
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-accelerated “on-water” hydroacylation of dialkyl azodicarboxylates†
Naya A.
Stini
 ,
Efthymios T.
Poursaitidis
,
Nikolaos F.
Nikitas‡
,
Michail
Kartsinis‡
,
Nikoleta
Spiliopoulou
,
Phoebe
Ananida-Dasenaki
and
Christoforos G.
Kokotos
*
,
Efthymios T.
Poursaitidis
,
Nikolaos F.
Nikitas‡
,
Michail
Kartsinis‡
,
Nikoleta
Spiliopoulou
,
Phoebe
Ananida-Dasenaki
and
Christoforos G.
Kokotos
*
Laboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis 15771, Athens, Greece. E-mail: ckokotos@chem.uoa.gr
First published on 4th January 2023
Abstract
The hydroacylation of dialkyl azodicarboxylates has received a lot of attention lately due to the great importance of acyl hydrazides in organic chemistry. Herein, we report an inexpensive and green photochemical approach, where light irradiation (390 nm) significantly accelerates the reaction between dialkyl azodicarboxylates and aldehydes, while water is employed as the solvent. A variety of aromatic and aliphatic aldehydes were converted into their corresponding acyl hydrazides in good to excellent yields in really short reaction times (15–210 min) and the reaction mechanism was also studied. Applications of this reaction in the syntheses of Vorinostat and Moclobemide were demonstrated.
Introduction
Over the years, organic chemistry has provided appropriate tools to scientists in order to rapidly and in a reliable fashion synthesize molecules of significance. Modern organic synthetic chemistry is on a continuous lookout for the development of novel, green and sustainable organic transformations, and at the same time, it targets the improvement of traditional organic reactions, providing environmentally friendly alternatives. Among them, organic transformations that provide the formation of new C–N bonds are widely studied due to the importance of the target molecules, such as natural products, drugs or materials.1 There are numerous ways one could envisage the retrosynthetic analysis to forge a new C–N bond; however, a reaction that is not so often discussed, but in the last few years has received a lot of attention, is the use of dialkyl azodicarboxylates and aldehydes for the formation of acyl hydrazides.2 The unambiguous and environmentally friendly formation of acyl hydrazides is of great importance due to the wide range of products that may be obtained from them, such as the drugs Vorinostat and Moclobemide.3 At the same time, the starting materials of this reaction are aldehydes, which are feedstock reagents with applications in medicinal chemistry and agrochemistry,4 and dialkyl azodicarboxylates. The synthetic utility of dialkyl azodicarboxylates is based on their electrophilicity5 since they possess both a strong electron-withdrawing ability and an unoccupied pi-orbital,6 which make them appropriate reagents for a number of named organic reactions, such as the Mitsunobu reaction or variations of the Baylis–Hillman reaction.1,7Historically, the first reported approach, as well as most of the synthetic strategies in the literature, for the construction of acyl hydrazides involves the use of transition metals as catalysts (Scheme 1A).2 Among the metals employed, rhodium acetate,8 zinc acetate,9 copper acetate,10 copper oxide nanoparticles11 or magnetite impregnated with cobalt12 have been described as the catalyst of choice depending on the reaction conditions (Scheme 1A). In 2014, Xu and coworkers described the synergistic effect of a Lewis acid (CoCO3) with a Brønsted acid, namely TFA, for the promotion of the reaction between aromatic or alkyl aldehydes and dialkyl azodicarboxylates (Scheme 1B).13 This method reported the cooperative action of both catalysts, where the Lewis acid activates the aldehyde, while the Brønsted acid activates the dialkyl azodicarboxylate partner. In 2009, researchers turned their attention to developing greener alternatives to these reactions. Along these lines, Headley and coworkers employed imidazolium-based ionic liquids for the promotion of the reaction (Scheme 1C),14 while Mariappan and coworkers employed microwave irradiation (Scheme 1D).15 In the former case, heating at 40 °C was necessary and the reaction time varied from 1–168 h depending on the aldehyde counterpart.14 In the latter case, pyridine was utilized in catalytic amounts and a short reaction time (10 min) was employed along with the microwave irradiation; however, a high temperature (140 °C) and DMF were utilized.15 Around the same period, in two different reports, one by Caddick and coworkers16 and the other by Headley, Ni and coworkers,17 water was utilized as the solvent (Scheme 1E). In the former case, a slight excess of the aldehyde (vs. dialkyl azodicarboxylate) was employed, leading to good to high yields with a reaction time that varied from 24–96 h,16 while in the latter case, a higher reagent ratio (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) led to similar yields, spanning from 3 to 360 h as the reaction time.17
:1) led to similar yields, spanning from 3 to 360 h as the reaction time.17
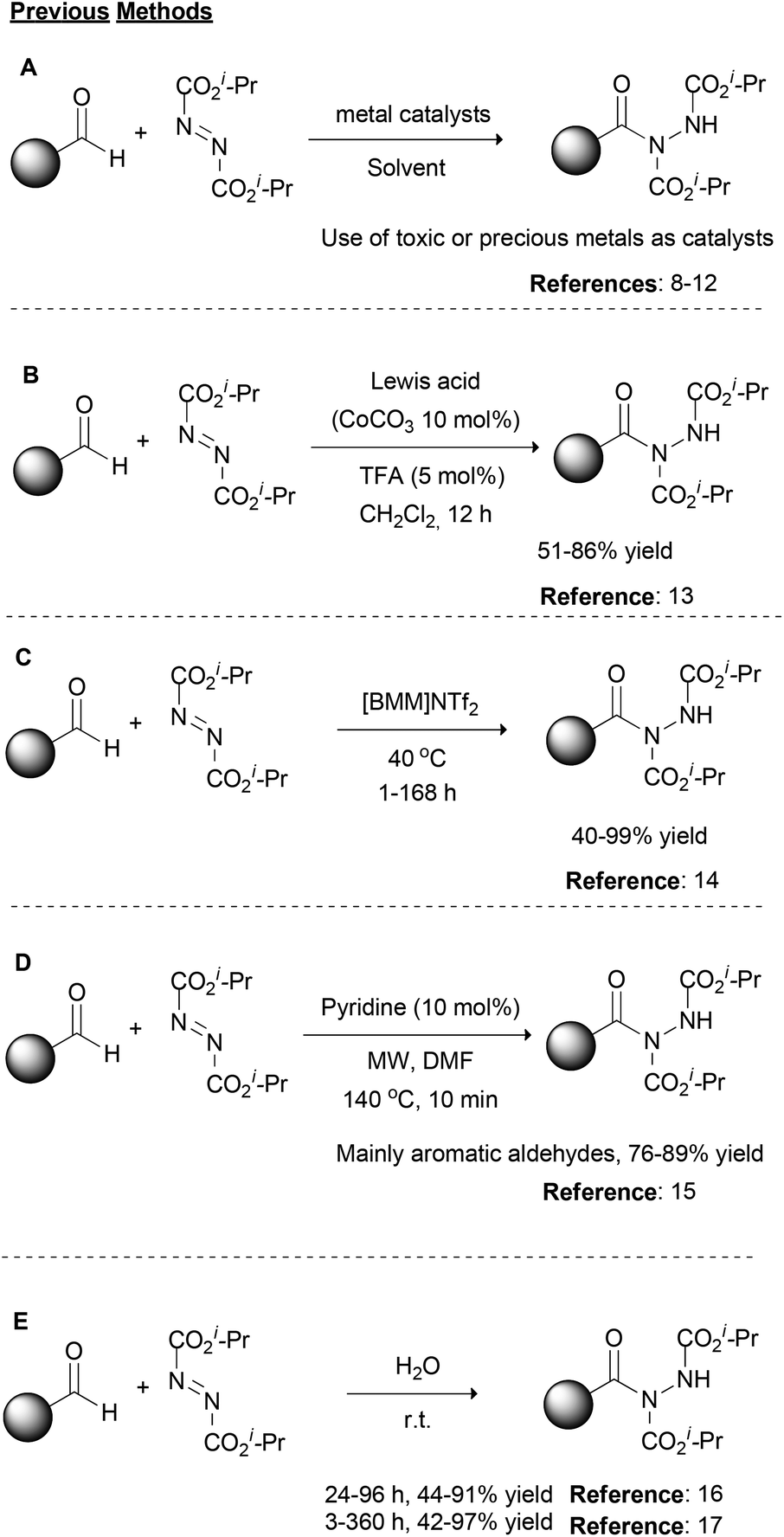 | ||
| Scheme 1 Common synthetic approaches for the hydroacylation of dialkyl azodicarboxylates reported in the literature. | ||
Photochemistry, the use of light to promote organic reactions, has been known since the 1900s.18 For many years, however, organic chemists abstained from the use of photochemistry in organic synthesis. In recent years, photoredox catalysis has brought a revolution in how organic chemists embrace photochemistry.19 In most cases, metal-based complexes are employed as photocatalysts since their properties can be tuned via ligand manipulation. Photoorganocatalysis is an even newer field, which employs small organic molecules as photocatalysts.20,21 As expected, the field of photochemistry soon realized the potential in catalyzing the hydroacylation of dialkyl azodicarboxylates (Scheme 2). In a pioneering work in 2013, Ryu, Fagnoni and coworkers reported the use of TBADT as the photocatalyst for the hydroacylation of dialkyl azodicarboxylates, using a Hg lamp or a solar box as the irradiation source (Scheme 2A).22 This work proved to be inspirational for our group and this reaction was among the first we studied in the field of photochemistry.3,23 Later in 2017, we also proposed the use of graphite flakes as the heterogeneous catalyst for the daylight-mediated hydroacylation of dialkyl azodicarboxylates (Scheme 2B),24 while in 2014, we employed PhCOCOOH as the photoinitiator for the hydroacylation of dialkyl azodicarboxylates (Scheme 2C).23a,i This protocol proved to be very general and we used this to introduce a one-pot protocol for the synthesis of hydroxamic acids and amides, which found application in the synthesis of two pharmaceuticals.3 Having a long experience in photochemistry and the hydroacylation of dialkyl azodicarboxylates and in an effort to further comply with the principles of green and sustainable chemistry,25 we decided to apply our knowledge to this reaction and combine it with different irradiation sources in order to achieve shorter reaction times and better yields (Scheme 2D). Herein, we were able to introduce the use of LED irradiation (390 nm) for the hydroacylation of dialkyl azodicarboxylates in the fast, green and light-accelerated synthesis of acyl hydrazides, using water as the solvent (Scheme 2D). The reaction time in all cases was quite short (15–210 min) versus all literature reports, while high to excellent yields were obtained. Finally, the application of the protocol in the synthesis of the two drugs, Vorinostat and Moclobemide, is demonstrated.
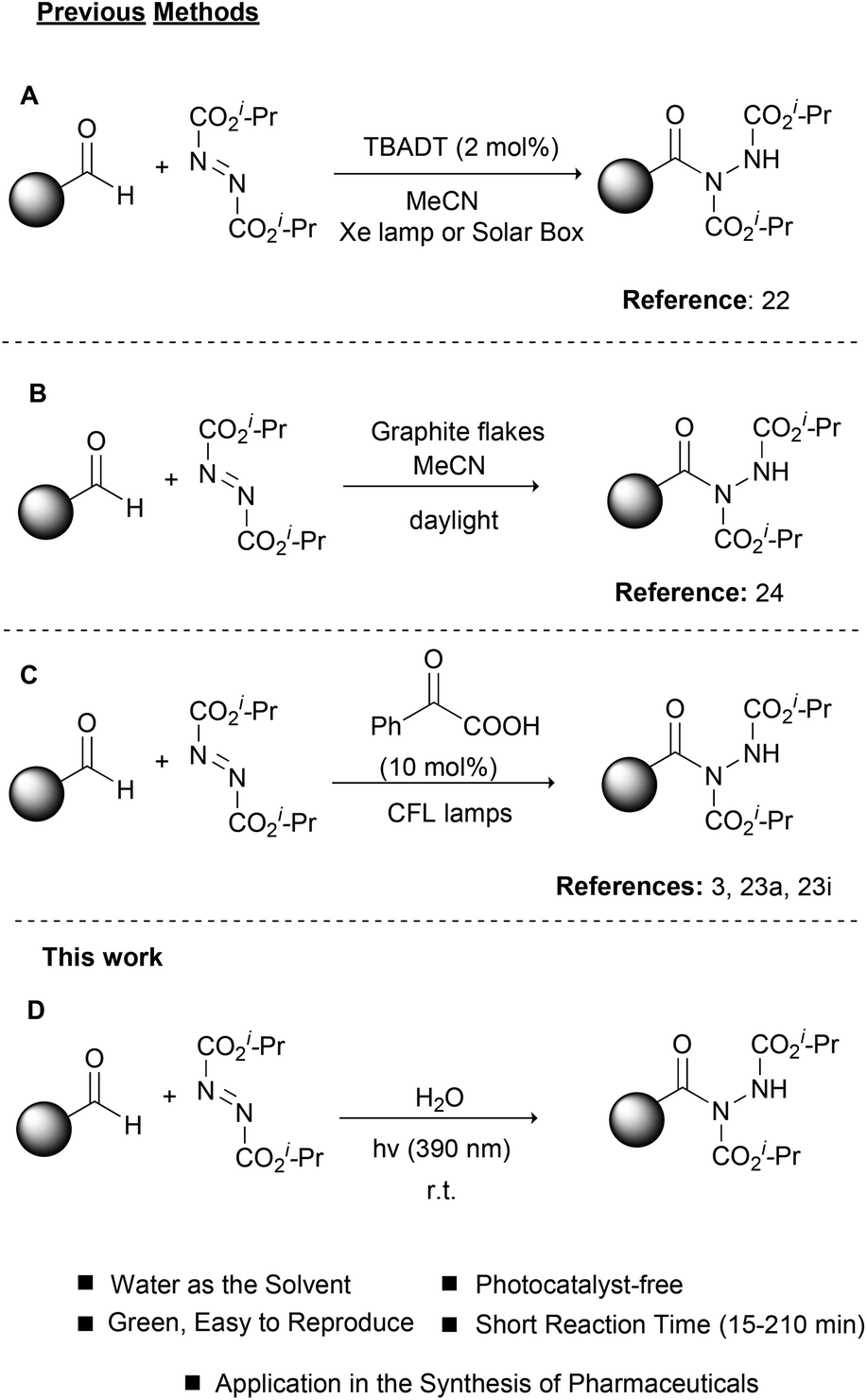 | ||
| Scheme 2 Photochemical approaches for the hydroacylation of dialkyl azodicarboxylates. | ||
Results and discussion
Our recent studies have shown that the nature of wavelength irradiation can have a huge impact on the outcome of a given reaction,26 in accordance with the literature. We began our investigation by evaluating how different wavelengths of irradiation affect the reaction between heptanal and diisopropyl azodicarboxylate (DIAD) (2a) with MeCN as the solvent (Table 1).27 The reaction completion was determined by TLC and NMR and the results showed that 390 nm and 440 nm were equally successful, since DIAD was fully consumed after three hours, whereas, in other cases, longer reaction times were required (Table 1). When we employed CFL lamps, which were employed in our previous studies,3,23a,i the reaction reached completion after 3.5–4 h of irradiation, depending on the lamps employed (Table 1, entry 7). We also performed the experiment in the dark to highlight the acceleration outcome due to irradiation.27 Indeed, after 3 h, only 5% of the starting material was consumed. Having opted for the wavelength irradiation of 440 nm, we evaluated a variety of solvents (Table 2). When the reaction was performed in MeCN, the desired product 3a was isolated as the sole product (Table 2, entry 1). However, when the reaction was performed in other organic solvents, 3a was not the only product observed (Table 2, entries 2–4 and 6–10). Interestingly, except from the desired product, the oligomerization of DIAD had also occurred, indicating the existence of a parallel reaction. In most of the solvents employed, even when the consumption of DIAD was full, the formation of the oligomers (2a-dimer or 2a-trimer) was more favorable than the formation of the desired product (Table 2, entries 4 and 7–10). The structures of 2a-dimer and 2a-trimer were verified by isolating them and characterizing them independently, upon the irradiation of DIAD alone.27 When THF was used as the reaction medium, the obtained product was not acyl hydrazide 3a, but the product of the reaction between dialkyl azodicarboxylate and THF, in accordance with the studies of Lee and Otte and ours (Table 2, entry 5).8,23f,i Our focus then turned to examining the possibility of employing a greener solvent.28 Thus, we tested water, which is known to have a positive effect in some reactions29 and as it is mentioned in the literature, it is a quite successful solvent for the specific reaction.16,17 In particular, following the pioneering contributions by Caddick, Chudasama and coworkers, water is known to accelerate acyl radical formation and facilitates a number of reactions.30 Thus, in this work, we envisioned a combined activation, derived from the “on-water” effect29 and the acyl radical formation that is favoured in water.30 Indeed, when the reaction was performed in water, 3a was obtained as the sole product in a quantitative yield in just 50 min (Table 2, entry 11). Thus, irradiation seems to accelerate the process even more. This fact is also supported by a simple comparison of the literature data. Caddick and co-workers reported that the reaction between butanal and 2a in water required 16 h to provide 90% of the hydroacylation product,16 while Headley, Ni and coworkers reported that the reaction between nonanal and 2a in water required 20 h to provide 92% of the hydroacylation product.17 Herein, the reaction of heptanal with 2a in water and upon irradiation required 55 min to provide a quantitative yield of 3a (100% conversion, Table 2, entry 11). However, since irradiation at 390 nm was equally effective, we performed the reaction at 390 nm as well, and in this case, the results were even better, since the reaction was completed faster, within just 40 minutes (Table 2, entry 12). Herein, the reaction of heptanal with 2a in water required approx. 40 minutes to provide a quantitative yield of 3a (100% conversion, 92% yield of the isolated product, Table 2, entry 12). In the case where water was used as the reaction medium, no oligomers were formed, thus making water the optimum solvent for the reaction, concerning both the reaction speed and oligomer formation.|
|
|||
|---|---|---|---|
| Entry | Irradiation source (nm) | Reaction time (h) | Conversion (%) |
| a Conversion determined by 1H-NMR. The reaction was performed with heptanal (1a) (114 mg, 1.00 mmol) and di-isopropyl azodicarboxylate (2a) (101 mg, 0.50 mmol) in MeCN (2 mL) under irradiation. | |||
| 1 | 370 | 21 h | 100 |
| 2 | 390 | 3 h | 100 |
| 3 | 427 | 5 h | 100 |
| 4 | 440 | 3 h | 100 |
| 5 | 456 | 3.5 h | 100 |
| 6 | 525 | 24 h | 100 |
| 7 | CFL lamps | 4 h | 100 |
|
|
|||||
|---|---|---|---|---|---|
| Entry | Irradiation source (nm) | Solvent | Yielda (%) | 2a-Dimer (%) | 2a-Trimer (%) |
| a Yield determined by 1H-NMR. The reaction was performed with heptanal (1a) (114 mg, 1.00 mmol) and di-isopropyl azodicarboxylate (2a) (101 mg, 0.50 mmol) in solvent (2 mL) under irradiation. The yield of the product after isolation by column chromatography in parenthesis. b The reaction was completed in 55 min. c The reaction was completed in 40 min. | |||||
| 1 | 440 | MeCN | 100 | — | — |
| 2 | 440 | Et2O | 53 | 23 | 24 |
| 3 | 440 | EtOAc | 47 | 46 | 7 |
| 4 | 440 | Toluene | 46 | 54 | — |
| 5 | 440 | THF | 0 | — | — |
| 6 | 440 | CH2Cl2 | 75 | 10 | 15 |
| 7 | 440 | Pet. Eth. | 23 | 55 | 2 |
| 8 | 440 | CCl4 | 46 | 54 | — |
| 9 | 440 | MeOH | 9 | 64 | 27 |
| 10 | 440 | CHCl3 | 46 | 48 | 6 |
| 11 | 440 | H 2 O | 100 (87) | — | — |
| 12 | 390 | H 2 O | 100 (92) | — | — |
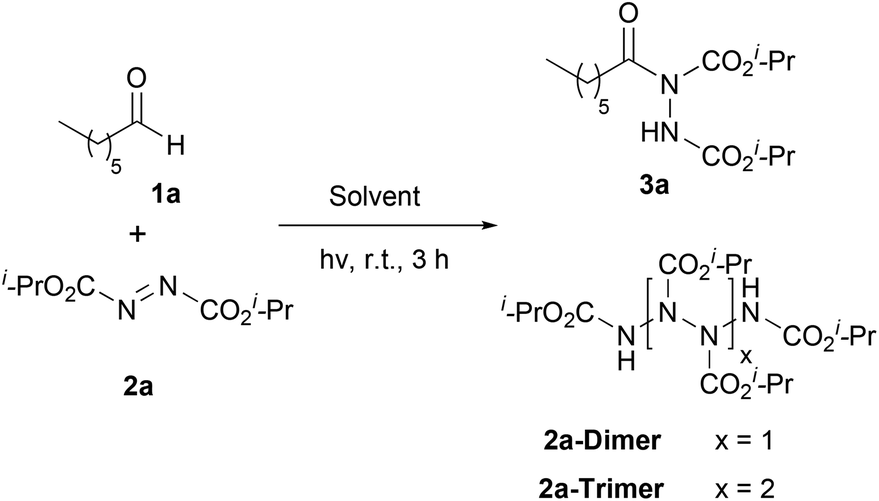
In an attempt to reduce the ratio of the employed aldehyde, we studied the behavior of the reaction in ratios of 1.5:1 and 1.1:1 of 1a:2a.27 We observed that as the amount of the aldehyde used tended to be stoichiometric, the reaction time increased. When 1.5 equiv. of aldehyde were employed (in H2O at 390 nm), only ten additional minutes (50 min) were required to reach completion; however, when 1.1 equiv. of 1a were employed (in H2O at 390 nm), the reaction was completed after approximately 2 h.27 Thus, we concluded that the optimum reaction ratio is 1.5:1. In order to ensure the effectiveness of the correct irradiation wavelength, we reperformed the reaction at different irradiation wavelengths, but this time by employing water as the solvent and using a 1.5:1 ratio.27 Irradiation at 390 nm provided the best results since the reaction was completed in just 50 minutes. However, the reaction time increased when the irradiation wavelength diverged from 400 nm, from 50 to 200 minutes. The diversity of the reaction times may be attributed to the wavelengths of the absorbance of DIAD, which will be discussed in the section dealing with the reaction mechanism.
To verify that the irradiation source is the main factor for the reaction outcome, we carried out the reaction under the optimum reaction conditions, but in the dark for 50 minutes and the desired product 3a was obtained in 17% yield,27 verifying the crucial role of water as the solvent29,30 and the acceleration provided by 390 nm irradiation. Additionally, we carried out the reaction under an argon atmosphere to verify that oxygen is essential for the reaction.27 To our surprise, only a slightly reduced 67% yield of 3a was obtained, indicating that the presence of oxygen accelerates, but does not play a crucial role in the reaction.27 We also checked the role of temperature in the progression of our reaction, and no differences were observed when a fan was used for the maintenance of the temperature at 27 °C.27 Furthermore, under the optimum reaction conditions, where reaction completion was observed, we attempted the isolation of the desired product by simple extractions (basic aqueous wash-extractions) and 3a can be obtained in a very pure form without the need for column chromatography.27
Having the optimum conditions in hand, the substrate scope of the methodology was evaluated, initially using a variety of aliphatic aldehydes and diisopropyl azodicarboxylate (2a) (Scheme 3). Aliphatic aldehydes bearing linear alkyl chains or alkyl chains with various branched parts, like 1a–e, afforded the desired products 3a–e in high to excellent yields, especially when 2 equivalents of the aldehyde were utilized, and in very short reaction times (3c, 3d, 3h and 3j). The presence of easily oxidized functionalities, like double or triple bonds (3f or 3g), did not alter the reaction outcome, leading to products in excellent yields in really short reaction times. In the case of α,α-disubstituted aldehydes, acyl radical formation can be coupled with a number of by-products, since the corresponding nucleophilic secondary aliphatic radical can be formed by CO extrusion. However, in the case of aldehydes 1j and 1k, the reaction was so fast (completion occurred in 15–60 min) that no such by-products were observed. Finally, a tertiary substituted aldehyde, pivaldehyde (1l), was employed, leading to the desired product 3l in 73% yield, after a reaction time of 80 min.
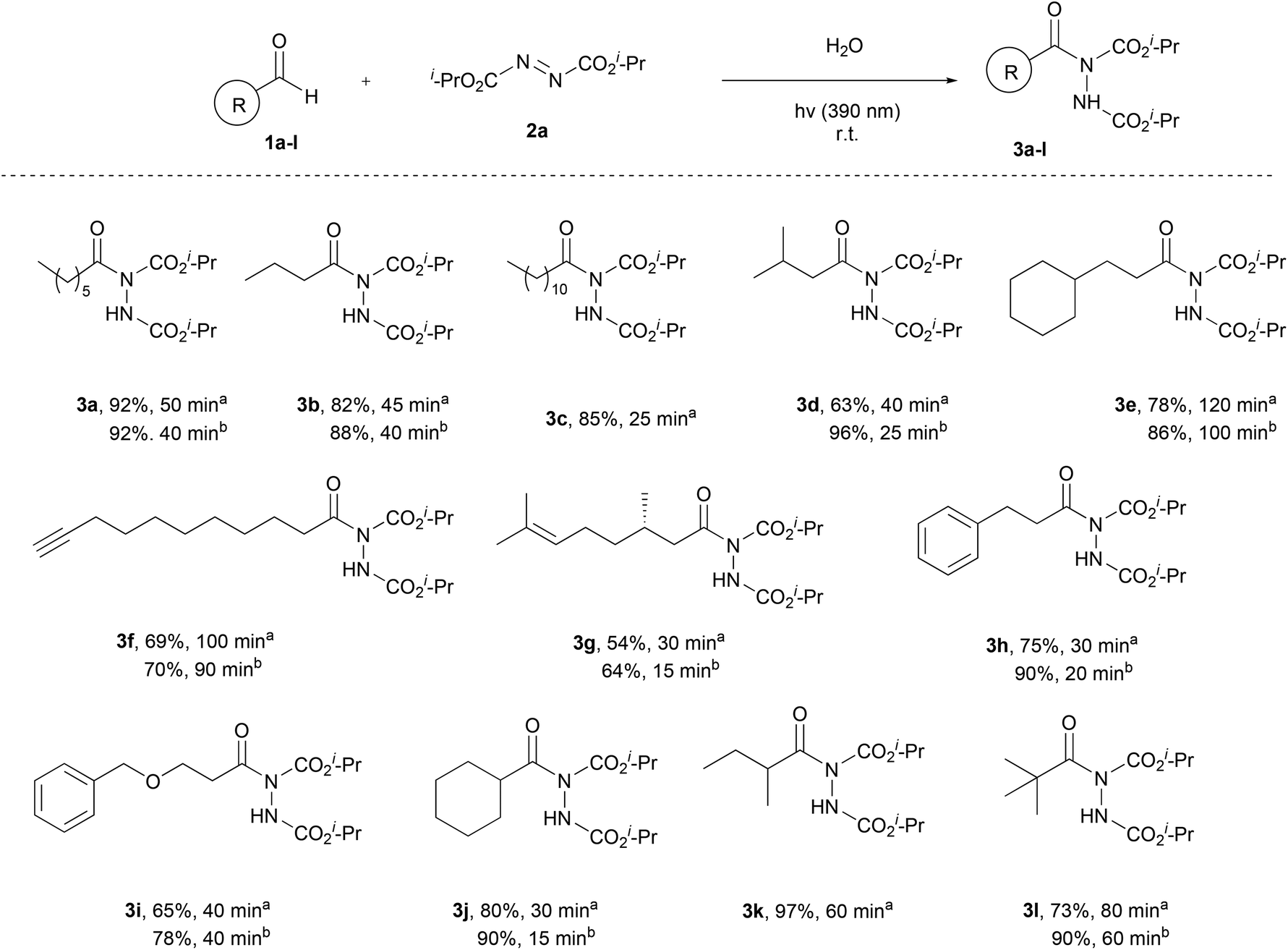 | ||
| Scheme 3 Substrate scope – aliphatic aldehydes. aReaction time and yield of the isolated product, when 1.5 equiv. of aldehyde were used. bReaction time and yield of the isolated product, when 2.0 equiv. of aldehyde were used. | ||
Once the aliphatic counterpart of the aldehyde was probed, we turned our attention to more difficult cased acyl radical formation, such as aromatic aldehydes and α,β-unsaturated aldehydes (Scheme 4). To our delight, aromatic aldehydes did not pose any problems concerning the yield and the reaction time, with some of them reacting in just 20 minutes (3o and 3p, 2 equiv. of aldehyde). Aromatic aldehydes substituted with electron-donating and electron-withdrawing groups required longer reaction times and the yields were mediocre to good (3n, 3q and 3t), even when 2 equivalents of aldehyde were used. Substitution at the ortho-position was also well tolerated, since 1r reacted significantly well, resulting in an excellent yield (94%). A heteroaromatic aldehyde (1u) was tested as well, which gave the desired product in a mediocre yield while requiring several hours for the reaction to be completed (3u). We also explored the use of α,β-unsaturated aldehydes, which afforded products 3v–x in mediocre to high yields, but longer reaction times were required. Finally, the scope of azodicarboxylates was probed by testing different dialkyl azodicarboxylates in reaction with heptanal (1a) and the desired products were obtained in very good yields (3y and 3z). The experiments were performed using a 1.5:1 and a 2:1 ratio of aldehyde:azodicarboxylate, in order to obtain results that can be compared with the literature and to understand better the mechanism of the reaction, which will be further analyzed in the section dealing with the mechanism. The results showed that when employing more equivalents of the aldehyde, better yields were obtained. In some cases, a significant increase in the yield was noted (3d, 3m, 3s and 3w), whilst in other cases, the obtained yields were similar (3f, 3g and 3p). In the cases of substrates where lower yields were obtained, the main byproduct that occurred was the dimer of diisopropyl azodicarboxylate (2a-dimer), whilst no decarboxylation in the α-substituted aldehydes was noted.
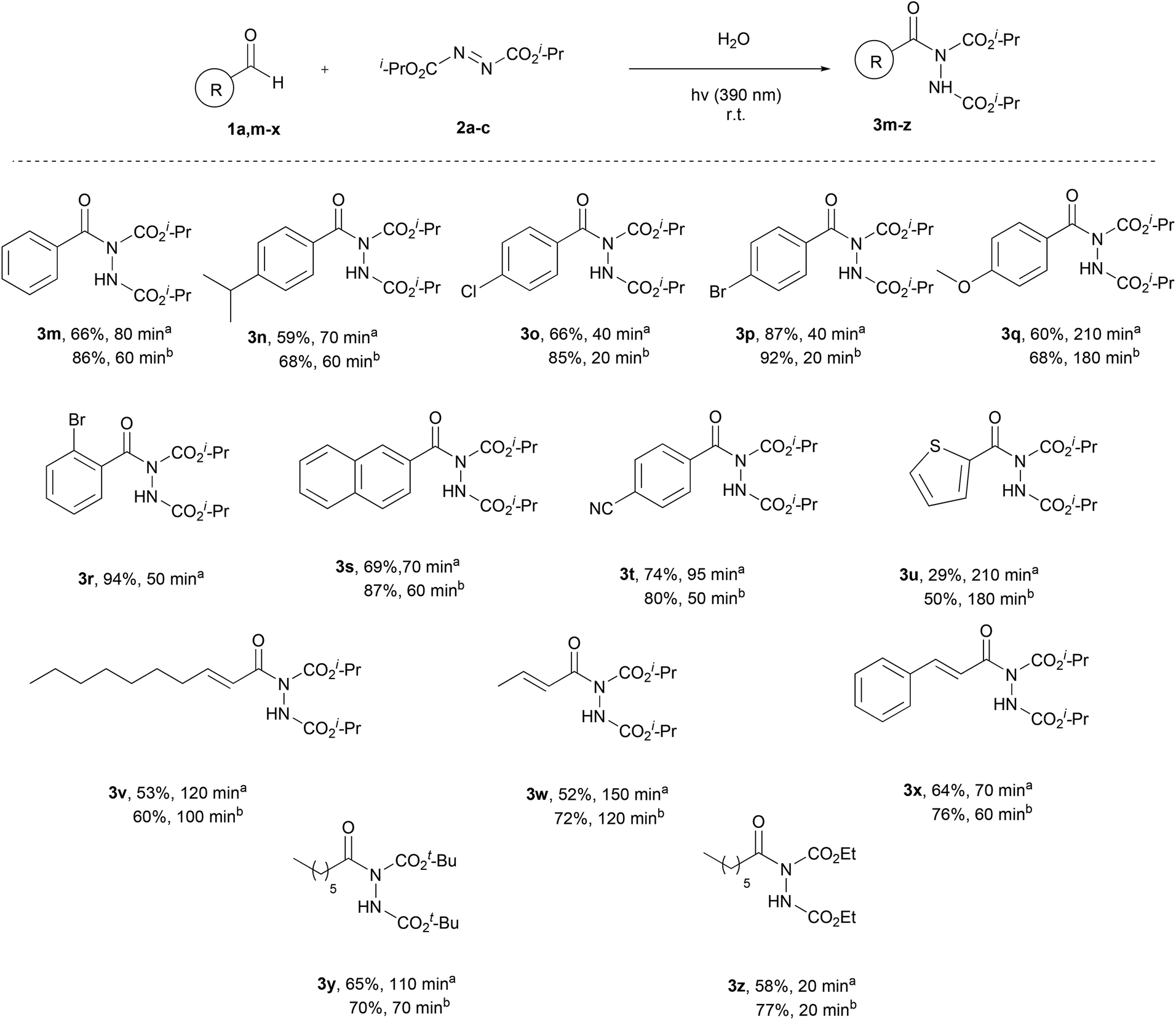 | ||
| Scheme 4 Substrate scope – aromatic aldehydes, α,β-unsaturated aldehydes, naturally-occurring aldehydes or different dialkyl azodicarboxylates. aReaction time and yield of the isolated product, when 1.5 equiv. of aldehyde were used. bReaction time and yield of the isolated product, when 2.0 equiv. of aldehyde were used. | ||
Moreover, we applied our method to the synthesis of two pharmaceuticals, the antidepressant Moclobemide and the anticancer drug Vorinostat (Scheme 5). We were able to synthesize both of these drugs in good yields, having excellent yields and short reaction times in the steps where our protocol was employed. Starting from suberic acid 4, aldehyde 7 was obtained via a three-step process. Following the developed method herein and reacting the intermediate acyl hydrazide with hydroxylamine, Vorinostat 8 was obtained. Similarly, starting from aldehyde 1o and following the developed method and the literature, Moclobemide 10 was obtained.
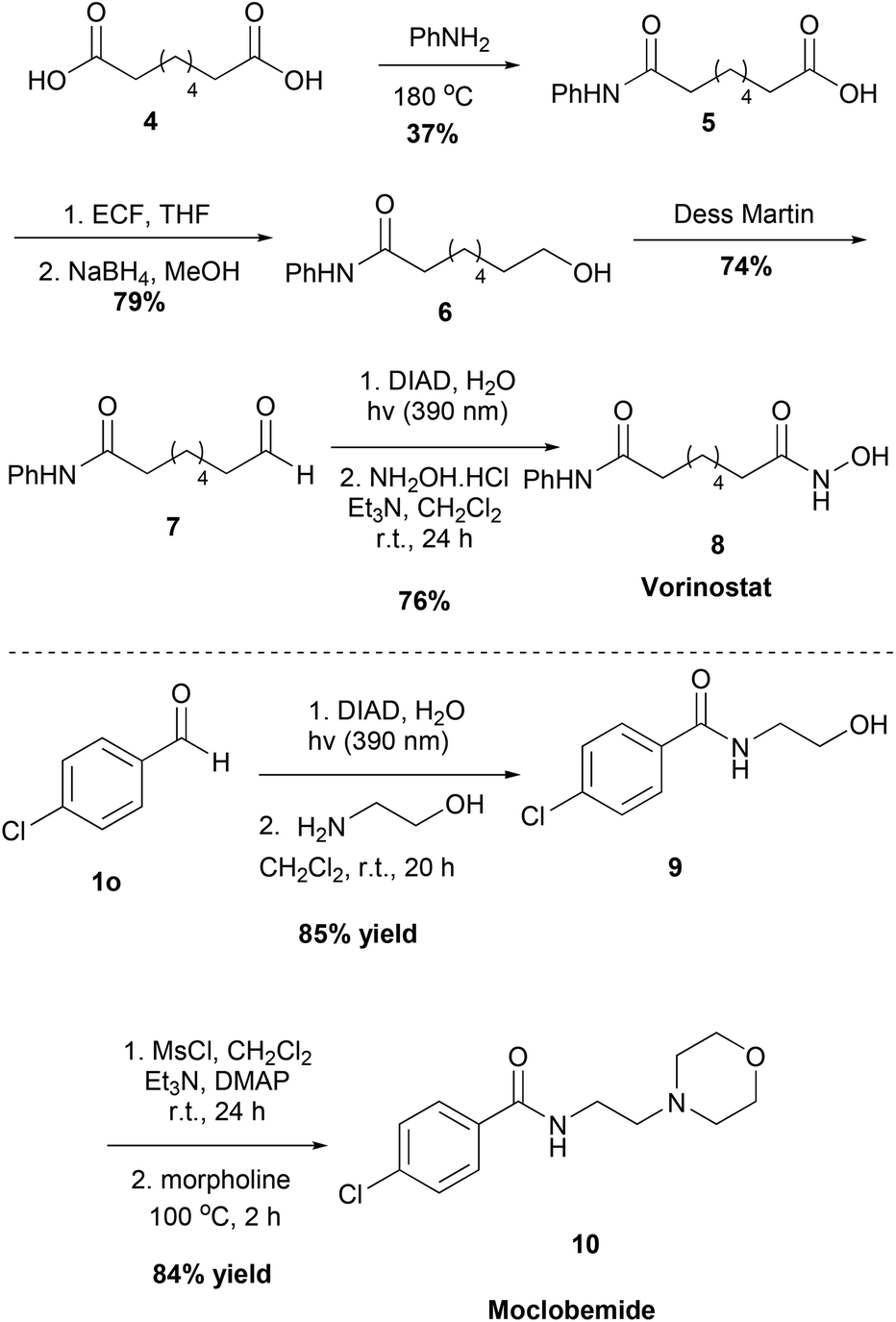 | ||
| Scheme 5 Moclobemide and Vorinostat synthesized using this protocol. | ||
Hence, having evaluated the substrate scope of the reaction and knowing the strengths and weaknesses of this protocol, we diverted our attention to study the reaction mechanism. We initiated our investigation by carrying out UV-Vis absorbance studies.27 According to the literature, when an electron donor is coupled with an electron acceptor in a ground state, an electron donor–acceptor (EDA complex) or Charge Transfer Complex is formed, which can be easily identified by a bathochromic shift in the UV-Vis absorbance spectrum when the two reagents are mixed.31 Very recently, EDA complexes were employed in order to initiate organic transformations.32 When heptanal or benzaldehyde was used, there was no difference in the absorbance in the UV spectrum, when mixed with 2a, indicating that no EDA is formed. The studies were performed in MeCN, in which the reagents can be fully dissolved. An obvious absorbance of DIAD was present around the 400 nm and 440 nm regions.
Some of the control experiments that were carried out enhanced our suspicions of the existence of 3 parallel reactions that take place simultaneously. Apart from the desired reaction between dialkyl azodicarboxylate and the aldehyde for the formation of the desired acyl hydrazides, the aldehydes are also oxidized to carboxylic acids upon irradiation30,33 and at the same time, dialkyl azodicarboxylates get oligomerized. More specifically, when the reaction was performed in the dark, after 50 minutes, only a small conversion to 3a was noted, however, 2a-dimer was formed. When the reaction was performed under an argon atmosphere, again a good conversion of 3a was observed.27 So, the presence of oxygen is not essential although it is known to accelerate the formation of acyl radicals from aldehydes.30 So, it seems that oxygen promotes two of the three reactions (the oxidation of the aldehyde to the acid and the formation of the desired product).
Bringing together the literature knowledge and our studies, we can conclude by proposing the following mechanism (Scheme 6). In the literature, the mechanism for the hydroacylation of dialkyl azodicarboxylates is mostly proposed involving radical intermediates,2 although some ionic paths have also been reported.2 In the literature, air and/or water are well-accepted in being able to generate acyl radical I from aldehydes 1, a process which is highly dependent on air, but water is known and proposed to accelerate this process (Scheme 6A, Pathway II).16,17,29b,30a–c,34b It is also well accepted that acyl radical I can react with different electrophiles, like DIAD, leading to the desired products (Scheme 6C).16,17,30c Moreover, we have more interest in photocatalysis and have been involved in the use of aldehydes as potential photoinitiators (Scheme 6A, up).20f,34a Under irradiation, aldehydes can lead to radicals I and II, which are known to take part in various processes, usually involving Hydrogen Atom Transfer (HAT) events, either with different reagents or with oxygen (Scheme 6A).20f Indeed, our observations are in accordance with these notions, however, acyl radicals and the desired product are produced in the absence of air, since the reaction under argon (no oxygen) led to diminished yields (67%), while solvents other than water led to slower reactions (if any).27 Thus, combining all these data, we can suggest that irradiation promotes the self-excitation of diisopropyl azodicarboxylate forming 2a*, due to its significant absorbance near 390 nm (Scheme 6B, Pathway III).272a* can be used as a sacrificial agent and promote HAT from the aldehyde to form acyl radical I and intermediate III. However intermediate III can also be formed from 2a* in the presence of water, which then oligomerizes forming dimers and trimers. 390 nm irradiation of DIAD (2a) in water for 80 min led to the consumption of 2a and the isolation, after column chromatography, of the same DIAD oligomers 2a-dimer in 42% yield and 2a-trimer in 20% yield, respectively.27 Although we have a long tradition of studying this hydroacylation reaction,3,23a,i,24 this is the first time that such oligomers (oligomerization of DIAD via HAT from III) have been detected by our group (and in the literature). These kinds of oligomers can be produced from III, followed by a second (and third) HAT with 2a (Scheme 6B).
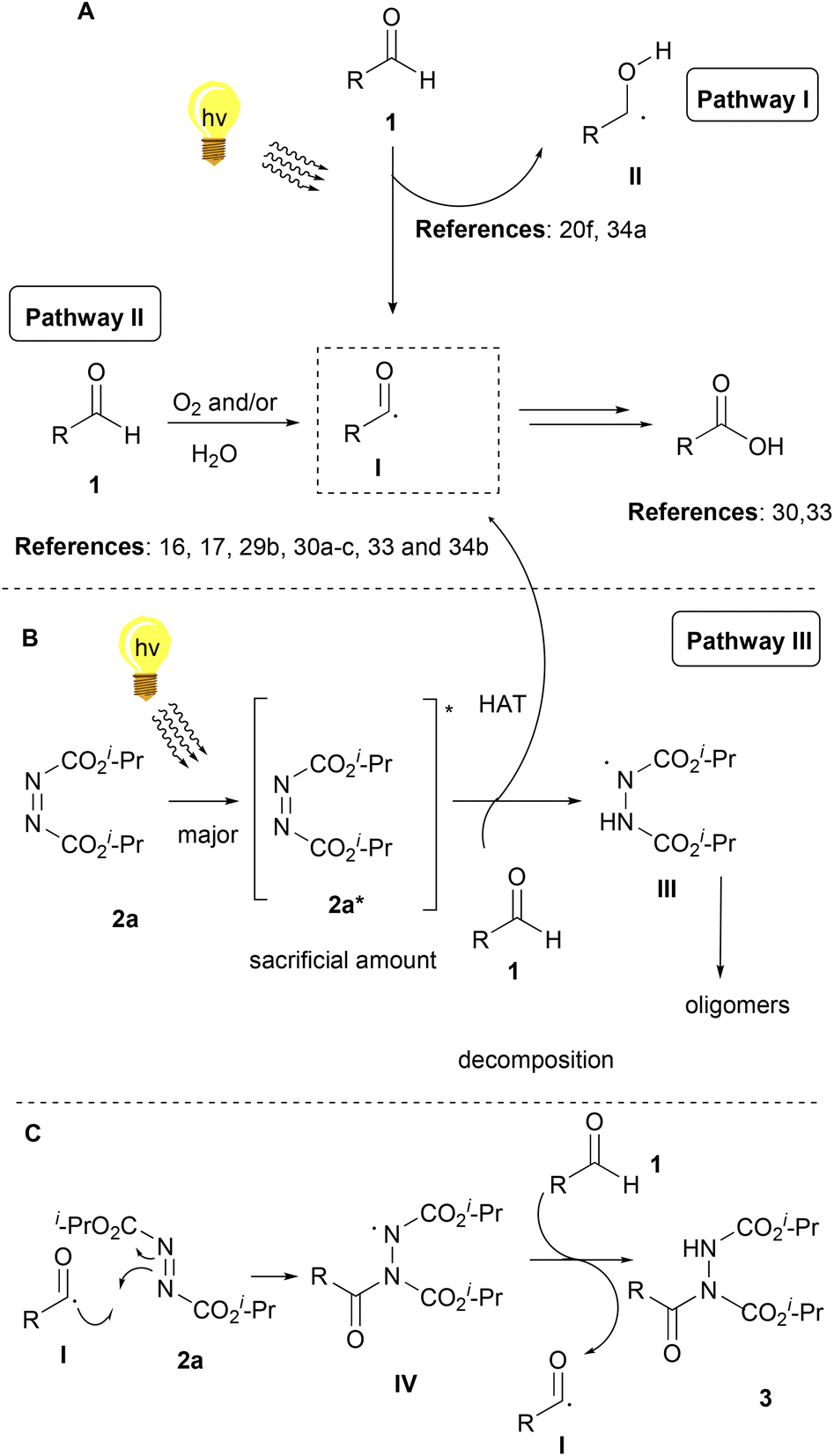 | ||
| Scheme 6 Proposed reaction mechanism. | ||
To summarize, acyl radical I can be formed in three ways. Firstly, by the aldehyde itself with the help of oxygen and/or with the help of water (Pathway II, minor pathway, irradiation is known to accelerate this pathway33) and secondly, by hv irradiation (Pathway I, minor pathway). Thirdly, by intermediate 2a* (Pathway III, major pathway), which will form intermediate III by hydrogen atom transfer from the aldehyde (pathway not requiring oxygen for acyl radical formation). Pathway III is the major pathway since when the control experiment under argon was carried out,273a was obtained in a high yield. If pathways I and II were major pathways, acyl radical I would have been formed with great difficulty, resulting in low yields. Nonetheless, in our case, the yield was just slightly diminished (67%), indicating that oxygen is not essential for the reaction, but plays an assisting role in enabling additional pathways for the formation of the acyl radical (Scheme 6A). Then, the acyl radical can react in two ways. Firstly, reacting in the presence of oxygen (Scheme 6A) to form the corresponding carboxylic acid.33,34 The second pathway (Scheme 6C) is the desired pathway, in which acyl radical I reacts with 2a to form intermediate IV, which promotes a HAT from the aldehyde (generating again acyl radical I), leading to the desired product 3. Hence, the mechanism proposed in Scheme 6B is believed to be dominant, however, the mechanisms in Scheme 6A contribute to an extent to the reaction outcome. This hypothesis of the mechanisms strengthens, by identifying the by-products that formed, in the cases where lower yields were obtained. In these cases, 2a-dimer formation was observed, as well as the remaining aldehyde and the corresponding carboxylic acid of the aldehyde. Last but not least, the enhancement of the obtained yields and the diminished reaction times when 2 equivalents of aldehyde 1 were used, indicate the significance of acyl radical formation. When larger amounts of the aldehyde exist in the reaction mixture, larger amounts of acyl radical I can be formed and react efficiently with 2a, resulting in the desired product 3a.
Conclusions
In conclusion, a green, inexpensive, fast and environmentally friendly synthetic protocol for the aerobic photochemical synthesis of acyl hydrazides 3 from aldehydes 1 and dialkyl azodicarboxylates 2 was developed. Using 390 nm Kessil lamps as the irradiation source and water as the solvent, light accelerates the formation of the acyl radical from the corresponding aldehyde, which reacts with dialkyl azodicarboxylate 2, leading to the desired products. A variety of aldehydes can be used in this protocol, including aromatic, aliphatic, α,β-unsaturated and naturally occurring aldehydes, affording the desired product in high yields and in short reaction times (15–210 min). Based on the literature precedent and our mechanistic investigations, three proposed mechanistic pathways are presented, while the pathway where dialkyl azodicarcoxylate is the initiator was proven to be the dominant. Finally, the synthesis of two commercially available drugs, Moclobemide and Vorinostat, was also demonstrated using our protocol.Experimental
General procedure for the synthesis of acyl hydrazides
In a test tube, aldehyde (0.75–1.00 mmol, 1.5–2.0 equiv.), dialkylazodicarboxylate (0.50 mmol, 1.0 equiv.) and H2O (2 mL) were added consecutively. The test tube was left stirring under Kessil lamp irradiation (390 nm) for 15–210 min depending on the substrate, until reaction completion, determined by TLC and NMR. After reaction completion, the reaction mixture was concentrated in vacuo. If the product is not of the desired purity, the desired product can be further purified by column chromatography.Author contributions
C. G. K. conceived the study, N. A. S., E. T. P. and C. G. K. designed the experiments and N. A. S. and E. T. P. analyzed the data. N. A. S., E. T. P., N. F. N., M. K., N. S. and P. A. D. performed the experiments. N. A. S. and C. G. K. prepared the draft of the manuscript and C. G. K. performed the final editing. The manuscript was written through the contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Hellenic Foundation for Research and Innovation (HFRI) for financial support through a grant, which is financed by 1st Call for H. F. R. I. Research Projects to Support Faculty Members & Researchers and the procurement of high-cost research equipment grant (grant number 655). N. S. would like to thank the Hellenic Foundation for Research and Innovation (HFRI) for financial support through a doctoral by the Hellenic Foundation for Research and Innovation (HFRI) under PhD Fellowship grant (Fellowship Number: 721).References
- For a recent review, see: V. Nair, A. T. Biju, S. C. Mathew and B. P. Babu, Chem. – Asian J., 2008, 3, 810–820 CrossRef CAS PubMed.
- For recent reviews, see: (a) A. Shamsabadi and V. Chudasama, Org. Biomol. Chem., 2017, 15, 17–33 RSC; (b) N. Spiliopoulou, C. T. Constantinou, I. Triandafillidi and C. G. Kokotos, Synthesis, 2020, 3219–3230 CAS.
- (a) G. N. Papadopoulos and C. G. Kokotos, Chem. – Eur. J., 2016, 22, 6964–6967 CrossRef CAS PubMed; (b) G. N. Papadopoulos and C. G. Kokotos, J. Org. Chem., 2016, 81, 7023–7028 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. Hubert, Z. Munzbergova and A. Santino, Pest Manag. Sci., 2007, 64, 57–64 CrossRef PubMed; (b) C. Gampe and V. A. Verma, J. Med. Chem., 2020, 63, 14357–14381 CrossRef CAS PubMed.
- T. Kanzian and H. Mayr, Chem. – Eur. J., 2010, 16, 11670–11677 CrossRef CAS PubMed.
- F. D. Marsh and M. E. Hermes, J. Am. Chem. Soc., 1965, 87, 1819–1820 CrossRef CAS.
- For selected reviews, see: (a) D. L. Hughes and R. A. Reamer, J. Org. Chem., 1996, 61, 2967–2971 CrossRef CAS PubMed; (b) V. Nair, R. S. Menon, A. R. Sreekanth, N. Abhilash and A. T. Biju, Acc. Chem. Res., 2006, 39, 520–530 CrossRef CAS PubMed; (c) P. Singh and M. Mritunjay, Asian J. Org. Chem., 2021, 10, 964–979 CrossRef CAS.
- D. Lee and R. D. Otte, J. Org. Chem., 2004, 69, 3569–3571 CrossRef CAS PubMed.
- Y. Qin, D. Zhou and M. Li, Lett. Org. Chem., 2012, 9, 267–272 CrossRef CAS.
- Y. Qin, Q. Peng, J. Song and D. Zhou, Tetrahedron Lett., 2011, 52, 5880–5883 CrossRef CAS.
- (a) S. M. Inamdar, V. K. More and S. K. Mandal, Chem. Lett., 2012, 41, 1484–1486 CrossRef CAS; (b) K. Azizi and A. Heydari, Synlett, 2017, 189–192 Search PubMed.
- J. M. Perez and D. J. Ramon, Adv. Synth. Catal., 2014, 356, 3039–3047 CrossRef CAS.
- H.-B. Zhang, Y. Wang, Y. Gu and P.-F. Xu, RSC Adv., 2014, 4, 27796–27799 RSC.
- B. Ni, Q. Zhang, S. Garre and A. D. Headley, Adv. Synth. Catal., 2009, 351, 875–880 CrossRef CAS.
- A. Mariappan, K. Rajaguru, S. Muthusubramarian and N. Bhuvanesh, Tetrahedron Lett., 2015, 56, 338–341 CrossRef CAS.
- V. Chudasama, M. J. Ahern, D. V. Dhokia, R. J. Fitzmaurice and S. Caddick, Chem. Commun., 2011, 47, 3269–3271 RSC.
- Q. Zhang, E. Parker, A. D. Headley and B. Ni, Synlett, 2010, 2453–2456 CAS.
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) K. L. Scubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef PubMed; (c) M. D. Kärkäs, J. A. Porco Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (d) D. Cambie, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noel, Chem. Rev., 2016, 116, 10276–10341 CrossRef CAS PubMed; (e) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzera and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (f) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed; (b) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (c) D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232–2242 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–11116 CrossRef CAS PubMed; (e) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC; (f) M. A. Theodoropoulou, N. F. Nikitas and C. G. Kokotos, Beilstein J. Org. Chem., 2020, 16, 833–857 CrossRef CAS PubMed; (g) N. F. Nikitas, P. L. Gkizis and C. G. Kokotos, Org. Biomol. Chem., 2021, 19, 5237–5253 RSC; (h) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- For selected examples, see: (a) T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198–6201 CrossRef CAS PubMed; (b) E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064–12068 CrossRef CAS PubMed; (c) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652–13655 CrossRef CAS PubMed; (d) D. Mazzarela, G. E. M. Crisenza and P. Melchiorre, J. Am. Chem. Soc., 2018, 140, 8439–8443 CrossRef PubMed; (e) T. P. Nicholls, D. Leonori and A. C. Bissember, Nat. Prod. Rep., 2016, 33, 1248 RSC; (f) Y. Xiong, J. Großkopf, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2022, 61, e202200555 CAS.
- I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Org. Lett., 2013, 15, 2554–2557 CrossRef CAS PubMed.
- For PhCOCOOH-mediated processes, see: (a) G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Chem. – Eur. J., 2014, 20, 13811–13814 CrossRef CAS PubMed; (b) D. Limnios and C. G. Kokotos, Adv. Synth. Catal., 2017, 359, 323–328 CrossRef CAS; (c) N. Kaplaneris, A. Bisticha, G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Green Chem., 2017, 19, 4451–4456 RSC; (d) G. N. Papadopoulos, E. Voutyritsa, N. Kaplaneris and C. G. Kokotos, Chem. – Eur. J., 2018, 24, 1726–1731 CrossRef CAS PubMed; (e) E. Voutyritsa and C. G. Kokotos, Angew. Chem., Int. Ed., 2020, 59, 1735–1741 CrossRef CAS PubMed; (f) G. N. Papadopoulos, M. G. Kokotou, N. Spiliopoulou, N. F. Nikitas, E. Voutyritsa, D. I. Tzaras, N. Kaplaneris and C. G. Kokotos, ChemSusChem, 2020, 13, 5934–5944 CrossRef CAS PubMed; (g) E. Voutyritsa, M. Garreau, M. G. Kokotou, I. Triandafillidi, J. Waser and C. G. Kokotos, Chem. – Eur. J., 2020, 26, 14453–14460 CrossRef CAS PubMed; (h) N. Spiliopoulou and C. G. Kokotos, Green Chem., 2021, 23, 546–551 RSC; (i) N. Spiliopoulou, P. L. Gkizis, I. Triandafillidi, N. F. Nikitas, C. S. Batsika, A. Bisticha and C. G. Kokotos, Chem. – Eur. J., 2022, 28, e202200023 CrossRef CAS PubMed For other photocatalysts, see: (j) N. F. Nikitas, I. Triandafillidi and C. G. Kokotos, Green Chem., 2019, 21, 669–674 RSC; (k) N. F. Nikitas, D. I. Tzaras, I. Triandafillidi and C. G. Kokotos, Green Chem., 2020, 22, 471–477 RSC; (l) N. Spiliopoulou, N. F. Nikitas and C. G. Kokotos, Green Chem., 2020, 22, 3539–3545 RSC.
- G. S. Koutoulogenis, M. G. Kokotou, E. Voutyritsa, D. Limnios and C. G. Kokotos, Org. Lett., 2017, 19, 1760–1763 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, University Press, Oxford, New York, 1998 Search PubMed.
- For recent studies from our group, where different irradiation wavelengths is the key for the reaction outcome, see: (a) N. F. Nikitas, E. Voutyritsa, P. L. Gkizis and C. G. Kokotos, Eur. J. Org. Chem., 2021, 96–101 CrossRef CAS; (b) I. Triandafillidi, N. F. Nikitas, P. L. Gkizis, N. Spiliopoulou and C. G. Kokotos, ChemSusChem, 2022, 15, e202102441 CrossRef CAS PubMed.
- For full reaction conditions, optimization and mechanistic studies, see the ESI.†.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef PubMed.
- For leading references on “on-water” catalysis, see: (a) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed For related reactions accelerated using aqueous media, see: (b) N. Shapiro, M. Kramer, I. Goldberg and A. Vigalok, Green Chem., 2010, 12, 582–584 RSC; (c) G. F. P. De Souza, J. A. Bonacin and A. G. Salles Jr., J. Org. Chem., 2018, 83, 8331–8340 CrossRef CAS PubMed.
- For the initial contribution on aerobic non-photochemical acyl radical formation from aldehydes, see: (a) V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592–596 CrossRef CAS PubMed For original contributions employing this idea, see: (b) V. Chudasama, R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Chem. Commun., 2010, 46, 133–135 RSC; (c) V. Chudasama, A. R. Akhbar, K. A. Bahou, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2013, 11, 7301–7317 RSC; (d) A. R. Akhbar, V. Chudasama, R. J. Fitzmaurice, L. Powell and S. Caddick, Chem. Commun., 2014, 50, 743–746 RSC; (e) V. Chudasama, RSC Adv., 2015, 5, 44423–44426 RSC; (f) A. Maruani, M. T. W. Lee, G. Watkins, A. R. Akhbar, H. Baggs, A. Shamsabadi, D. A. Richards and V. Chudasama, RSC Adv., 2016, 6, 3372–3376 RSC; (g) A. Shamsabadi, J. Ren and V. Chudasama, RSC Adv., 2017, 7, 27608–27611 RSC; (h) A. Shamsabadi and V. Chudasama, Chem. Commun., 2018, 54, 11180–11183 RSC; (i) A. Shamsabadi, A. Maruani, N. Ahmed and V. Chudasama, Org. Biomol. Chem., 2020, 18, 6258–6264 RSC.
- (a) R. S. Mulliken, J. Phys. Chem., 1952, 56, 801–822 CrossRef CAS; (b) I. R. Gould and S. Farid, Acc. Chem. Res., 1996, 29, 522–528 CrossRef CAS; (c) S. Farid, J. P. Dinnocenzo, P. B. Merkel, R. H. Young, D. Shukla and G. Guirado, J. Am. Chem. Soc., 2011, 133, 11580–11587 CrossRef CAS PubMed; (d) M. Koch, G. Licari and E. Vaunthey, J. Phys. Chem. B, 2015, 119, 11846–11857 CrossRef CAS PubMed.
- For selected examples, see: (a) E. Arceo, I. D. Junberg, A. Alvarez and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (b) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassini and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS PubMed; (c) M. L. Spell, K. Devenaux, C. G. Bresnahan, B. L. Bernard, W. Sheffield, R. Kumar and J. R. Ragains, Angew. Chem., Int. Ed., 2016, 55, 6515–6519 CrossRef CAS PubMed; (d) R. Wang, L. Wang, Q. Xu, B. Y. Ren and F. Liang, Org. Lett., 2019, 21, 3072–3076 CrossRef CAS PubMed.
- C. S. Batsika, C. Koutsilieris, G. S. Koutoulogenis, M. G. Kokotou and C. G. Kokotos, Green Chem., 2022, 24, 6224–6231 RSC.
- (a) N. F. Nikitas, M. A. Theodoropoulou and C. G. Kokotos, Eur. J. Org. Chem., 2021, 1168–1173 CrossRef CAS; (b) I. Triandafillidi, M. G. Kokotou, D. Lotter, C. Sparr and C. G. Kokotos, Chem. Sci., 2021, 12, 10191–10196 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental data, 1H and 13C NMR data, UV-Vis, optimization and mechanistic studies. See DOI: https://doi.org/10.1039/d2ob02204d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |