
Electrochemically enhanced deoxygenative cross-coupling of aryl ketones with heteroarenes through in situ generated benzyl carbocations†
Yiyi
Zhang
ab,
Jianxin
Hou
b,
Hui
Yang
b,
Shengdong
Wang
*a and
Kedong
Yuan
 *a
*a
aGuangzhou Municipal and Guangdong Provincial Key Laboratory of Molecular Target & Clinical Pharmacology, the NMPA and State Key Laboratory of Respiratory Disease, School of Pharmaceutical Sciences and the Fifth Affiliated Hospital, Guangzhou Medical University, Guangzhou 511436, China. E-mail: wangshengdong@gzmu.edu.cn; kedongyuan@gzhmu.edu.cn
bTianjin Key Laboratory of Advanced Functional Porous Materials, Institute for New Energy Materials and Low-Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin, 300384, P.R. China
First published on 22nd November 2022
Abstract
Triflic acids/silanes as cooperative reductants enable the convenient transformation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds through a multistep reaction pathway in one pot. Electrolysis of the acidic reaction mixture significantly improved carbonyl reduction and thus facilitated the generation of benzyl carbocations, which show high reactivity towards electron-rich heteroarenes for C–C bond formation.
O bonds through a multistep reaction pathway in one pot. Electrolysis of the acidic reaction mixture significantly improved carbonyl reduction and thus facilitated the generation of benzyl carbocations, which show high reactivity towards electron-rich heteroarenes for C–C bond formation.
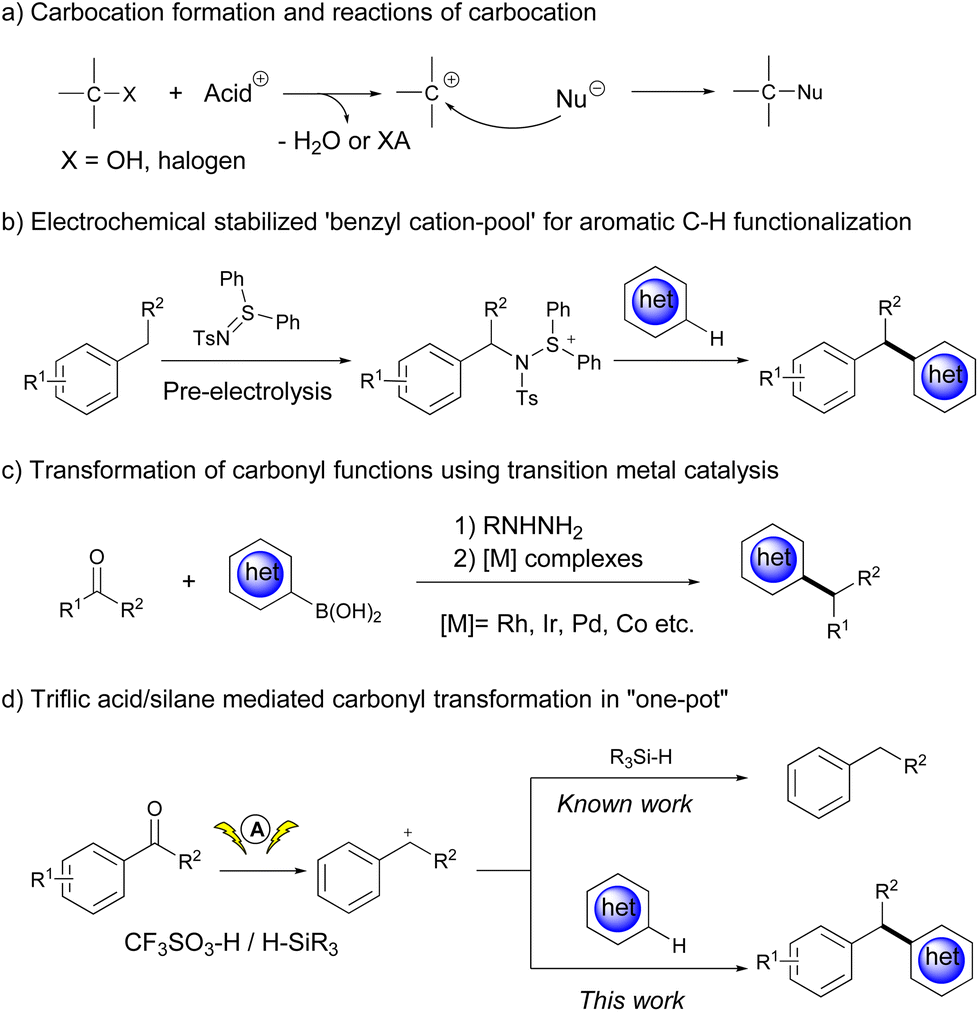
Although carbocation intermediates have been widely used as reactive electrophilic species in the design of organic reactions,1 the scope of the carbocation precursors is rather limited. The major concern about carbocations arises from their short life span and instability; therefore, benzyl carbocations and the most substituted alkyl carbocations generated from the corresponding alcohols or alkyl halides in the presence of acids have been extensively investigated thus far (Scheme 1a).2 To solve this problem, Yoshida et al. developed a “cation pool” strategy, which enables the accumulation of carbocations by oxidation of benzylic sp3 C–H bonds in the absence of nucleophiles, thus expanding the scope of the SN1 reaction to a diverse array of nucleophiles.3 A recent effort from the same group improved the synthetic utility of benzyl carbocations by introducing some stabilizing agents (Scheme 1b).4 In spite of these achievements, it would be of great importance to expand the scope of carbocation precursors.
 | ||
| Scheme 1 Research background. | ||
Aryl ketones are ubiquitous in nature and have many applications. If the carbonyl functional group in aryl ketones is deoxygenatively converted, it will provide a broad spectrum of aromatic molecules useful in organic synthesis.5 However, such a transformation is challenging, and stoichiometric reducing agents are generally required to transform the carbonyl group to alcohols or other convertible functional groups (Scheme 1c).5b,6 To our delight, aryl ketones could be temporarily transformed into the corresponding benzyl carbocations in a Brønsted acid/silane reduction system.7 Even so, the intermolecular reaction between benzyl carbocations and nucleophiles might face competition from hydride species that reduce benzyl carbocations to deliver hydrocarbons (Scheme 1d, top). An interesting phenomenon from our previous research showed that the electrochemical activation of carbonyl derivatives could significantly promote the nucleophilic addition reaction between aldehydes and various nucleophiles, including heteroarenes, alcohols, thiols and silane derivatives.8 Driven by curiosity, we wonder if ketones could be directly activated as carbocation precursors to react with heteroarenes under electrochemical conditions, which also provides an alternative route for diarylalkane synthesis.9
Herein, we report triflic acid/silane mediated cross-coupling reactions between aryl ketones and (hetero)arenes, which were significantly promoted under electrochemical conditions (Scheme 1d, bottom).
Initially, diphenyl ketone 1a and benzofuran 2a were selected as the model substrates to explore potential reactions. Surprisingly, no reaction occurred with several Brønsted acids, such as CF3COOH, H2SO4, and p-tolylsulfonic acid (Table 1, entry 1. See also Table S1†), while the use of triflic acid dramatically activated the substrates, leading to the formation of 2-benzhydrylbenzofuran 3a in 68% yield with complete C2 selectivity (entry 2). Increasing the temperature did boost the reactivity of the transformation, and part of 1a remained unreacted after 3 hours of refluxing (entries 3 and 4). However, the presence of electricity significantly improved the reactivity, and the reaction was completed after constant current electrolysis at 10 mA for 3 hours, affording 3a in quantitative yield (entry 6). It is obvious that electrolysis is a much more efficient approach than the use of thermal conditions (entry 7). A decreased loading of CF3SO3H resulted in a low conversion of 1a (entry 8). Several silanes were investigated and there was no obvious reactivity difference for the electrochemically promoted deoxygenative cross-coupling reaction (entries 9–11); thus, H(Me)2SiOSi(Me)2H (TMDS) as a more economical silane source was used for the reaction. It is worth mentioning that the absence of silane would shut off the reaction (entry 11). The electrode materials did not significantly affect the reactivity of the deoxygenative transformation (entries 12 and 13). The experiment showed that the electrochemical reaction was less efficient in a divided cell than in an undivided cell (entry 14). Therefore, using stoichiometric triflic acid/TMDS in 0.1 M CH3CN under constant current electrolysis at 10 mA for 3 hours was selected as the optimal conditions, which drive the complete conversion of 1a and afford 3a in 82% isolated yield with exclusive C2 regioselectivity on benzofuran. Notably, the reaction also worked quite well on a 1.0 mmol scale without compromising the reactivity and selectivity (entry 15).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Variation from standard conditions | Yield of 3ab (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a 1a, 0. 5 mmol; 2a, 1.0 mmol; CF3SO3H, 0.9 mmol; H(Me)2SiOSi(Me)2H, 0.25 mmol; CH3CN2 (5 mL) in an undivided IKA ElectraSyn 2.0 cell with a graphite plate as the anode and a Cu plate as the cathode. Constant current 10 mA, rt, 3 h. Yields were determined by using 50 μL of dodecane as the internal standard. b Isolated yield was given in the parentheses. c Room temperature without electrolysis. d Reaction performed in a sealed Schlenk tube without inert atmosphere protection. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | CF3COOH in lieu of CF3SO3H | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2c | Room temp. | 68 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3c | 60 °C | 71 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4c,d | 90 °C | 81 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Electrolysis under 5 mA | 75 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Electrolysis under 10 mA | 100 (82) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Electrolysis under 10 mA for 1 h | 83 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 1.0 equiv. of CF3SO3H | 68 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Ph2SiH2 | 100 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | Ph3SiH | 100 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | No silane | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | Glass carbon was used as the anode | 94 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | Graphite was used as the cathode | 100 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | Reaction in a divided cell | 78 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | Reaction on a 1.0 mmol scale | 100 (83) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
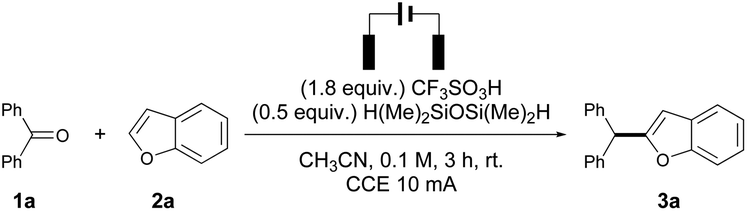
With the optimal conditions in hand, the reaction scope of aryl ketones was investigated to understand the functional group compatibility. When the reaction was performed using 4,4′-dimethylbenzophenone and 2a, a competing reaction occurred to generate the corresponding hydrocarbon side-product 4,4-dimethyldiphenylmethane, which suppressed the yield of cross-coupling product 3b to 32% with complete C2 selectivity. A similar phenomenon was observed on using electron-donating group (–OMe, –NMe2) substituted benzophenones as substrates; either diarylmethane products were formed or poor conversion was observed under the standard conditions. Thus, 3c and 3d were obtained in 36% and 33% isolated yields, respectively. In contrast, fluoro-substituted diaryl ketone 1e and nitro substituted diaryl ketone 1f showed higher reactivities towards 2a under the optimized conditions, affording 3e–f in good isolated yields, though some C3 regioisomer was observed with 4-fluorobenzophenone. 1-Tetralone reacted with 2a under electrochemical conditions to give 3g in 75% isolated yield with lower C2 regio-selectivity. Diarylalkane bearing cyclopropyl (3h) was obtained only in 19% isolated yield, with 45% of phenyl cyclopropyl ketone unconverted, which was in line with the previous experimental result for carbonyl reduction using CF3COOH/Et3SiH,7 while diarylalkane bearing cyclopentyl (3i) was obtained in good isolated yields and with C2 regio-selectivity without suffering from the steric hindrance effect. 1,2-Diphenylethan-1-one reacted with 2a to give the desired product 3j in good yield and selectivity. Compound 3k was formed in a moderate yield from trans-chalcone and 2a in the presence of an excess of silane (1.0 equiv.), with the CC double bond reduced. The alkyl halide could be tolerated under the reaction conditions to form 3l in 27% isolated yield, and debromination product 3m was also isolated in 31% yield from the same reaction. The reactivity of 2-methyl-1-phenylpropan-1-one was similar to that of phenyl cyclopropyl ketone, and 3n was produced in low yield (25% aryl ketone remain unreacted and 15% hydrocarbon product formed). Acetophenone derivatives showed high reactivity with 2a under electrochemical conditions to give 3o–q in excellent yields, while the regioselectivities were generally lower than those of the abovementioned aryl ketones. Several aliphatic ketones were also investigated and no reactivity was observed in these cases, apparently due to the absence of stable cation species (Table 2).
| a Aryl ketone 1, 0.5 mmol; 2a, 1.0 mmol; CF3SO3H, 0.9 mmol; H(Me)2SiOSi(Me)2H, 0.25 mmol; CH3CN (5 mL) in an undivided IKA ElectraSyn 2.0 cell with a graphite plate as the anode and a Cu plate as the cathode. Constant current 10 mA, rt. for 3 h. Isolated yields were presented and regioselectivities were determined by GC and GC-MS analyses. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
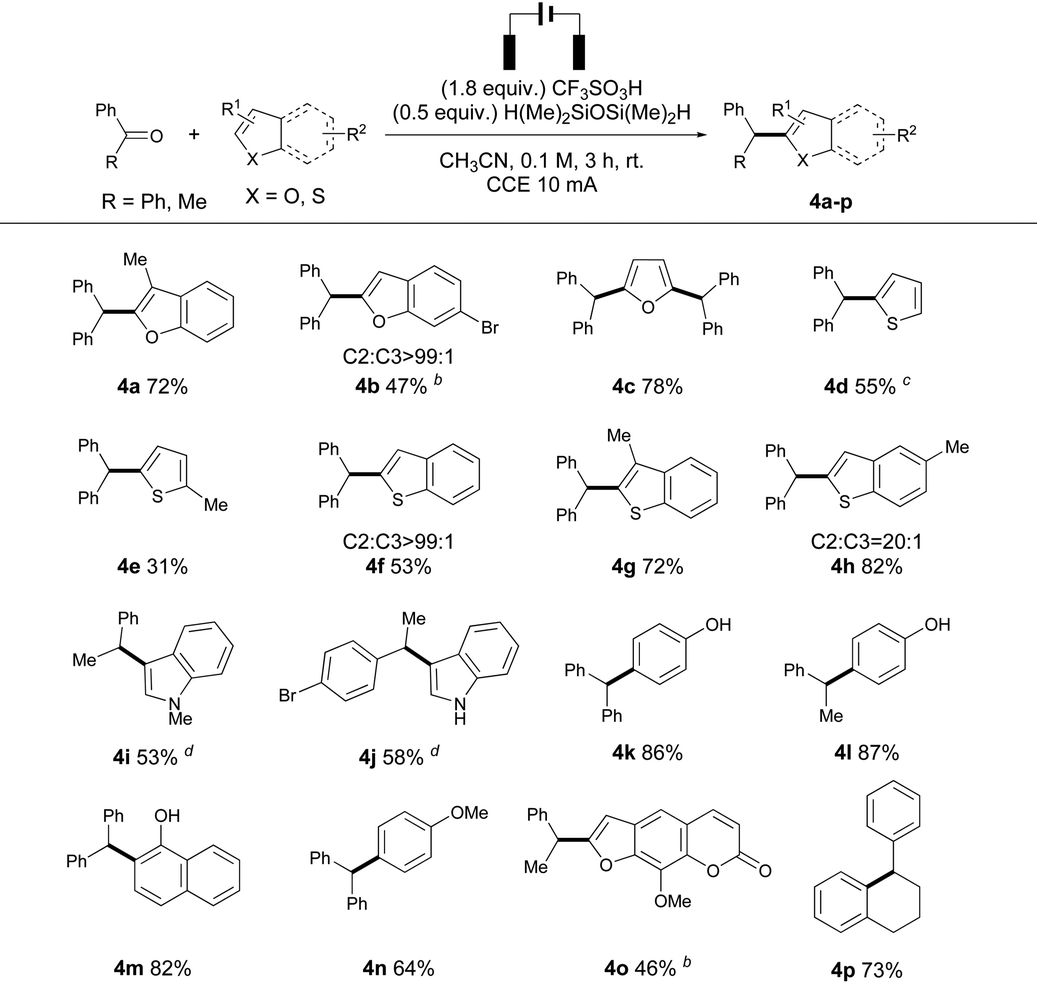
Next, we set out to inspect the reactivity between a diverse array of heteroarenes. Though similar products could be prepared using benzyl alcohols, the transformation was not successful from aryl ketones and heteroarenes. 3-Methyl benzofuran reacted with 1a to afford 4a in high yield, and the presence of an electron withdrawing group (Br) on benzofuran decreased the nucleophilicity of heteroarenes, leading to a low yield of 4b. A disubstituted furan product 4c was isolated in good yield, which is similar to the product formation using the “cation pool” strategy,6a owing to the enhanced nucleophilicity after monoalkylation. However, the reactions of thiophenes and 1a were less effective due to the negative effects of anodic oxidation, producing 4d–e in moderate to low yields. Benzothiophenes exhibited higher reactivities, and 4f–h were produced in good yields and regioselectivities. Electron-rich N-heteroarenes were prone to oxidation on the anode surface, and hence C3 substituted indoles (4i–j) were obtained in moderate yield by decreasing the constant current to 2 mA. Phenol derivatives could react with both benzophenone and acetophenone to produce 4k and 4l in 86% and 87% isolated yields, respectively. 1-Naphthol reacted with 1a under electrolysis to form 4m in 82% isolated yield. Electron-rich arenes reacted with benzophenone to give the desired product 4n in good yield and regioselectivity. Surprisingly, no other regioisomers were observed in these reactions (4k–n). The post-functionalization of a natural product was also successful, and product 4o was prepared from acetophenone and 8-methoxypsoralen in 46% isolated yield. The intramolecular deoxygenative cyclization of 1,4-diphenylbutan-1-one (1p) was successful, affording 4p in 73% isolated yield. Notably, almost no reaction occurred without electricity in the cases of 4o and 4p (Table 3).
| a Aryl ketone 1, 0.5 mmol; (hetero)arene 2, 1.0 mmol; CF3SO3H, 0.9 mmol; H(Me)2SiOSi(Me)2H, 0.25 mmol; CH3CN (5 mL) in an undivided IKA ElectraSyn 2.0 cell with a graphite plate as the anode and a Cu plate as the cathode. Constant current 10 mA, rt, 3 h. b Reaction for 8 h. c 2,5-Disubstituted product was observed. d Electrolysis under 2 mA for 3 hours. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Several control experiments were performed to gain some insights into the reaction pathways. As suggested in Scheme 2a, deoxygenation of 1a under standard conditions proceeded smoothly to afford diphenylmethane in quantitative yield, while a similar reaction in the absence of electrolysis gave the same product only in 51% yield. Such a result suggests that the occurrence of electrolysis would promote the deoxygenative reduction process. The reaction of diphenylmethanol and 2a gave a quantitative yield of 3a, with the same result compared to the reaction of 1a (Scheme 2b). Therefore, silane was not necessary in this step, which was consistent with previous reports that triflic acid can promote the dehydrative cross-coupling of diarylmethanols and heteroarenes.10 Surprisingly, no alcohols were observed in all reactions, and a side product generated from TMDS and triflic acid (R3SiOTf in Scheme 3)10a,11 was detected by GC-MS in all cases, indicating that the protonated ketones were directly reduced to alcohols via hydride transfer from silane. Reduction of ketones to alcohols was possibly the key step and dehydration proceeded rapidly to generate benzyl carbocations, which have been known in Brønsted acid/silane catalyzed reactions,12 and it also could be intercepted by using stoichiometric alcohol nucleophiles (CD3OD, see Scheme S1 in the ESI†). Additionally, cyclic voltammetry (CV) measurements showed that the anodic current obviously increased from the mixture of 1a and 2a compared to the respective study of each substrate, which corresponded to the enhanced reactivity of cross-coupling reactions between benzyl carbocations and heteroarenes (see Fig. S2 in the ESI†). The occurrence of electrolysis, together with the strong acid media, possibly improved the stability of benzyl carbocations,13 thereby increasing the reactivities of deoxygenative cross-coupling reactions.
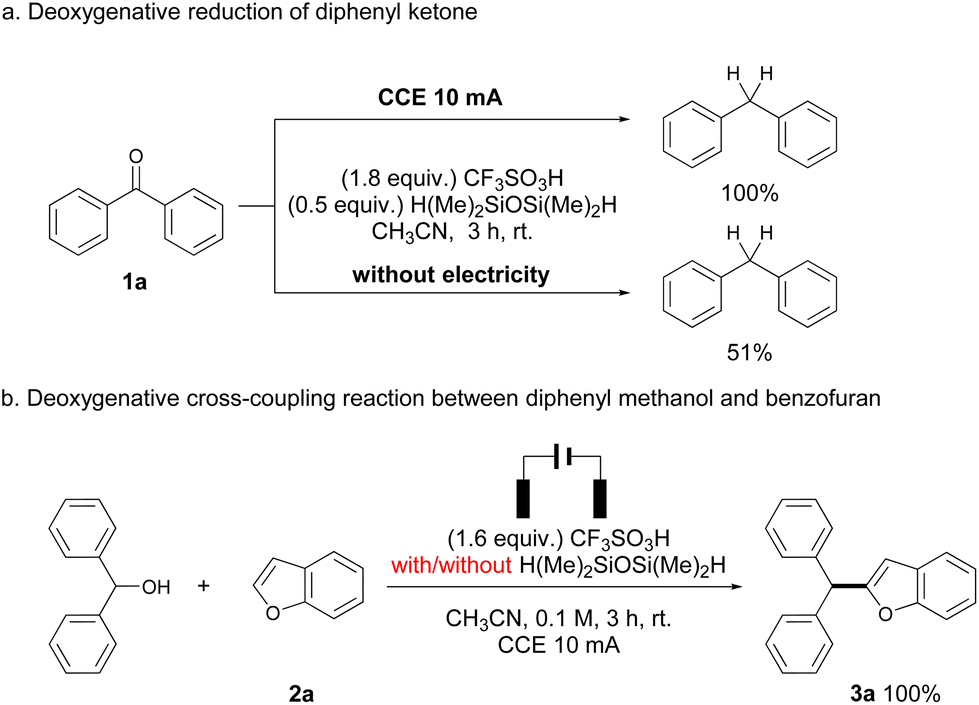 | ||
| Scheme 2 Control experiments. | ||
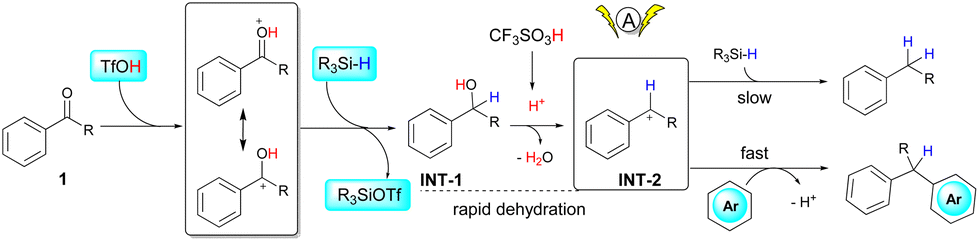 | ||
| Scheme 3 Proposed reaction pathway. | ||
Based on the above experiments, a multistep reaction pathway is proposed for the one-pot cross-coupling of aryl ketones and heteroarenes (Scheme 3). Initially, the proton from triflic acid and the hydride from silane work synergistically to reduce the carbonyl group from 1 to form alcohol INT-1. With excess triflic acid, fast dehydration of the hydroxyl group leads to the generation of benzyl carbocations INT-2, which then react rapidly with electron-rich heteroarenes to form cross-coupling products rather than reacting with silane to give hydrocarbon products, as shown in Scheme 2b. The electrolytic conditions significantly promote the process of carbonyl reduction and thus facilitate the generation of benzyl carbocations.
In conclusion, we developed a one-pot protocol for C(sp2)–C(sp3) bond formation from aryl ketones and (hetero)arenes using triflic acid and the H(Me)2SiOSi(Me)2H cooperative reduction system. The reaction tolerates a wide array of aryl ketones and heteroarenes to deliver diarylalkanes in high efficiency and selectivity. A multistep pathway involving in situ generated benzyl carbocations is proposed for the deoxygenative cross-coupling reactions. Notably, the presence of electricity and the super acidic conditions promote the generation of benzyl carbocations and thus enhance their reactivity toward heteroarene nucleophiles. This research brings new opportunities for carbonyl group transformations using the mechanism of cation chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the High-level University Construction Fund of Guangdong Province (02-412-2202-2119), the National Natural Science Foundation of China (21702145), and the Natural Science Foundation of Tianjin City (19JCYBJC17700) for providing funding for our research.References
- (a) G. A. Olah, Angew. Chem., Int. Ed. Engl., 1995, 34, 1395–1405 CrossRef; (b) G. A. Olah, J. Org. Chem., 2001, 66, 5943–5957 CrossRef CAS.
- (a) J. I. Yoshida and S. Suga, Chem. – Eur. J., 2002, 8, 2650 CrossRef CAS; (b) E. S. Stoyanov and I. V. Stoyanova, ChemistrySelect, 2020, 5, 9277–9280 CrossRef CAS.
- (a) J. I. Yoshida, Y. Ashikari, K. Matsumoto and T. Nokami, J. Synth. Org. Chem., 2013, 71, 1136–1144 CrossRef CAS; (b) J. I. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702–4730 CrossRef CAS; (c) M. Okajima, K. Soga, T. Nokami, S. Suga and J. I. Yoshida, Org. Lett., 2006, 8, 5005–5007 CrossRef CAS PubMed.
- R. Hayashi, A. Shimizu and J. I. Yoshida, J. Am. Chem. Soc., 2016, 138, 8400–8403 CrossRef CAS.
- (a) D. P. Todd, B. B. Thompson, A. J. Nett and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 12788–12791 CrossRef CAS PubMed; (b) S. Asako and L. Ilies, Chem. Lett., 2020, 49, 1386–1393 CrossRef CAS; (c) R. Xie, W.-H. Mao, H.-H. Jia, J.-L. Sun, G.-P. Lu, H.-F. Jiang and M. Zhang, Chem. Sci., 2021, 12, 13802–13808 RSC; (d) J.-B. Li, C.-Y. Huang and C.-J. Li, Angew. Chem., Int. Ed., 2022, 61, e202112770 CAS.
- For deoxygenative transformation of carbonyl groups via diazo derivatives, see: (a) J.-Y. Yu and R. Kuwano, Org. Lett., 2008, 10, 973–976 CrossRef CAS; (b) H. Wang, X. J. Dai and C. J. Li, Nat. Chem., 2017, 9, 374–378 CrossRef CAS PubMed; (c) S. Wang, B. Y. Cheng, M. Srsen and B. Konig, J. Am. Chem. Soc., 2020, 142, 7524–7531 CrossRef CAS PubMed; (d) Y. Xia and J. Wang, J. Am. Chem. Soc., 2020, 142, 10592–10605 CrossRef CAS.
- C. T. West, S. J. Donnelly, D. A. Kooistra and M. P. Doyl, J. Org. Chem., 1973, 38, 2675–2681 CrossRef CAS.
- (a) C. Liu, Y. Shen, Z. Xiao, H. Yang, X. Han, K. Yuan and Y. Ding, Green Chem., 2019, 21, 4030–4034 RSC; (b) H. Yang, Y. Shen, Z. Xiao, C. Liu, K. Yuan and Y. Ding, Chem. Commun., 2020, 56, 2435–2438 RSC; (c) C. Liu, Z. Xiao, S. Wu, Y. Shen, K. Yuan and Y. Ding, ChemSusChem, 2020, 13, 1997–2001 CrossRef CAS.
- (a) M. Dryzhakov, E. Richmond and J. Moran, Synthesis, 2016, 935–959 CAS; (b) R. Dada, G. Singh, A. Pareek, S. Kausar and S. Yaragorla, Tetrahedron Lett., 2016, 57, 3739–3742 CrossRef CAS; (c) V. D. Vukovic, E. Richmond, E. Wolf and J. Moran, Angew. Chem., Int. Ed., 2017, 56, 3085–3089 CrossRef CAS.
- (a) B. A. Gellert, N. Kahlcke, M. Feurer and S. Roth, Chem. – Eur. J., 2011, 17, 12203–12209 CrossRef CAS PubMed; (b) S. Estopiñá-Durán and J. E. Taylor, Chem. – Eur. J., 2021, 27, 106–120 CrossRef PubMed.
- Note: the formation of CF3SO3SiMe2OSiMe2H (M.W. 282) was detected by GC-MS in all reactions.
- (a) M. P. Doyle and C. T. West, J. Org. Chem., 1975, 40, 3835–3838 CrossRef CAS; (b) H. Mayr and B. Dogan, Tetrahedron Lett., 1997, 38, 1013–1016 CrossRef CAS; (c) M. Shibuya, S. Fujita, M. Abe and Y. Yamamoto, ACS Catal., 2017, 7, 2848–2852 CrossRef CAS.
- For the reaction between triflic acid and CH3CN, see: (a) G. E. Salnikov, A. M. Genaev, V. G. Vasiliev and V. G. Shubin, Org. Biomol. Chem., 2012, 10, 2282–2288 RSC; (b) B. D. McCarthy, D. J. Martin, E. S. Rountree, A. C. Ullman and J. L. Dempsey, Inorg. Chem., 2014, 53, 8350–8361 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob02065c |
| This journal is © The Royal Society of Chemistry 2023 |