
Molecular torsion springs: alteration of helix curvature in frustrated tertiary folds†
Friedericke S.
Menke
a,
Daniela
Mazzier
a,
Barbara
Wicher
 b,
Lars
Allmendinger
a,
Brice
Kauffmann
c,
Victor
Maurizot
d and
Ivan
Huc
*a
b,
Lars
Allmendinger
a,
Brice
Kauffmann
c,
Victor
Maurizot
d and
Ivan
Huc
*a
aDepartment of Pharmacy, Ludwig-Maximilians-University, Butenandstraße 5–13, 81377 Munich, Germany. E-mail: ivan.huc@cup.lmu.de
bDepartment of Chemical Technology of Drugs, Poznan University of Medical Sciences, Grunwaldzka 6, 60-780 Poznan, Poland
cInstitut Européen de Chimie et Biologie (UMS3011/US001), CNRS, Inserm, Université de Bordeaux, 2 rue Robert Escarpit, F-33600 Pessac, France
dCBMN (UMR 5248), Univ. Bordeaux, CNRS, Bordeaux INP, 2 rue Robert Escarpit, 33600 Pessac, France
First published on 4th January 2023
Abstract
The first abiotic foldamer tertiary structures have been recently reported in the form of aromatic helix–turn–helix motifs based on oligo-quinolinecarboxamides held together by intramolecular hydrogen bonds. Tertiary folds were predicted by computational modelling of the hydrogen-bonding interfaces between helices and later verified by X-ray crystallography. However, the prognosis of how the conformational preference inherent to each helix influences the tertiary structure warranted further investigation. Several new helix–turn–helix sequences were synthesised in which some hydrogen bonds have been removed. Contrary to expectations, this change did not strongly destabilise the tertiary folds. On closer inspection, a new crystal structure revealed that helices adopt their natural curvature when some hydrogen bonds are missing and undergo some spring torsion upon forming the said hydrogen bonds, thus potentially giving rise to a conformational frustration. This phenomenon sheds light on the aggregation behaviour of the helices when they are not linked by a turn unit.
Introduction
The functions of biopolymers are enabled by their shapes and folded structures. This structure–function relationship has inspired chemists to design foldamers, synthetic oligomers also able to adopt folded conformations.1–7 Eliciting secondary structures such as single helices or sheets in a great variety of synthetic oligomers has been successful when using aliphatic, aliphatic–aromatic, or aromatic amino acid building blocks. Useful properties of these secondary structures have been reported, including biological activity, and even their potential as therapeutic agents.8–16 Thus, short foldamers may be able to cross cell membranes efficiently while having low toxicity and immunogenicity.17–21 They may also resist proteolytic degradation.In biopolymers, numerous functions emerge only in tertiary and quaternary structures. Accordingly, many more functions of foldamers may be expected by reaching similar size and structural complexity.22 However, predicting folding conducive to tertiary structure formation is far more challenging. Two aspects that make tertiary structure prediction difficult are cooperativity and frustration. Cooperativity refers to the fact that individual secondary modules of a tertiary structure need not be inherently stable: α-helices found in proteins are more often than not unstable when isolated but are stabilised within the full sequence tertiary fold. Frustration refers to the fact that the overall stability gained in the tertiary fold allows non-ideal – energetically costly – conformations to be present: strain is acceptable to a certain extent and may be beneficial to functions.23 Despite the fact that the energy terms associated with cooperativity and frustration are difficult to estimate – they usually reflect multiple contributions that partly compensate for each other – the ab initio design of proteins has made great progress.24–31 Synthetically accessible mini-proteins have also attracted interest due to their potential applications in biotechnology and medicine.32 Using various approaches, steps were made away from purely α-peptidic backbones, and β-amino acids were introduced in tertiary structures.33–39 Some helix bundles from β-peptides40,41 and oligoureas42,43 have also been reported.
To access shapes and functions remote from (and beyond) those achieved by biopolymers, research in the field of abiotic foldamers is conducted. Such foldamers consist of units not seen in the natural world that nevertheless fold into conformationally ordered states stabilised by non-covalent interactions. A major class of abiotic foldamers are aromatic oligomers, i.e. oligomers that possess aryl rings in their main chain.6,7,44–46 A prototypical family of aromatic foldamers are the oligoamides of 8-amino-2-quinolinecarboxylic acid (Q in Fig. 1a), which adopt particularly stable helical conformations in essentially any solvent.47–49 Qn oligoamide helices are so stable that they tolerate a large proportion of more flexible 6-aminomethyl-2-pyridinecarboxylic acid units (P in Fig. 1a).50–53 Oligomers consisting of Q and P units are easily accessible by solid phase synthesis,52 and show a high propensity to crystallise. They may find applications in diverse areas, including circularly polarised luminescence,54,55 charge transport and metal coordination,56 and protein surface recognition.11,16,57–59
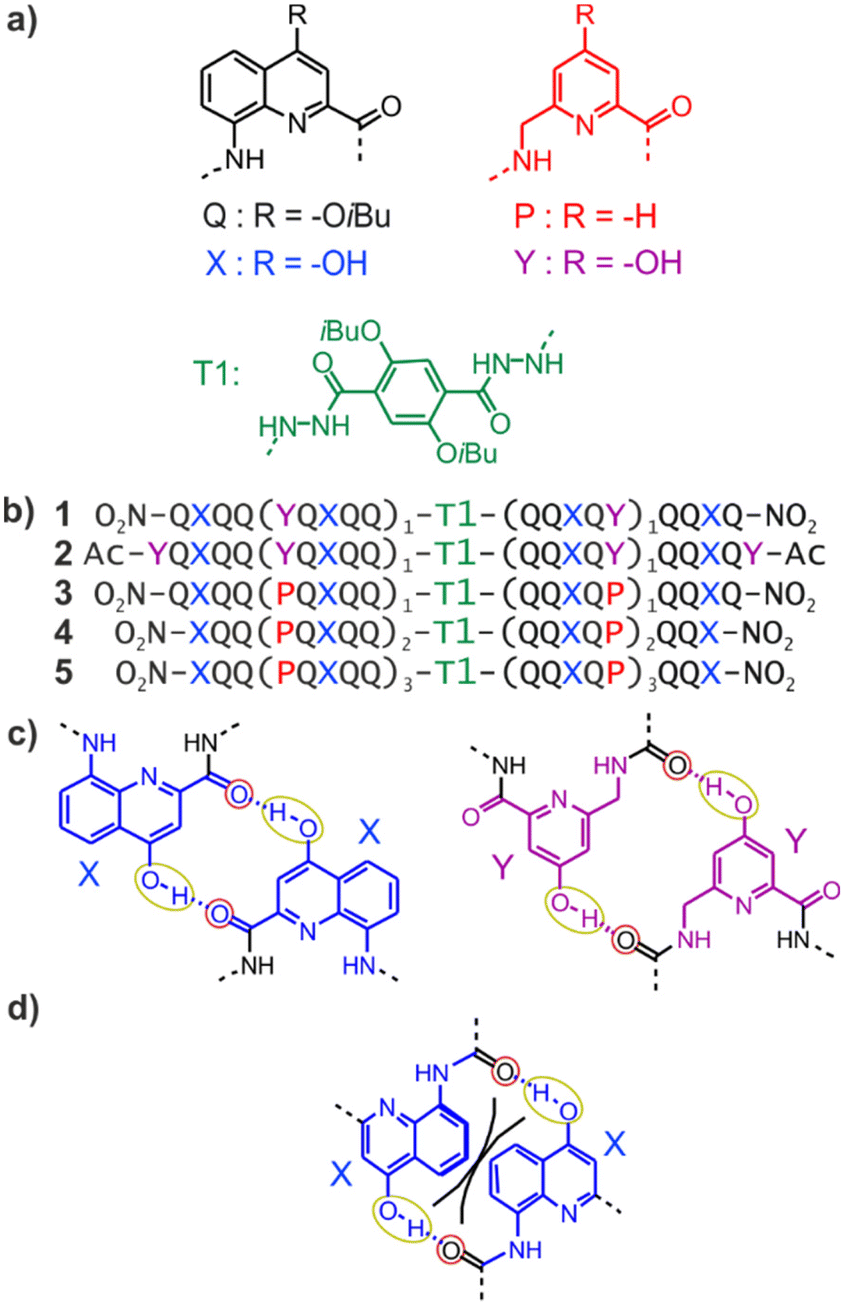 | ||
| Fig. 1 (a) Structures of units Q, X, Y, P, and T1. (b) Foldamer sequences. Note the inversion of C → N polarity on each side of the central T1 unit. In sequences ending with an 8-nitro group, “NO2” replaces the terminal amine. (c) Hydrogen-bonding patterns involving X and Y units as observed in the structures of 1 and 2. The yellow and red circles around the hydrogen bond donors and acceptors correspond to the yellow cups and red knobs in Fig. 2. (d) Expected steric clash (black lines) if Y units were replaced by X units.60 | ||
Early steps were made towards abiotic tertiary structures by connecting several Qn helices to various types of linkers.61–63 Recently, the first true abiotic tertiary foldamers were designed and characterised in the form of helix–turn–helix motifs composed of two helical oligomers connected at their C-terminus by a T1 turn unit (Fig. 1a) and in which inter-helix hydrogen bonds were mediated by units X and Y, the 4-hydroxy substituted analogues of Q and P, respectively.60,64,65 This design exploits the fact that all amide carbonyl groups, that is, good hydrogen bond acceptors, diverge from Qn helices. X and Y units were introduced so that each hydroxy group forms a hydrogen bond with a carbonyl group of the other helix as, for example, in sequences 1 and 2 (Fig. 1b). The outcome was inter-helix hydrogen bonding patterns involving pairs of X monomers and pairs of Y monomers (Fig. 1c). One may note that the hydrogen bonding pattern involving two Y units could not be achieved with X units because the benzenic rings of the latter would sterically hamper a short X-to-X distance (Fig. 1d).
As a consequence of inter-helix hydrogen bonding, the two helices are held parallel to each other at a close distance and with the same N-to-C orientation.‡60,65 In addition, the hydrogen bonds only form when the two helices have the same handedness. Schematic representations in Fig. 2a–c depict the resulting architectures, including the fact that a pair of hydrogen bonds form at every helix turn, involving alternatively X and Y units. Thus, six inter-helix hydrogen bonds formed in sequence 1 (Fig. 2a and b) and eight in sequence 2 (Fig. 2c). The resulting helix–turn–helix motifs are very stable in chlorinated solvents and may be disrupted upon adding DMSO, a hydrogen bonding competitor, leading to the emergence of a conformer with one P and one M helix (Fig. 2h).60 However, earlier investigations also revealed that, in the absence of turn units, independent helices aggregate through modes other than the parallel PP or MM head-to-head motif found in helix–turn–helix structures. Instead, trimeric parallel aggregates and tilted dimers were characterised.60 This outcome points non-ideal interactions or conformations within the tertiary motifs that are constrained by the geometry of the turn unit.
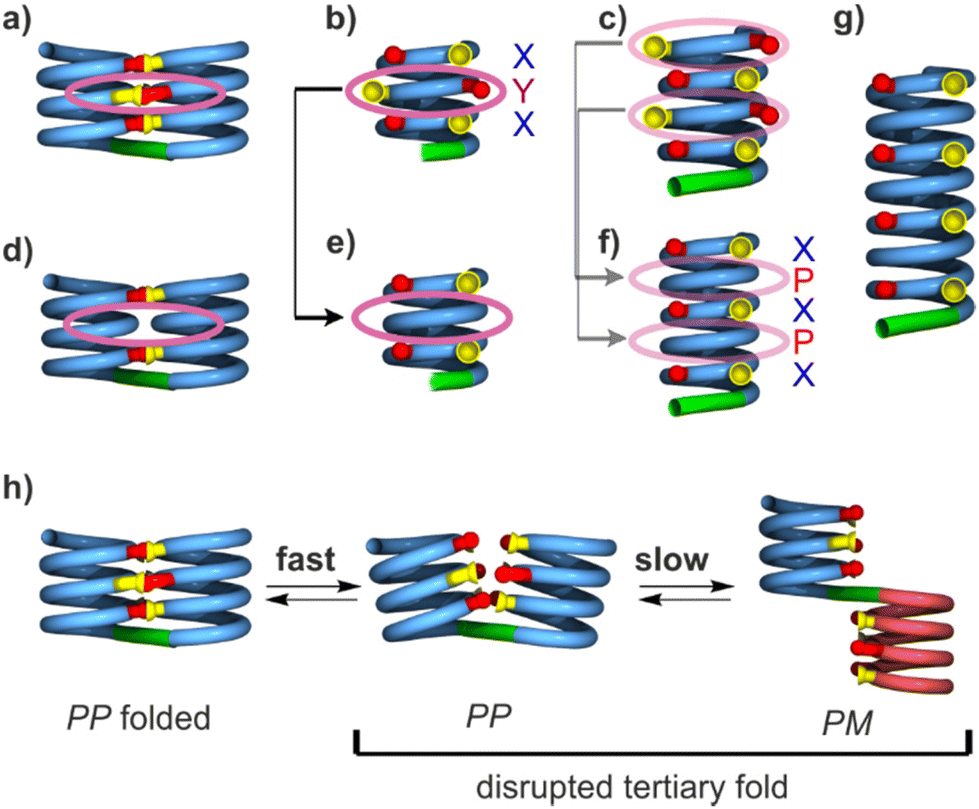 | ||
| Fig. 2 Schematic representation of helix-turn-helix tertiary motifs of different lengths with and without Y units. (a) Side view of a helix-turn-helix-motif with six hydrogen bonds formed by sequence 1. (b) Front view of the hydrogen-bonding interface involved in the structure shown in (a). (c) Front view of a hydrogen-bonding interface one turn longer than that of (b), i.e. with eight hydrogen bonding sites (two X and two Y units per helix), as it occurs in 2. (d–f) Structures analogous to those of (a–c), respectively, in which Y units have been replaced by P. Hydrogen bonding sites associated with X units are present at every other helix turn. (g) Hydrogen-bonding interface two turns longer than that shown in (f) with eight hydrogen bonding sites (four X units). (h) Schematic representation of the conformational equilibria involved when polar solvents disrupt a helix–turn–helix fold. The arrays of hydroxy protons and carbonyl oxygen atoms involved in hydrogen bonding are shown in yellow and red, respectively. Blue, red and green tubes represent P- and M-helical segments and turn units, respectively. Pink ovals indicate hydrogen bonding sites associated with Y units or the equivalent areas where Y units have been replaced by P. | ||
Here we show that, contrary to expectations, removing some hydrogen bonds within the helix–turn–helix structures, namely replacing Y by P (sequences 3–5, Fig. 1b), does not result in a strong destabilisation. A crystallographic structure of 3 showed that the missing hydrogen bonds permit a relaxation of the helix curvature, which must be somewhat spring-loaded by the Y-to-Y hydrogen bonding. These results thus shed light on subtle aspects of the interactions and tensions within abiotic tertiary folds that resemble those observed in proteins23 and provide guidelines to elaborate these designs further.
Results and discussion
Design, synthesis, and helix–turn–helix folding
Sequences 3, 4 and 5 were conceived as analogues of 1 and 2 containing P units instead of Y (Fig. 1b). In 3–5, inter-helix hydrogen bonding is mediated only by X units. It may occur at every other helix turn and not at every turn as in 1 or 2. Sequence 3 is identical to 1 but for the replacement of the two Y units by P. With a total of four X units, up to four inter-helix hydrogen bonds may form within a tertiary helix–turn–helix fold of 3 (Fig. 2d and e). Exploiting the fact that helices of Q/P sequences span two full turns every five units,47 compound 4 is composed of helical segments that have both been extended by one QQXQP pentad with respect to the helices of 3. Compound 4 thus has the potential for six inter-helix hydrogen bonds (Fig. 2f), as in 1, though its helices are two turns longer. Similarly, the helical segments of 5 have each been extended by one more QQXQP pentad with respect to the helices of 4. Compound 5 has the potential for eight inter-helix hydrogen bonds (Fig. 2g), as in 2, though the helices of 5 span over seven turns and are almost twice as long as the helices of 2.Compounds 3–5 were synthesised following the same approach (see ESI†). Helical segments spanning from the N-terminus to the unit before the linker were prepared on solid phase using Q, P, and X monomers with an Fmoc-amine protection, a free carboxylic acid, and a tBu-ether protection of the hydroxy group of X.52 Coupling was mediated by acid chloride activation, and Fmoc was removed after each coupling with piperidine. The last monomer introduced at the N-terminus was either X or Q with a nitro group in position 8. Synthesis was performed on an acid labile Sasrin® resin so that mild acid (hexafluoroisopropanol) allowed for resin cleavage while preserving tBu-ether protection of X units. Sequences were then purified by crystallisation in CH2Cl2/methanol before being coupled in solution to T1 units using PyBOP as a coupling agent. Finally, the protected precursors of 3, 4 and 5 were purified by crystallisation from CH2Cl2/methanol (for 3), or by recycling GPC (for 4 and 5).
Before tBu-ether cleavage at X units, helices cannot form hydrogen bonds. All PP/MM and PM conformers of these precursors are thus populated (Fig. 2h). Equilibrium between PP/MM and PM conformers is slow on the NMR time scale and NMR spectra show two sets of signals in different proportions, typically around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S1†), as was previously observed for the precursors of 1 and 2.60 A crystal structure of the protected precursor of 3 in its PM conformation is shown Fig. S2a,† which presumably corresponds to the major species in solution. The final products 3–5 were obtained after TFA deprotection of the X units without further purification. The 1H NMR spectra of 1–5 all show a single set of signals (Fig. 3), indicating conformational equilibria are no longer at play and that a single species is present in solution, thus hinting at the formation of helix–turn–helix motifs. This also applied to the longest sequence 5, even though it could not be isolated in a very pure form. Also, further investigations of 5 were not conducted because of its low solubility.
:1 (Fig. S1†), as was previously observed for the precursors of 1 and 2.60 A crystal structure of the protected precursor of 3 in its PM conformation is shown Fig. S2a,† which presumably corresponds to the major species in solution. The final products 3–5 were obtained after TFA deprotection of the X units without further purification. The 1H NMR spectra of 1–5 all show a single set of signals (Fig. 3), indicating conformational equilibria are no longer at play and that a single species is present in solution, thus hinting at the formation of helix–turn–helix motifs. This also applied to the longest sequence 5, even though it could not be isolated in a very pure form. Also, further investigations of 5 were not conducted because of its low solubility.
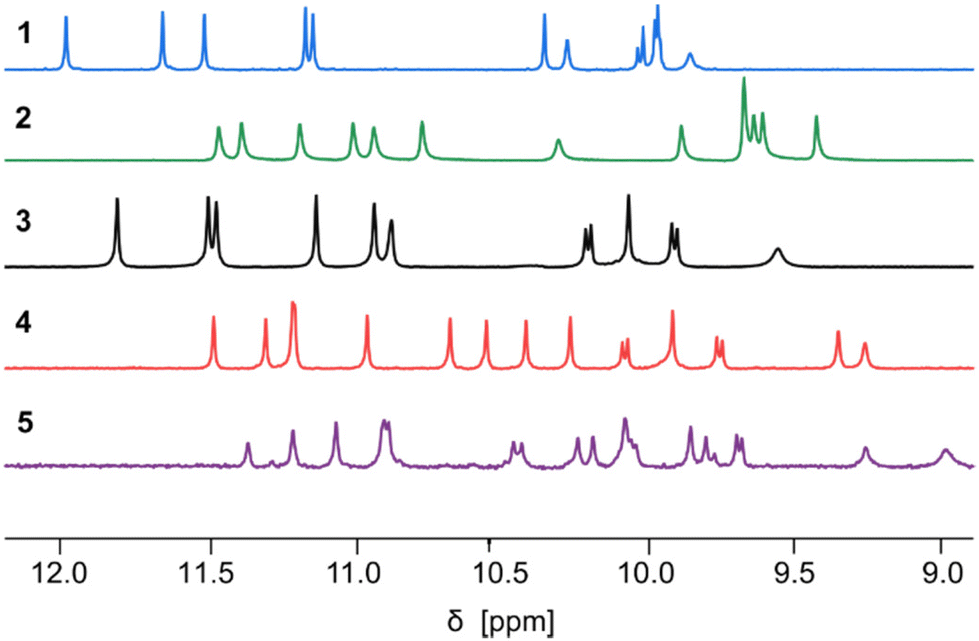 | ||
| Fig. 3 Part of the 500 MHz 1H NMR spectra of sequences 1–5 in CDCl3 at 25 °C showing the resonances of amide protons and hydrogen-bonded hydroxy protons. The spectra of 1 and 2 are from ref. 60. | ||
Helix–turn–helix folding was confirmed by a solid state structure of 3 (Fig. 4b and d) that closely resembles that of 1 (Fig. 4a and c). The molecular structure of 3 shows the expected head-to-head parallel arrangement and the same handedness of the two helices. The structure admits a pseudo-C2 symmetry axis pointed through the aromatic ring of the turn unit. The crystal lattice is centrosymmetric and contains both the PP and the MM conformers. Four inter-helix hydrogen bonds between X units form according to the expected pattern (Fig. 1c). As for the Y units of 1, the P units of 3 face each other but no hydrogen bonding, not even a C–H⋯O contact, is observed. In 1, the shortest C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯CAr distance is around 3.3 Å, while it is 3.9 Å in 3. The molecular structures 1 and 3 both show the same slight bending of the turn unit (Fig. 4a and b). The side views of 1 and 3 (Fig. 4a and b) display a minor difference: at a given helix turn, all four hydrogen bond donors and acceptors (yellow and red spheres belonging to a pair of hydrogen-bonded X or Y units) are in the same plane in the structure of 1 whereas they are slightly out of plane in the structure of 3. Furthermore, top views (Fig. 4c and d) showed an alignment of the hydrogen bond donors and acceptors of X units in the structure of 3, but not in the structure of 1. In summary, NMR and crystallographic data concur to show that, for P-containing sequence 3, four hydrogen bonds are sufficient to stabilise the tertiary fold.
O⋯CAr distance is around 3.3 Å, while it is 3.9 Å in 3. The molecular structures 1 and 3 both show the same slight bending of the turn unit (Fig. 4a and b). The side views of 1 and 3 (Fig. 4a and b) display a minor difference: at a given helix turn, all four hydrogen bond donors and acceptors (yellow and red spheres belonging to a pair of hydrogen-bonded X or Y units) are in the same plane in the structure of 1 whereas they are slightly out of plane in the structure of 3. Furthermore, top views (Fig. 4c and d) showed an alignment of the hydrogen bond donors and acceptors of X units in the structure of 3, but not in the structure of 1. In summary, NMR and crystallographic data concur to show that, for P-containing sequence 3, four hydrogen bonds are sufficient to stabilise the tertiary fold.
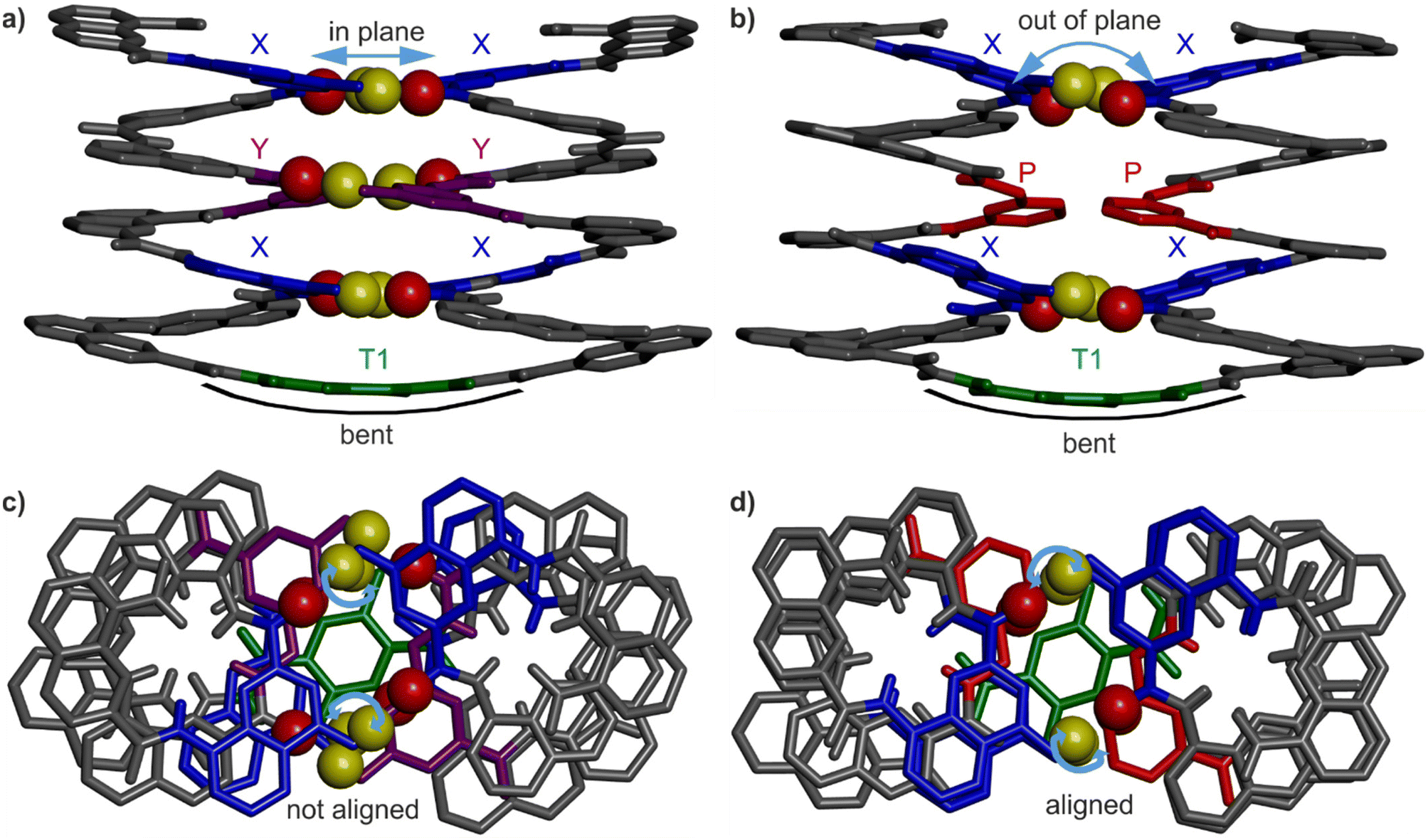 | ||
| Fig. 4 (a) And (b) show the side views of the crystal structures of 1 and 3, respectively. (c) And (d) show the top views of the crystal structures of 1 and 3, respectively. The hydroxy protons and carbonyl oxygen atoms of the hydrogen-bonding arrays are shown as yellow and red balls, respectively. The X units are shown in blue, the Y units in violet, the P-units in red and the turn units in green. Blue arrows point to notable differences between the two structures. Included solvent molecules, non-polar hydrogen atoms and side chains are omitted for clarity. | ||
Solution state studies reveal unexpected stability patterns
Polar solvents such as DMSO and DMF compete for hydrogen bonding and have the capacity to disrupt the hydrogen-bonded interface of helix–turn–helix motifs, giving rise to structures as those shown in Fig. 2h. For example, adding DMSO-d6 to a CDCl3 solution of 2 resulted in shifts of 1H NMR signals associated with a fast equilibrium between folded and disrupted conformations without helix handedness inversion, leading to an averaging of the 1H NMR signals.60 Adding DMSO to the protected precursor of 2, which is unable to fold, did not result in such changes. The chemical shift variations occur over a narrow range of solvent compositions suggesting a cooperative, all-or-nothing, disruption of the hydrogen-bonding interface. Concomitantly to the chemical shift variations, 1H NMR spectra show the emergence of a new species assigned to the disrupted PM conformers, in slow exchange with the PP and MM conformers. In the case of 2, a solid state structure of the disrupted PP/MM conformers was obtained from crystals grown from DMF.60Similar experiments, that is, 1H NMR monitoring of polar solvent-induced disruption of the tertiary fold, were carried out with compound 3, a priori the least stable helix–turn–helix motif of all since it contains only four inter-helix hydrogen bonds (Fig. 4b). Deuterated DMSO, pyridine, DMF, methanol, acetonitrile, acetone and tetrahydrofuran were tested to compare their ability to disrupt the four hydrogen bonds (Fig. S3–S12†). These experiments were important not only to assess the robustness of the tertiary folds but also for practical reasons, for example, to guide the choice of crystallisation solvents. Remarkably, relatively small chemical shift variations and no second set of 1H NMR signals, meaning no disruption of the tertiary turn, was observed even in pure acetone-d6 or tetrahydrofuran-d8. Similarly, up to 70% [vol/vol] of acetonitrile-d3 or 50% of methanol-d3 could be added to a chloroform solution of 3 without any disruption. For these two solvents, higher volume fractions could not be reached due to the precipitation of the sample. Precipitation also occurred upon adding 30% of DMF-d7. Nevertheless, chemical shift changes and the emergence of a new set of signals were observed in the course of solvent additions, indicating a certain level of helix–turn–helix disruption, with a transition above 30% of DMF-d7. In the case of DMSO-d6 and pyridine-d5, complete disruption was achieved without any precipitation. Chemical shift variations showed sharp cooperative transitions near 18.5% of DMSO-d6 and 27.5% of pyridine-d5, indicating the stronger ability of DMSO to interfere with intramolecular hydrogen bonding among all tested polar solvents. Conversely, a comparison of the 1H NMR spectra of 3 in CDCl3 containing 16% of either DMSO-d6, pyridine-d5 or DMF-d7 revealed different proportions of the PM conformer in these three solvents: 30%, 20% and <10%, respectively (Fig. 5a). Altogether, these results establish a clear ranking of the disrupting ability of these three solvents, alongside the other polar solvents in which non disruption was observed. As a final control experiment, the addition of pyridine and DMF to the protected precursor of 3, which is unable to fold, did not result in significant changes of the chemical shifts (Fig. S13–S15†).
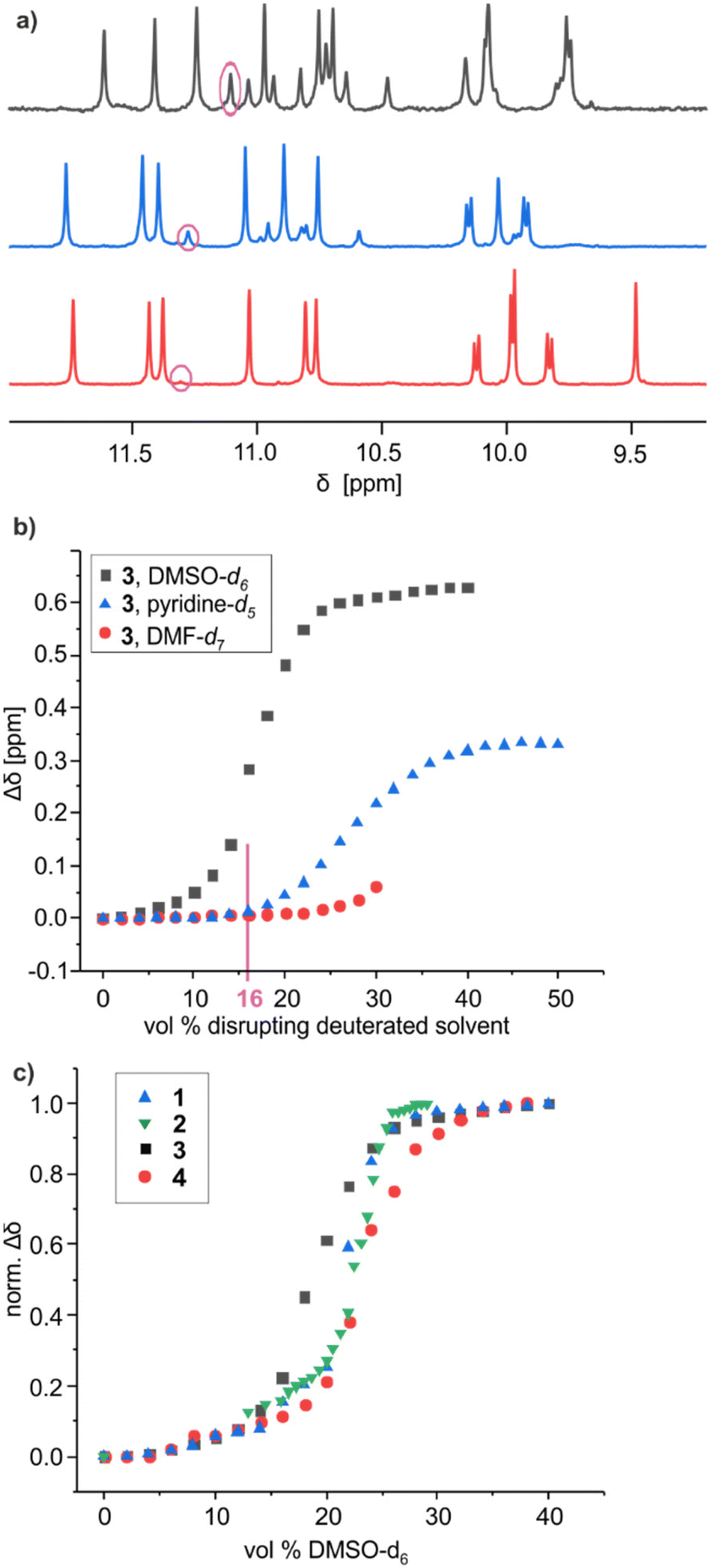 | ||
| Fig. 5 (a) Part of the 1H NMR spectra (500 MHz, 25 °C) of 3 in a 84:16 [vol/vol] mixture of CDCl3 and a disrupting solvent like DMF-d7 (red), pyridine-d5 (blue) and DMSO-d6 (black), showing the amide and hydrogen-bonded hydroxy proton resonances. Signals encircled in pink belong to the disrupted structures. (b) Variations of the chemical shift of selected 1H NMR signals of 3 upon adding disruptive solvents to CDCl3 solutions (see ESI†). Precipitation occurred above 32% of DMF-d7. (c) Normalised variations (Δδ/Δδmax) of the chemical shift of selected 1H NMR signals of 1, 2, 3, and 4 upon adding DMSO-d6 to CDCl3 solutions. Normalisation facilitates comparison even when the amplitude and sign of the chemical shift variation vary. | ||
In order to assess the stability of the tertiary folds as a function of the number and nature of the hydrogen bonds involved, we next compared the ability of DMSO to disrupt the folds of 1, 2, 3 and 4 (Fig. S16–S19†). Chemical shift variations (Δδ) of a representative proton of these species upon adding DMSO-d6 to a solution in CDCl3 are shown in Fig. 5c. As explained above, these variations reflect the rapid equilibrium between the folded and disrupted helix–turn–helix without inversion of helix handedness. Unsurprisingly, the transition between the folded and disrupted conformations of 3 occurred with the smallest amount of DMSO (inflexion near 18.5%), since this compound involves only four hydrogen bonds at its helix–helix interface. However, the trend for the other compounds turned out to deviate from what could be expected. For instance, the transition between folded and disrupted states occurs at similar amounts of DMSO for 1 (21.5%) and 2 (22.5%), as if the additional two hydrogen bonds associated with the peripheral Y units in the structure of 2 had no significant effects on the tertiary structure stability. Furthermore, compound 4 proved the most stable of all: about 24% of DMSO is required to reach the transition. Compound 4 has six hydrogen bonds at its helix–helix interface, that is, as many as 1 and two less than 2, but hydrogen bonds in 4 only involve X units. To enhance the stability of 3, it is thus more beneficial to add two X units (as in 4) than to replace two P with two Y units (as in 1). These results suggest that the hydrogen bonds involving Y units provide less stabilisation to the tertiary folds than those involving the X units.
The solid state structures point to helix curvature strain
That pairs of hydrogen-bonded X units and pairs of hydrogen-bonded Y do not contribute to the same extent to the stability of helix–turn–helix motifs invited a closer look at the solid state structures. As mentioned above, Qn helices span exactly five units per two turns.47 The same applies when Q and P units are mixed.51 Thus, when looking at a helix from the top, down the helix axis, the inner rim of the backbone typically has the shape of a 15-crown-5. For the solid state structures of 1 and 3, the helix inner rim of 3 does show a 15-crown-5-ether shape (Fig. 6b), whereas the helix inner rim of 1 does not (Fig. 6a). This means that, in the case of 1, helix curvature deviates from its preferred form. To identify where this deviation occurs, the structures of 1 and 3 were overlaid so that the turn units and the C-terminal Q units perfectly match. Then, the extent of their overlap was compared at every helix turn (Fig. 7). It was found that the hydrogen-bonded pairs of X units closest to T1 overlap almost perfectly (Fig. 7d). One turn further away from T1, the Y units (in 1) and the P units (in 3) also overlap well. However, the subsequent Q units that are hydrogen bonded to Y in 1 and not to P in 3 are significantly offset in the two structures (Fig. 7c). Removing the hydroxy groups of Y thus results in a change of curvature, allowing the conformation to match more closely five units per two turns. In other words, hydrogen bonding between Y units is acceptable but it proceeds at the cost of a certain strain (spring loading) of helix curvature. The differences between the structures of 1 and 3 extend beyond Y (or P) units all the way to the next helix turn. Thus, the peripheral pair of X units form hydrogen bonds in the same manner in the two structures, but their position as well as the position of the N-terminal Q units differ (Fig. 7b).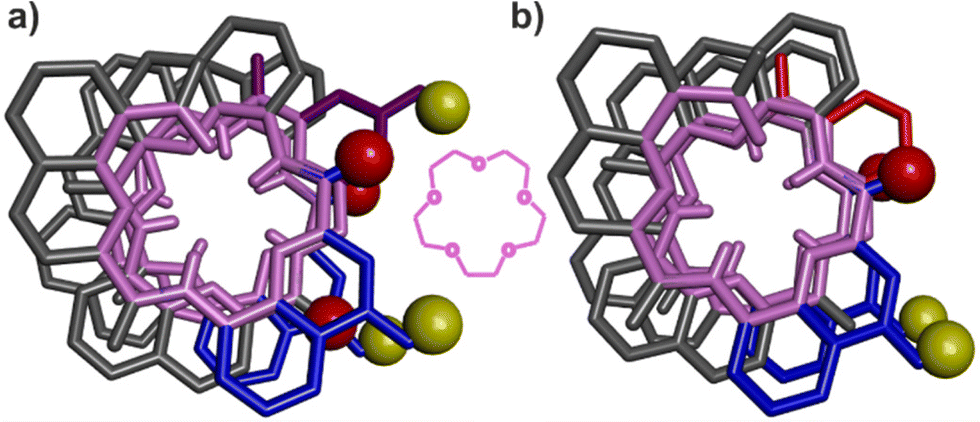 | ||
| Fig. 6 Top view of one helix of the crystal structures of 1 (a) and of 3 (b). The inner rim of each helix is highlighted in pink. The hydroxy protons and carbonyl oxygen atoms involved in helix-to-helix hydrogen bonding are shown as yellow and red balls, respectively. The X units are shown in blue, the Y units in violet and the P units in red. Included solvent molecules, non-polar hydrogen atoms and side chains are omitted for clarity. For comparison to the inner rims, a 15-crown-5 is drawn between (a) and (b). | ||
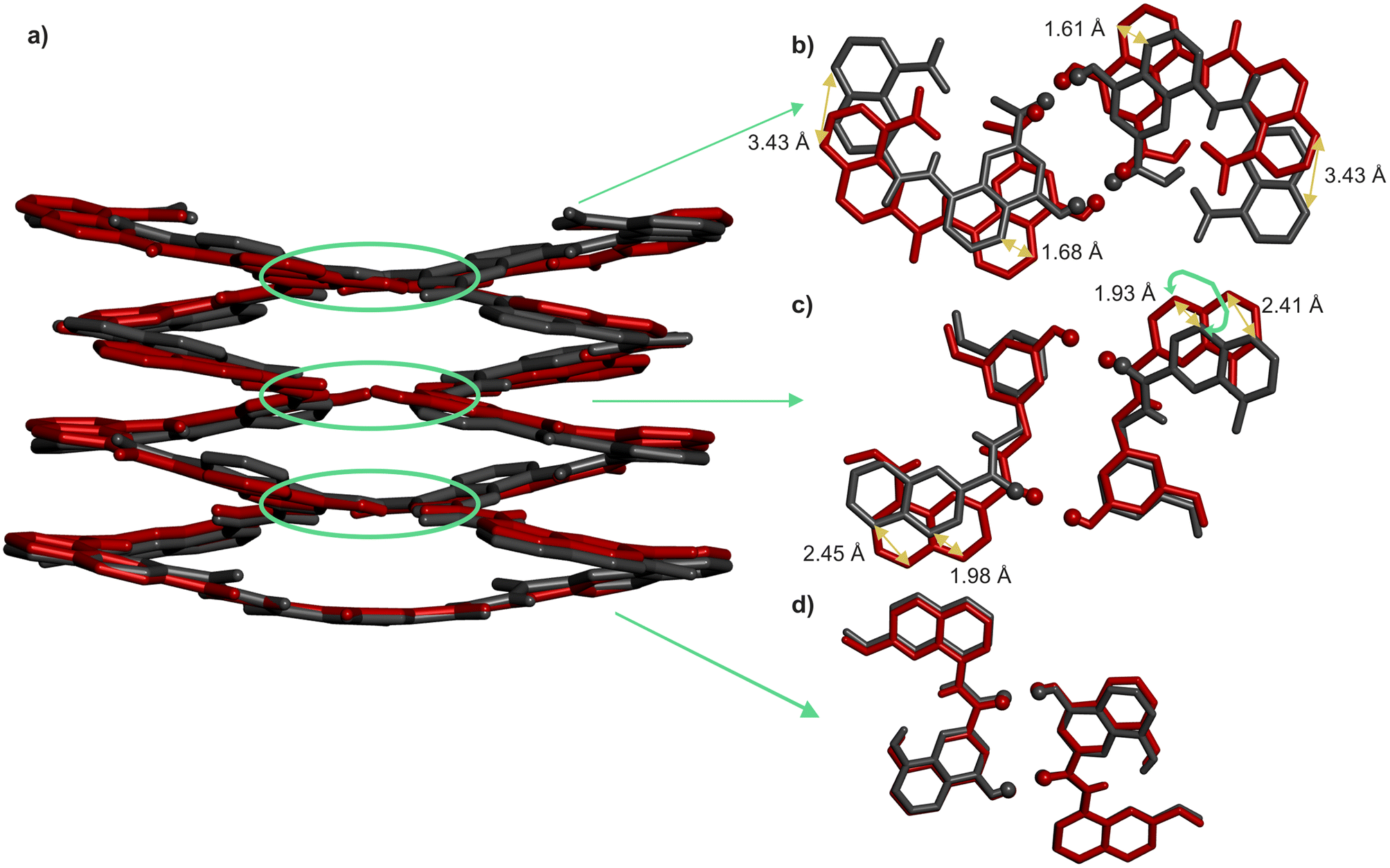 | ||
| Fig. 7 (a) Side view of the overlay of the crystal structures of 1 (red) and 3 (grey) in tube representation. The turn units and C-terminal Q units of the two structures have been overlaid and match almost perfectly. (b–d) Slices of the overlaid helix-turn-helix structures at different helix-helix contacts (marked with green circles and arrows) showing poor overlap in areas closer to the N-termini. Included solvent molecules, non-polar hydrogen atoms and side chains are omitted for clarity. | ||
We thus theorise that inter-helix hydrogen bonding involving the hydroxy groups of Y units requires a deviation of helix curvature from its preferred conformation. Put simply, a disfavoured secondary fold was induced by tertiary folding. This interpretation was further validated by the crystal structures of the PM conformers of the synthetic precursors of 2 and 3, in which X and Y units are protected as tBu-ethers and within which no hydrogen bond can form (Fig. S2†). In both cases, the inner rims of the helices have the preferred 15-crown-5 shape, indicating that no strain in helix curvature occurred in the absence of inter-helix hydrogen bonds.
These insights also shed light on previous, not-well explained observations, namely the self-assembly behaviour of the X- and Y-containing individual helical segments of 1 and 2 when they are not connected by a T1 unit.60 We reported before that for such helices, X and Y units mediate intermolecular inter-helix hydrogen bonding, that is, a sort of helix bundling. However, the intramolecular hydrogen-bonding interface characteristic of 1 and 2 is never observed within aggregates: no head-to-head parallel PP or MM dimer form. Instead, tilted dimers and parallel trimers were characterised (Fig. 8).60 In a tilted dimer, the helix axes are oriented at an angle of +120° or −120°. In a trimer, the helix axes are parallel but the relative orientation of X and Y units differ from those of the helix–turn–helix motif. Both the tilted dimer and trimer configurations cannot occur intramolecularly within 1 and 2 due to the rigidity and geometry of T1. In all motifs, all X and Y hydroxy groups are involved in hydrogen bonding to an amide carbonyl group. We previously assigned the formation of tilted dimers and parallel trimers to potentially stronger, e.g. better oriented or shorter, hydrogen bonds. Now we might connect this behaviour to the fact that all helices adopt their preferred curvature in the aggregates. The views down the helix axis in Fig. 8 clearly show the 15-crown-5 shape of the inner rim. Tilted dimer and parallel trimer formation might thus also be guided by the absence of curvature strain. Parallel dimer formation is disfavoured in this respect.
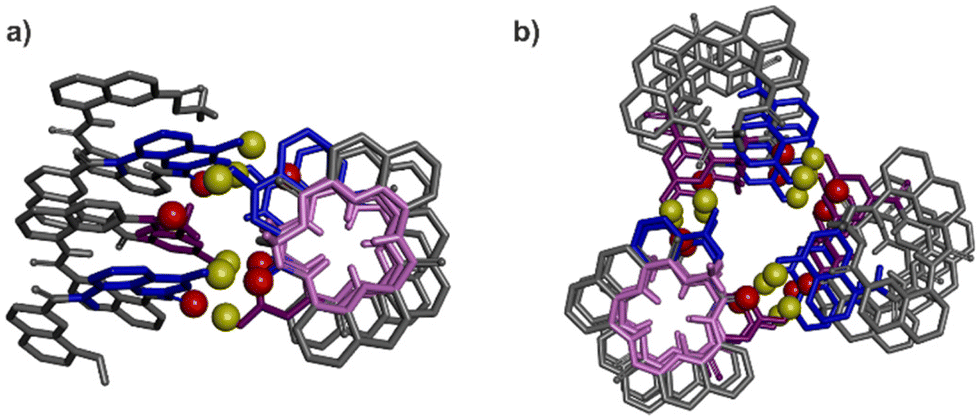 | ||
| Fig. 8 Previously reported crystal structures of a tilted dimer (a) and a parallel trimer (b).60 Views have been oriented to see one helix down its axis. The corresponding inner rim is highlighted in pink, showing a 15-crown-5 shape. The other helices all have the same conformation. The hydroxy protons and carbonyl oxygen atoms involved in helix–helix hydrogen bonding are shown as yellow and red balls, respectively. The X units are shown in blue and the Y units in violet tubes. Included solvent molecules, non-polar hydrogen atoms and side-chains are omitted for clarity. | ||
Conformational frustration is ubiquitous in tertiary protein structures.23 For example, burying a hydrophobic residue in the hydrophobic core of a protein will generate frustration if the adjacent residue is polar and not involved in hydrogen bonding or salt bridging. Conversely, bringing this polar residue to the protein surface will generate frustration if it leads to exposure of the adjacent hydrophobic residue to water. The consequences, including the benefits, of frustration in protein folding, have been extensively investigated. For instance, the co-existence of multiple conflicting conformations and sub-optimal energetic state shapes the energy landscape and facilitates protein folding, i.e. it makes it more likely to reach a conformation close to the ground state.66 In addition, redistributing local frustration facilitates conformational transitions as they occur, for example, during enzyme catalysis.67–70 In short, if a tertiary protein fold would be deprived of conformational frustration, it would turn into a rigid body lacking functionality.71,72 The discovery of non-ideal folding in abiotic tertiary folds thus represents an important step towards the future design not only of structures, but also of functions associated to dynamics.
Conclusions
The design and synthesis of large and complex abiotic folded tertiary structures similar to proteins represent ambitious and exciting challenges for chemistry. Developing structures that fold like biological macromolecules in media other than water is intriguing and will undoubtedly enable us to explore functions that would be unthinkable in living organisms. The successful prediction and synthesis of aromatic oligoamide helix–turn–helix motifs stabilised by hydrogen bonds is a first step towards this goal. We have presented a detailed analysis of the solid state structures of such tertiary folds stabilised by hydrogen-bonded interfaces based on X units only, or on both X and Y units, as well as a solution study of the susceptibility of these interfaces towards disruption in the presence of polar solvents. This, together with the structures of aggregated helices not linked by turn units highlighted possible deviations of the helices from their preferred curvature upon interacting with one another. Such deviations amount to a sort of torsional spring loading. They occur only under the constraint of relatively rigid T1 turn units in the absence of which helices generally find different ways to interact that do not impair their curvature. It is thus the tertiary folding that alters the secondary fold. In this respect it would be interesting to explore the folding behaviour of sequences in which other, less rigid, turns replace T1 unit.The conformational frustration associated with non-ideal helix curvature is analogous to frustration as it occurs in protein tertiary folds. In enzymes, local conformational destabilisation may promote conformational changes and mediate catalysis.67–69 In view of the future development of abiotic tertiary folds with enzyme-like function, the deliberate introduction of tensions and destabilisation is a worthy endeavor.
Finally, X- and Y-containing helices have been shown to form hydrogen-bonded assemblies that differ from the helix–turn–helix structures mediated by turn T1 in part because helices are able to adopt their preferred curvature in these assemblies and not in the tertiary structures. The self-assembly behaviour of helices in which Y has been replaced by P warrants further investigation. Tertiary structures are less strained in this case, as we have shown here, but this does not prevent individual helices from finding yet other ways to interact than that imposed by rigid T1 units. Research along these lines is currently conducted in our laboratory and will be reported in due course.
Author contributions
FM and DM performed syntheses. FM and LA performed solution studies. BW and BK performed crystallographic studies. All authors contributed to experiment design and interpretation. IH supervised the research. FM and IH wrote the manuscript. All authors proofread and improved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the DFG (Excellence Cluster 114, CIPSM). M. Palchyk is gratefully acknowledged for contributing synthetic precursors, S. Kwon for monomer synthesis, A. Schmidt for mass measurement and C. Glas for assistance with NMR measurements. We are grateful to F. Borel for beam time and technical assistance during data collection on the FIP beamline at ESRF (Grenoble).References
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS PubMed.
- R. V. Nair, K. N. Vijayadas, A. Roy and G. J. Sanjayan, Eur. J. Org. Chem., 2014, 7763–7780 CrossRef CAS.
- I. Saraogi and A. D. Hamilton, Chem. Soc. Rev., 2009, 38, 1726–1743 RSC.
- I. Huc, Eur. J. Org. Chem., 2004, 17–29 CrossRef CAS.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- J. W. Checco and S. H. Gellman, Curr. Opin. Struct. Biol., 2016, 39, 96–105 CrossRef CAS PubMed.
- J. P. Saludes, J. B. Ames and J. Gervay-Hague, J. Am. Chem. Soc., 2009, 131, 5495–5505 CrossRef CAS PubMed.
- J. W. Checco, E. F. Lee, M. Evangelista, N. J. Sleebs, K. Rogers, A. Pettikiriarachchi, N. J. Kershaw, G. A. Eddinger, D. G. Belair, J. L. Wilson, C. H. Eller, R. T. Raines, W. L. Murphy, B. J. Smith, S. H. Gellman and W. D. Fairlie, J. Am. Chem. Soc., 2015, 137, 11365–11375 CrossRef CAS PubMed.
- K. Ziach, C. Chollet, V. Parissi, P. Prabhakaran, M. Marchivie, V. Corvaglia, P. P. Bose, K. Laxmi-Reddy, F. Godde, J.-M. Schmitter, S. Chaignepain, P. Pourquier and I. Huc, Nat. Chem., 2018, 10, 511–518 CrossRef CAS PubMed.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. Degrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- S. Abdulkadir, C. Li, W. Jiang, X. Zhao, P. Sang, L. Wei, Y. Hu, Q. Li and J. Cai, J. Am. Chem. Soc., 2022, 144, 270–281 CrossRef CAS PubMed.
- J. Niu, A. J. Cederstrand, G. A. Eddinger, B. Yin, J. W. Checco, C. A. Bingman, V. K. Outlaw and S. H. Gellman, J. Am. Chem. Soc., 2022, 144, 9610–9617 CrossRef CAS PubMed.
- R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury and E. Valeur, J. Med. Chem., 2016, 59, 9599–9621 CrossRef CAS PubMed.
- J. Ahmed, T. C. Fitch, C. M. Donnelly, J. A. Joseph, T. D. Ball, M. M. Bassil, A. Son, C. Zhang, A. Ledreux, S. Horowitz, Y. Qin, D. Paredes and S. Kumar, Nat. Commun., 2022, 13, 2273 CrossRef CAS PubMed.
- M. Rueping, Y. Mahajan, M. Sauer and D. Seebach, ChemBioChem, 2002, 3, 257–259 CrossRef CAS PubMed.
- J. Iriondo-Alberdi, K. Laxmi-Reddy, B. Bouguerne, C. Staedel and I. Huc, ChemBioChem, 2010, 11, 1679–1685 CrossRef CAS PubMed.
- T. B. Potocky, A. K. Menon and S. H. Gellman, J. Am. Chem. Soc., 2005, 127, 3686–3687 CrossRef CAS PubMed.
- M. Oba, ChemBioChem, 2019, 20, 2041–2045 CrossRef CAS PubMed.
- M. Pasco, C. Dolain and G. Guichard, in Comprehensive supramolecular chemistry II, ed. J. L. Atwood, G. W. Gokel and L. J. Barbour, Elsevier, Amsterdam, Netherlands, 2017, pp. 89–125 Search PubMed.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331–337 CrossRef CAS PubMed.
- D. U. Ferreiro, E. A. Komives and P. G. Wolynes, Q. Rev. Biophys., 2014, 47, 285–363 CrossRef PubMed.
- D. Hilvert, Annu. Rev. Biochem., 2013, 82, 447–470 CrossRef CAS PubMed.
- N. H. Joh, T. Wang, M. P. Bhate, R. Acharya, Y. Wu, M. Grabe, M. Hong, G. Grigoryan and W. F. DeGrado, Science, 2014, 346, 1520–1524 CrossRef CAS PubMed.
- S. E. Boyken, Z. Chen, B. Groves, R. A. Langan, G. Oberdorfer, A. Ford, J. M. Gilmore, C. Xu, F. DiMaio, J. Henrique Pereira, B. Sankaran, G. Seelig, P. H. Zwart and D. Baker, Science, 2016, 352, 680–687 CrossRef CAS PubMed.
- P.-S. Huang, S. E. Boyken and D. Baker, Nature, 2016, 537, 320–327 CrossRef CAS PubMed.
- B. Dang, H. Wu, V. K. Mulligan, M. Mravic, Y. Wu, T. Lemmin, A. Ford, D.-A. Silva, D. Baker and W. F. Degrado, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10852–10857 CrossRef CAS PubMed.
- W. M. Dawson, G. G. Rhys and D. N. Woolfson, Curr. Opin. Chem. Biol., 2019, 52, 102–111 CrossRef CAS PubMed.
- J. Dou, A. A. Vorobieva, W. Sheffler, L. A. Doyle, H. Park, M. J. Bick, B. Mao, G. W. Foight, M. Y. Lee, L. A. Gagnon, L. Carter, B. Sankaran, S. Ovchinnikov, E. Marcos, P.-S. Huang, J. C. Vaughan, B. L. Stoddard and D. Baker, Nature, 2018, 561, 485–491 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- E. G. Baker, G. J. Bartlett, K. L. Porter Goff and D. N. Woolfson, Acc. Chem. Res., 2017, 50, 2085–2092 CrossRef CAS PubMed.
- K. Ożga, M. Drewniak-Świtalska, E. Rudzińska-Szostak and Ł. Berlicki, ChemPlusChem, 2021, 86, 646–649 CrossRef PubMed.
- M. Drewniak-Świtalska, B. Barycza, E. Rudzińska-Szostak, P. Morawiak and Ł. Berlicki, Org. Biomol. Chem., 2021, 19, 4272–4278 RSC.
- M. Bejger, P. Fortuna, M. Drewniak-Świtalska, J. Plewka, W. Rypniewski and Ł. Berlicki, Chem. Commun., 2021, 57, 6015–6018 RSC.
- Z. E. Reinert and W. S. Horne, Org. Biomol. Chem., 2014, 12, 8796–8802 RSC.
- K. L. George and W. S. Horne, Acc. Chem. Res., 2018, 51, 1220–1228 CrossRef CAS PubMed.
- T. L. Raguse, J. R. Lai, P. R. Leplae and S. H. Gellman, Org. Lett., 2001, 3, 3963–3966 CrossRef CAS PubMed.
- J. W. Checco and S. H. Gellman, ChemBioChem, 2017, 18, 291–299 CrossRef CAS PubMed.
- E. J. Petersson, C. J. Craig, D. S. Daniels, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 5344–5345 CrossRef CAS PubMed.
- D. S. Daniels, E. J. Petersson, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 1532–1533 CrossRef CAS PubMed.
- G. W. Collie, K. Pulka-Ziach, C. M. Lombardo, J. Fremaux, F. Rosu, M. Decossas, L. Mauran, O. Lambert, V. Gabelica, C. D. Mackereth and G. Guichard, Nat. Chem., 2015, 7, 871–878 CrossRef CAS PubMed.
- G. W. Collie, R. Bailly, K. Pulka-Ziach, C. M. Lombardo, L. Mauran, N. Taib-Maamar, J. Dessolin, C. D. Mackereth and G. Guichard, J. Am. Chem. Soc., 2017, 139, 6128–6137 CrossRef CAS PubMed.
- V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS PubMed.
- Y. Hamuro, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1996, 118, 7529–7541 CrossRef CAS.
- J. Zhu, R. D. Parra, H. Zeng, E. Skrzypczak-Jankun, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2000, 122, 4219–4220 CrossRef CAS.
- H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448–3449 CrossRef CAS PubMed.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337–6339 RSC.
- F. Devaux, X. Li, D. Sluysmans, V. Maurizot, E. Bakalis, F. Zerbetto, I. Huc and A.-S. Duwez, Chem, 2021, 7, 1333–1346 CAS.
- N. Delsuc, F. Godde, B. Kauffmann, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2007, 129, 11348–11349 CrossRef CAS PubMed.
- D. Sánchez-García, B. Kauffmann, T. Kawanami, H. Ihara, M. Takafuji, M.-H. Delville and I. Huc, J. Am. Chem. Soc., 2009, 131, 8642–8648 CrossRef PubMed.
- B. Baptiste, C. Douat-Casassus, K. Laxmi-Reddy, F. Godde and I. Huc, J. Org. Chem., 2010, 75, 7175–7185 CrossRef CAS PubMed.
- M. Vallade, P. Sai Reddy, L. Fischer and I. Huc, Eur. J. Org. Chem., 2018, 5489–5498 CrossRef CAS.
- D. Zheng, L. Zheng, C. Yu, Y. Zhan, Y. Wang and H. Jiang, Org. Lett., 2019, 21, 2555–2559 CrossRef CAS PubMed.
- E. Merlet, K. Moreno, A. Tron, N. D. McClenaghan, B. Kauffmann, Y. Ferrand and C. Olivier, Chem. Commun., 2019, 55, 9825–9828 RSC.
- J. Wang, B. Wicher, A. Méndez-Ardoy, X. Li, G. Pecastaings, T. Buffeteau, D. M. Bassani, V. Maurizot and I. Huc, Angew. Chem., 2021, 133, 18609–18614 CrossRef.
- S. Kumar, M. Birol, D. E. Schlamadinger, S. P. Wojcik, E. Rhoades and A. D. Miranker, Nat. Commun., 2016, 7, 11412 CrossRef CAS PubMed.
- S. Kumar, A. Henning-Knechtel, I. Chehade, M. Magzoub and A. D. Hamilton, J. Am. Chem. Soc., 2017, 139, 17098–17108 CrossRef CAS PubMed.
- J. Buratto, C. Colombo, M. Stupfel, S. J. Dawson, C. Dolain, B. Langlois D'Estaintot, L. Fischer, T. Granier, M. Laguerre, B. Gallois and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 883–887 CrossRef CAS PubMed.
- S. De, B. Chi, T. Granier, T. Qi, V. Maurizot and I. Huc, Nat. Chem., 2018, 10, 51–57 CrossRef CAS PubMed.
- V. Maurizot, C. Dolain, Y. Leydet, J.-M. Léger, P. Guionneau and I. Huc, J. Am. Chem. Soc., 2004, 126, 10049–10052 CrossRef CAS PubMed.
- N. Delsuc, S. Massip, J.-M. Léger, B. Kauffmann and I. Huc, J. Am. Chem. Soc., 2011, 133, 3165–3172 CrossRef CAS PubMed.
- C. Dolain, J.-M. Léger, N. Delsuc, H. Gornitzka and I. Huc, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16146–16151 CrossRef CAS PubMed.
- D. Mazzier, S. De, B. Wicher, V. Maurizot and I. Huc, Chem. Sci., 2019, 10, 6984–6991 RSC.
- D. Mazzier, S. De, B. Wicher, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2020, 59, 1606–1610 CrossRef CAS PubMed.
- C. Clementi, H. Nymeyer and J. N. Onuchic, J. Mol. Biol., 2000, 298, 937–953 CrossRef CAS PubMed.
- B. K. Shoichet, W. A. Baase, R. Kuroki and B. W. Matthews, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 452–456 CrossRef CAS PubMed.
- E. M. Meiering, L. Serrano and A. R. Fersht, J. Mol. Biol., 1992, 225, 585–589 CrossRef CAS PubMed.
- N. Tokuriki and D. S. Tawfik, Curr. Opin. Struct. Biol., 2009, 19, 596–604 CrossRef CAS PubMed.
- H. Im, H.-Y. Ahn and M.-H. Yu, Protein Sci., 2000, 9, 1497–1502 CrossRef CAS PubMed.
- A. M. Figueiredo, S. B.-M. Whittaker, S. E. Knowling, S. E. Radford and G. R. Moore, Protein Sci., 2013, 22, 1722–1738 CrossRef CAS PubMed.
- S. B.-M. Whittaker, N. J. Clayden and G. R. Moore, J. Mol. Biol., 2011, 414, 511–529 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures, detailed experimental protocols and characterisation of new compounds as well as crystallographic data. CCDC 2213460 and 2213461. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob02109a |
| ‡ Helix–turn–helix motifs with opposite N-to-C orientation of the helices, i.e. head-to-tail, were also produced. See ref. 65. |
| This journal is © The Royal Society of Chemistry 2023 |