
Metal-free synthesis of N-sulfonyl imines from benzyl alcohol derivatives and iminoiodinanes via mechanochemistry†
Souvik
Guha
 a,
Ritwik
Bhattacharya
a,
Jesni
Jacob
b,
Mahesh
Ravva
b and
Subhabrata
Sen
*a
a,
Ritwik
Bhattacharya
a,
Jesni
Jacob
b,
Mahesh
Ravva
b and
Subhabrata
Sen
*a
aDepartment of Chemistry, School of Natural Sciences, Shiv Nadar Institution of Eminence Deemed to be University, Chithera, Dadri, Gautam Buddha Nagar, UP 201314, India. E-mail: subhabrata.sen@snu.edu.in
bDepartment of Chemistry, SRM-AP, Amaravati, AP, India
First published on 15th June 2023
Abstract
An expedient and operationally convenient mechanochemical synthesis of aryl/heteroaryl N-sulfonyl imines is reported by reacting iminoiodinanes with numerous aryl/heteroaryl benzyl alcohols in ball milling apparatus (RETSCH 400™) with three 5 mm stainless steel (ss) balls in a 5 mL stainless steel (ss) reaction jar. CHCl3 (η = 0.2–0.4 μL mg−1) was used as a liquid assisted grinding (LAG) auxiliary. This metal catalyst- and base- free synthesis with nominal amounts of solvents (as LAGs) demonstrated an efficient N-sulfonyl transfer reaction from iminoiodinanes to afford the desired compounds in moderate to good yields. Substituted N-sulfonyl imines are crucial as standalone natural product building blocks and drug intermediates as well as precursors of sulfonamides which have been involved in potential small molecule therapy in many therapeutic programs. The putative mechanisms of the transformations are discussed based on control reactions and DFT calculations.
Introduction
Nitrenes, electrophilic nitrogen moieties, play a crucial role in organic synthetic chemistry.1 First defined by Tienmann in 1891, nitrene-based reactions typically involve C–H amination/amidation, aziridine formation, addition to alkynes/olefins, sulfamidation, etc.2,3 In recent times, iminoiodinane, a formal electrophilic nitrogen donor and a more environmentally tolerant nitrene, has been used in various N-transfer reactions such as metal-catalyzed and metal-free aziridination of olefins, metal-catalyzed reactions with ynamides, synthesis of 2,2-diarylenamides, conversion of aldehydes to N-sulfonylimines, etc. to facilitate N–C bonds and generate various nitrogenous products.4–12 We envisioned the synthesis of N-sulfonyl imines from iminoiodinanes and benzyl alcohol derivatives via mechanochemistry.In the last few decades, various organic transformations via mechanochemical ball milling have been increasingly reported. They are eco-friendly since they either are solvent-free or occur with microliter volumes of solvents as liquid assisted grinding (LAG) auxiliaries represented with η (μL mg−1) which is expected to be between 0 and 1.13–21 The unique selling point (USP) of mechanochemistry is facilitating organic reactions via milling or grinding, thereby eliminating the need for bulk dissolution of reactants.22 These are substantial improvements from performing organic reactions with round bottom flasks, stirrers, heaters and solvents, which are replaced with automated ball mills containing jars, balls and milling media as either solids or liquids in microliter quantities. Furthermore, shorter reaction times, lower reaction temperature and improved “green” metrics compared to solution-based strategies are additional advantages. IUPAC has selected it as one of the 10 innovations that could change the world.23a
By virtue of their presence in numerous bioactive compounds exhibiting various therapeutic effects, sulfonamide moieties are regarded as crucial pharmaceutically relevant building blocks.23–26 In a bid to access them, sulfonyl imines that are either electron-deficient or electron-rich have garnered considerable attention as suitable substrates.27–36 Additionally, these imines are involved in numerous organic transformations viz. C–H functionalization, nucleophilic addition, cycloaddition and many more.37–53
The biological utility of N-sulfonyl imines has prompted many synthetic efforts to generate them. Conventional strategies to synthesize N-sulfonyl imines involve condensation of sulfonamides with carbonyl moieties (condensation of aldehydes with sulfonamides) and non-dehydrative coupling of isocyanate derivatives with aldehydes (Scheme 1a), and only two oxidative reactions of primary benzyl alcohols with sulfonamides or chloramine T (Scheme 1b).54–67 Since the in situ oxidation of benzyl alcohol derivatives to the corresponding aldehydes and subsequent imine formation is attractive, a catalyst/base free synthesis of sulfonyl imines would be interesting yet elusive.
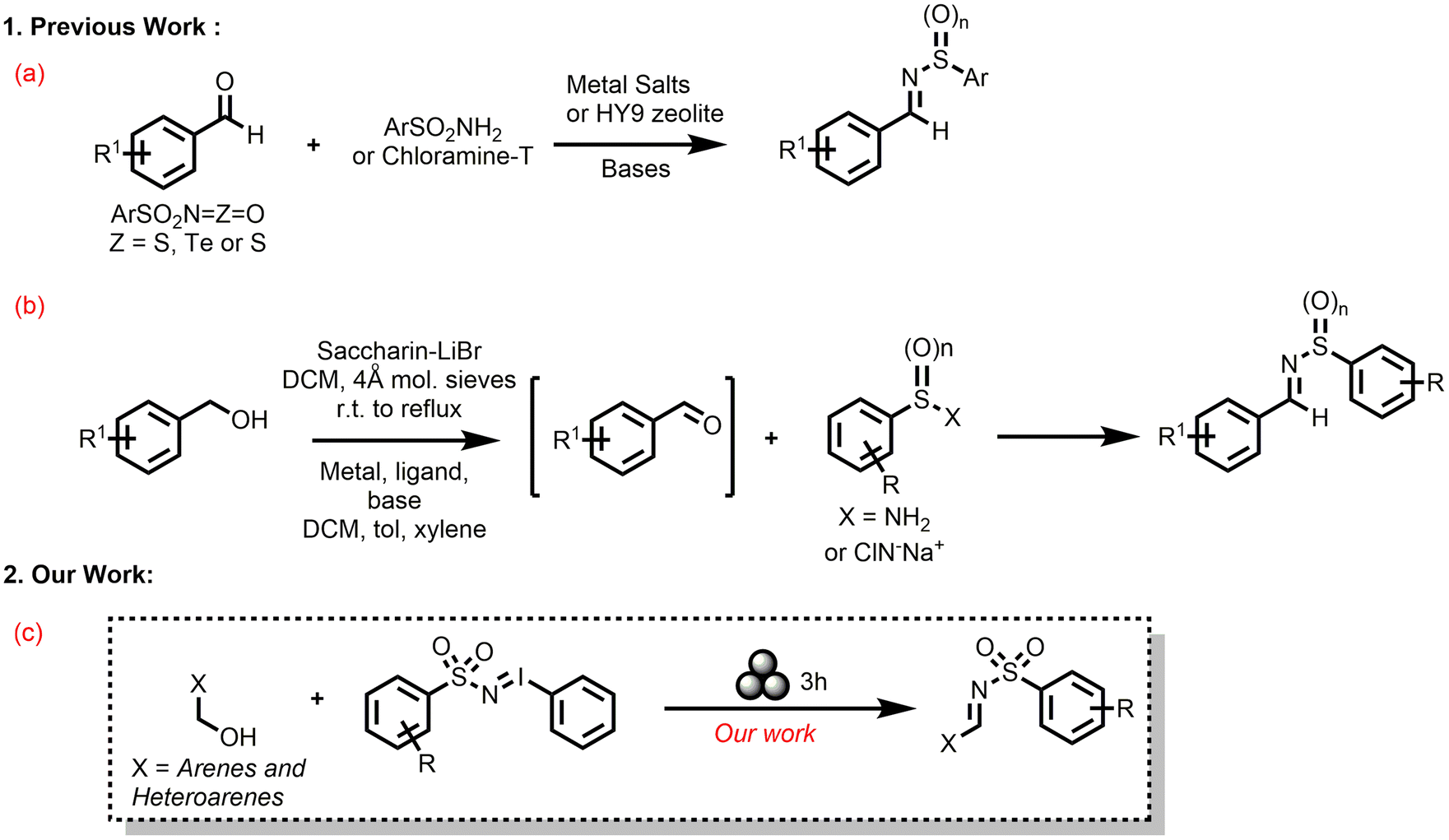 | ||
| Scheme 1 (a)–(b) Literature reports and (c) our mechanochemical strategy for the formation of N-sulfonyl imines. | ||
A close scrutiny of these solvent-based syntheses of N-sulfonyl imines that are reported in the literature revealed the usage of copious amounts of organic solvents, Lewis acids/bases, metal catalysts, high temperature, and pressure. In a bid for a direct improvement from all these existing procedures for the synthesis of N-sulfonyl imines, herein we report a mechanochemical N-sulfonyl transfer reaction with numerous iminoiodinanes (Scheme 1c). The desired N-sulfonyl imines were synthesized under ball-milling conditions from various aryl and hetero aryl 1°-benzyl alcohols with 3 stainless-steel balls (∅ = 5 mm, mtot = 0.94 g) in a ball milling jar (stainless steel, 5 mL) under air at 30 Hz for 3 h in moderate to excellent yields. CHCl3 (η = 0.2–0.4 μL mg−1) was used as a liquid assisted grinding auxiliary. No bases, acids, and catalysts were used. Control experiments and DFT calculations helped in elucidating the reaction mechanisms. The procedures are also scalable to gram quantities.
It is noteworthy that the solvents (in our case chloroform) which are used in our protocol are only at ∼0.2–0.4 μL mg−1, whereas under heterogeneous reaction conditions, they are used at ∼500 μL to 1 mL mg−1 at the least. For our scale-up reaction at ∼1 g, we have used 350 μL of CHCl3. This is substantially lower than that in a standard heterogeneous reaction (average requirement could be from 10 to 30 mL), which further highlights the sustainable nature of our protocol.
Results and discussion
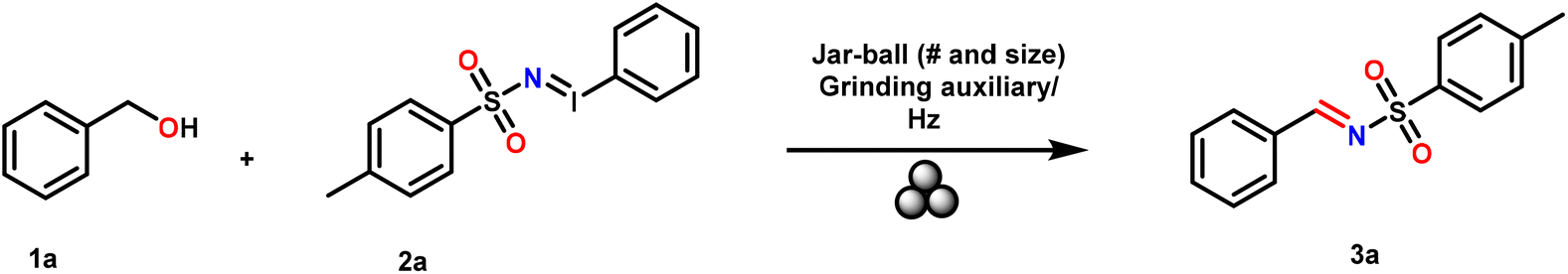
At the outset, the synthesis of N-sulfonyl imines was optimized with benzyl alcohol 1a (1 eq.) and N-tosyl iminoiodinane 2a (1 eq.) as reaction partners in a variety of solvents such as tetrahydrofuran, ethyl acetate, acetonitrile, 1,4-dioxane, dichloromethane and chloroform (reaction scale: 50 mg of 1a in 5 mL of solvent [0.092 M solution concentration]). The reaction was only feasible in chlorinated solvents; however, the yields were poor (5 to 12%). The reaction under neat conditions worked better with a yield of 21%. It was observed that despite the improvement in the yield due to a heterogeneous mixture, a substantial amount of starting materials was scattered at the surface of the reaction vessel, which could rationalize the yield of 21%. Since under homogeneous conditions the chlorinated solvents worked better than the others, we used dichloromethane and chloroform in 1/20th of the volume used under homogeneous conditions. The reaction yield improved remarkably to 40%. That is when we envisaged the application of mechanochemistry.Next, when 1a and 2a were reacted at 30 Hz for 60 minutes under mechanochemical ball milling in a Retsch MM 400 mixer mill with solid state grinding in a stainless steel (ss) jar (5 mL) with three ss balls of 5 mm diameter, the desired product 3a was obtained in 13% yield (Table 1, entry 1). Under the same conditions with 2 equivalents of 2a, there was no improvement in the yield (Table 1, entry 2). In the next two reactions (with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 1a and 2a) for an increased reaction time from 120 to 180 minutes, there was a slight improvement in the yield to 18 and 23%, respectively (Table 1, entries 3 and 4). At 30 Hz for 180 minutes when SiO2 was added as a grinding auxiliary, there was a further increment of the yield of 3a to 28% (Table 1, entry 5). However, the same reaction with 2 eq. of 2a did not improve the yield further (Table 1, entry 6). The reaction (1:1 of 1a and 2a) with sea sand and 4 Å molecular sieves as grinding auxiliaries (Table 1, entries 7 and 8) was also not helpful in improving the yield of 3a. Interestingly at 30 Hz for 300 minutes with 4 Å molecular sieves, the reaction yield improved considerably to 36% (Table 1, entry 9). Here, it is noteworthy that alumina (Al2O3) as a grinding auxiliary could not improve the yield any further (Table 1, entry 10). These moderate results prompted us to explore liquid assisted grinding (LAG) (Table 1, entries 11–19) and it was gratifying to observe that at 30 Hz for 180 minutes with chloroform (CHCl3) at η = 0.2 μL mg−1 of 1a generated the desired product in the best yield of 70% (Table 1, entry 19). Next the optimization was performed with the 2 eq. of 2a (Table 1, entry 20), 7 mm and 10 mm ss balls (2 and 3) (Table 1, entries 21 and 22) and zirconium or Teflon balls and jars (Table 1, entries 23 and 24). However, the yields in those reactions were not better than that of the one performed under the conditions depicted in entry 19 (Table 1). The average reaction time ranged from 1 to 5 h. However, it is noteworthy that aliphatic alcohols such as methanol, ethanol, isopropanol and n-butanol failed to show any significant product formation.
:1 1a and 2a) for an increased reaction time from 120 to 180 minutes, there was a slight improvement in the yield to 18 and 23%, respectively (Table 1, entries 3 and 4). At 30 Hz for 180 minutes when SiO2 was added as a grinding auxiliary, there was a further increment of the yield of 3a to 28% (Table 1, entry 5). However, the same reaction with 2 eq. of 2a did not improve the yield further (Table 1, entry 6). The reaction (1:1 of 1a and 2a) with sea sand and 4 Å molecular sieves as grinding auxiliaries (Table 1, entries 7 and 8) was also not helpful in improving the yield of 3a. Interestingly at 30 Hz for 300 minutes with 4 Å molecular sieves, the reaction yield improved considerably to 36% (Table 1, entry 9). Here, it is noteworthy that alumina (Al2O3) as a grinding auxiliary could not improve the yield any further (Table 1, entry 10). These moderate results prompted us to explore liquid assisted grinding (LAG) (Table 1, entries 11–19) and it was gratifying to observe that at 30 Hz for 180 minutes with chloroform (CHCl3) at η = 0.2 μL mg−1 of 1a generated the desired product in the best yield of 70% (Table 1, entry 19). Next the optimization was performed with the 2 eq. of 2a (Table 1, entry 20), 7 mm and 10 mm ss balls (2 and 3) (Table 1, entries 21 and 22) and zirconium or Teflon balls and jars (Table 1, entries 23 and 24). However, the yields in those reactions were not better than that of the one performed under the conditions depicted in entry 19 (Table 1). The average reaction time ranged from 1 to 5 h. However, it is noteworthy that aliphatic alcohols such as methanol, ethanol, isopropanol and n-butanol failed to show any significant product formation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Eq. of 2a | Jar-ball (# and size) | Frequency with reaction time | Grinding auxiliary | Yield of 3aa,b (%) |
| a Unless until mentioned, all the reactions were carried out on a (1 equiv.) 0.92 mmol scale with respect to 1a. b Yield based on the isolated amount by column chromatography. c Grinding auxiliary taken was 200 mg. d η = 0.2 μL mg−1. n.d. = not determined. | |||||
| 1 | 1 | ss (3 and 5 mm) | 30 Hz for 60 min | Neat | 13 |
| 2 | 2 | ss (3 and 5 mm) | 30 Hz for 60 min | Neat | 14 |
| 3 | 1 | ss (3 and 5 mm) | 30 Hz for 120 min | Neat | 18 |
| 4 | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | Neat | 23 |
| 5c | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | SiO2 | 28 |
| 6c | 2 | ss (3 and 5 mm) | 30 Hz for 180 min | SiO2 | 29 |
| 7c | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | Sea sand | 26 |
| 8c | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | 4 Å MS | 23 |
| 9c | 1 | ss (3 and 5 mm) | 30 Hz for 300 min | 4 Å MS | 36 |
| 10c | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | Al2O3 | 17 |
| 11 | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | EtOAcd | 32 |
| 12 | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | THFd | 24 |
| 13 | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | ACNd | 40 |
| 14 | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | Toluened | n.d. |
| 15 | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | DCMd | 62 |
| 16 | 2 | ss (3 and 5 mm) | 30 Hz for 180 min | DCMd | 60 |
| 17 | 2 | ss (3 and 5 mm) | 30 Hz for 120 min | DCMd | 56 |
| 18 | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | DCEd | 64 |
| 19 | 1 | ss (3 and 5 mm) | 30 Hz for 180 min | CHCl 3 | 70 |
| 20 | 2 | ss (3 and 5 mm) | 30 Hz for 180 min | CHCl3d | 68 |
| 21 | 1 | ss (3 and 7 mm) | 30 Hz for 120 min | CHCl3d | 65 |
| 22 | 1 | ss (2 and 10 mm) | 30 Hz for 120 min | CHCl3d | 62 |
| 23 | 1 | Zr (3 and 5 mm) | 30 Hz for 180 min | CHCl3d | 52 |
| 24 | 1 | Teflon (3 and 5 mm) | 30 Hz for 180 min | CHCl3d | 56 |
After the optimization, the substrate scope was explored with various aromatic and hetero-aromatic benzyl alcohols 1a–z and iminoiodinanes 2a–f (Scheme 2a) and it was observed that the reaction tolerated these diversely substituted substrates and rapidly provided various N-sulfonylimines 3a–3ag (Scheme 2a) in 45% to 85% yields. It was noticed that neutral or electron-rich aromatic benzyl alcohols such as 1a, 1c, 1d, 1g–j, 1l, 1o–p, and 1r–v favored the reaction slightly better (yields range from 61% to 85%) than the corresponding electron-poor benzyl alcohols, such as 1b, 1e, 1f, 1k, 1m–n and 1q. Reactions of furan-2-ylmethanol 1w, thiophen-2-ylmethanol 1x and tert-butyl 3-(hydroxymethyl)-1H-indole-1-carboxylate 1y with numerous iminoiodinanes 2a, 2c, 2d and 2f afforded the corresponding sulfonylimines 3w–y and 3ab–3ag in 66 to 85% yields (Scheme 2a). It is noteworthy that the reaction with liquid alcohols i.e., 1a–f, 1l, 1n, 1q, 1w, 1x, and 1z required even less CHCl3 in the optimized procedure to generate the desired products. To demonstrate the scalability of this strategy under the optimized solvent, catalyst and base free mechanochemical conditions, the reaction of 1s (0.006 mol) with 2s (0.006 mol) was performed with 2 stainless-steel balls (∅ = 10 mm, mtot = 2.7 g) in a ball milling jar (stainless steel, 10 mL). Using CHCl3 (as a LAG) (η = 0.35 μL mg−1) at 30 Hz, 3s was obtained in 1.26 g within 200 minutes. The reaction was completed in 3.5 h (Scheme 2b).
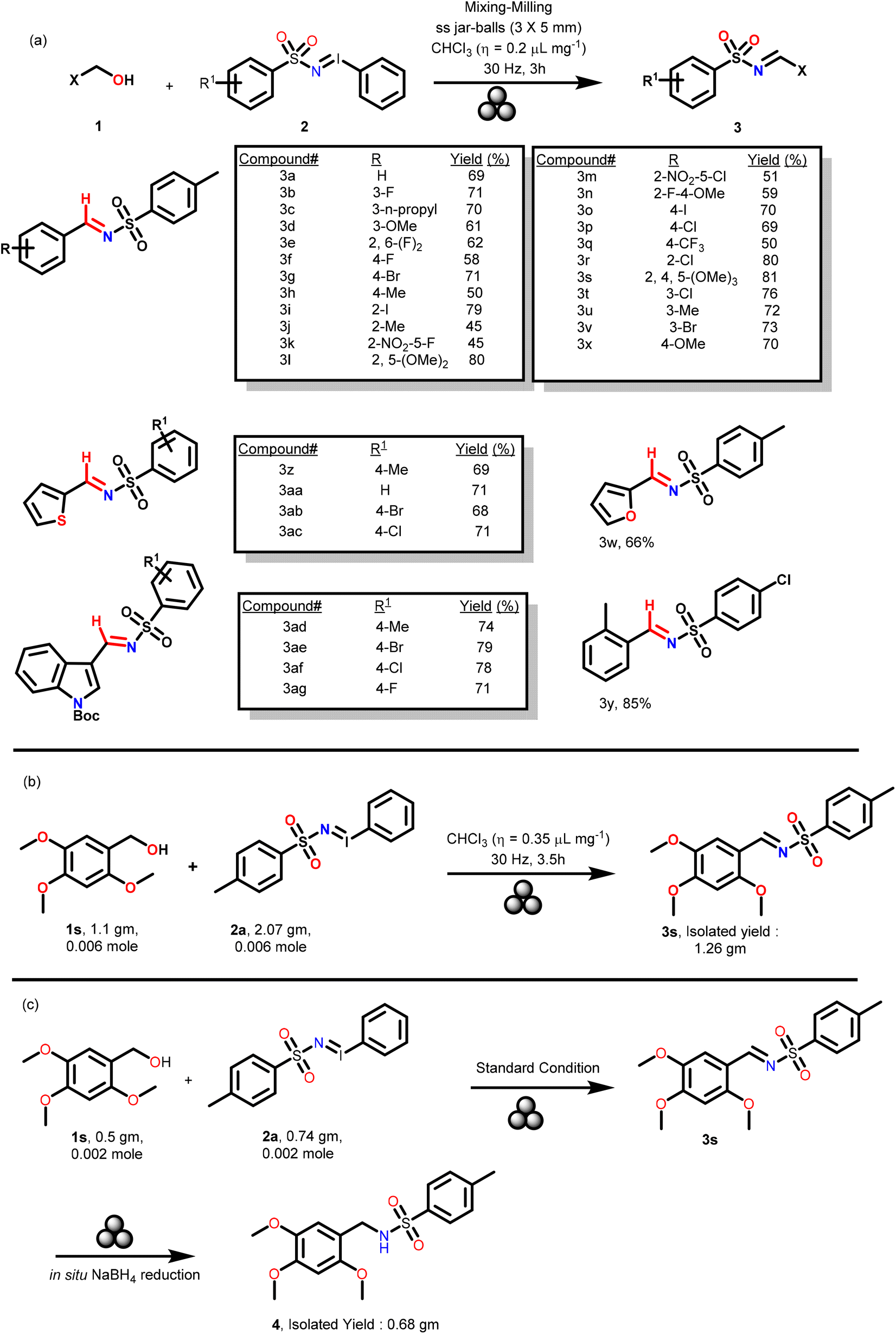 | ||
| Scheme 2 (a) Exploration of the scope of diversely substituted benzylic alcohols 1 as substrates and iminoiodinanes 2 as aminating agents. (b) Gram scale reaction of 1s to generate 1.26 g of 3s. (c) Reduction of imine 3s to generate 4 under ball milling conditions. | ||
Sulfonyl imines are appropriate substrates for sulfonamides. Sulfonamides are an important class of compounds as they are ubiquitously present in numerous bioactive natural products and active pharmaceutical ingredients.23–26 Hence, in a bid to demonstrate the utility of our sulfonyl imine compounds, in the crude mechanochemical reaction mixture of the product 3s, sodium borohydride (NaBH4) and methanol (η = 0.2 μL mg−1) as a LAG were added (refer the ESI† for details) (Scheme 2c). Compound 3s underwent facile reduction in a quantitative amount under the mechanochemical conditions to afford the corresponding sulfonamide 4 (Scheme 2c).
To gain further insight into the reaction mechanism benzyl alcohol 1a was reacted with N-tosyl iminoiodinane 2a in the presence of the radical scavenger BHT (Scheme 3). The fact that the desired product was obtained in 62% yield which was comparable to the yield obtained under the standard reaction conditions indicated that the reaction could have proceeded through an ionic pathway without formation of any radicals. Next, the reaction of benzaldehyde with para-tolylsulfonamide 2a′ under the optimized conditions did not yield any product (Scheme 3b). This demonstrated that the iminoiodinane 2a indeed plays a crucial role in this reaction.
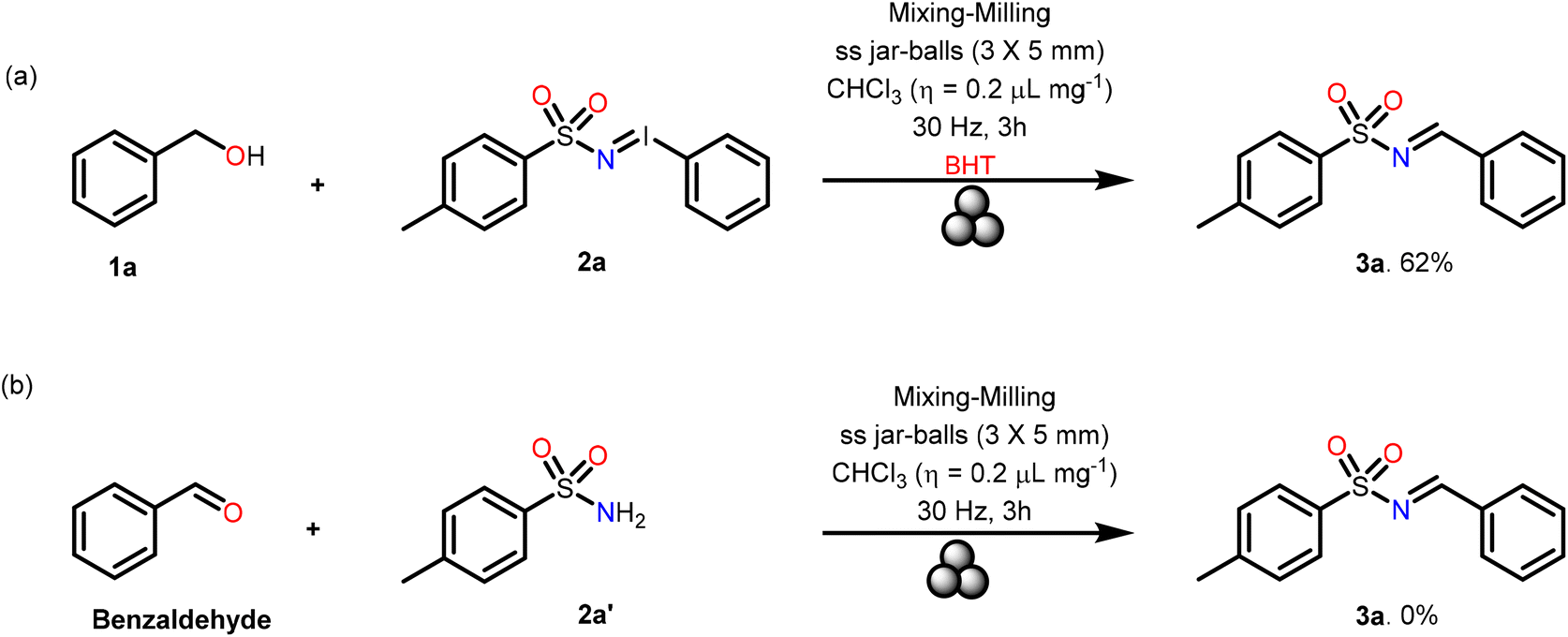 | ||
| Scheme 3 (a and b) Control reactions to understand the mechanism of formation of 3a. | ||
For deeper understanding of the reaction mechanism, density functional theory (DFT) calculations were performed (Scheme 4). Molecular geometries of all structures involved in the reaction mechanism (reactants, transition states, intermediates, and products) were optimized using the M06-2X method without any symmetry constraints in the gas phase.68,69 Two different basis sets were used, viz., the MIDIX and the 6-31G** basis sets were employed for the iodine (I) atom and for all the other atoms (C, H, N, O, and S), respectively. All calculations were carried out using the Gaussian 16 code.70
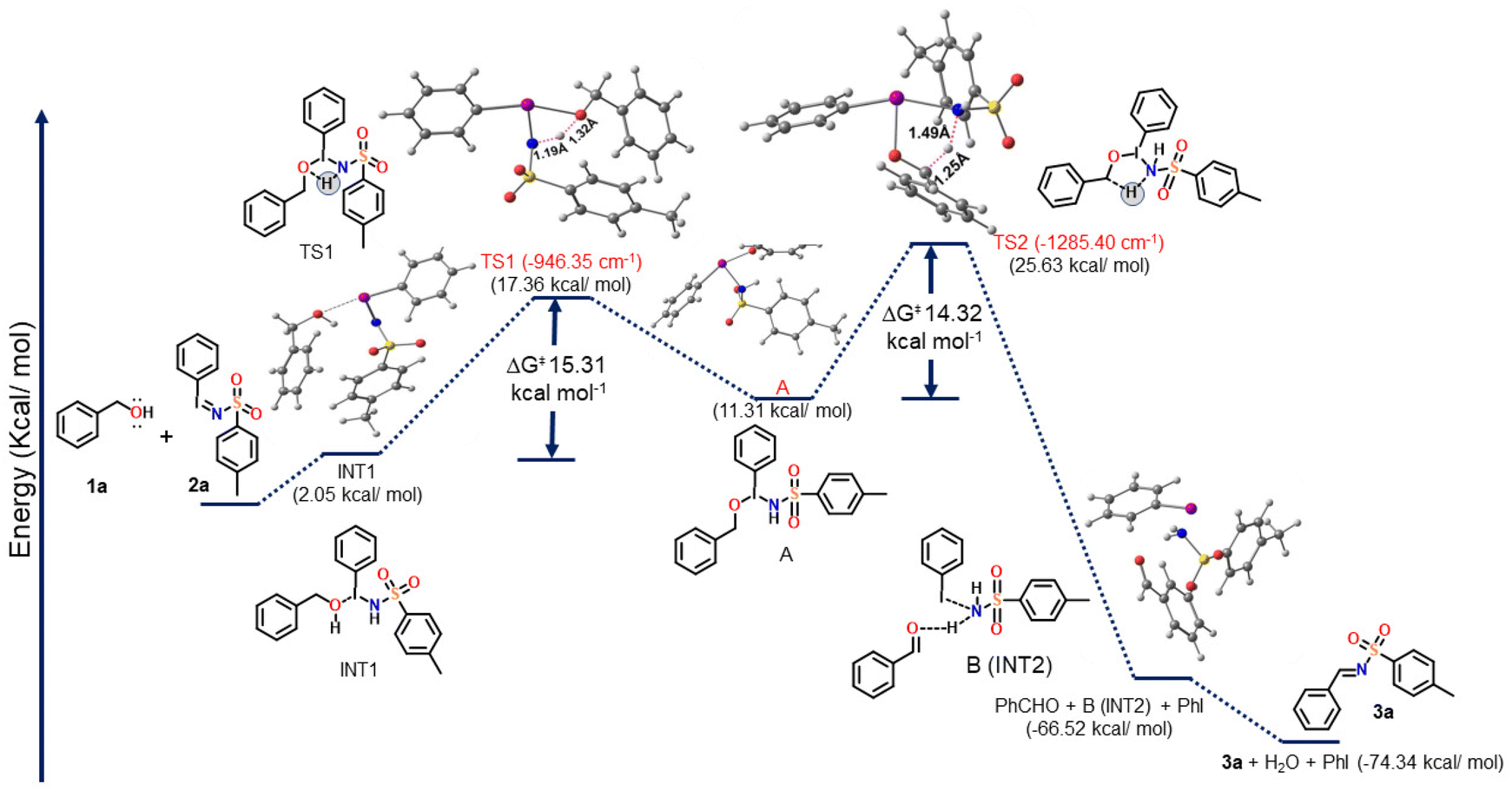 | ||
| Scheme 4 Calculated energy profile diagram of the proposed reaction as determined by M06-2X methods. Optimized geometries of the reactants, transition states, intermediates and products obtained at the M06-2X/6-31G** (C,N,O,S,H)/MIDIX (I) level of theory. Purple, gray, red, blue, yellow, and white colours represent I, C, O, N, S and H, respectively. | ||
Scheme 4 depicts the proposed mechanism, the optimized geometry of all the structures involved in the putative mechanism at the M06-2X/6-31G** (C, N, O, S, H)/MIDIX (I) level of theory (all the Cartesian coordinates of the optimized structures are provided in the ESI†) and the calculated free energy profile for the proposed mechanism. The Gibbs free energy (ΔG) of the reactants (1a and 2a) was taken as a reference and set to zero in the energy profile diagram (Scheme 4). The relative Gibbs free energies (ΔG in kcal mol−1) of all the remaining structures, including two intermediates (INT1 and INT2), two transition states (TS1 and TS2), and the products, were then calculated and plotted in comparison with that of the reactants (1a + 2a).
From DFT calculations, it shows that the formation of INT1via the reaction between 1a and 2a, with a free energy penalty of ∼2.05 kcal mol−1. The intermediate A was then formed via the transition state TS1 with a barrier of 15.31 kcal mol−1, and it involved the proton transfer from the O atom of benzyl alcohol to the imine group of 2a. The geometric parameters of TS1 include the bond lengths of O–H and N–H, which are 1.45 Å and 1.10 Å, respectively. Next, the intermediate INT2 was generated via the second transition state (TS2) with a reaction energy barrier of 14.32 kcal mol−1. The reaction that produced INT2viaTS2 is exergonic (−66.52 kcal mol−1). TS2 had an imaginary frequency of −1285 cm−1 and bond lengths of 1.38 Å (C–H) and 1.34 Å (N–H). Finally, the desired product 3a formed from INT2 with the release of PhI and H2O.
The substituent effect on, both in the alcohol substrates 1 and the iminoiodinanes 2, on the rate of formation of the reaction intermediates and the corresponding products, kinetics experiments were performed using 4-n-propoxy benzyl alcohol 1s (an electron-rich system) and 4-fluorobenzyl alcohol 1f (an electron-poor system) with 2a. The competing substrates were taken in stoichiometric amounts and the reaction mixtures were subjected to milling under the standard conditions for 3 h. The desired pure products were isolated by flash column chromatography. From the isolated yields (Scheme 5), it could be understood that the substituent effect is quite significant at the benzene ring of the benzyl alcohol derivatives 1f and 1s, as the n-propoxy analog 3c was obtained in a 3:1 ratio over the fluoro derivative 3f with an overall yield of 72% (Scheme 5). In contrast, the substituent effect was not much significant for the iminoiodinane derivatives as the reaction of 2a and 2c with 1y afforded 3ad and 3ag in a 1:1 ratio with an overall 54% yield (Scheme 5). Hence nominal product selectivity was observed with electron-rich (2a) over electron-poor (2c) iminoiodinanes.
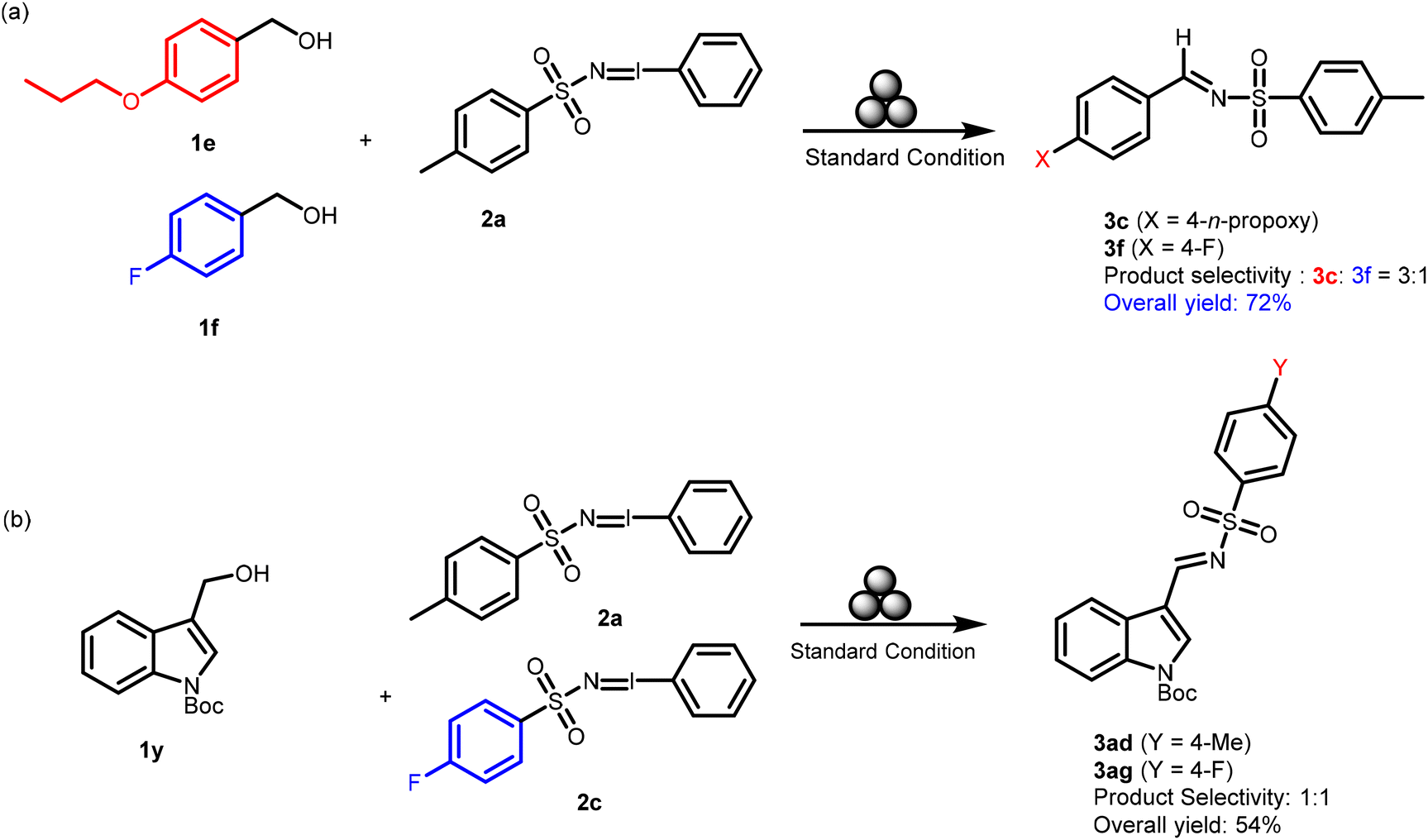 | ||
| Scheme 5 Competition experiments to compare the rate of reaction of (a) electron-rich and electron-poor benzyl alcohol derivatives with an iminoiodinane and (b) electron-rich and electron-poor iminoiodinanes with a benzyl alcohol derivative. | ||
Finally, green metrics were calculated to assess the environmental impact, effectiveness, and sustainability of our mechanochemical protocol (Table 2, for details, see the ESI†). The evaluation of the findings reported in the table indicated a nearly ten-fold lower E-factor (a crucial green metric defined as the ratio of the mass of waste to the mass of the product) for our mechanochemically promoted metal-free N-sulfonyl transfer reactions with iminoiodinanes for the synthesis of N-sulfonyl imines, and hence it is advantageous over heterogeneous solvent based strategies for the synthesis of the same N-sulfonyl imines 3a and 3g. However, the eco-scale values (which quantify the greener aspect of a synthetic strategy based on the (i) yield of the isolated product, (ii) cost of the chemicals, (iii) safety issues, (iv) technical set-up, (v) conditions of the reactions viz. temperature and time, and (vi) reaction work-up/purification methods) were comparable.65 Looking beyond the insightful numerical results of the green metrics, it is worth emphasizing that our procedure does not require additional heating, which prevents any potential degradation of heat-labile compounds. This methodology avoids using large amounts of solvents, which in turn is beneficial for the environment.
Conclusion
In summary, we have reported a metal- and base-free N–C coupling reaction of aryl and hetero-aryl benzyl alcohols 1a–z with diversely substituted N-sulfonyl iminoiodinanes 2a–f in 1:1 and 2:1 stoichiometric ratios, respectively, by mechanochemical mixing-milling in an RETSCH mixer mill (MM 400) at ambient temperature and with 0.2 to 0.4 μL mg−1 CHCl3 as a LAG, to generate N-sulfonyl imines 3. These facile and ecofriendly N-sulfonyl transfer reactions proceeded to completion in nearly 3 h. The gram scale protocol for sulfonyl imines is also reported here. The in-depth proof-of-concept study, control experiments and density functional theory calculations assisted in deducing the reaction mechanisms. The competition reactions of electron-rich and electron-poor substrates (1e/f respectively) with 2a indicated that electron-rich substrates favor the reaction more, whereas the same between electron-rich and electron-poor iminoiodinanes (2a/c) with a benzyl alcohol derivative 1y indicated no influence of the electronics on the product formation. Hence, our sustainable mechanochemical protocol described herein contributes significantly to a not so rich repertoire of strategies to generate N-sulfonyl imines. Since they are important building blocks and precursors of sulfonamides our strategy would have wide usage in the pharmaceutical, chemical and agrochemical industries to access such compounds.
Experimental procedures
Resource availability
Representative experimental procedure for the preparation of aryl/heteroaryl sulfonyl imines
:5 to 80:20) Rf = 0.2].
:30 to 50:50) Rf = 0.3] to afford 3s in 66% yield.
Author contributions
S. G. and R. B. synthesized the compounds; M. R. and J. J. did the computational study; S. S. conceptualized the project and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Shiv Nadar Institute of Eminence Deemed to be University.References
- Nitrenes, ed. W. Lwowski, Interscience, New York, 1970 Search PubMed.
- F. Tiemann, Ber. Dtsch. Chem. Ges., 1891, 24, 4162–4167 CrossRef.
- Y. C. Wang, X. J. Lai, K. Huang, S. Yadav, G. Qiu, L. Zhang and H. Zhou, Org. Chem. Front., 2021, 8, 1677–1693 RSC.
- B. Darses, R. Rodrigues, L. Neuville, M. Mazurais and P. Dauban, Chem. Commun., 2017, 53, 493–508 RSC.
- A. Bal, S. Maiti and P. Mal, Chem. – Asian J., 2020, 15, 624–635 CrossRef CAS PubMed.
- I. Tornieporth-Oetting and T. Klapötke, Angew. Chem., Int. Ed. Engl., 1990, 29, 677–679 CrossRef.
- T. Kumar Achar and P. Mal, Adv. Synth. Catal., 2015, 357, 3977–3985 CrossRef CAS.
- J. A. Bull, L. Degennaro and R. Luisi, Synlett, 2017, 2525–2538 CrossRef CAS.
- M. Andresini, A. Tota, L. Degennaro, J. A. Bull and R. Luisi, Chem. – Eur. J., 2021, 27, 17293–17321 CrossRef CAS PubMed.
- M. Zenzola, R. Doran, L. Degennaro, R. Luisi and J. A. Bull, Angew. Chem., 2016, 128, 7319–7323 CrossRef.
- G. B. Craven, E. L. Briggs, C. M. Zammit, A. McDermott, S. Greed, D. P. Affron, C. Leinfellner, H. R. Cudmore, R. R. Tweedy, R. Luisi and J. A. Bull, J. Org. Chem., 2021, 86, 7403–7424 CrossRef CAS PubMed.
- S. Chaabouni, J. F. Lohier, A. L. Barthelemy, T. Glachet, E. Anselmi, G. Dagousset, P. Diter, B. Pegot, E. Magnier and V. Reboul, Chem. – Eur. J., 2018, 24, 17006–17010 CrossRef CAS PubMed.
- For Reviews: (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett, W. Jones and A. Krebs, Chem. Soc. Rev., 2012, 41, 413–447 RSC; (b) X. Liu, Y. Li, L. Zeng, X. Li, N. Chen, S. Bai, H. He, Q. Wang and C. Zhang, Adv. Mater., 2022, 34, 2108327 CrossRef CAS PubMed; (c) R. T. O'Neill and R. Boulatov, Nat. Rev. Chem., 2021, 5, 148–167 CrossRef PubMed; (d) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC; (e) J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (f) X. L. Wu, J. J. Xia and G. W. Wang, Org. Biomol. Chem., 2008, 6, 548–553 RSC; (g) G. W. Wang and X. L. Wu, Adv. Synth. Catal., 2007, 349, 1977–1982 CrossRef CAS; (h) D. Virieux, F. Delogu, A. Porcheddu, F. García and E. Colacino, J. Org. Chem., 2021, 86, 13885–13894 CrossRef CAS PubMed; (i) G. W. Wang, Chin. J. Chem., 2021, 39, 1797–1803 CrossRef CAS; (j) Y. Gao, C. Feng, T. Seo, K. Kubota and H. Ito, Chem. Sci., 2022, 13, 430–438 RSC; (k) A. C. Jones, W. I. Nicholson, J. A. Leitch and D. L. Browne, Org. Lett., 2021, 23, 6337–6341 CrossRef CAS PubMed; (l) L. Li, C. Niu and G. W. Wang, Chin. J. Chem., 2022, 40, 2539–2545 CrossRef CAS; (m) R. K. Fang, Z. C. Yin, J. S. Chen and G. W. Wang, Green Chem. Lett. Rev., 2022, 15, 519–528 CrossRef CAS.
- L. Takacs, Chem. Soc. Rev., 2013, 42, 7649–7659 RSC.
- G. W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- V. Šepelák, A. Düvel, M. Wilkening, K. D. Becker and P. Heitjans, Chem. Soc. Rev., 2013, 42, 7507–7520 RSC.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14, 2350–2362 RSC.
- A. L. Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846–855 RSC.
- P. Baláž, M. Achimovičová, M. Baláž, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor and R. Kumar, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- (a) S. Ni, M. Hribersek, S. K. Baddigam, F. J. Ingner, A. Orthaber, P. J. Gates and L. T. Pilarski, Angew. Chem., Int. Ed., 2021, 60, 6660–6666 CrossRef CAS PubMed; (b) C. Ballatore, J. H. Soper, F. Piscitelli, M. James, L. Huang, O. Atasoylu, D. M. Huryn, J. Q. Trojanowski, V. M. Y. Lee, K. R. Brunden and A. B. Smith III, J. Med. Chem., 2011, 54, 6969–6983 CrossRef CAS PubMed.
- S. R. Malwal, D. Sriram, P. Yogeeswari, V. B. Konkimalla and H. Chakrapani, J. Med. Chem., 2012, 55, 553–557 CrossRef CAS PubMed.
- C. T. Supuran, A. Scozzafava and B. W. Clare, Med. Res. Rev., 2002, 22, 329–372 CrossRef CAS PubMed.
- M. V. Papadopoulou, W. D. Bloomer, H. S. Rosenzweig, E. Chatelain, M. Kaiser, S. R. Wilkinson, C. McKenzie and J. R. Loset, J. Med. Chem., 2012, 55, 5554–5565 CrossRef CAS PubMed.
- J. P. Bégué, D. Bonnet-Delpon, B. Crousse and J. Legros, Chem. Soc. Rev., 2005, 34, 562–572 RSC.
- S. M. Weinreb and R. K. Orr, Synthesis, 2005, 1205–1207 CrossRef CAS.
- C. H. Senananake, D. Krishnamurthy, Z. H. Lu, Z. Han and I. Gallou, Aldrichimica Acta, 2005, 38, 93–104 Search PubMed.
- P. Zhou, B. C. Chen and F. A. Davis, Tetrahedron, 2004, 60, 8003–8030 CrossRef CAS.
- M. Gohain, Synlett, 2003, 2097–2098 CrossRef CAS.
- J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984–995 CrossRef CAS PubMed.
- J. A. Ellman, Pure Appl. Chem., 2003, 75, 39–46 CrossRef CAS.
- F. A. Davis and B. C. Chen, Chem. Soc. Rev., 1998, 27, 13–18 RSC.
- R. Bloch, Chem. Rev., 1998, 98, 1407–1438 CrossRef CAS PubMed.
- D. Enders and U. Reinhold, Tetrahedron: Asymmetry, 1997, 8, 1895–2073 CrossRef CAS.
- T. Ooi, Y. Uematsu and K. Maruoka, J. Am. Chem. Soc., 2006, 128, 2548–2549 CrossRef CAS PubMed.
- H. F. Duan, Y. X. Jia, L. X. Wang and Q. L. Zhou, Org. Lett., 2006, 8, 2567–2569 CrossRef CAS PubMed.
- H. Fujisawa, E. Takahashi and T. Mukaiyama, Chem. – Eur. J., 2006, 12, 5082–5093 CrossRef CAS PubMed.
- M. Shi, L. H. Chen and C. Q. Li, J. Am. Chem. Soc., 2005, 127, 3790–3800 CrossRef CAS PubMed.
- T. Soeta, M. Kuriyama and K. Tomioka, J. Org. Chem., 2005, 70, 297–300 CrossRef CAS PubMed.
- T. Hayashi, M. Kawai and N. Tokunaga, Angew. Chem., 2004, 116, 6251–6254 CrossRef.
- H. K. Yim and H. N. C. Wong, J. Org. Chem., 2004, 69, 2892–2895 CrossRef CAS PubMed.
- P. Wipf, C. Kendall and C. R. J. Stephenson, J. Am. Chem. Soc., 2003, 125, 761–768 CrossRef CAS PubMed.
- V. K. Aggarwal, E. Alonso, M. Ferrara and S. E. Spey, J. Org. Chem., 2002, 67, 2335–2344 CrossRef CAS PubMed.
- K. I. Yamada, H. Fujihara, Y. Yamamoto, Y. Miwa, T. Taga and K. Tomioka, Org. Lett., 2002, 4, 3509–3511 CrossRef CAS PubMed.
- D. K. Wang, Y. G. Zhou, Y. Tang, X. L. Hou and L. X. Dai, J. Org. Chem., 1999, 64, 4233–4237 CrossRef CAS.
- O. G. Mancheno, R. G. Arrayas and J. C. Carretero, J. Am. Chem. Soc., 2004, 126, 456–457 CrossRef CAS PubMed.
- P. E. Morgan, R. McCague and A. Whiting, J. Chem. Soc., Perkin Trans. 1, 2000, 515–526 RSC.
- S. Yao, M. Johannsen, R. G. Hazell and K. A. Jørgensen, Angew. Chem., Int. Ed., 1998, 37, 3121–3124 CrossRef CAS PubMed.
- T. Bauer, S. Szymanski, A. Jezewski, P. Gluzinski and J. Jurczak, Tetrahedron: Asymmetry, 1997, 8, 2619–2625 CrossRef CAS.
- J. Sisko and S. M. A. Weinreb, Tetrahedron Lett., 1989, 30, 3037–3040 CrossRef CAS.
- D. L. Boger, W. L. Corbett, T. T. Curran and A. M. Kasper, J. Am. Chem. Soc., 1991, 113, 1713–1729 CrossRef CAS.
- J. H. Wynne, S. E. Price, J. R. Rorer and W. M. Stalick, Synth. Commun., 2003, 33, 341–352 CrossRef CAS.
- A. Khalafi-Nezhad, A. Parhami, A. Zare, A. N. Shirazi, A. R. M. Zare and A. Hassaninejad, Can. J. Chem., 2008, 86, 456–461 CrossRef CAS.
- X. F. Wu, C. Vovard-Le Bray, L. Bechki and C. Darcel, Tetrahedron, 2009, 65, 7380–7384 CrossRef CAS.
- J. W. W. Chang, T. M. U. Ton, S. Tania, P. C. Taylor and P. W. H. Chan, Chem. Commun., 2010, 46, 922–924 RSC.
- R. Chawla, A. K. Singh and L. D. S. Yadav, Tetrahedron Lett., 2014, 55, 3553–3556 CrossRef CAS.
- S. Morales, F. G. Guijarro, J. L. Garcia Ruano and M. B. Cid, J. Am. Chem. Soc., 2014, 136, 1082–1089 CrossRef CAS PubMed.
- J. T. Reeves, M. D. Visco, M. A. Marsini, N. Grinberg, C. A. Busacca, A. E. Mattson and C. H. Senanayake, Org. Lett., 2015, 17, 2442–2445 CrossRef CAS PubMed.
- D. Huang, X. Wang, X. Wang, W. Chen, X. Wang and Y. Hu, Org. Lett., 2016, 18, 604–607 CrossRef CAS PubMed.
- B. M. Trost and C. Marrs, J. Org. Chem., 1991, 56, 6468–6470 CrossRef CAS.
- J. Sisko and S. M. Weinreb, J. Org. Chem., 1990, 55, 393–395 CrossRef CAS.
- R. Patel, V. P. Srivastava and L. D. S. Yadav, Adv. Synth. Catal., 2010, 352, 1610–1614 CrossRef CAS.
- G. Zhang, S. Xu, X. Xie, C. Ding and S. Shan, RSC Adv., 2017, 7, 9431–9435 RSC.
- A. C. Brueckner, E. N. Hancock, E. J. Anders, M. M. Tierney, H. R. Morgan, K. A. Scott and A. A. Lamar, Org. Biomol. Chem., 2016, 14, 4387–4392 RSC.
- M. D. Hopkins, K. A. Scott, B. C. DeMier, H. R. Morgan, J. A. Macgruder and A. A. Lamar, Org. Biomol. Chem., 2017, 15, 9209–9216 RSC.
- Y. Zhao and D. G. Truhlar, Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc, Wallingford, CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00791j |
| This journal is © The Royal Society of Chemistry 2023 |