,1 finding widespread application as fluorescence sensors,3 emitters for light-emitting devices,4 organic field effect transistors,5 photovoltaic materials,6 two-photon absorption chromophores,7 and second harmonic generation materials.8The pyrimidine ring is a six membered aromatic heterocycle with two nitrogen atoms at positions 1 and 3. Due to the π-deficient character of this heterocycle, the pyrimidinyl fragments act as electron-withdrawing groups. In the case of pyrimidin-2-yl, pyrimidin-4-yl, and pyrimidin-6-yl derivatives, the two nitrogen atoms are in a conjugated position relative to the substituent, making them stronger electron-withdrawing groups than pyridinyl or other diazinyl rings.9 This electron-withdrawing character can be tuned by engaging the electron lone pairs of the nitrogen atoms upon protonation, complexation, alkylation, or hydrogen bonding. Pyrimidine derivatives substituted with electron-donating fragments through π-conjugated linkers are highly fluorescent and show emission properties highly sensitive to such external stimuli. As a consequence, pyrimidine chromophores are suitable candidates for various optoelectronic applications.10
In this context, arylvinylpyrimidine chromophores were intensively studied in the early 2000s. Since then, more than one-hundred linear 2-arylvinyl and 4-arylvinyl, V-shaped 4,6-bis(arylvinyl) and 2,4-bis(arylvinyl), and more recently Y-shaped 2,4,6-tris(arylvinyl)pyrimidines have been designed.9 In these structures, substituents on the aryl moieties or at different positions on the pyrimidine ring have a significant impact on their photophysical behavior (Fig. 1).
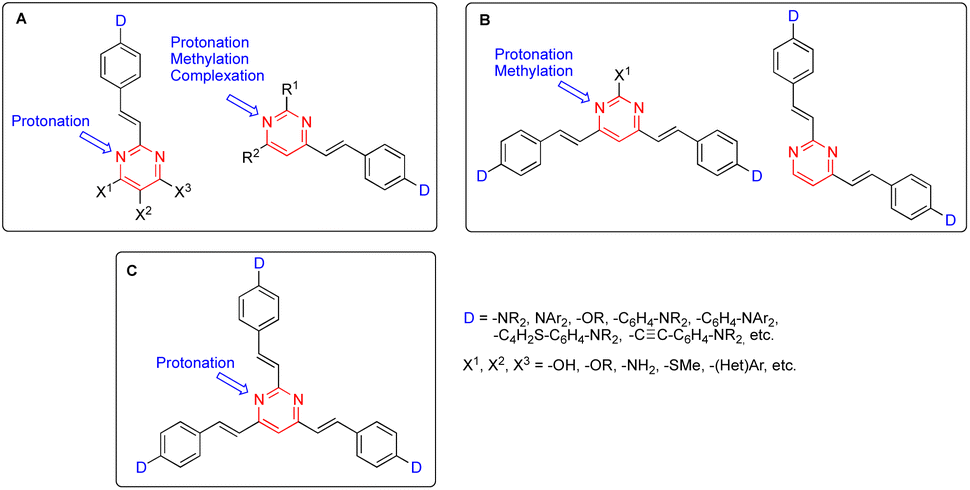 | ||
| Fig. 1 General structures of 2-arylvinyl and 4-arylvinyl (A), 4,6-bis(arylvinyl) and 2,4-bis(arylvinyl) (B), and 2,4,6-tris(arylvinyl)pyrimidines (C). | ||
In this review, we will give a general outline of the synthetic methodologies, photophysical properties, and applications of arylvinylpyrimidine chromophores. Coverage of the large number of reported structures is beyond the scope of this paper and, therefore, special emphasis will be placed on those compounds with the greatest potential.
2. Synthetic approaches
The arylvinylpyrimidine scaffold is usually obtained by functionalizing previously formed pyrimidine rings. Although the Suzuki cross-coupling reaction of chloropyrimidines with potassium alkenyltrifluoroborates has been used for their synthesis,11 the main methodology is the Knoevenagel condensation of aromatic aldehydes with methylpyrimidine derivatives (Scheme 1).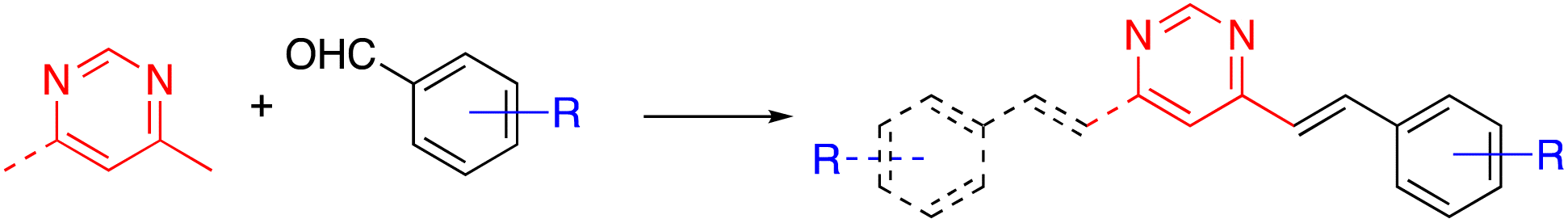 | ||
| Scheme 1 Synthesis of arylvinylpyrimidines from methylpyrimidines by the Knoevenagel condensation. | ||
In particular, Vanden Eynde and coworkers developed a methodology based on the use of concentrated aqueous NaOH solution as the solvent and Aliquat 336® as the phase transfer catalyst.12 The main advantages of this experimental protocol include the wide range of starting materials available in the market, the use of more environmentally friendly conditions in most cases, and the simple process of treatment and purification of the final products, which often consists of a simple recrystallization method. The method is not only straightforward but also rather economical due to the absence of organic solvents.
This approach initially allowed the synthesis of 4-arylvinyl and 4,6-bis(arylvinyl)pyrimidines,13–15 although it was later extended to the preparation of 2-arylvinylpyrimidines.16 The products were formed exclusively as E-stereoisomers. The stereochemistry of the double bonds was unambiguously determined from the coupling constant of the vinylic proton in the 1H NMR spectra (3J(H–H) ≈ 16 Hz).13,14 This method tolerated a large variety of substituents on the benzaldehyde precursor. When 4-trifluoromethylbenzaldehyde was used, the reaction required a less concentrated NaOH solution to prevent the Cannizzaro side reaction of the aldehyde.14 On the other hand, formyl-substituted arylvinylpyrimidines could be obtained from 4-(diethoxymethyl)benzaldehyde followed by deprotection of the acetal group.12b Bromoderivatives 1 and 2 (Fig. 2) were the key intermediates that could be used for further functionalization by palladium-catalyzed Suzuki–Miyaura or Sonogashira cross coupling reactions.13,14,17
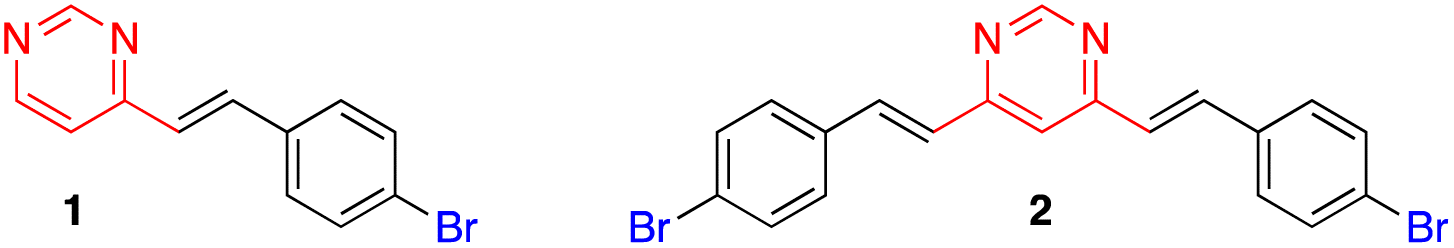 | ||
| Fig. 2 Structures of bromo-substituted arylvinylpyrimidines 1 and 2. | ||
Depending on the substituent of the benzaldehyde derivative, it may be necessary to use another base/solvent pair for the Knoevenagel condensation, such as KButO in THF,18 KButO in DMF,19 KButO without solvent (ground solid),20 KOH in DMSO,21 or NaH in THF.22 In some cases, this reaction was carried out under acidic conditions (HCl in EtOH).23
In general, it is difficult to obtain mono-arylvinylpyrimidines starting from 4,6-dimethylpyrimidine.14,20b Nevertheless, when the dimethylpyrimidine fragment was embedded in a poly(methyl methacrylate)-based copolymer, surprisingly only monocondensation products were obtained.24 On the other hand, the Knoevenagel condensation of 4,6-dimethylpyrimidines with terephthalaldehyde or other dialdehydes allowed access to poly(arylvinylpyrimidine) macromolecules.25,26
While 2-methyl-, 4-methyl- and 4,6-dimethylpyrimidine are commercially available and cost-effective chemicals, the accessibility of 2,4-dimethyl- and 2,4,6-trimethylpyrimidine is much more limited. Thus, the preparation of 2,4-bis(arylvinyl) and 2,4,6-tris(arylvinyl)pyrimidines has been approached from 2-chloro-4-methylpyrimidine and 2-chloro-4,6-dimethylpyrimidine, respectively, by means of a combination of Suzuki–Miyaura cross coupling and Knoevenagel condensation reactions (Scheme 2).27 2,4,6-Tris(arylvinyl)pyrimidines with three different arms could be obtained starting from 2,4-dichloro-6-methylpyrimidine. The higher reactivity of the C4 over the C2 carbon allowed to carry out two Suzuki–Miyaura cross coupling reactions in a sequential manner with excellent selectivity, followed by the Knoevenagel condensation with the corresponding aromatic aldehyde in the methyl group at the C6 position.
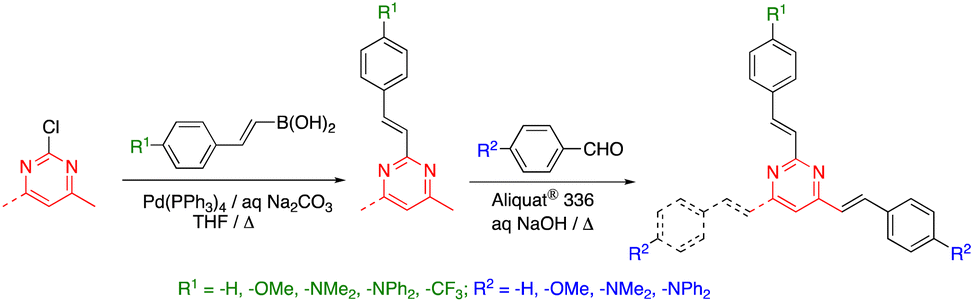 | ||
| Scheme 2 Synthesis of 2,4-bis(arylvinyl) and 2,4,6-tris(arylvinyl)pyrimidines. | ||
Arylvinylpyrimidine derivatives are bench stable compounds in the solid state. Nevertheless, partial reversible protonation could be observed in chlorinated solvents as a result of photogeneration of HCl. Furthermore, some Z/E isomerization was also reported when in the solution was left for a few days.13,14 Solid state photodimerization has also been reported to give rise to cyclobutane derivatives.22
3. Structure–emission property relationships
Due to their more extended π-conjugated linkers, arylvinylpyrimidines exhibit red-shifted absorption and emission relative to their phenylpyrimidine analogues, although the photoluminescence quantum yield (PLQY) is usually lower in moderately polar solvents.9 They generally must be substituted with strong electron-donating amino groups to show significant emission. Nevertheless, alkoxy-substituted arylvinylpyrimidines are also highly emissive.9 The emission properties of a series of dimethylamino-substituted styrylpyrimidines are presented in Fig. 3. 4-(p-N,N-Dimethylaminostyryl)pyrimidine 3 displays cyan emission with moderate PLQY (ΦF = 0.15) in CH2Cl2. When the pyrimidine was substituted at position 2 (compound 4) instead of position 4, a blue shift of emission was observed with a dramatic decrease in the PLQY. In contrast, a bathochromic shift of emission to green with a significant increase of the PLQY was observed for the 4,6-disubstituted pyrimidine 5. The emission of 2,4,6-stryrylpyrimidine 6 was further red-shifted but with a slightly lower PLQY than compound 5. The extension of the π-conjugated linker between the pyrimidin-4-yl fragment and the dimethylamino group on chromophores 7, 8, and 9 caused a significant bathochromic shift in the emission and an increase in the PLQY relative to 3.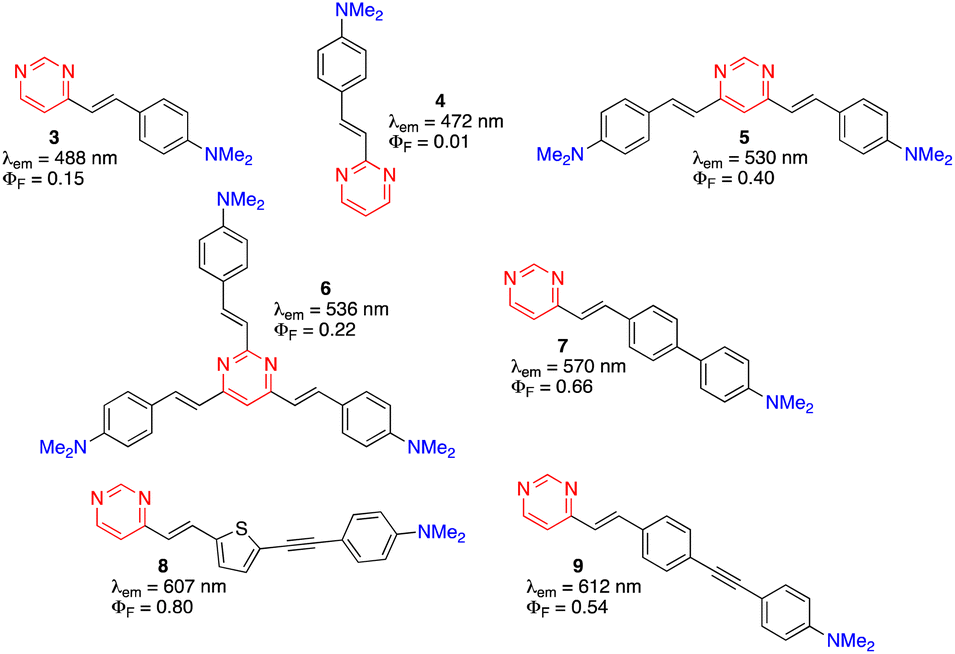 | ||
| Fig. 3 Structures and emission properties of dimethylamino-substituted styrylpyrimidines 3–9 in CH2Cl2.13,14,17,27,28 | ||
The PLQY of 2,4,6-tris(arylvinyl)pyrimidines is highly dependent on the nature of the substituent on the C2 arm.27 In the diphenylamino series (Fig. 4), compound 9a exhibited similar photoluminescence properties (λem, PLQY) as 4,6-distyrylpyrimidine 10 in CH2Cl2. When the C2 arm contained a stronger electron-donating dimethylamino group (9b), a red-shifted emission was observed with a dramatic decrease in the PLQY (in this case the ICT occurs mainly along the C2 arm). A bathochromic shift was also achieved when the C2 arm was left unsubstituted (9c) or substituted with an electron-withdrawing trifluoromethyl group (9d), albeit with a significant increase in the PLQY up to 1.00. In this case, the ICT occured mainly in the C4 and C6 arms and the electron-attracting character of the pyrimidine nucleus was reinforced by the substituent on the C2 arm.
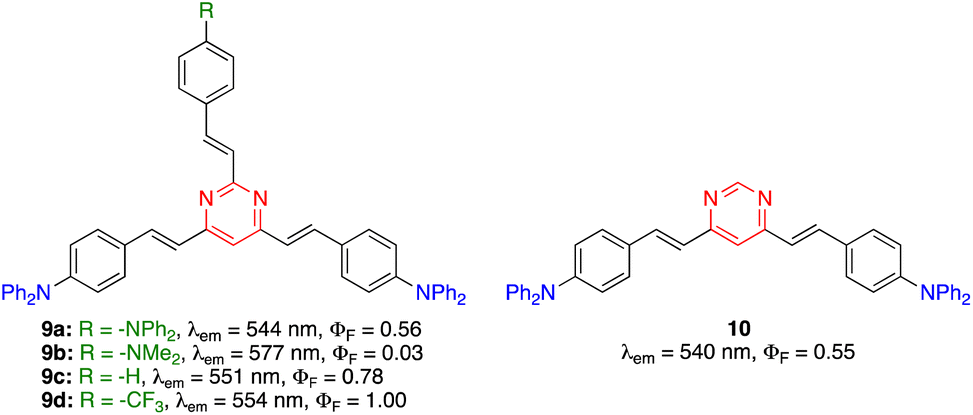 | ||
| Fig. 4 Structures and photophysical properties of diphenylamino-substituted styrylpyrimidines 9 and 10 in CH2Cl2.27 | ||
Some arylvinylpyrimidines are also highly luminescent in the solid state, in particular the diphenylamino derivatives. For example, compound 10 exhibits green emission in a thin film (ΦF = 0.18)29 and compound 11 emits yellow light in the KBr matrix (Fig. 5).30
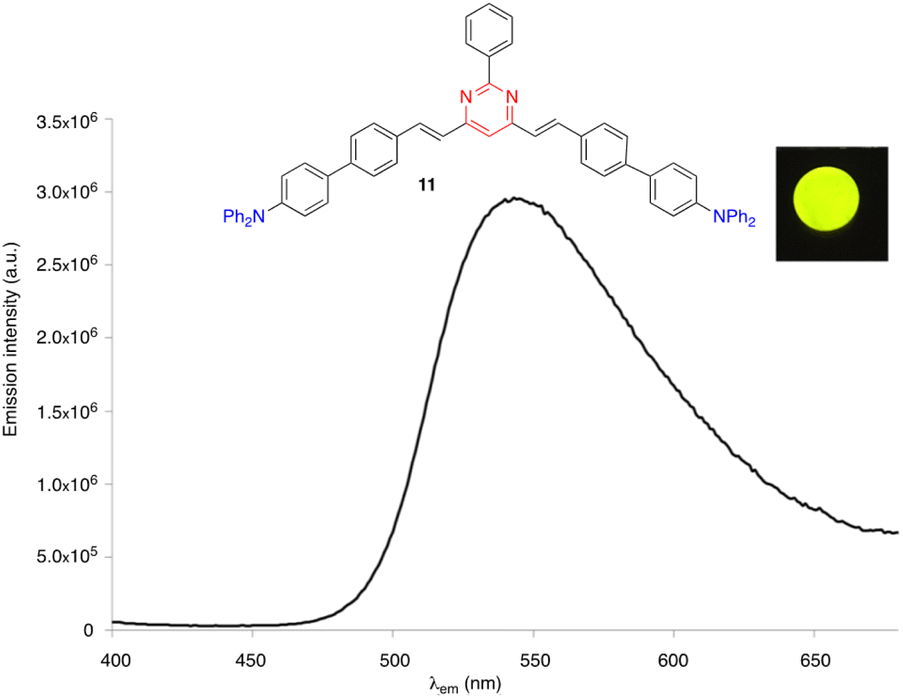 | ||
| Fig. 5 The solid state emission spectrum of compound 11 embedded into a KBr pellet (3 wt%). Inset: Picture taken in the dark of the KBr pellet upon irradiation with a UV hand-held lamp (λexc = 366 nm).30 | ||
4. Sensitivity of emission to external stimuli. Applications as fluorescent (bio)sensors
As mentioned above, the photophysical properties of arylvinylpyrimidines have been demonstrated to be particularly sensitive to environmental stimuli, such as changes in polarity, pH, and the presence of metal cations. In this sense, the ease of protonation, complexation, and hydrogen bonding of the electron lone pairs of the nitrogen atoms provides an excellent tool for developing new (bio)sensing applications. We will briefly describe some of the most typical of these approaches throughout this section.4.1. Polarity
Push–pull luminescent materials are generally characterized by a slight positive absorption solvatochromism and a pronounced positive emission solvatochromism. This red shift of the emission band with increasing solvent polarity can be explained by the stabilization of a highly polar excited state.31 However, the emission intensity is significantly reduced in more polar solvents. Fig. 6 shows the emission solvatochromism observed for compound 11.30 The magnitude of solvatochromism increases considerably with more extended π-conjugated linkers, such as biphenylenevinylene.32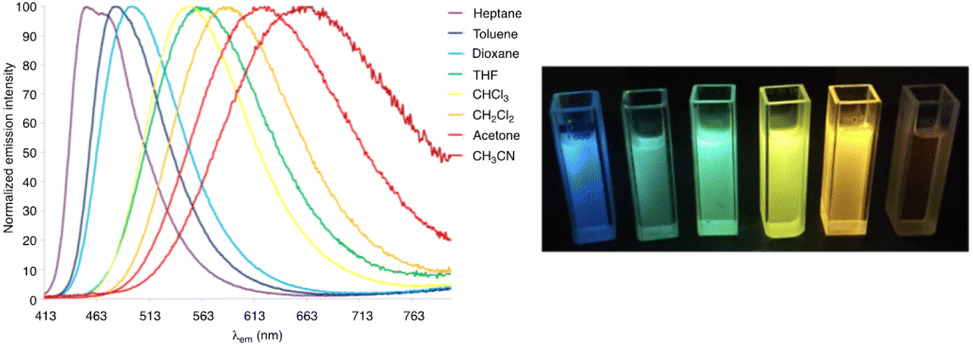 | ||
| Fig. 6 Left: The normalized emission spectra of compound 11 in a series of aprotic solvents (λexc = 403 nm). Right: Fluorescence color changes experienced by 11 in various solvents (from left to right: n-heptane, toluene, 1,4-dioxane, chloroform, CH2Cl2, and MeCN). The picture was taken in the dark upon irradiation with a UV hand-held lamp (λem = 366 nm).30 | ||
4.2. pH
The pyrimidine ring generally acts as a monobasic compound with pKa ≈ 1.1 in water.33 Therefore, it is a weaker base than pyridine (pKa ≈ 5.2). After protonation, the electron-withdrawing inductive effect of the resulting quaternary nitrogen drastically reduces the basicity of the second nitrogen atom (second pKa ≈ −6.3).34 For 4-(arylvinyl)pyrimidines, theoretical calculations have shown that protonation occurs almost exclusively at the N1 atom, both in the ground and excited states.35,36 The protonation of the pyrimidine ring increases its electron-withdrawing character, which enhances the ICT in the push–pull structure and causes a bathochromic shift of the charge transfer absorption band. Fig. 7 illustrates the color change of CH2Cl2 solutions of different compounds 10 and 12 upon the addition of trifluoroacetic acid (TFA). The process is fully reversible.14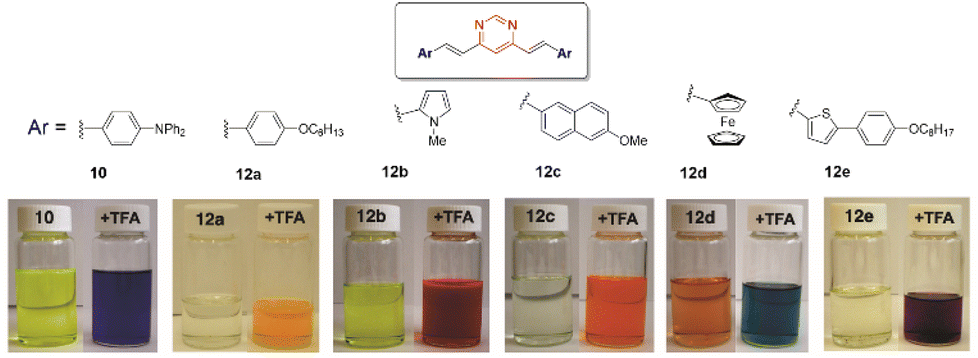 | ||
| Fig. 7 Digital photographs showing the color change experienced by CH2Cl2 solutions of various bis(arylvinyl)pyrimidines 10 and 12 in the presence of 10−2 M TFA. Adapted from ref. 14, (copyright 2009, American chemical Society). | ||
The protonation of amino-substituted derivatives generally leads to emission quenching,13,14,16,30,37,38 although in rare exceptions diphenylamino derivatives remain luminescent.39 Compounds substituted with moderate electron-donating groups, such as methoxy, methyl, or thiomethyl groups, also remain emissive after protonation.13,14,16,27,37 The gradual addition of acid leads to the progressive disappearance of both the absorption and emission bands and the appearance of new red-shifted bands corresponding to the protonated forms (Fig. 8). The visible/emission color change is fully reversible by neutralization with a base.
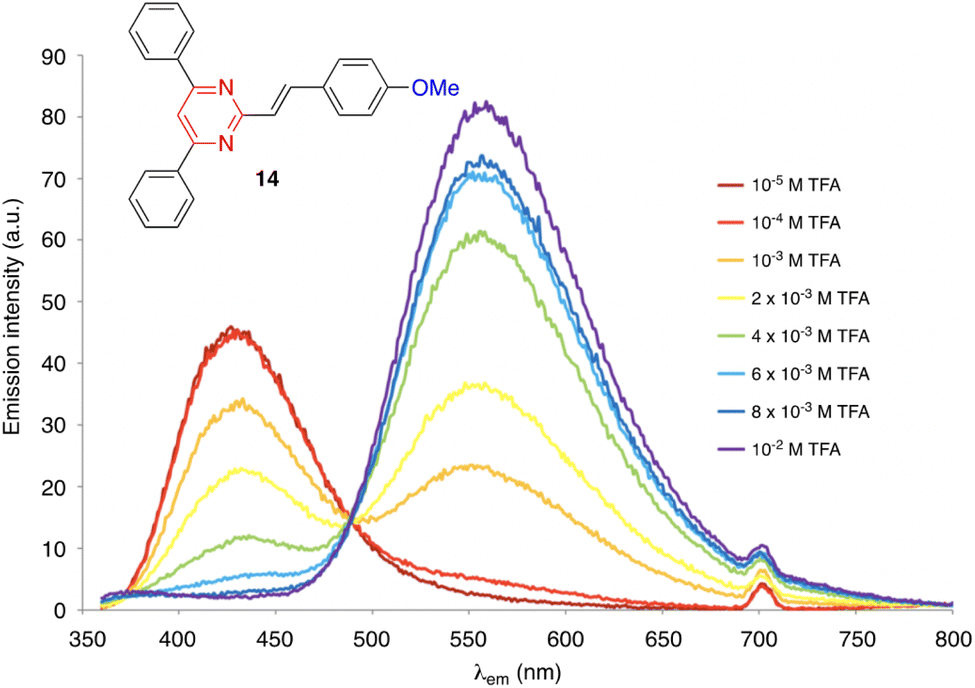 | ||
| Fig. 8 Evolution of the emission spectrum of compound 14 (c = 2 × 10−6 M) in CH2Cl2 solution upon increasing the addition of TFA, excitation at 350 nm. Adapted with permission from ref. 16 (Copyright 2013, Elsevier). | ||
On the other hand, derivatives 15 and 16 (Fig. 9) show null or very little fluorescence in CH2Cl2 solution, respectively. However, the presence of acid induces a substantial enhancement in the luminescence response. The absence of emission for 15 can be explained by an excited-state intramolecular proton transfer (ESIPT) from the OH group to the nitrogen atom of the pyrimidine ring.40 The protonation of the pyrimidine ring by the addition of TFA inhibits the ESIPT process, causing a reversible switch on fluorescence response that results in the progressive appearance of a green-yellow emission band (λem = 550 nm). This acidochromic behavior has also been demonstrated in the solid state by exposure to acid–base vapors. The protonated pyranylidene derivative 16 exhibits a much more intense, blue-shifted emission band compared to the neutral species (λem = 450 nm, ΦF = 0.13), which is attributed to the formation of a pyrylium cation after the addition of TFA.41 Both compounds appear as interesting candidates for anticounterfeiting applications.
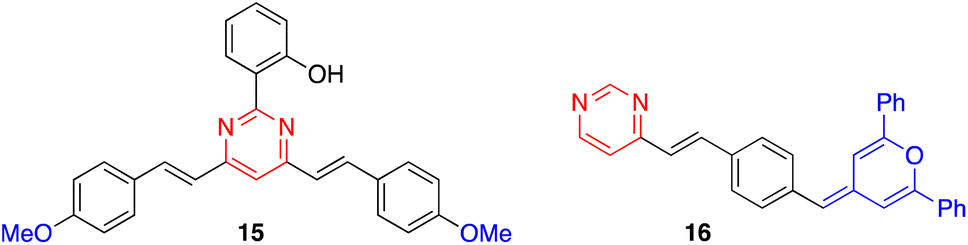 | ||
| Fig. 9 Structure of derivatives 15 and 16. | ||
Mi and coworkers have recently designed a series of 4,6-distyrylpyrimidine derivatives 17–22 with carbazole substituents (Fig. 10).42 These compounds self-assemble into organogels (in toluene/xylene/mesitylene for 17, 19, and 20, and DMF/1,4-dioxane for 18, 21, and 22) and exhibit an intense emission. The addition of TFA destroys the organogel structure and quenches the emission. A bluish green color emitting xerogel-based film of 18 was described as a TFA vapor sensor with a decay time of 0.6 s and a detection limit of 95 ppb. These high-performance properties are explained by the high surface-to-volume ratio of the nanomaterial and the large space in the 3D network.
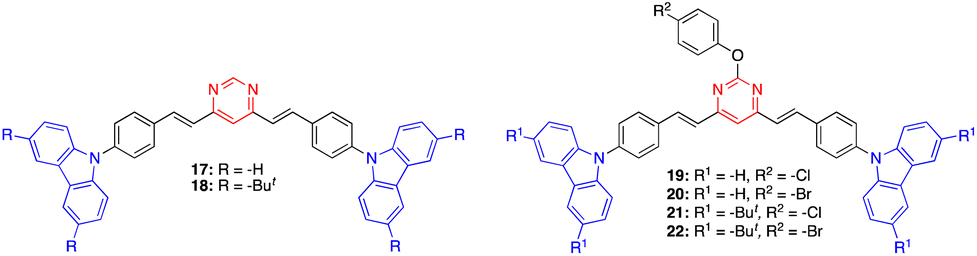 | ||
| Fig. 10 Structure of pyrimidines 17–21. | ||
Pablos and coworkers have designed polymer membranes that exhibit gel behavior, incorporating proton-sensitive 4,6-distyrylpyrimidine units for the visual detection of the acidity of water at pH below 4 and beyond the pH scale (Fig. 11).43 The pKa values of the membranes range from 2.7 to −6.5 in water. Polymer coatings of cotton and other fibers produce smart fabrics that respond colorimetrically to an acid environment, paving the way for fully sensory apparels and smart tags.
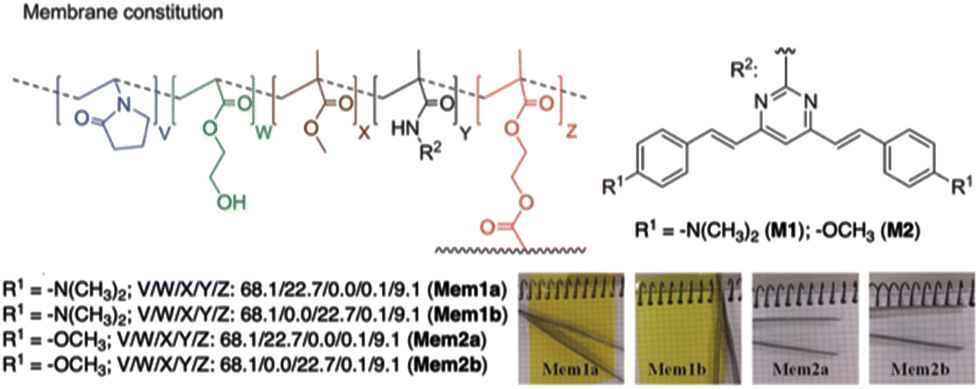 | ||
| Fig. 11 Structure of the polymer membrane designed by Pablos and coworkers. Reproduced with permission from ref. 43 (copyright 2014, Royal Chemical Society). | ||
4.3. Metal cations
The presence of electron lone pairs on the nitrogen atoms of the pyrimidine ring allow it to complex with metal cations. In this context, arylvinylpyrimidines appear as attractive platforms for the development of fluorescence cation sensors.Thus, some of us designed a series of 4-arylvinyl-2,6-di(pyridin-2-yl)pyrimidine chromophores that act as tridentate ligands, coordinating a wide variety of metal ions in a similar way to the well-known terpyridine unit.44 A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry was determined for the Zn2+ and Sn2+ complexes with a remarkably high affinity for Zn2+ (greater than terpyridines) (Scheme 3). The coordination leads to a marked bathochromic effect in the absorption spectra and various responses in the emission spectra (i.e., fluorescence quenching or fluorescence intensity enhancement) depending on the metal cation and the arylvinyl moiety. For instance, a noteworthy increase in the emission intensity is observed when Sn2+ is added to a THF solution of compound 22 (Fig. 12, left). The use of a water-soluble ligand with long polyethylene glycol chains (compound 23), as well as filter paper disks pretreated with the ligands are various strategies employed for the detection of Zn2+, Ca2+, and Sn2+ in aqueous media. In this manner, the Zn2+ cation can be detected visually at a concentration as low as 0.6 ppm (10−5 M) (Fig. 12, right). Some of these ligands were also incorporated into pluronic nanodots, but this strategy dramatically decreases their sensitivity.45
:1 stoichiometry was determined for the Zn2+ and Sn2+ complexes with a remarkably high affinity for Zn2+ (greater than terpyridines) (Scheme 3). The coordination leads to a marked bathochromic effect in the absorption spectra and various responses in the emission spectra (i.e., fluorescence quenching or fluorescence intensity enhancement) depending on the metal cation and the arylvinyl moiety. For instance, a noteworthy increase in the emission intensity is observed when Sn2+ is added to a THF solution of compound 22 (Fig. 12, left). The use of a water-soluble ligand with long polyethylene glycol chains (compound 23), as well as filter paper disks pretreated with the ligands are various strategies employed for the detection of Zn2+, Ca2+, and Sn2+ in aqueous media. In this manner, the Zn2+ cation can be detected visually at a concentration as low as 0.6 ppm (10−5 M) (Fig. 12, right). Some of these ligands were also incorporated into pluronic nanodots, but this strategy dramatically decreases their sensitivity.45
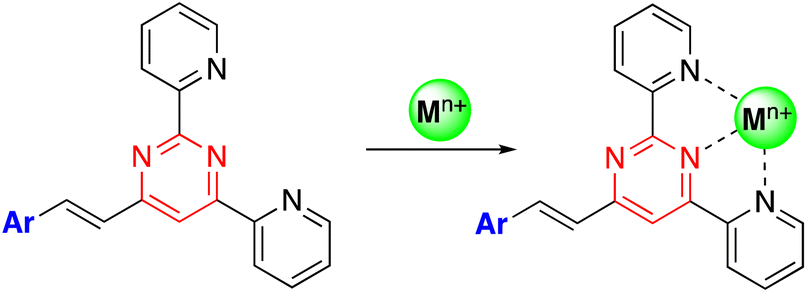 | ||
| Scheme 3 Preparation of metal complexes from of 4-arylvinyl-2,6-di(pyridin-2-yl)pyrimidines. | ||
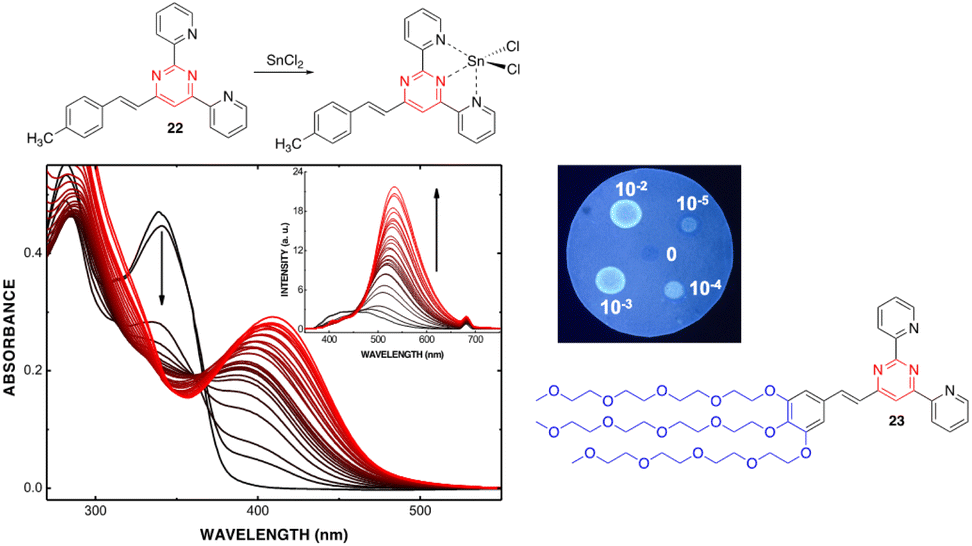 | ||
| Fig. 12 Left: Changes in the UV/Vis spectrum of 22 (2.0 × 10−5 M in THF) upon addition of SnCl2 (from 0 to 7.8 × 10−3 M). Inset: Changes in the emission spectrum (excitation at 341 nm). Top right: Spot test to detect Zn2+ in aqueous media (10−2–10−5 M) using a filter paper disk pretreated with 22. Bottom right: Water soluble 4-styryl-2,6-di(pyridin-2-yl)pyrimidine 23. Reproduced with permission from ref. 44 (copyright 2013, Elsevier Limited). | ||
Other exciting derivatives that our group designed and studied were the dipicolylamine (DPA)-substituted styrylpyrimidines 24 and 25 (Fig. 13).18a In this type of structure, both the pyrimidine and DPA units can act as metal cation binding sites. In the styrylpyrazine and styrylquinoxaline analogues, the presence of Zn2+, Cd2+ or Hg2+ leads to a blue shift in the emission. This indicates a decreased ICT in the molecules due to coordination of the metal cation on the DPA electron-donating group. In contrast, a red shift associated with a substantial quenching of the fluorescence intensity was observed for pyrimidines 24 and 25, which may be explained by an extra binding of the metal cation on the electron-withdrawing pyrimidine.
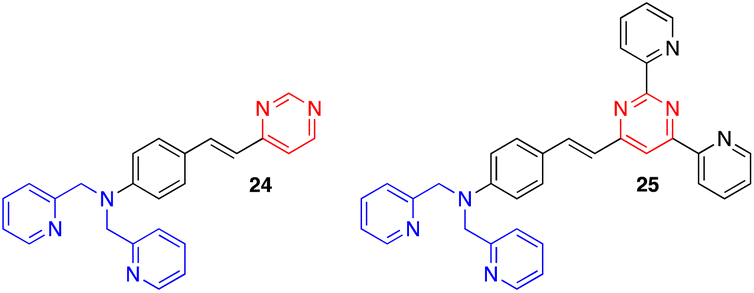 | ||
| Fig. 13 Structure of dipicolylamine styrylpyrimidines 24 and 25. | ||
Kaur and coworkers have designed chromophores 26 and 27 as colorimetric and highly selective fluorescent probes for the dual detection of Hg2+ and Cu2+ in aqueous solution (Fig. 14).46,47 In both systems, the detection of Hg2+ is based on the Kucherov reaction on the terminal alkyne grafted at position 2 of the pyrimidine ring, and the detection of Cu2+ on the coordination of the metal cation with different heteroatoms and π-π bonds. While compound 26 exhibits a red shift in the absorption spectra with significant visual color changes and quenching of the fluorescence, compound 27 works as both a colorimetric and ratiometric fluorescence probe (Fig. 15). The detection limits (18 × 10−9 M for Hg2+ and 21 × 10−9 M for Cu2+) are sufficiently low to allow fluorogenic detection at nanomolar concentrations. In addition, solid-state sensors on silica and filter paper were also developed.
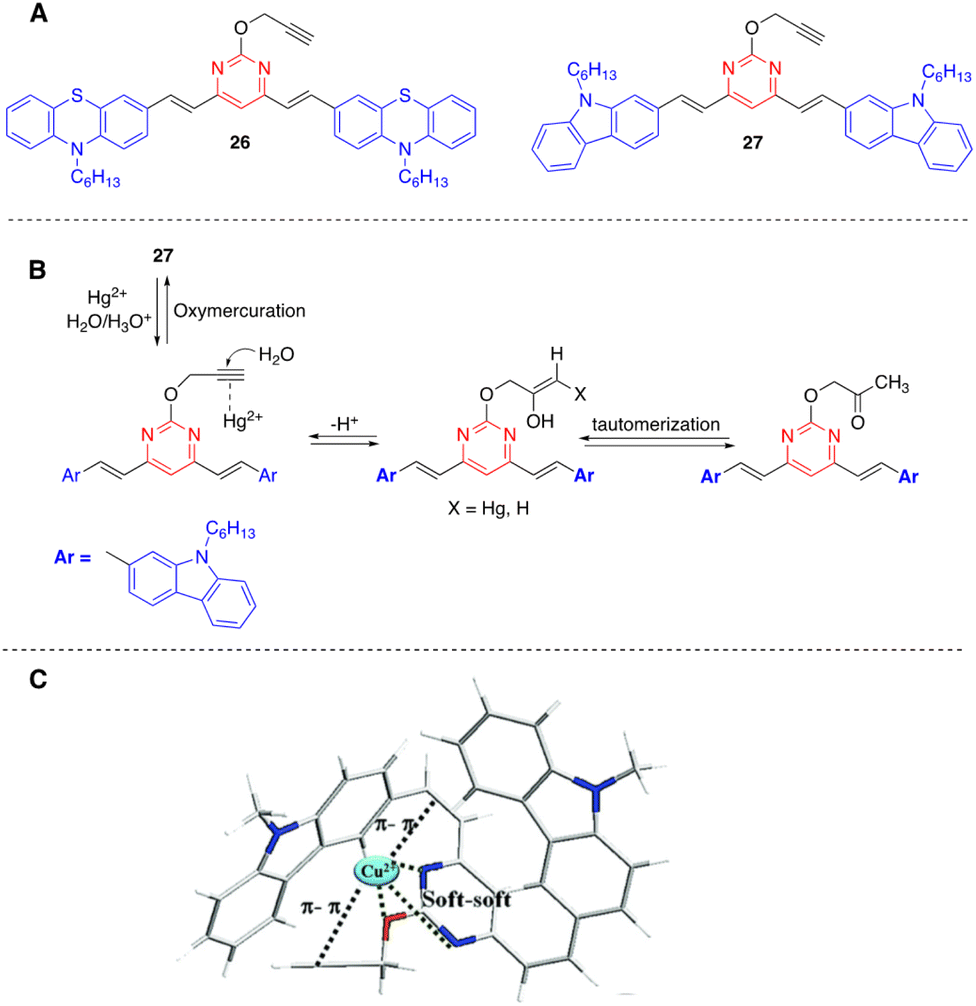 | ||
| Fig. 14 Structure of Hg2+/Cu2+ fluorescence sensors 26 and 27 (A), proposed mechanism for the hydration of probe 27 in the presence of Hg2+ (B), and proposed coordinating sites in probe 27 for Cu2+ (C). Adapted with permission from ref. 47 (Copyright 2015, Royal Chemical Society). | ||
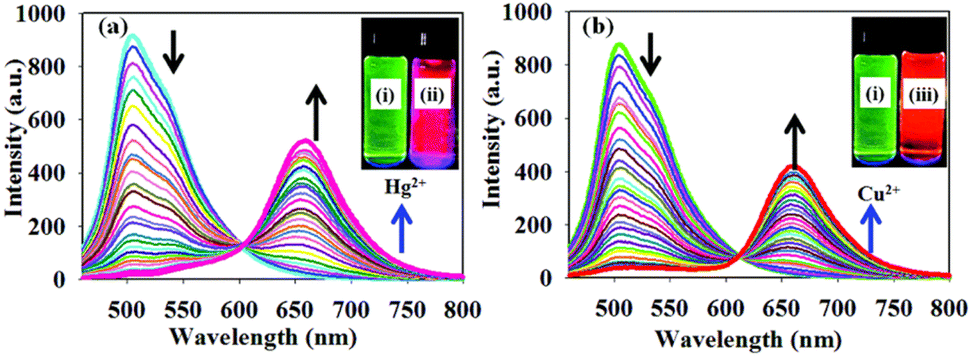 | ||
| Fig. 15 Fluorescence spectra of probe 27 (1.0 μM) THF–HEPES buffer (v/v, 7:3, pH 7.4) upon incremental addition of (a) Hg2+ (0.01–2 equivalents) and (b) Cu2+ (0.01–2 equivalents). Inset: Images showing the emission colors of (i) probe 27, (ii) probe 27 with Hg2+, and (iii) probe 27 with Cu2+ under UV light irradiation. Adapted with permission from ref. 47 (Copyright 2015, Royal Chemical Society). | ||
Zhang and coworkers reported that styrylpyrimidines 28 and 29 show strong fluorescence quenching and good ratiometric responses towards Cu2+ and Fe3+, respectively. The two-photon fluorescence is reversible by the subsequent addition of ethylenediaminetetraacetic acid (EDTA), which allow the use of these pyrimidines as sensitive probes to selectively detect Cu2+ and Fe3+ in live cells without interference from other metal ions (Fig. 16).48 Previously, the same team had described another styrylpyrimidine (compound 30) that could also be used as an ON–OFF fluorescent probe for the in vivo detection of Cu2+ (Fig. 16).49
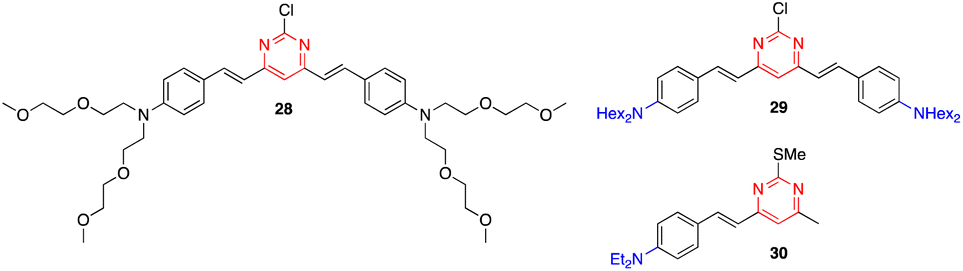 | ||
| Fig. 16 Structure of styrylpyrimidines 28–30 used as Cu2+/Fe3+ fluorescence sensors. | ||
4.4. Nitroaromatic explosives
The detection of nitroaromatic explosives by fluorescence quenching has been extensively developed in the context of homeland security and different azaheterocyclic push–pull systems have been designed for such an application.50The detection of low volatile 2,4,6-trinitrotoluene (TNT) is typically based on the contamination of this material by the more volatile 2,4-dinitrotoluene (DNT). Compounds 10 and 31 (Fig. 17, left) are strongly emissive molecules in solution that exhibit a similar moderate PLQY in spin-cast films (ΦF = 0.18 for 10, ΦF = 0.22 for 31). Exposure to DNT vapors leads to a strong decrease in the emission response of the film.29 However, the real-time sensing dynamics are strikingly different. While the initial fluorescence signal of 10 decreases by 50% after 430 s, it only takes 13 s for compound 31 (more than one order of magnitude faster) (Fig. 17, right). The incorporation of a long flexible chain into the structure creates a more porous thin film that allows a better penetration of DNT vapors into the material, which explain the faster kinetics observed.
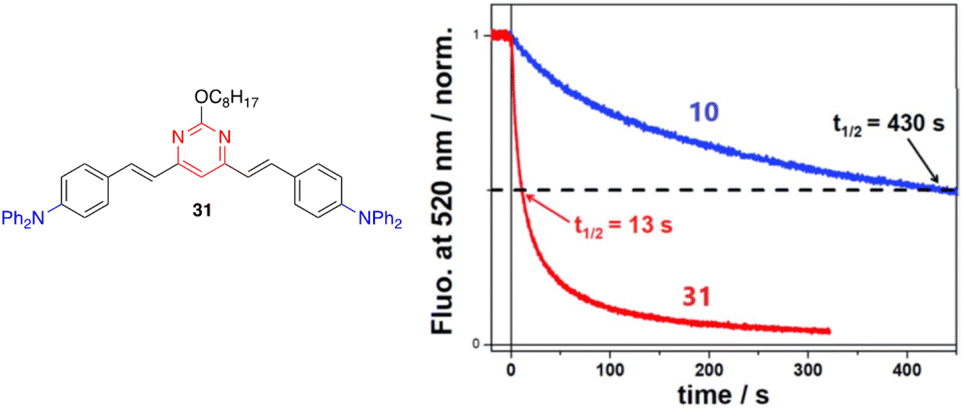 | ||
| Fig. 17 Left: Structure of styrylpyrimidine 31. Right: Time-dependent fluorescence intensity of compounds 10 and 31 in films of similar thicknesses (40 ± 5 Å) upon exposure to a N2 flow of 300 mL min−1 with DNT at a vapor pressure of ∼60 ppb. The half-time for fluorescence decay is also indicated for each film. Reproduced with permission from ref. 29 (Copyright 2019, Royal Chemical Society). | ||
4.5. Hydrogen sulfide
Wang and coworkers developed the biocompatible, non-luminescent styrylpyrimidine probe 32 for the recognition of exogenous/endogenous H2S at the cellular level.51 Upon the addition of H2S, compound 32 underwent thiolytic cleavage to generate the highly luminescent derivative 33 with excellent selectivity, high sensitivity, and a low detection limit of 3.81 μM in buffered media (Scheme 4). More importantly, 32 can precisely accumulate in the endoplasmic reticulum and holds great potential for tracking endogenous H2S in various physiological and pathological processes.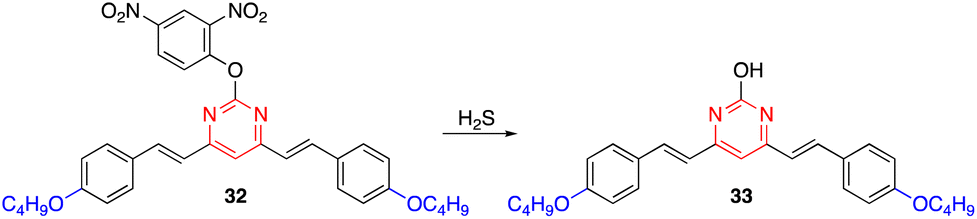 | ||
| Scheme 4 Thiolytic cleavage of probe 32 to generate luminescent chromophore 33 upon exposure to H2S. | ||
4.6. Biomolecules
Aryvinylpyrimidine fluorophores have also found application in the detection of biomolecules. The N-methylpyrimidinium derivative 34 is not fluorescent in aqueous media but shows good affinity for double-stranded DNA (DrewAT). Binding to DNA results in a strong bathochromic shift and increased emission (F/F0 = 35) (Fig. 18).39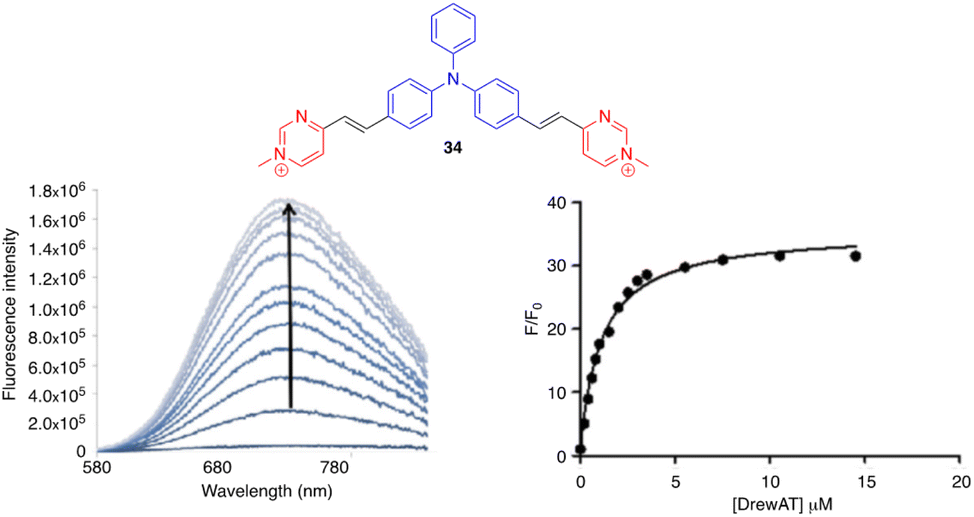 | ||
| Fig. 18 Left: Fluorometric titration of DrewAT with compound 34 (c = 2 μM in 10 mM lithium cacodylate buffer pH 7.2, 100 mM NaCl, [drewAt] = 0–15 μM). Right: Fluorescence enhancement of 34 upon the addition of drewAT. Reproduced with permission from ref. 39 (Copyright 2013, Elsevier Limited). | ||
On the other hand, a series of amino-substituted 4,6-distyrylpyrimidine derivatives 5 and 35–38 have been designed for the in vitro detection of a disease-associated protein deposit in the human brain tissue by fluorescence microscopy (Fig. 3 and 19).19 Compounds 5 and 38 showed significantly higher selectivity for aggregated tau protein over β-amyloid peptides, allowing the selective imaging of tau. Moreover, the ability of 5 to pass the blood–brain barrier was demonstrated and used to detect tau aggregates in Bowman's glands of the olfactory epithelium.
 | ||
| Fig. 19 Structure of 4,6-distyrylpyrimidines 35–38. | ||
Yang and coworkers designed the OFF–ON fluorescent probe 39, which exhibits a highly sensitive and selective fluorescence response to NAD(P)H quinone oxidoreductase-1 (NQO1, also known as DT-diaphorase). In the presence of NQO1, a metabolizing enzyme overexpressed in many solid tumors, the quinone propionic acid group is cleaved, giving rise to the luminescent chromophore 40 (Scheme 5).52 Probe 39 was successfully applied in detecting endogenous NQO1 in living cancer cells.
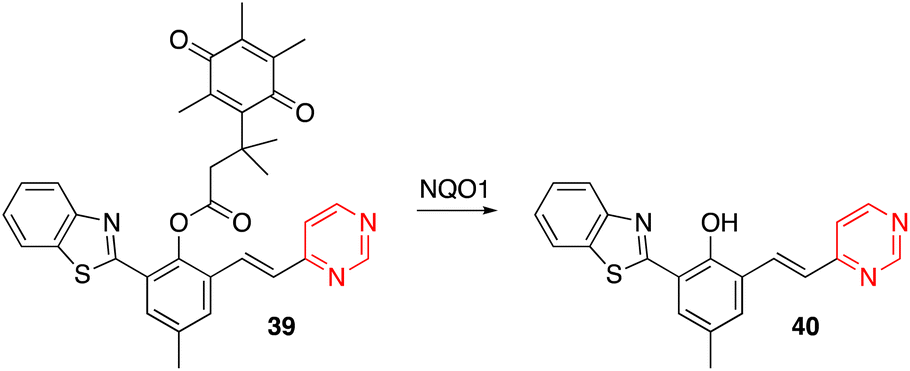 | ||
| Scheme 5 Cleavage of the quinone propionic acid group of 39 in the presence of NQO1 leading to the luminescent arylvinylpyrimidine 40. | ||
5. Dual panchromatic emission
Significant differences in the photophysical behavior of acridan-substituted arylvinylpyrimidines 41 and 42 have been revealed (Fig. 20, top).53 Diphenylamino derivatives 41c and 42c showed classic positive emission solvatochromism, whereas dual emission was observed for methoxy derivatives 41a–b and 42a–b in polar solvents. This dual emission consisted of a broad red-shifted band emitted from a state with ICT or twisted ICT nature and a blue-shifted band attributed to a locally excited state. The acridan moiety is completely isolated and does not affect the fundamental absorption properties. The methoxy derivatives also exhibit aggregation-induced emission in mixtures of THF/water, as well as white-light dual emission in thin films (Fig. 20, bottom), rendering these materials highly promising for the development of white organic light-emitting diodes (WOLEDs). Likewise, the controlled protonation of some blue-emitting arylvinylpyrimidine derivatives leads to dual panchromatic emission with chromaticity coordinates close to pure white light, both in solution and in the solid state.34,36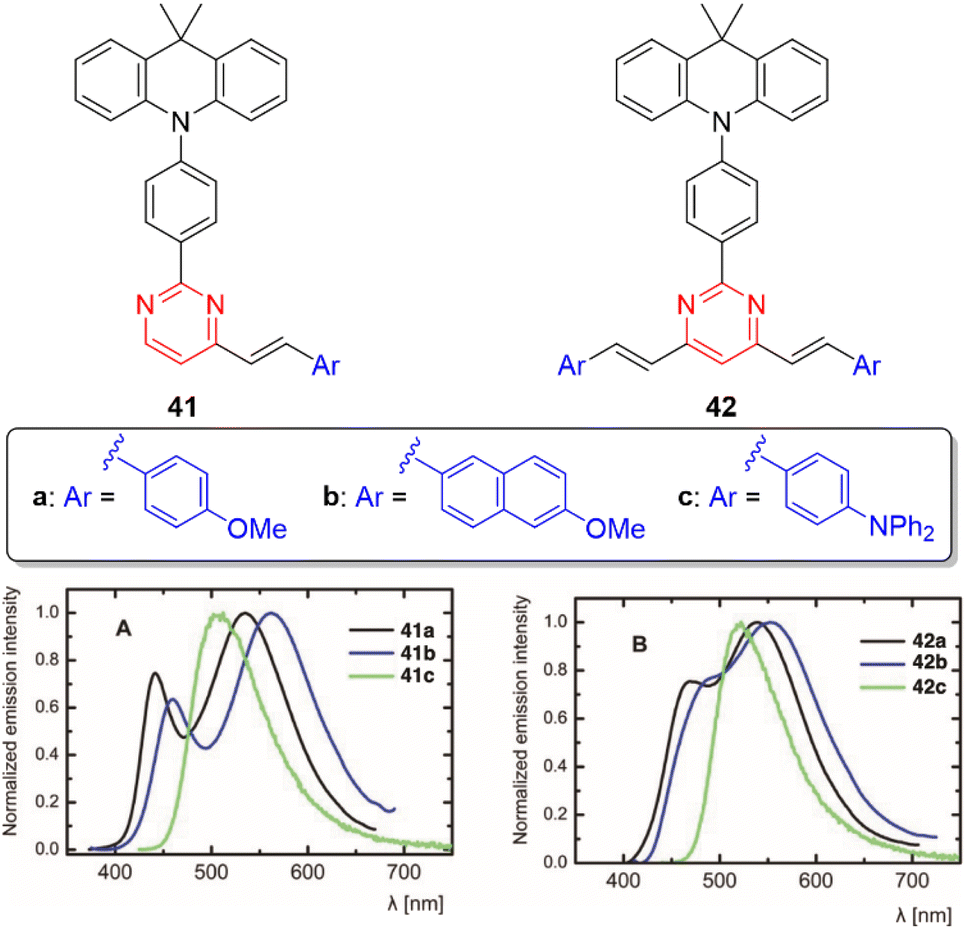 | ||
| Fig. 20 Top: Structure of the acridan-substituted arylvinylpyrimidines 41 and 42. Bottom: Emission spectra of neat thin film of 41 and 42. Adapted with permission from ref. 53 (Copyright 2021, Wiley). | ||
6. Multimodal switches
The diphenylamino-substituted styrylpyrimidine 43 has been designed as a bimodal molecular switch (Scheme 6).54 Upon addition of HCl or an oxidizing agent such as NOSbF6, a progressive decrease in the absorption band at 399 nm associated with the appearance of a new red-shifted band at 503 nm was observed. One equivalent of oxidizing agent and 10 equivalents of HCl were required for the complete transformation of the system, which can be reversed by two different stimuli. The initial spectrum could be restored by adding a well-known reducing agent such as hydrazine hydrate or a base such as triethylamine. On the other hand, a redox potential of ∼0.3 V (vs. Fc/Fc+) also induced the same changes in the absorption spectrum as chemical oxidation. This result may be explained by the pathway proposed in Scheme 6, which consists of an oxidative cascade initiated with the formation of a radical cation that after abstraction of a hydrogen atom from the surrounding medium gives the corresponding protonated form 43+H+ in an irreversible process. When a potential higher than 0.5 V (vs. Fc/Fc+) was applied, a second anodic process assigned to the reversible oxidation of the triphenylamino core was observed. Analogously, the chemical oxidation of compound 44 (Fig. 21) led successively to the corresponding monoprotonated and diprotonated forms, allowing its absorption maximum to switch from the near-UV to the visible, and finally to almost the near-IR region.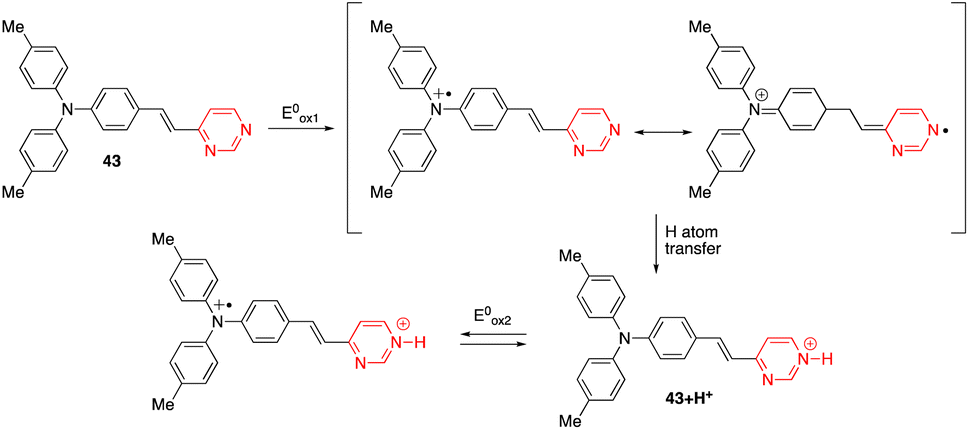 | ||
| Scheme 6 Proposed route to explain the formation of the protonated form of compound 43 under an electrochemical stimulation. | ||
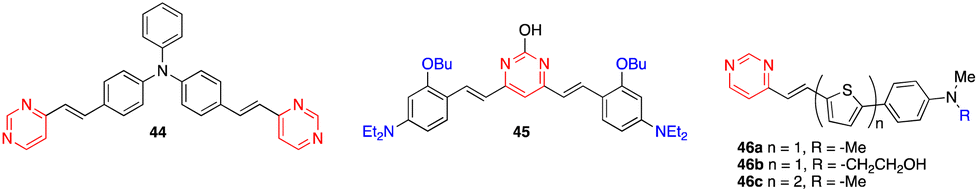 | ||
| Fig. 21 Structures of chromophores 44–46. | ||
7. Photocatalysis
Zhuang and coworkers designed 4,6-arylvinylpyrimidine 45/graphitic carbon nitride (g-C3N4) nanocomposites for photocatalysis (Fig. 21).55 The 45/g-C3N4 photocatalyst with 1 wt% of 45 exhibits the highest photocatalytic activity for the degradation of rhodamine solution under irradiation with visible light. It was reported that the introduction of the arylvinylpyrimidine derivative 45 into the nanomaterial structure promoted interfacial charge transfer and decreased photoinduced electron–hole recombination, thus improving the photocatalytic performance.8. Dye-sensitized solar cells
Azines, including pyrimidine, have been used as alternative anchoring groups for dye-sensitized solar cells.56 In this context, the thienylenevinylene pyrimidine derivatives 46 were recently described as light harvesting units (Fig. 21).57 In this series, the chromophore 46c exhibits the highest power conversion efficiency with η = 6.4%.9. Two-photon absorption
Two-photon absorption (TPA) is a third order nonlinear optical (NLO) phenomenon where two photons are absorbed simultaneously upon laser excitation, each with half of the corresponding one-photon absorption energy. TPA is effectively restricted to the focal volume of the laser beam and allows excitation in the biological window of tissues between 700 and 1100 nm. The TPA efficiency is quoted as a two-photon cross section (δ), mostly presented in Göppert-Mayer units (1 GM = 10−50 cm4 s per photon). A large library of NLO chromophores based on pyrimidine have been reported and fundamental structure–property relationships have been recently reviewed and discussed.58The effect of branching on the TPA properties has been investigated in the diphenylamino-substituted styrylpyrimidine series.59 An increase of the TPA cross section was observed when going from monostyrylpyrimidine 47, to distyrylpyrimidine 10, and tristyrylpyrimidine 9a (Fig. 22). Nevertheless, the regions of linear and non-linear optical spectra of the two and three branched chromophores are similar. Unlike the vast majority of existing branching systems, compound 9a does not have a C3 symmetry, which enhances the TPA response while maintaining the spectral position.
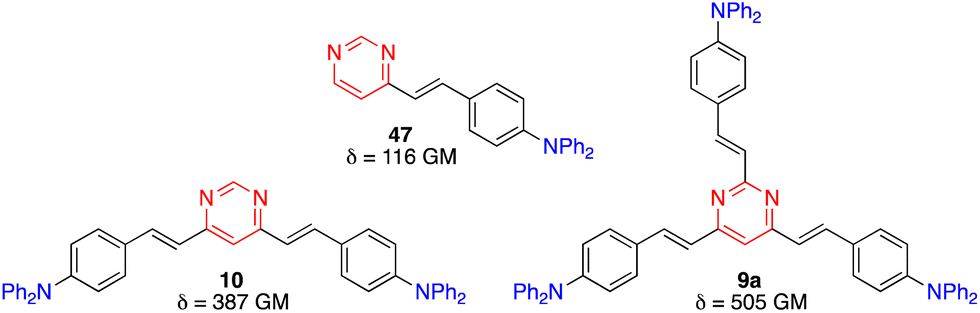 | ||
| Fig. 22 TPA cross sections in CH2Cl2 of diphenylamino-substituted styrylpyrimidine derivatives 9a, 10, and 47. | ||
On the other hand, the octupolar D(–π-A)3 arylvinylpyrimidines 48 and 49 with a triphenylamine central core (Fig. 23) were characterized by a moderate TPA cross section (256 GM at 760 nm and 427 GM at 800 nm in CH2Cl2, respectively) and PLQY > 0.5.60
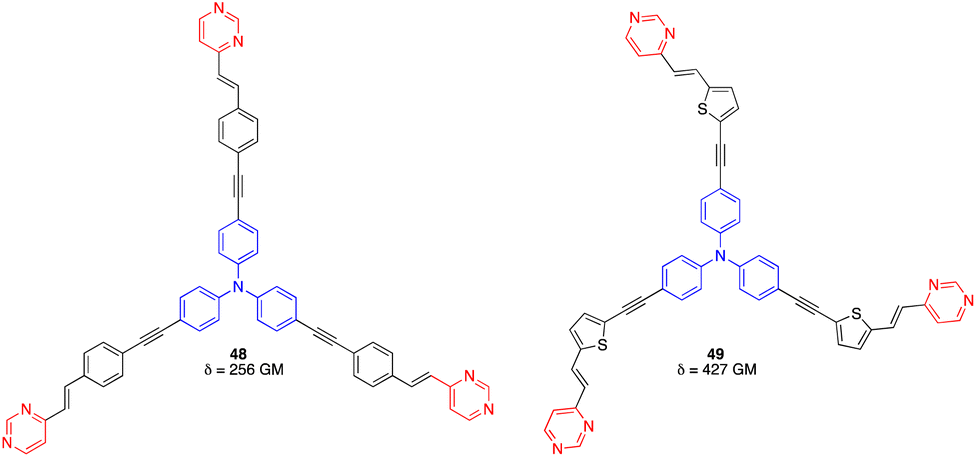 | ||
| Fig. 23 Structures of octupolar arylvinylpyrimidines 48 and 49. | ||
Tetrastyrylpyrimidines 50 and 51 and the fluorene analogue 52 were characterized by TPA cross sections ranging from 460 to 1022 GM in THF at 775–790 nm (Fig. 24).15 Complexation with Zn2+ with 50 and 51 induces a planarization of the structure with a decrease and red shift of TPA cross sections. In contrast, a divergent effect was observed for the complex of the fluorene analogue 52 as the 3D structure is maintained, inducing a marked increase in the TPA cross section up to 2000 GM.
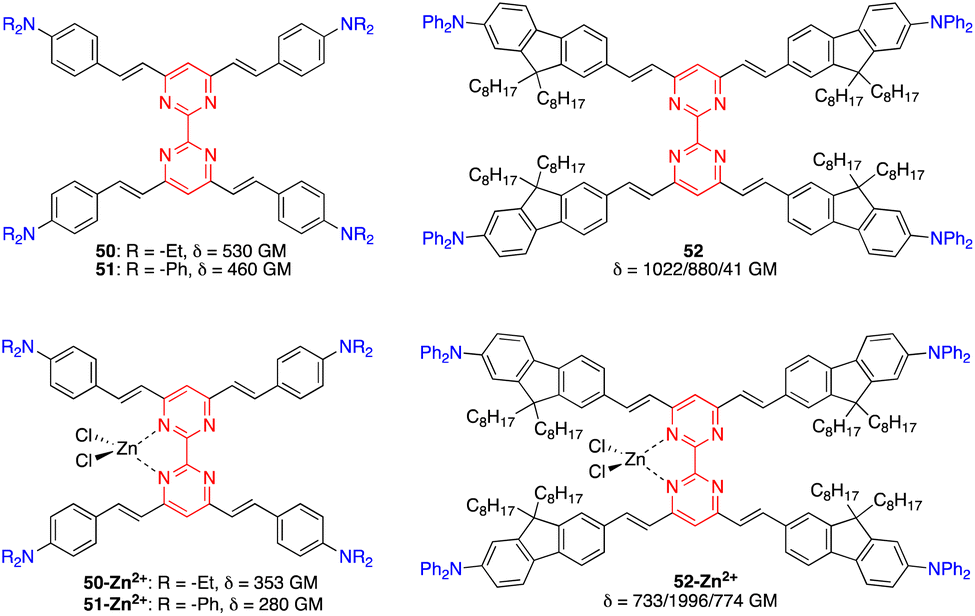 | ||
| Fig. 24 Structures of bipyrimidine chromophores 50–52. | ||
In the following subsections, we will highlight some key applications of selected materials.
9.1. Bioimaging
Due to their significant TPA cross sections (several hundred GM) and high PLQYs, many amino-substituted 4,6-distyrylpyrimidines have been used for bioimaging applications. The required water solubility, if necessary, can be provided by functionalizing the styrylpyrimidine scaffold. Various 4,6-distyrylpyrimidines TPA fluorophores, such as 53–57, stain the cytoplasmic region of cells (Fig. 25, top).23a,b,61 On the other hand, chromophores 58 with hexafluorophosphate water-solubilizing groups revealed to be mitochondrial targeting probes,62 while chromophores 59 and 60 target the endoplasmic reticulum (Fig. 25, bottom).63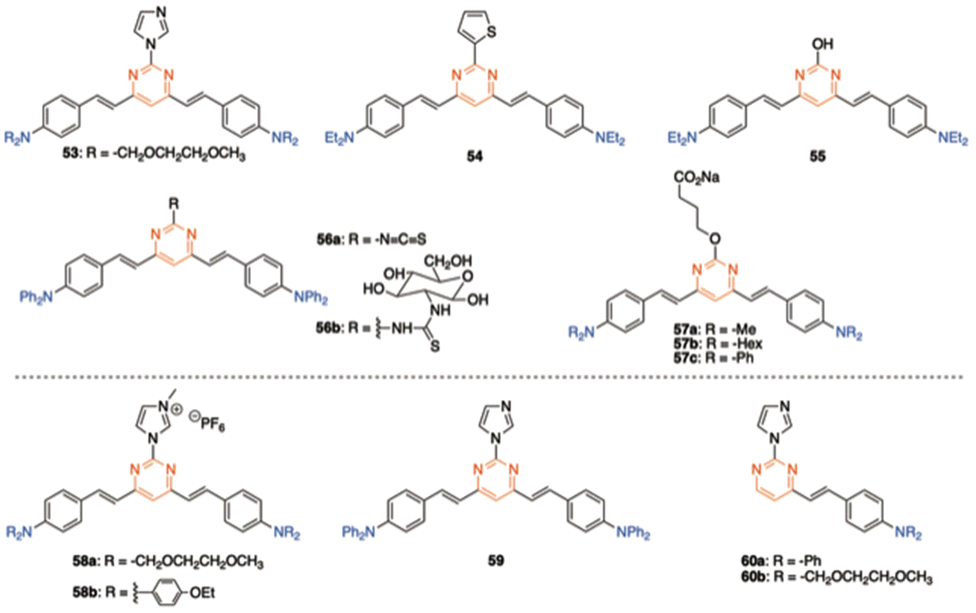 | ||
| Fig. 25 Structures of different amino-substituted 4,6-distyrylpyrimidines used in bioimaging. | ||
The 4,6-distyrylpyrimidine 61 was designed as a TPA OFF–ON fluorescent probe to monitor the phosphatase activity in cells and tissues (Scheme 7).64 The hydroxyl group at the C2 position of the pyrimidine ring can be used for conjugation with different cell-penetrating peptides, allowing organelle- and tumor cell-specific imaging of phosphatase activities in both live mammalian cells and Drosophila brains.
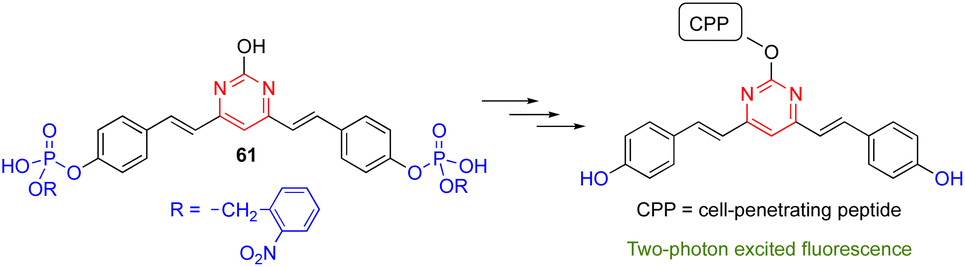 | ||
| Scheme 7 Distyrylpyrimidine 61 as the OFF–ON probe to monitor phosphatase activity. | ||
9.2. Data storage
Li and coworkers reported the 4,6-distyrylpyrimidine 62, which combines a noteworthy TPA cross section, broad TPA band throughout the whole 700–900 nm range, and high fluorescence quantum yield (Fig. 26). Such compound is of particular interest for optical storage in the visible and NIR regions when it is used as a dopant photoinitiator in poly(methyl methacrylate) films.20b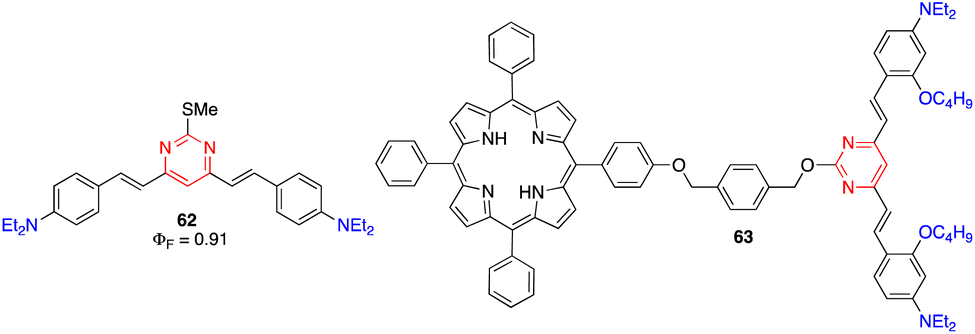 | ||
| Fig. 26 Structures of arylvinylpyrimidines 62 and 63 used for data storage and optical power limiting applications, respectively. | ||
9.3 Optical limiting
Wang and coworkers designed compound 63 for optical power limiting, in which a cooperative effect between the porphyrin and the di(arylvinyl)pyrimidine fragment was demonstrated (Fig. 26).65 The enhanced optical limiting properties were ascribed to a combination of excited-state absorption, reverse saturable absorption, non-linear refraction, and fluorescence resonance energy transfer from the pyrimidine chromophore to the porphyrin core.9.4. Microfabrication
Malval and coworkers described a new conjugated oligomer 64, which has alternating vinylpyrimidine and triphenylamine subunits (Fig. 27).26 Size exclusion chromatography, NMR analyses, and DFT calculations have shown that this oligomer consists of 8 to 10 units and exhibits a highly distorted geometry, resulting in a weak interchromophore coupling within the oligomer and strong reduction of the effective conjugation length. This explains why the absorption and emission properties resemble those measured for its 4,6-distyrylpyrimidine monomer 10. Nevertheless, 61 shows a larger TPA cross section (δ = 5093 GM at 800 nm) than 10 (δ = 360 GM). This very high TPA ability has been aimed to improve the TPA-induced polymerization efficiency of a bicomponent photoinitiator system for microfabrication (Fig. 27).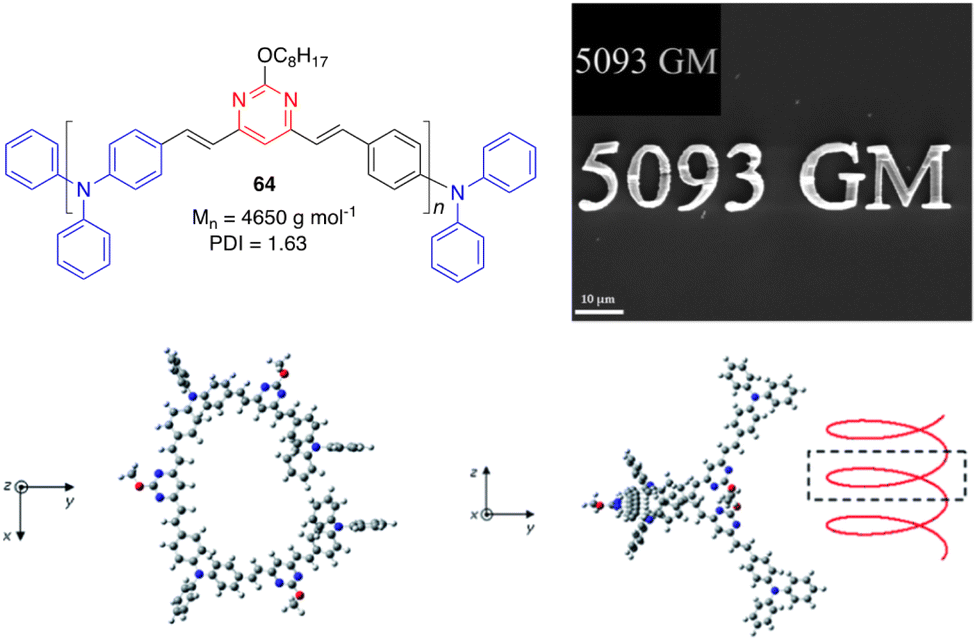 | ||
| Fig. 27 Top left: Structure of the oligomer 64. Top right: SEM image of a μ-structure fabricated upon excitation at 800 nm of a diacrylate resin with 64 as the TPA sensitizer. Inset: 2D model master. Bottom: Optimized geometries of 64 associating three monomer units (PBE0/6-31G(d) level). Adapted with permission from ref. 26 (Copyright 2013, Royal Chemical Society). | ||
10. Second order NLO chromophores
Another common feature of non-centrosymmetric organic push–pull derivatives is their second-order NLO properties that specifically lead to sum frequency generation, including second harmonic generation (SHG),66 Pockels effect, and optical rectification,67 all of which have found application in optical signal processing and integrated optics.68 The second-order NLO properties can be estimated in solution using the electric-field induced second harmonic generation (EFISH) method at a non-resonant incident wavelength of 1907 nm. The EFISH method is based on the application of a high external electric field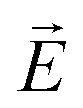 on the analytical cell to align the chromophores in the same direction. With this method, the NLO response is determined as the scalar product between the permanent dipole moment of the molecule
on the analytical cell to align the chromophores in the same direction. With this method, the NLO response is determined as the scalar product between the permanent dipole moment of the molecule 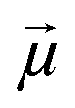 and the vector component β// of its hyperpolarizability tensor β. The results can be influenced by resonance phenomena and a corrected value of μβ0 according to the two-state model better representing the real NLO response.69
and the vector component β// of its hyperpolarizability tensor β. The results can be influenced by resonance phenomena and a corrected value of μβ0 according to the two-state model better representing the real NLO response.69
Due to the modest values shown by classical arylvinylpyrimidines such as 47 (μβ0 = 150 × 10−48 esu),13,58 diverse strategies have been employed to enhance the NLO responses. One of them involves the alteration of the π-conjugated linker. Thus, whereas a biphenylene linker does not significantly improve the NLO response (65), probably because the conjugation is limited by torsion between the two phenyl rings,13,70 the incorporation of a triple bond (66) and the replacement of one phenylene by a thienylene moiety (67) enhance the NLO response (Fig. 28).17 The use of proaromatic pyranylidene groups in the structures of the styrylpyrimidine 16 (μβ0 = 300 × 10−48 esu) and the 4,6-distyrylpyrimidine 68 (μβ0 = 480 × 10−48 esu) positively influences the NLO response.71 Nevertheless, the most efficient way to increase the value of μβ0 is to reinforce the electron-withdrawing character of the pyrimidine ring by complexation with tungsten pentacarbonyl (69) or methylation at the N1 atom (70).72 The latter leads to a value of μβ0 around 9 times higher (μβ0 = 1394 × 10−48 esu) than that of the reference chromophore 47. The chromophore 71, with a 2,4-disubstituted methylated pyrimidinium core, exhibits the highest μβ0 value in the series (2500 × 10−48 esu).73
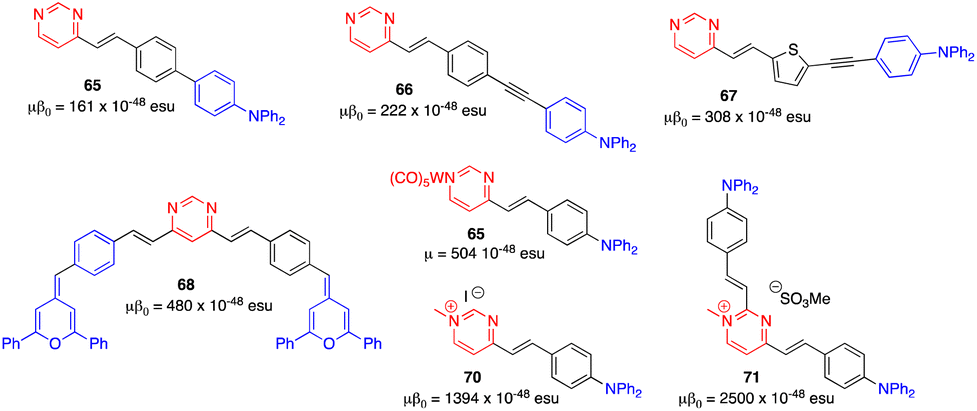 | ||
| Fig. 28 Structures and NLO responses of compounds 65–71. | ||
Organometallic derivatives 72–74, in which a metallic center (Pt or Ru) is incorporated into the π-conjugated structure, show particularly high μβ0 values of up to 6000 × 10−48 esu for the ruthenium complex 74 (Fig. 29).74,75
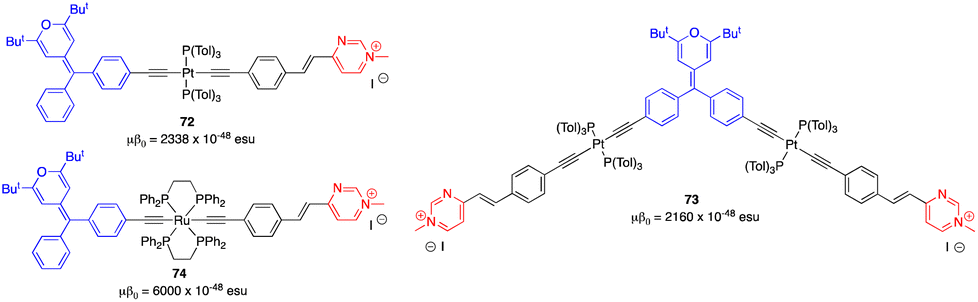 | ||
| Fig. 29 Structures and NLO responses of organometallic compounds 72–74. | ||
Akdas-Kiliç and coworkers have designed a series of tetra(arylvinyl)bipyrimidines 75 with long alkoxyl chains (Fig. 30).76,77 Compounds 75a and 75b with C12 and C16 chains exhibit mesomorphic as well as second-order NLO properties in solution (β0 = 38–42 × 10−30 esu, measured by hyper-Rayleigh scattering in CH2Cl2). Nevertheless, they are SHG silent in their liquid crystalline phases due to the formation of columnar mesophases with hexagonal symmetry. In contrast, grafting of branched chiral alkyl chains (75c) leads to right- or left-handed twisted columnar structures depending on the chirality of the side chains. This strategy allows translation of the NLO response from molecular level to the bulk level as shown by SHG measurements.
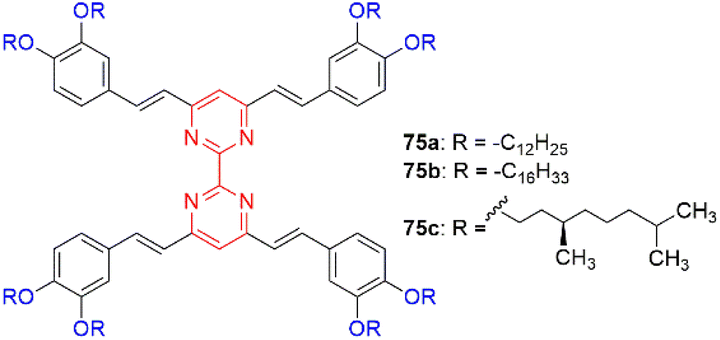 | ||
| Fig. 30 Structures of tetra(arylvinyl)bipyrimidines 75. | ||
11. Conclusions
This article has provided a general overview of the main strategies for the synthesis of arylvinylpyrimidines, as well as highlighting their interesting photophysical properties. Specifically, these compounds are typically prepared by the Knoevenagel condensation of aromatic aldehydes with methylpyrimidine derivatives. This methodology is the most suitable because of a wide range of commercially available starting materials, the use of environmentally friendly conditions in most cases, the easy treatment of crude reaction mixtures, and the high control of the stereochemistry.All these molecules display extraordinary optical properties, although they generally must be substituted with electron-donating amino or alkoxy groups to show significant emission. The final luminescence properties strongly depend on the substitution at pyrimidine and aryl moieties and are particularly sensitive to environmental stimuli, which provides an excellent tool for developing new sensing and optoelectronic devices. Nowadays, the main interest in this area is focused on addressing the needs of the industry and overcoming the difficulties of implementing the material in real devices, which is still a major challenge.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the Junta de Comunidades de Castilla-La Mancha/FEDER is gratefully acknowledged by S. A. and J. R.-L. (project SBPLY/21/180501/000042).References
- F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC.
- L. Beverina and G. A. Pagani, Acc. Chem. Res., 2014, 47, 319–329 CrossRef CAS.
- (a) A. Pal, M. Karmakar, S. R. Bhatta and A. Thakur, Coord. Chem. Rev., 2021, 448, 214167 CrossRef CAS; (b) E. V. Verbitskiy, G. L. Rusinov, O. N. Chupakhin and V. N. Charushin, Dyes Pigm., 2020, 180, 108414 CrossRef CAS.
- (a) M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef; (b) J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2013, 1, 4599–4604 RSC.
- H. Moon, W. S. Jahng and M. D. Curtis, J. Mater. Chem., 2008, 18, 4856–4863 RSC.
- (a) J.-M. Ji, H. Zhou and H. K. Kim, J. Mater. Chem. A, 2018, 6, 14518–14545 RSC; (b) V. Malytskyi, J.-J. Simon, L. Patrone and J.-M. Raimundo, RSC Adv., 2015, 5, 354–397 RSC; (c) W. Li, L. Ye, S. Li, H. Yao, H. Ade and J. Hou, Adv. Mater., 2018, 30, 1707170 CrossRef.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- J. Liu, C. Ouyang, F. Huo, W. He and A. Cao, Dyes Pigm., 2020, 181, 108509 CrossRef CAS.
- S. Achelle, J. Rodríguez-López and F. Robin-le Guen, ChemistrySelect, 2018, 3, 1852–1886 CrossRef CAS.
- (a) S. Achelle and N. Plé, Curr. Org. Synth., 2012, 9, 163–187 CrossRef CAS; (b) G. N. Lipunova, E. V. Nosova, V. N. Charushin and O. N. Chupakhin, Curr. Org. Synth., 2018, 15, 793–814 CrossRef CAS; (c) R. Komatsu, H. Sasabe and J. Kido, J. Photonics Energy, 2018, 8, 032108 Search PubMed.
- G. A. Molander and C. R. Bernardi, J. Org. Chem., 2002, 67, 8424–8429 CrossRef CAS.
- (a) J.-J. Vanden Eynde, L. Pascal, Y. Van Haverbeke and P. Dubois, Synth. Commun., 2001, 31, 3167–3173 CrossRef CAS; (b) L. Pascal, J.-J. Vanden Eynde, Y. Van Haverbeke, P. Dubois, A. Michel, U. Rant, E. Zojer, G. Leising, L. O. Van Dorn, N. E. Gruhn, J. Cornil and J.-L. Brédas, J. Phys. Chem. B, 2002, 106, 6442–6450 CrossRef CAS.
- S. Achelle, A. Barsella, C. Baudequin, B. Caro and F. Robin-le Guen, J. Org. Chem., 2012, 77, 4087–4096 CrossRef CAS PubMed.
- S. Achelle, I. Nouira, B. Pfaffinger, Y. Ramondenc, N. Plé and J. Rodríguez-López, J. Org. Chem., 2009, 74, 3711–3717 CrossRef CAS PubMed.
- P. Savel, H. Akdas-Kiliç, J.-P. Malval, A. Spangenberg, T. Roisnel and J.-L. Fillaut, J. Mater. Chem. C, 2014, 2, 295–305 RSC.
- S. Achelle and F. Robin-le Guen, Tetrahedron Lett., 2013, 54, 4491–4496 CrossRef CAS.
- S. Achelle, A. Barsella, B. Caro and F. Robin-le Guen, RSC Adv., 2015, 5, 39218–39227 RSC.
- (a) S. Achelle, J. Rodríguez-López, F. Bureš and F. Robin-le Guen, Dyes Pigm., 2015, 121, 305–311 CrossRef CAS; (b) G. Khalil, C. Orvain, L. Fang, L. Barloy, A. Chaumont, C. Gaiddon, M. Henry, N. Kyritsakas and P. Mobian, Dalton Trans., 2016, 45, 19072–19085 RSC; (c) H. Akdas-Kiliç, T. Roisnel, I. Ledoux and H. le Bozec, New J. Chem., 2009, 33, 1470–1473 RSC.
- A. Boländer, D. Kieser, C. Voss, S. Bauer, C. Schön, S. Burgold, T. Bittner, J. Hölzer, R. Heyny-von Haußen, G. Mall, V. Goetschy, C. Czech, H. Knust, R. Berger, J. Herms, I. Hilger and B. Schmidt, J. Med. Chem., 2012, 55, 9170–9180 CrossRef.
- (a) F.-A. Martin, C. Baudequin, C. Fiol-Petit, M. Darabantu, Y. Ramondenc and N. Plé, Tetrahedron, 2014, 70, 2546–2555 CrossRef CAS; (b) L. Li, Y.-P. Tian, J.-X. Yang, P.-P. Sun, J.-Y. Wu, H.-P. Zhou, S.-Y. Zhang, B.-K. Jin, X.-J. Xing, C.-K. Wang, M. Li, G.-H. Cheng, H.-H. Tang, W.-H. Huang, X.-T. Tao and M.-H. Jiang, Chem. – Asian J., 2009, 4, 668–680 CrossRef CAS PubMed.
- M. Klikar, P. le Poul, A. Růžička, O. Pytela, A. Barsella, K. D. Dorkenoo, F. Robin-le Guen, F. Bureš and S. Achelle, J. Org. Chem., 2017, 82, 9435–9451 CrossRef CAS PubMed.
- A. A. Parent, D. H. Ess and J. A. Katzenellenbogen, J. Org. Chem., 2014, 79, 5448–5462 CrossRef CAS PubMed.
- (a) H. Wang, Q. Zhang, J. Zhang, L. Li, Q. Zhang, S. Li, S. Zhang, J. Wu and Y. Tian, Dyes Pigm., 2014, 102, 263–272 CrossRef CAS; (b) B. Liu, H.-L. Zhang, J. Liu, Y.-D. Zhao, Q.-M. Luo and Z.-L. Huang, J. Mater. Chem., 2007, 17, 2921–2929 RSC.
- S. Achelle, L. Bodiou, J. Charrier and F. Robin-le Guen, C. R. Chim., 2016, 19, 279–285 CrossRef.
- S. S. Gunathilake, H. D. Magurudeniya, P. Huang, H. Nguyen, E. A. Rainbolt, M. C. Stefan and M. C. Biewer, Polym. Chem., 2013, 4, 5216–5219 RSC.
- J.-P. Malval, S. Achelle, L. Bodiou, A. Spangenberg, L. C. Gomez, O. Soppera and F. Robin-le Guen, J. Mater. Chem. C, 2014, 2, 7869–7880 RSC.
- M. Fecková, P. le Poul, F. Robin-le Guen, T. Roisnel, O. Pytela, M. Klikar, F. Bureš and S. Achelle, J. Org. Chem., 2018, 83, 11712–11726 CrossRef PubMed.
- C. Denneval, S. Achelle, C. Baudequin and F. Robin-le Guen, Dyes Pigm., 2014, 110, 49–55 CrossRef CAS.
- J.-P. Malval, M. Cranney, S. Achelle, H. Akdas-Kiliç, J.-L. Fillaut, N. Cabon, F. Robin-le Guen, O. Soppera and Y. Molard, Chem. Commun., 2019, 55, 14331–14334 RSC.
- L. Diarra, F. Robin-le Guen and S. Achelle, Molbank, 2021, M1278 CrossRef CAS.
- R. Lartia, C. Allain, G. Bordeau, F. Schmidt, C. Fiorini-Debuisschert, F. Charra and M.-P. Teulade-Fichou, J. Org. Chem., 2008, 73, 1732–1744 CrossRef CAS PubMed.
- S. Achelle and F. Robin-le Guen, J. Photochem. Photobiol., A, 2017, 348, 281–286 CrossRef CAS.
- R. Milcent, Chimie Organique Hétérocyclique, EDP Science, Les Ulis, 2003, p. 605 Search PubMed.
- S. Achelle, J. Rodríguez-López, F. Bureš and F. Robin-le Guen, Chem. Rec., 2020, 20, 440–451 CrossRef CAS.
- M. Hodée, A. Lenne, J. Rodríguez-López, F. Robin-le Guen, C. Katan, S. Achelle and A. Fihey, New J. Chem., 2021, 45, 19132–19144 RSC.
- S. Achelle, J. Rodríguez-López, C. Katan and F. Robin-le Guen, J. Phys. Chem. C, 2016, 120, 26986–26995 CrossRef CAS.
- C. Hadad, S. Achelle, J. C. García-Martínez and J. Rodríguez-López, J. Org. Chem., 2011, 76, 3837–3845 CrossRef CAS.
- F. Kournoutas, I. K. Kalis, M. Fecková, S. Achelle and M. Fakis, J. Photochem. Photobiol., A, 2020, 391, 112398 CrossRef CAS.
- A. I. Aranda, S. Achelle, F. Hammerer, F. Mahuteau-Betzer and M.-P. Teulade-Fichou, Dyes Pigm., 2012, 95, 400–407 CrossRef CAS.
- R. Plaza-Pedroche, M. P. Fernández-Liencres, S. B. Jiménez-Pulido, N. A. Illán-Cabeza, S. Achelle, A. Navarro and J. Rodríguez-López, ACS Appl. Mater. Interfaces, 2022, 14, 24964–24979 CrossRef CAS.
- S. Achelle, S. Kahlal, J.-Y. Saillard, N. Cabon, B. Caro and F. Robin-le Guen, Tetrahedron, 2014, 70, 2804–2815 CrossRef CAS.
- W. Mi, Z. Qu, J. Sun, J. Sung, F. Zhang and K. Ye, Dyes Pigm., 2018, 150, 207–215 CrossRef CAS.
- J. L. Pablos, P. Estévez, A. Muñoz, S. Ibeas, F. Serna, F. C. García and J. M. García, J. Mater. Chem. A, 2015, 3, 2833–2843 RSC.
- C. Hadad, S. Achelle, I. López-Solera, J. C. García-Martínez and J. Rodríguez-López, Dyes Pigm., 2013, 97, 230–237 CrossRef CAS.
- L. Vurth, C. Hadad, S. Achelle, J. C. García-Martínez, J. Rodríguez-López and O. Stéphan, Colloid Polym. Sci., 2012, 290, 1353–1359 CrossRef CAS.
- M. Kaur, M. J. Cho and D. H. Choi, Dyes Pigm., 2016, 125, 1–7 CrossRef CAS.
- M. Kaur, Y.-H. Ahn, K. Choi, M. J. Cho and D. H. Choi, Org. Biomol. Chem., 2015, 13, 7149–7153 RSC.
- Q. Zhang, X. Tian, H. Wang, Z. Hu, J. Wu, H. Zhou, S. Zhang, J. Yang, Z. Sun, Y. Tian and K. Uvdal, Sens. Actuators, B, 2016, 222, 574–578 CrossRef CAS.
- Q. Zhang, L. Li, M. Zhang, Z. Liu, J. Wu, H. Zhou, J. Yang, S. Zhang and Y. Tian, Dalton Trans., 2013, 42, 8848–8853 RSC.
- E. V. Verbitskiy, G. L. Rusinov, O. N. Chupakhin and V. N. Charushin, Dyes Pigm., 2020, 180, 108414 CrossRef CAS.
- H. Wang, L. Hu, B. Xu, H. Chen, F. Cai, N. Yang, Q. Wu, K. Uvdal, Z. Hu and L. Li, Dyes Pigm., 2020, 179, 108390 CrossRef CAS.
- Q. Yang, Y. Wen, J. Xu and S. Shao, Talanta, 2020, 216, 120982 CrossRef CAS.
- M. Fecková, I. K. Kalis, T. Roisnel, P. le Poul, O. Pytela, M. Klikar, F. Robin-le Guen, F. Bureš, M. Fakis and S. Achelle, Chem. – Eur. J., 2021, 27, 1145–1159 CrossRef PubMed.
- O. Alévêque, S. Achelle and L. Sanguinet, ChemPhotoChem, 2022 DOI:10.1002/cptc.202200201 , in press.
- J. Zhuang, J. Zhang, J. Pang, A. Wang, X. Wang and W. Zhu, Dyes Pigm., 2019, 163, 634–640 CrossRef CAS.
- E. V. Verbitskiy, G. L. Rusinov, O. N. Chupakhin and V. N. Charushin, Dyes Pigm., 2021, 194, 109650 CrossRef CAS.
- I. Duerto, S. Sarasa, D. Barrios, J. Orduna, B. Villacampa and M.-J. Blesa, Dyes Pigm., 2022, 203, 110310 CrossRef CAS.
- M. Fecková, P. le Poul, F. Bureš, F. Robin-le Guen and S. Achelle, Dyes Pigm., 2020, 182, 108659 CrossRef.
- F. Kournoutas, A. Fihey, J.-P. Malval, A. Spangenberg, M. Fecková, P. le Poul, C. Katan, F. Robin-le Guen, F. Bureš, S. Achelle and M. Fakis, Phys. Chem. Chem. Phys., 2020, 22, 4165–4176 RSC.
- D. Cvejn, S. Achelle, O. Pytela, J.-P. Malval, A. Spangenberg, N. Cabon, F. Bureš and F. Robin-le Guen, Dyes Pigm., 2016, 124, 101–109 CrossRef CAS.
- (a) Q. Zhang, L. Luo, H. Xu, Z. Hu, C. Brommesson, J. Wu, Z. Sun, Y. Tian and K. Uvdal, New J. Chem., 2016, 40, 3456–3463 RSC; (b) C. Tang, Q. Zhang, D. Li, J. Zhang, P. Shi, S. Li, J. Wu and Y. Tian, Dyes Pigm., 2013, 99, 20–28 CrossRef CAS; (c) Q. Zhang, J. Luo, L. Ye, H. Wang, B. Huang, J. Zhang, J. Wu, S. Zhang and Y. Tian, J. Mol. Struct., 2014, 1074, 33–42 CrossRef CAS; (d) J. Yang, W. Hu, H. Li, H. Hou, Y. Tu and B. Liu, Photochem. Photobiol. Sci., 2018, 17, 474–481 CrossRef CAS.
- Q. Zhang, R. Guan, X. Tian, L. Luo, H. Zhou, S. Li, J. Wu and Y. Tian, RSC Adv., 2017, 7, 20068–20075 RSC.
- (a) L. Hu, S. Hussain, T. Liu, Y. Yue, J. Liu, Y. Tian and X. Tian, New J. Chem., 2018, 42, 14725–14728 RSC; (b) Q. Zhang, X. Tian, Z. Hu, C. Brommesson, J. Wu, H. Zhou, J. Yang, Z. Sun, Y. Tian and K. Uvdal, Dyes Pigm., 2016, 126, 286–295 CrossRef CAS.
- L. Li, J. Ge, H. Wu, Q.-H. Xu and S. Q. Yao, J. Am. Chem. Soc., 2012, 134, 12157–12167 CrossRef CAS PubMed.
- A. Wang, L. Long, S. Meng, X. Li, W. Zhao, Y. Song, M. P. Cifuentes, M. G. Humphrey and C. Zhang, Org. Biomol. Chem., 2013, 11, 4250–4257 RSC.
- A. Morita, Theory of sum frequency generation spectroscopy, Springer Nature Singapore Pte Ltd., 2018 Search PubMed.
- M. Bass, P. A. Franken, J. F. Ward and G. Weinreich, Phys. Rev. Lett., 1962, 9, 446–448 CrossRef CAS.
- (a) Y. Qi and Y. Li, Nanophotonics, 2020, 9, 1287–1320 CAS; (b) O.-S. Prelipceanu, M. Prelipceanu, N. Paraschiv, A. Popa and O. Geman, Symmetry, 2019, 11, 60 CrossRef CAS; (c) C. Koos, P. Vorreau, T. Vallaitis, P. Dumon, W. Bogaerts, R. Baets, B. Esembeson, I. Biaggio, T. Michinobu, F. Diederich, W. Freude and J. Leuthold, Nat. Photonics, 2009, 3, 216–219 CrossRef CAS; (d) D. Cotter, R. J. Manning, K. J. Blow, A. D. Ellis, A. E. Kelly, D. Nesset, I. D. Phillips, A. J. Poustie and D. C. Rogers, Science, 1999, 286, 1523–1528 CrossRef CAS; (e) B. Jalali and S. Fathpour, J. Lightwave Technol., 2006, 24, 4600–4615 CAS.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
- F. Castet, A. Pic and B. Champagne, Dyes Pigm., 2014, 110, 256–260 CrossRef CAS.
- S. Achelle, J.-P. Malval, S. Aloïse, A. Barsella, A. Spangenberg, L. Mager, H. Akdas-Kiliç, J.-L. Fillaut, B. Caro and F. Robin-le Guen, ChemPhysChem, 2013, 14, 2725–2736 CrossRef CAS PubMed.
- S. Achelle, S. Kahlal, A. Barsella, J.-Y. Saillard, X. Che, J. Vallet, F. Bureš, B. Caro and F. Robin-le Guen, Dyes Pigm., 2015, 113, 562–570 CrossRef CAS.
- S. Achelle, E. V. Verbitskiy, M. Fecková, F. Bureš, A. Barsella and F. Robin-le Guen, ChemPlusChem, 2021, 86, 758–762 CrossRef CAS.
- (a) R. J. Durand, S. Gauthier, S. Achelle, S. Kahlal, J.-Y. Saillard, A. Barsella, L. Wojcik, N. le Poul and F. Robin-le Guen, Dalton Trans., 2017, 46, 3059–3069 RSC; (b) R. J. Durand, S. Achelle, F. Robin-le Guen, E. Caytan, N. le Poul, A. Barsella, P. Guevara Level, D. Jacquemin and S. Gauthier, Dalton Trans., 2021, 50, 4623–4633 RSC.
- R. J. Durand, S. Gauthier, S. Achelle, T. Groizard, S. Kahlal, J.-Y. Saillard, A. Barsella, N. le Poul and F. Robin-le Guen, Dalton Trans., 2018, 47, 3965–3975 RSC.
- H. Akdas-Kiliç, M. Godfroy, J.-L. Fillaut, B. Donnio, B. Heinrich, P. Kędziora, J.-P. Malval, A. Spangenberg, S. van Cleuvenbergen, K. Clays and F. Camerel, J. Phys. Chem. C, 2015, 119, 3697–3710 CrossRef.
- S. van Cleuvenbergen, P. Kędziora, J.-L. Fillaut, T. Verbiest, K. Clays, H. Akdas-Kiliç and F. Camerel, Angew. Chem., Int. Ed., 2017, 56, 9546–9550 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |