Electrochemical direct C–H mono and bis-chalcogenation of indolizine frameworks under oxidant-free conditions†
Received
11th July 2023
, Accepted 1st September 2023
First published on 4th September 2023
Abstract
Herein, we disclosed a sustainable electrochemical approach for site-selective C–H mono and bis-chalcogenation (sulfenylation or selenylation) of indolizine frameworks. Diversely functionalized disulfides and diselenides possessing EDGs and EWGs were successfully reacted with a variety of indolizines to directly access sulfenylated/selenylated indolizines in 40–96% yields. A mechanistic radical pathway was also validated with control experiments and cyclic voltammogram data.
Introduction
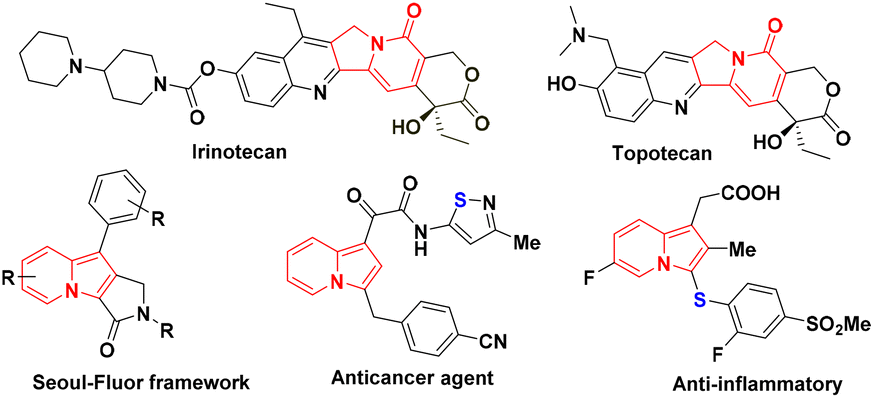
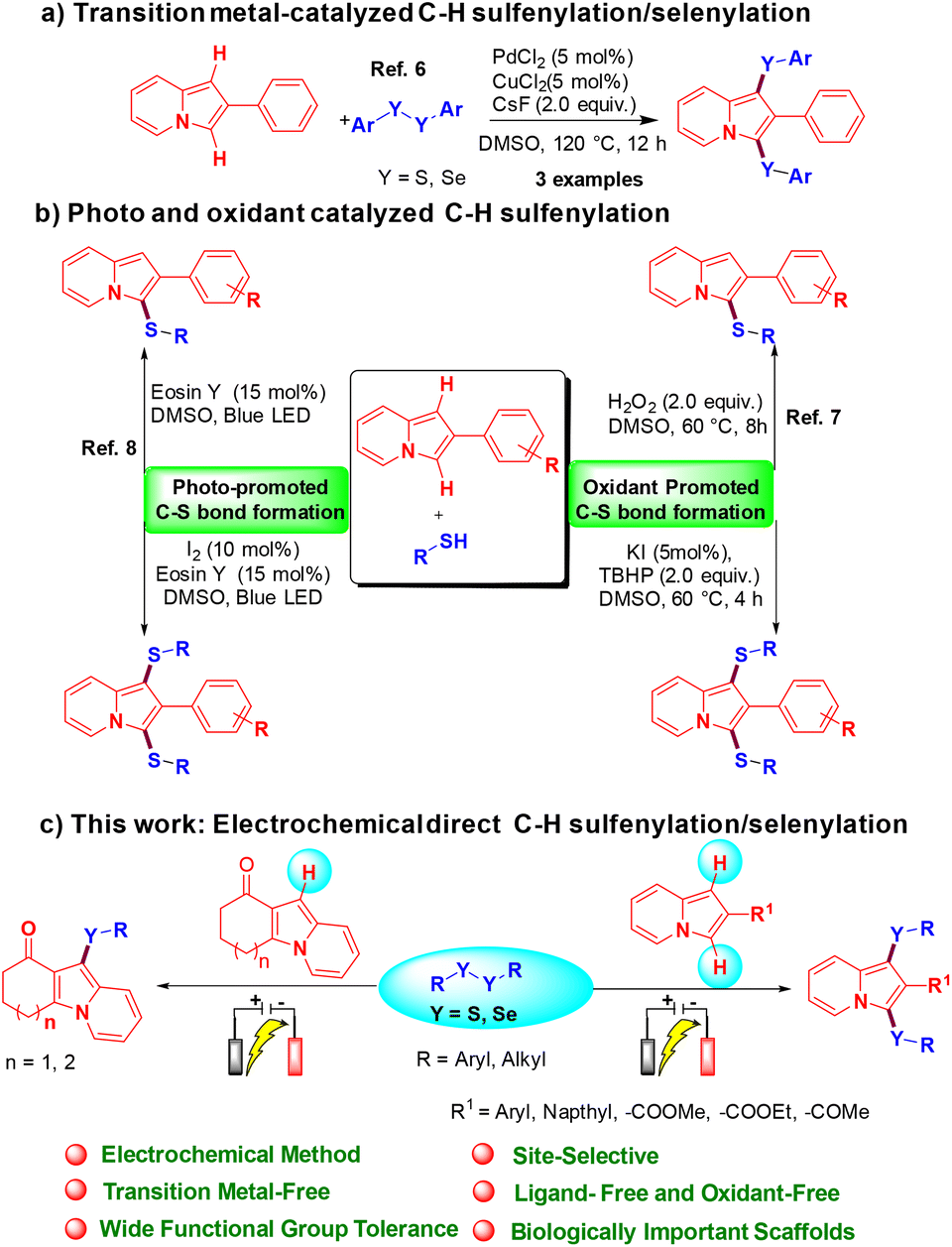
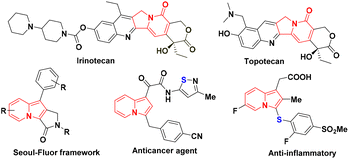
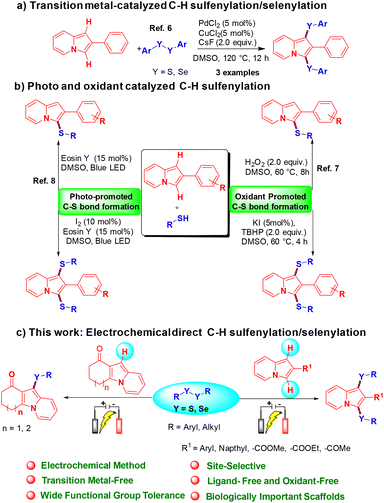
Over the past few decades, indolizine frameworks have attracted considerable attention from the scientific community because of their photosensitive properties and biological significance.1 A variety of functionalized indolizine core moieties have been explored for their pharmaceutical applications such as antitubercular, antioxidant, antimicrobial, anticancer, and anti-inflammatory activities.2 As shown in Fig. 1, the Seoul-Fluor framework is in the limelight with its intrinsic fluorescence properties,1a commercial drugs Irinotecan and Topotecan are being used for the treatment of ovarian, colon, and lung cancers, etc.3 Meanwhile, sulfur-decorated molecules have been used widely as important intermediates and catalysts for the synthesis of complex molecules.4a–h Also, selenium-possessing organic frameworks play a significant role in chemical biology, asymmetric catalysis, cross-coupling reactions, organic synthesis, materials science, and natural products.4i,j Interestingly, indolizines with sulfur frameworks have shown promising biological activities such as efficient antagonism of CRTH2 and anti-inflammatory properties.5 The use of indolizines in the photosensitive material and drug development program always encourages the synthetic community for their efficient synthesis in an atom-economical and sustainable manner. Transition metal-catalyzed,6 photocatalyzed7 and oxidant-promoted8 protocols have been reported for the direct sulfenylation/selenylation of indolizine derivatives (Scheme 1). Despite their utility, these procedures suffer from some drawbacks such as limited substrate scope, requirement of transition metal catalysts, oxidants, high temperature, photocatalysts, blue LED light, etc. In addition, the unpleasant odour of thiols limits the use of these methodologies in the large-scale production of particular compounds in industries. Moreover, oxidant and photocatalysis are limited to only C–H sulfenylations.7,8 Therefore, the development of a simple, odorless, efficient and sustainable synthetic route for the direct C–H chalcogenation of indolizine scaffolds is highly desirable.
 |
| | Fig. 1 Indolizine-based photosensitive and bioactive molecules. | |
 |
| | Scheme 1 C–H sulfenylation/selenylation of indolizine derivatives. | |
In recent years, synthetic chemists who regularly pursue green chemistry have promoted the use of the electro-organic technique for the synthesis of value-added molecules.9 Electrochemical synthesis is a powerful and greener approach for direct chalcogenation of various chemical entities by obviating the use of stoichiometric amounts of oxidants, catalysts, and ligands.10 In sharp contrast, we expand our research program on sustainable electrochemical synthesis of value-added molecules,11 and we herein for the first time report a transition metal-free, regioselective direct C–H mono and bis-chalcogenation protocol for indolizine scaffolds using disulfides or diselenides as reaction partners via an electro-organic synthetic strategy under oxidant-free conditions.
Results and discussion
We initiated our electrochemical investigation with model substrates indolizine 1a and diphenyl disulfide (2a) in an undivided cell equipped with a graphite anode and a graphite cathode of an IKA ElectraSyn 2.0 instrument. Gratifyingly, the reaction of indolizine 1a and diphenyl disulfide (2a) under a 10 mA electric constant current with the electrolyte LiClO4 and the additive KI (50 mol%), in acetonitrile solvent afforded the desired product 3aa with a 70% yield in only 6 h (Table 1, entry 1). Next, various reaction parameters were screened and the results are depicted in Table 1. During control experiments, the desired product 3aa was not observed which evidences that electricity and an iodo source are essential for this interesting C–H sulfenylation protocol (entries 2 and 3). Furthermore, we checked the compatibility of solvents such as DMSO, EtOH, DMF, DCE, and EtOAc which afforded 3aa in the range of 48–65% yields and suggested that acetonitrile is the best choice of solvent for this protocol (entries 4–8). To enhance the yield of 3aa, various iodo sources such as NaI, TBAI, and NH4I were also examined, providing 3aa in 45–64% yields (entries 9–11). Various electrode combinations such as C/Ni, C/RVC, C/Pt, and RVC/Ni were also tried, and among them, C/Ni and C/Pt were found to be superior and provided 3aa in 80% and 81% yields, respectively (entries 12–15). Various electrolytes such as LiBr, KPF6, nBu4NPF6, and CH3COONa were also screened; among them, nBu4NPF6 was found to be superior to others providing 3aa in 85% yield (entries 16–19). Increasing the current intensity to 20 mA provided 3aa in 75% yield along with unidentified side-products, whereas on decreasing it to 5 mA, 3aa was obtained in 66% yield along with unreactive starting precursors (entry 20). It was also observed that a reduced equimolar concentration of 2a (0.75 equiv.) gave only a 73% yield of 3aa whereas an increased equimolar concentration (1.5 equiv.) of 2a provided 3aa in 84% yield (entry 21). On the basis of these experiments, the reaction conditions of entry 18 were considered as the optimized reaction conditions for further study of this interesting electrochemical C–H sulfenylation protocol.
Table 1 Optimization of the reaction conditionsa
|
|
| Entry |
Electrolyte |
Electrode (+)/(−) |
Additive |
Solvent |
Yield of 3aab (%) |
|
Reaction conditions: indolizine 1a (0.5 mmol), diphenyl disulfide (2a, 0.5 mmol), additive (50 mol%), and electrolyte (50 mol%) in a solvent was electrolyzed with a constant current of 10 mA at RT in the ElectraSyn 2.0 instrument for 6 h.
Isolated yields of 3aa are based on 1a.
20 mA current was used instead of 10 mA.
5 mA current was used instead of 10 mA.
0.38 mmol of 2a was used instead of 0.5 mmol.
0.75 mmol of 2a was used instead of 0.5 mmol.
|
| 1 |
LiClO4 |
C/C |
KI |
CH3CN |
70 |
| 2 |
LiClO4 |
— |
KI |
CH3CN |
0 |
| 3 |
LiClO4 |
C/C |
— |
CH3CN |
0 |
| 4 |
LiClO4 |
C/C |
KI |
DMF |
48 |
| 5 |
LiClO4 |
C/C |
KI |
DMSO |
56 |
| 6 |
LiClO4 |
C/C |
KI |
EtOH |
52 |
| 7 |
LiClO4 |
C/C |
KI |
DCE |
65 |
| 8 |
LiClO4 |
C/C |
KI |
EtOAc |
62 |
| 9 |
LiClO4 |
C/C |
NaI |
CH3CN |
64 |
| 10 |
LiClO4 |
C/C |
TBAI |
CH3CN |
45 |
| 11 |
LiClO4 |
C/C |
NH4I |
CH3CN |
48 |
| 12 |
LiClO4 |
C/Ni |
KI |
CH3CN |
80 |
| 13 |
LiClO4 |
C/RVC |
KI |
CH3CN |
67 |
| 14 |
LiClO4 |
C/Pt |
KI |
CH3CN |
81 |
| 15 |
LiClO4 |
RVC/Ni |
KI |
CH3CN |
78 |
| 16 |
LiBr |
C/Ni |
KI |
CH3CN |
70 |
| 17 |
KPF6 |
C/Ni |
KI |
CH3CN |
70 |
| 18 |
n
Bu4NPF6 |
C/Ni |
KI |
CH3CN |
85 |
| 19 |
CH3COONa |
C/Ni |
KI |
CH3CN |
70 |
| 20 |
n
Bu4NPF6 |
C/Ni |
KI |
CH3CN |
75c, 66d |
| 21 |
n
Bu4NPF6 |
C/Ni |
KI |
CH3CN |
73e, 84f |
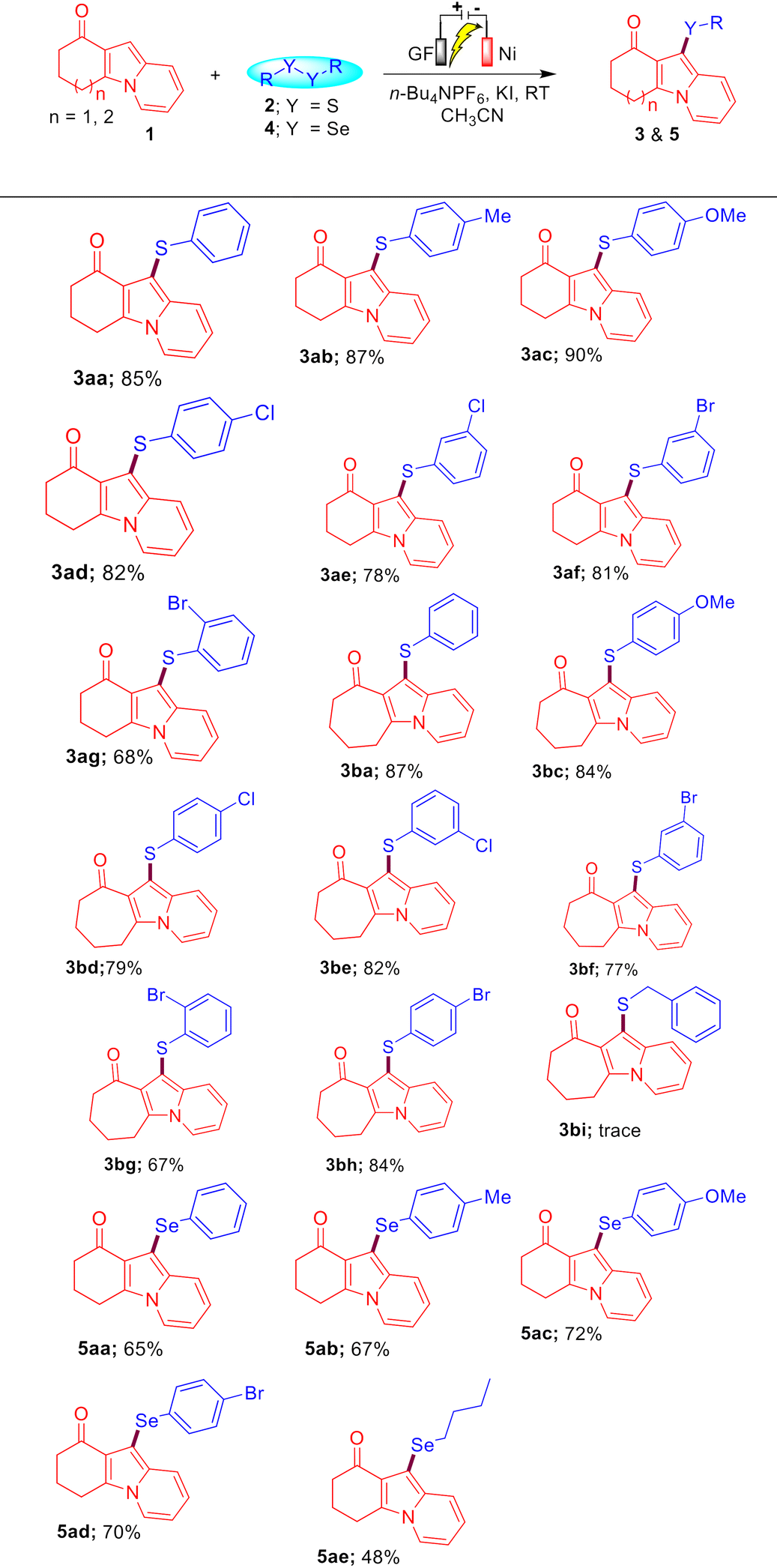
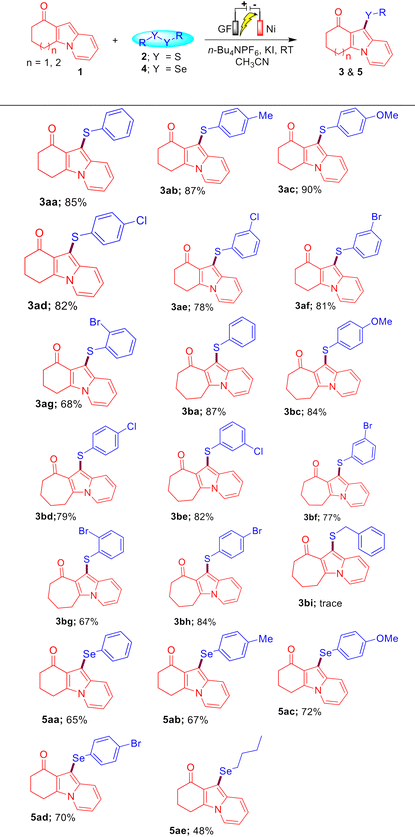
We next investigated the generality of this protocol with the optimized reaction conditions, under which a wide array of sulfenylated/selenylated indolizines 3 and 5 were synthesized as shown in Table 2. A variety of indolizines 1a and 1b successfully reacted with diversely functionalized disulfides 2a–2h and diselenides 4a–4e under the optimized reaction conditions to provide the corresponding sulfenylated and selenylated indolizines 3aa–3bh and 5aa–5ae in 67–90% and 48–72% yields, respectively. Indolizine 1a reacted well with disulfides 2b and 2c having EDGs (4-Me and 4-OMe) to provide the corresponding sulfenylated indolizines 3ab and 3ac in 87% and 90% yields, respectively. Diverse disulfides having o/m/p-halo-substitutions (–Br and –Cl) 2d–2g were also tolerated in this protocol and afforded the sulfenylated indolizines 3ad–3ag in 68–82% yields, which can be further functionalized under transition metal-catalysis to access value-added compounds.
Table 2 Substrate scope for electrochemical C–H mono-chalcogenation of indolizines 1a,b
|
Reaction conditions: indolizines 1 (0.5 mmol), disulfides 2 (0.5 mmol)/diselenides 4 (0.5 mmol), KI (50 mol%) and n-Bu4NPF6 (50 mol%) in acetonitrile were electrolyzed with a continuous current of 10 mA at RT in the ElectraSyn 2.0 instrument for 6 h.
Isolated yields of 3 are based on 1.
|
|
The sterically hindered ortho-bromo-substituted disulfide 2g was found to be less reactive compared to the meta/para bromo-substitution to access the sulfenylated indolizine 3ag in 68% yield. The cycloheptyl-fused indolizine 1b also reacted well with disulfides 2a–2h possessing EDGs (–OMe) as well as EWGs (–Cl and –Br) to provide the sulfenylated indolizines 3ba–3bh in 67–87% yields. It is worth mentioning here that aliphatic disulfides were not suitable for this electrochemical C–H sulfenylation of indolizines. When we treated indolizine 1b with disulfide 2i under the optimized reaction conditions, only a trace amount of the corresponding product 3bi was obtained. To further expand the synthetic utility of the developed protocol, a diverse variety of diselenides 4a–4e were also examined with indolizine 1a under the standard conditions. It was found that aromatic as well as aliphatic diselenides reacted well with indolizines 1a to access the corresponding selenylated-indolizines 5aa–5ae in 48–72% yields. The low yield of product 5ae may be attributed to the less reactivity of alkyl diselenides.
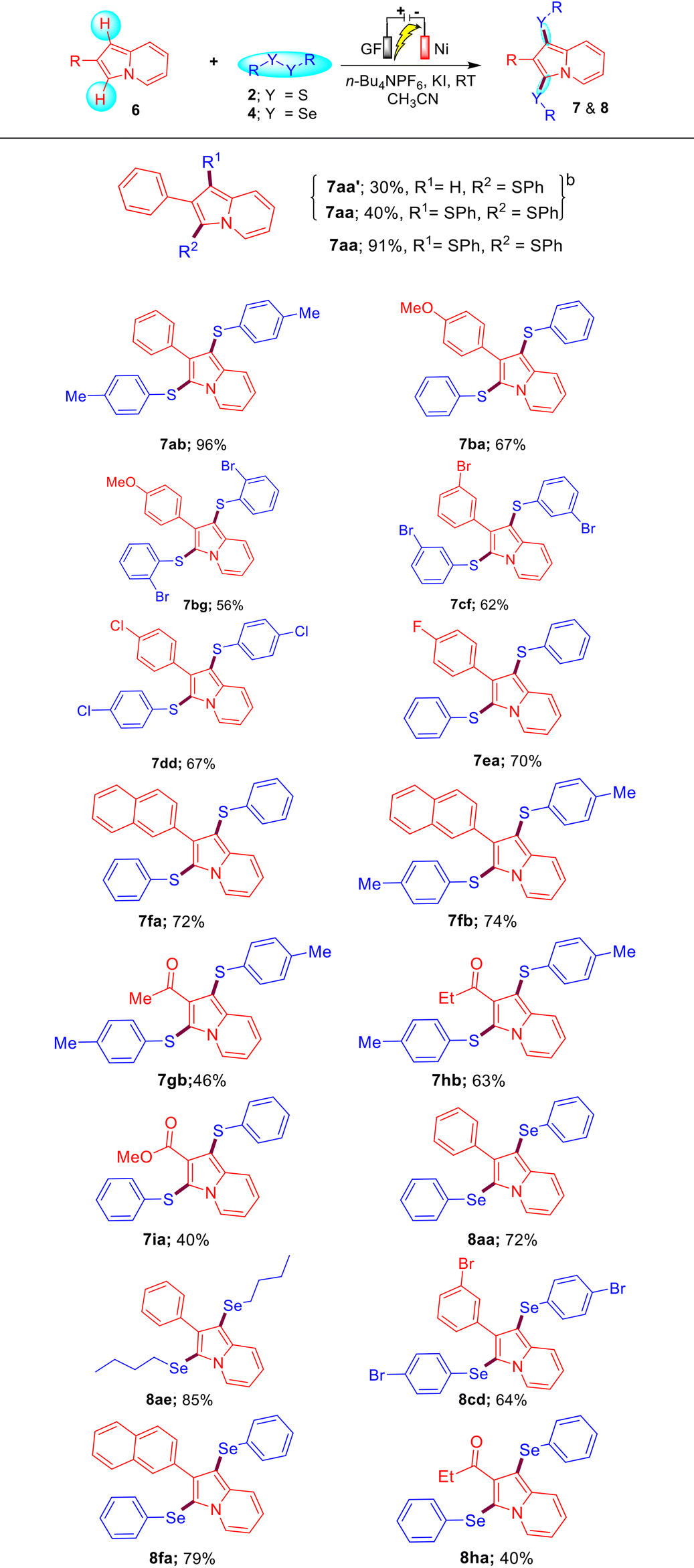
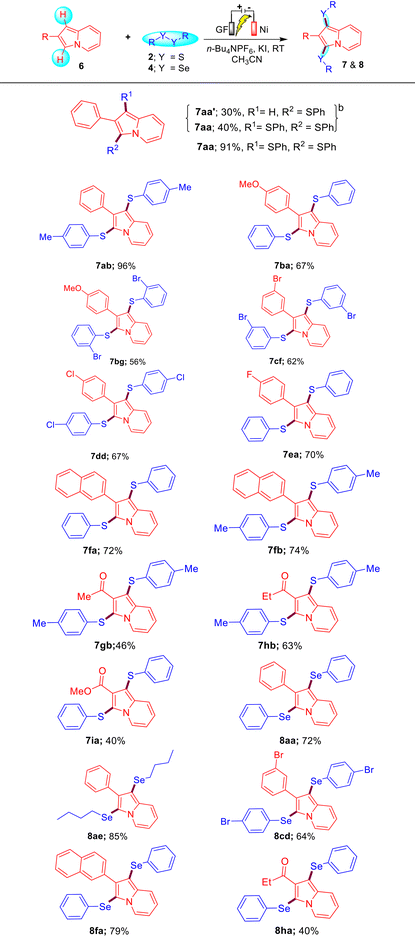
To further extend the generality of this protocol, we carried out a reaction between substrates 6a and 2a under the optimized reaction conditions, which resulted in the formation of a mixture of compounds 7aa′ and 7aa in 30% and 40% yields, respectively. Gratifyingly, on increasing the amount of 2a (0.75 mmol), KI (100 mol%), and time (12 h), we obtained compound 7aa as the sole product in 91% yield. Encouraged by this result, we then synthesized a diverse variety of bis-sulfenylated indolizines 7aa–7ia and bis-selenylated indolizines 8aa–8ha in 40–96% isolated yields under the optimized reaction conditions as shown in Table 3. It was found that the reaction of indolizine 6a and substituted indolizines 6b–6e (4-OMe, 4-F, 4-Cl, and 3-Br) with a variety of disulfides (4-Me, 4-Cl, 2-Br, and 3-Br) proceeded well under the optimized reaction conditions and gave the desired sulfenylated-indolizine molecules 7aa–7ea in 56–96% yields. Naphthyl-substituted indolizine 6f also reacted smoothly with disulfides 2a and 2b to afford 7fa and 7fb in 72% and 74% yields, respectively. Interestingly, indolizines possessing ketonic groups (–COMe and –COEt) and an ester group (–COOMe) showed less reactivity towards the reaction with disulfides 2a and 2b and afforded the corresponding 7gb–7ia in 40–63% yields. Remarkably, aliphatic as well as aromatic diselenides also reacted well with diverse indolizines 8a, 8c, and 8f and afforded bis-selenylated indolizines 8aa–8ha in 40–85% yields. The low yield of product 8ha may be attributed to the COEt (EWG) group of indolizine.
Table 3 Substrate scope for electrochemical C–H bis-chalcogenation of indolizines 6a,c
|
Reaction conditions: indolizines 6 (0.5 mmol), disulfides 2 (0.75 mmol)/diselenides 4 (0.75 mmol), KI (100 mol%) and n-Bu4NPF6 (50 mol%) in acetonitrile were electrolyzed with a constant current of 10 mA at RT in the ElectraSyn 2.0 instrument for 12 h.
Indolizines 6 (0.5 mmol), disulfides 2 (0.5 mmol), KI (50 mol%) and n-Bu4NPF6 (50 mol%) in acetonitrile were electrolyzed with a continuous current of 10 mA at RT for 12 h.
Isolated yields of 7 and 8 are based on 6.
|
|
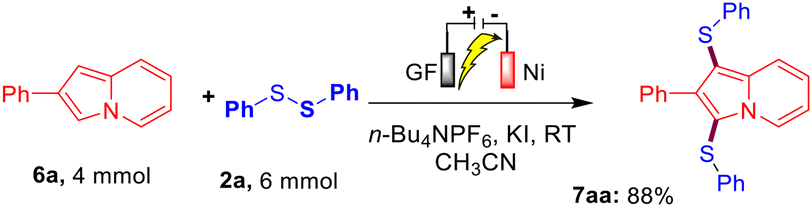
Furthermore, to establish the synthetic utility of this interesting electrochemical direct C–H bis-sulfenylation protocol, a large-scale experiment was performed. When we reacted 4.0 mmol of indolizine 6a with 2a ( 6.0 mmol) under the optimized reaction conditions, the corresponding bis-sulfenylated indolizine 7aa was obtained in 88% yield as shown in Scheme 2.
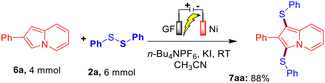 |
| | Scheme 2 Large-scale experiment between 6a and 2a. | |
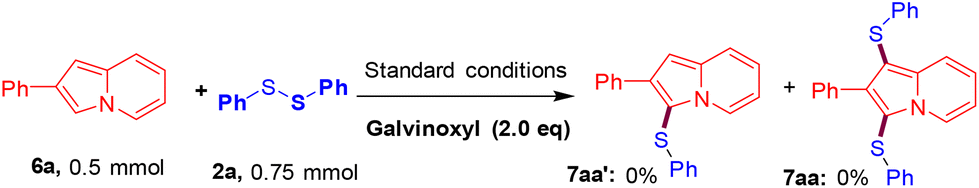
To obtain the intrinsic reaction pathway of this electrochemical C–S/C–Se bond-forming protocol, a single electron transfer (SET) pathway is an obvious consideration. The roles of electricity and KI were already established in optimization studies (Table 1, entries 2 and 3). Therefore, a radical trapping experiment was carried out by employing a galvinoxyl free radical. The reaction between 6a and 2a in the presence of the galvinoxyl free radical (2.0 equiv.) under standard reaction conditions completely aborted the formation of the corresponding mono-sulfenylated product 7aa′ as well as bis-sulfenylated product 7aa (Scheme 3). The results of this radical trapping experiment suggested a radical pathway.
 |
| | Scheme 3 Control experiment. | |
Furthermore, cyclic voltammetry (CV) experiments were also performed to establish the plausible mechanistic pathway for this interesting C–S/C–Se cross-coupling methodology (Fig. S1–S6 in the ESI†). The CV of n-Bu4NPF6 (0.1 M) showed no oxidation peak (graph A). The CV of n-Bu4NPF6 (0.1 M) and KI (10 mM) showed an oxidation peak at +1.28 V which indicates that the iodide ion gets oxidized into an iodine radical or iodine (graph A). The CV of 2a (5 mM), KI (10 mM), and n-Bu4NPF6 (0.1 M) showed an apparent oxidation peak at +2.27 V (graph B) and a reduction peak at −1.33 V (graph C). The CV of 1a (5 mM) and n-Bu4NPF6 (0.1 M) showed two apparent oxidation peaks at +1.48 V and +1.92 V (graph D). The CV of 6a (5 mM) and n-Bu4NPF6 (0.1 M) showed two apparent oxidation peaks at +1.17 V and +1.72 V (graph E). The CV of the mixture of 1a, 2a, KI (10 mM) and n-Bu4NPF6 (0.1 M) showed apparent oxidation peaks at +2.05 and +2.49 V (graph F), due to the possible chemical interaction between compounds 1a and 2a. The CV of the mixture of 6a, 2a, KI (10 mM) and n-Bu4NPF6 (0.1 M) showed apparent oxidation peaks at +1.42 V and +2.33 V (graph G), due to the possible chemical interaction between compounds 6a and 2a.
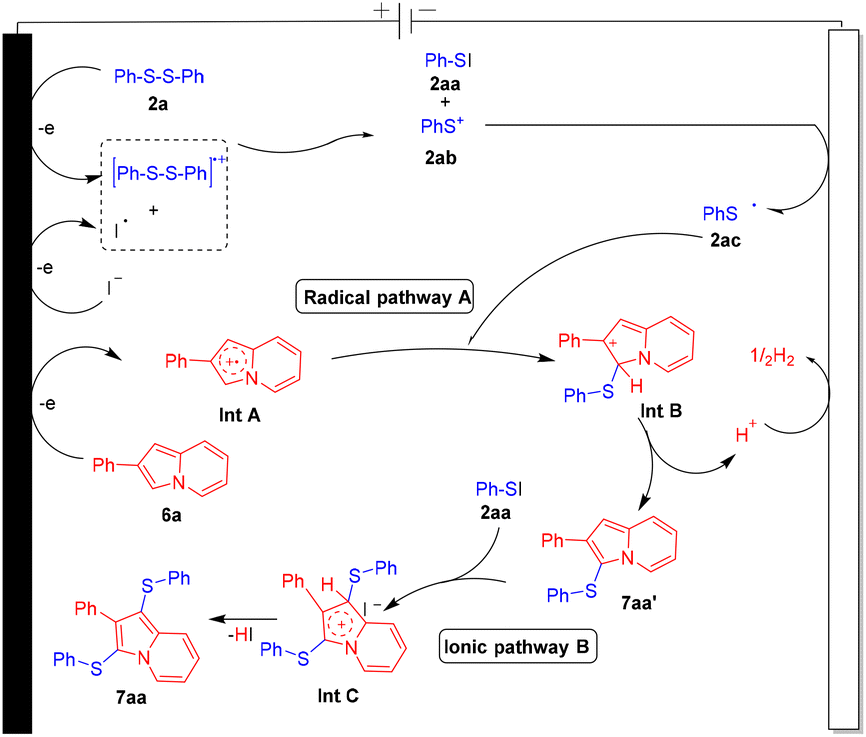
Based on previous literature reports,7,8,12a,b cyclic voltammogram data and the above-mentioned control experiment, a plausible reaction pathway is depicted for this protocol by using 6a and 2a as reacting substrates (Fig. 2). 2a initially oxidized at an anodic position to form the disulfide radical cationic species which on interaction with an iodine radical forms PhSI (2aa) and phenyl sulfide cation species 2ab. Phenyl sulfide cation species 2ab upon cathodic reduction transforms into phenyl sulfide radical species 2ac. Upon anodic oxidation, substrate 6a converts into radical cationic species Int A which on reaction with thiyl radical 2ac converts into the cationic species Int B which further on elimination of a H+ ion transforms into mono-sulfenylated product 7aa′. Furthermore, in the ionic pathway, PhSI interacts with 7aa′ and forms the cationic intermediate species Int C which further on elimination of HI converts into product 7aa. H2 was liberated at the cathodic position. A similar mechanistic hypothesis is also applicable for the oxidative selenylation of indolizine molecules.
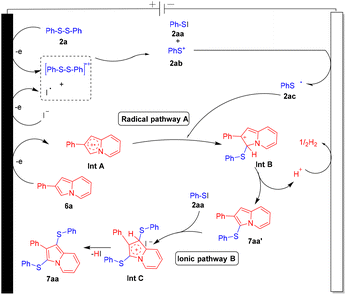 |
| | Fig. 2 Plausible reaction mechanism. | |
Experimental
General information
All the chemicals and reagents were purchased from commercial suppliers and used as received. Column chromatography was performed over silica-gel (particle size: 100–200 mesh) using hexanes and ethyl acetate as eluents. The aluminium-supported silica plate Si 60 F254 was used for thin layer chromatography. 1H NMR, 13C NMR, and HRMS techniques were used for the analysis of the synthesized compounds. 1H NMR and 13C NMR spectra were recorded on a JEOL ECS-400 instrument in CDCl3 solvent. Chemical shifts are reported in ppm with reference to TMS at 0.00 ppm and coupling constants (J) are given in hertz. 1H NMR peak signals were reported as s (singlet), br s (broad singlet), d (doublet), t (triplet), quint (quintet), sext (sextet), dd (double doublet), dt (doublet of triplet), td (triplet of doublet), ddd (doublet of double doublet), and m (multiplet). In the 13C NMR spectra, chemical shifts are reported in ppm with reference to the center line of a triplet of chloroform-d at 77.10 ppm. High-resolution mass spectra (HRMS) were recorded on an Agilent technologies Q-TOF B.06.01 mass spectrometer. All electrocatalytic reactions were carried out on an IKA ElectraSyn 2.0 instrument. CV experiments were performed on an IKA ElectraSyn 2.0 pro package using commercially available (IKA) glassy carbon (3.00 mm diameter) as the working electrode, a platinum electrode (platinum plated on copper; 8 × 52.5 × 2 mm) as the counter electrode and Ag wire as the reference electrode. The starting precursors 1a, 1b, 6g–6i and 6a–6f were synthesized according to the reported literature.8,13
General procedure for Table 2
An undivided ElectraSyn 2.0 cell equipped with a graphite anode and a nickel cathode was charged with indolizines 1 (0.5 mmol), disulfides 2 (0.5 mmol)/diselenides 4 (0.5 mmol), KI (50 mol%), and n-Bu4NPF6 (50 mol%) in acetonitrile (4 mL) solvent. The reaction mixtures were stirred and electrolyzed at a constant current of 10 mA at room temperature for 6 h via the manual programming of the IKA ElectraSyn 2.0 instrument. After the completion of the reactions, the acetonitrile solvents were evaporated under reduced pressure to obtain crude products, which were further purified through column chromatography using EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexanes as an eluent.
:hexanes as an eluent.
10-(Phenylthio)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (3aa).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and diphenyl disulfide 2a (0.5 mmol, 0.109 g), and after column chromatography (18–20% EtOAc/hexanes), 3aa was obtained as a dark brown solid; yield: 0.125 g, 85%; M.P.: 166 °C; 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 7.2 Hz, 1H), 7.55 (d, J = 8.8 Hz, 1H), 7.08–7.00 (m, 4H), 6.95 (td, J = 6.8 and 1.6 Hz, 1H), 6.75 (t, J = 6.4 Hz, 1H), 6.62 (t, J = 6.8 Hz, 1H), 2.93 (t, J = 6.4 Hz, 2H), 2.54 (J = 5.6 Hz, 2H), 2.22 (quint, J = 6.4 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 194.8, 139.4, 136.5, 133.5, 128.5, 126.5, 124.9, 123.1, 122.7, 120.3, 119.3, 113.3, 95.9, 39.3, 23.1, 21.3; HRMS (ESI) exact mass calcd for C18H15NOS + H (M + H), 294.0947; found: 294.0953.
10-(p-Tolylthio)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (3ab).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(4-methylphenyl) disulfide 2b (0.5 mmol, 0.123 g), and after column chromatography (15–20% EtOAc/hexanes), 3ab was obtained as a green solid; yield: 0.134 g, 87%; M.P.: 117 °C; 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 7.2 Hz, 1H), 7.66 (d, J = 8.8 Hz, 1H), 7.05 (d, J = 8.4 Hz, 2H), 6.96 (d, J = 8.4 Hz, 2H), 6.82 (t, J = 6.8 Hz, 1H), 6.69 (t, J = 6.8 Hz, 1H), 3.00 (t, J = 6.4 Hz, 2H), 2.64–2.60 (m, 2H), 2.30 (t, J = 6.4 Hz, 2H), 2.23 (s, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 194.8, 136.4, 135.7, 134.8, 133.3, 129.4, 127.3, 123.1, 122.6, 120.1, 119.5, 113.3, 96.9, 39.4, 23.2, 21.4, 20.9; HRMS (ESI) exact mass calcd for C19H17NOS + H (M + H), 308.1104; found: 308.1112.
10-((4-Methoxyphenyl)thio)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (3ac).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(4-methoxyphenyl) disulfide 2c (0.5 mmol, 0.139 g), and after column chromatography (18–20% EtOAc/hexanes), 3ac was obtained as a dark green solid; yield: 0.146 g, 90%; M.P.: 125 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 6.8 Hz, 2H), 7.22 (d, J = 8.8 Hz, 2H), 6.84–6.65 (m, 4H), 3.71 (s, 3H), 2.97 (t, J = 6.4 Hz, 2H), 2.61 (t, J = 6.4 Hz, 2H), 2.28 (quint, J = 6.4 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 194.8, 158.0, 136.1, 133.2, 130.1, 129.8, 122.8, 122.6, 120.1, 119.4, 114.3, 113.2, 98.3, 55.2, 39.4, 23.1, 21.3; HRMS (ESI) exact mass calcd for C19H17NO2S + Na (M + H), 346.0872; found: 346.0884.
10-((4-Chlorophenyl)thio)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (3ad).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(4-chlorophenyl) disulfide 2d (0.5 mmol, 0.144 g), and after column chromatography (18–20% EtOAc/hexanes), 3ad was obtained as a green solid; yield: 0.135 g, 82%; M.P.: 134 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 7.2 Hz, 1H), 7.55 (d, J = 9.2 Hz, 1H), 7.04–6.94 (m, 4H), 6.81–6.77 (m, 1H), 6.65 (td, J = 6.8 and 1.2 Hz, 1H), 2.95 (t, J = 6.4 Hz, 2H), 2.54 (t, J = 6.4 Hz, 2H), 2.26 (quint, J = 6.4 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 194.8, 138.1, 136.6, 133.7, 128.6, 127.8, 123.0, 122.8, 120.6, 119.2, 113.4, 95.4, 39.3, 23.1, 21.3; HRMS (ESI) exact mass calcd for C18H14ClNOS + H (M + H), 328.0558; found 328.0547.
10-((3-Chlorophenyl)thio)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (3ae).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(3-chlorophenyl) disulfide 2e (0.5 mmol, 0.144 g), and after column chromatography (18–20% EtOAc/hexanes), 3ae was obtained as a dark green solid; yield: 0.128 g, 78%; M.P.: 141 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (dt, J = 7.2 and 1.2 Hz, 1H), 7.51 (dt, J = 9.2 and 1.2 Hz, 1H), 6.99–6.95 (m, 1H), 6.91–6.88 (m, 3H), 6.77 (ddd, J = 9.2, 6.8 and 1.2 Hz, 1H), 6.63 (td, J = 6.8 and 1.2 Hz, 1H), 2.93 (t, J = 6.4 Hz, 2H), 2.54–2.51 (m, 2H) 2.22 (quint, J = 6.4 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 194.7, 141.9, 136.7, 134.4, 133.8, 129.5, 125.6, 124.8, 124.3, 122.9, 122.8, 120.8, 118.9, 113.4, 94.2, 39.2, 23.1, 21.2; HRMS (ESI) exact mass calcd for C18H14ClNOS + H (M + H), 328.0558; found: 328.0567.
10-((3-Bromophenyl)thio)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (3af).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(3-bromophenyl) disulfide 2f (0.5 mmol, 0.188 g), and after column chromatography (12–15% EtOAc/hexanes), 3af was obtained as a green solid; yield: 0.151 g, 81%; M.P.: 140 °C; 1H NMR (400 MHz, CDCl3) δ 7.69 (dt, J = 7.2 and 1.2 Hz, 1H), 7.53 (dt, J = 9.2 and 1.2 Hz, 1H), 7.08–7.06 (m, 2H), 6.97–6.90 (m, 2H), 6.80 (ddd, 6.8 and 1.2 Hz, 1H), 6.66 (td, J = 6.8 and 1.2 Hz, 1H), 2.95 (t, J = 6.4 Hz, 2H), 2.55 (t, J = 6.4 Hz, 2H) 2.25 (quint, J = 6.4 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 194.7, 142.2, 136.7, 133.8, 129.9, 128.6, 127.8, 124.8, 123.0, 122.8, 122.7, 120.8, 119.1, 113.5, 94.4, 39.3, 23.1, 21.3; HRMS (ESI) exact mass calcd for C18H14BrNOS + H (M + H), 372.0052; found: 372.0065.
10-((2-Bromophenyl)thio)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (3ag).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(2-bromophenyl) disulfide 2g (0.5 mmol, 0.188 g), and after column chromatography (20–22% EtOAc/hexanes), 3ag was obtained as a green solid; yield: 0.127 g, 68%; M.P.: 172 °C; 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 6.8 Hz, 1H), 7.54 (d, J = 9.2 Hz, 1H), 7.38 (dd, J = 8.0 and 1.6 Hz, 1H), 6.89 (td, J = 7.6 and 1.2 Hz, 1H), 6.81–6.75 (m, 2H), 6.65 (td, J = 6.8 and 1.6 Hz, 1H), 6.47 (dd, J = 7.6 and 1.6 Hz, 1H), 2.96 (t, J = 6.4 Hz, 2H), 2.55–2.52 (m, 2H), 2.24 (quint, J = 6.4 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 194.5, 140.8, 136.8, 133.8, 132.5, 127.3, 126.5, 125.6, 123.4, 122.8, 120.7, 120.1, 119.3, 113.5, 94.5, 39.3, 23.2, 21.4; HRMS (ESI) exact mass calcd for C18H14BrNOS + H (M + H), 372.0052; found: 372.0058.
11-(Phenylthio)-6,7,8,9-tetrahydro-10H-cyclohepta[b]indolizin-10-one (3ba).
The title compound was prepared by following the general procedure for Table 2 using 6,7,8,9-tetrahydro-10H-cyclohepta[b]indolizin-10-one 1b (0.5 mmol, 0.100 g) and diphenyl disulfide 2a (0.5 mmol, 0.109 g), and after column chromatography (12–15% EtOAc/hexanes), 3ba was obtained as a dark green solid; yield: 0.134 g, 87%; M.P.: 113 °C; 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 7.2 Hz, 1H), 7.50 (d, J = 9.2 Hz, 1H), 7.04–7.01 (m, 2H), 6.96–6.88 (m, 3H), 6.71–6.67 (m, 1H), 6.57 (td, J = 6.8 and 1.6 Hz, 1H), 2.97 (t, J = 6.4 Hz, 2H), 2.69 (t, J = 6.4 Hz, 2H), 1.97–1.92 (m, 2H), 1.87–1.81 (m, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 198.7, 140.1, 135.9, 128.8, 128.4, 128.1, 125.9, 124.4, 122.5, 119.6, 119.1, 113.1, 97.2, 44.3, 25.3, 25.1, 22.5; HRMS (ESI) exact mass calcd for C19H17NOS + H (M + H), 308.1104; found 308.1117.
11-((4-Methoxyphenyl)thio)-6,7,8,9-tetrahydro-10H-cyclohepta[b] indolizin-10-one (3bc).
The title compound was prepared by following the general procedure for Table 2 using 6,7,8,9-tetrahydro-10H-cyclohepta[b]indolizin-10-one 1b (0.5 mmol, 0.100 g) and bis(4-methoxyphenyl) disulfide 2c (0.5 mmol, 0.139 g), and after column chromatography (20–22% EtOAc/hexanes), 3bc was obtained as a dark brown solid; yield: 0.142 g, 84%; M.P.: 76 °C; 1H NMR (400 MHz, CDCl3) δ 7.79 (dt, J = 7.2 and 1.2 Hz, 1H), 7.68 (dt, J = 9.2 and 1.2 Hz, 1H), 7.14 (dt, J = 9.2 and 2.4 Hz, 2H), 6.82 (ddd, J = 9.2, 6.4 and 1.2 Hz, 1H), 6.75–6.72 (m, 2H), 6.69 (ddd, J = 7.2, 6.4 and 1.6 Hz, 1H), 3.73 (s, 3H), 3.08 (t, J = 6.0 Hz, 2H), 2.82 (t, J = 6.4 Hz, 2H), 2.11–2.04 (m, 2H), 1.99–1.93 (m, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 199.1, 157.7, 135.7, 130.6, 129.2, 128.6, 128.1, 122.42, 119.50, 114.31, 113.19, 99.4, 55.3, 44.5, 25.5, 24.9, 22.6; HRMS (ESI) exact mass calcd for C20H19NO2S + H (M + H), 338.1209; found: 338.1193.
11-((4-Chlorophenyl)thio)-6,7,8,9-tetrahydro-10H-cyclohepta[b] indolizin-10-one (3bd).
The title compound was prepared by following the general procedure for Table 2 using 6,7,8,9-tetrahydro-10H-cyclohepta[b]indolizin-10-one 1b (0.5 mmol, 0.100 g) and bis(4-chlorophenyl) disulfide 2d (0.5 mmol, 0.144 g), and after column chromatography (12–15% EtOAc/hexanes), 3bd was obtained as a brown solid; yield: 0.135 g, 79%; M.P.: 101 °C; 1H NMR (400 MHz, CDCl3) δ 7.75 (dd, J = 7.2 and 1.2 Hz, 1H), 7.54 (dd, J = 8.8 and 1.2 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.90 (d, J = 9.6 Hz, 2H), 6.80–6.75 (m, 1H), 6.65 (td, J = 7.2 and 1.2 Hz, 1H), 3.04 (t, J = 6.0 Hz, 2H), 2.74 (t, J = 6.8 Hz, 2H), 2.05–1.99 (m, 2H), 1.92–1.86 (m, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 198.8, 138.9, 136.1, 130.3, 129.1, 128.5, 128.1, 127.3, 122.6, 120.0, 119.1, 113.3, 97.1, 44.5, 25.6, 25.2, 22.6; HRMS (ESI) exact mass calcd for C19H16ClNOS + H (M + H), 342.0714; found: 342.0723.
11-((3-Chlorophenyl)thio)-6,7,8,9-tetrahydro-10H-cyclohepta[b] indolizin-10-one (3be).
The title compound was prepared by following the general procedure for Table 2 using 6,7,8,9-tetrahydro-10H-cyclohepta[b]indolizin-10-one 1b (0.5 mmol, 0.100 g) and bis(3-chlorophenyl) disulfide 2e (0.5 mmol, 0.144 g), and after column chromatography (12–15% EtOAc/hexanes), 3be was obtained as a dark green solid; yield: 0.140 g, 82%; M.P.: 98 °C; 1H NMR (400 MHz, CDCl3) δ 7.76 (dt, J = 7.2 and 1.2 Hz, 1H), 7.54 (dt, J = 9.2 and 1.2 Hz, 1H), 7.01–6.97 (m, 1H), 6.92–6.86 (m, 3H), 6.80–6.76 (m, 1H), 6.68–6.64 (m, 1H), 3.05 (t, J = 6.0 Hz, 2H), 2.75 (t, J = 6.4 Hz, 2H), 2.06–1.97 (m, 2H), 1.93–1.86 (m, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 198.7, 142.7, 136.2, 134.4, 129.5, 129.2, 128.1, 125.4, 124.6, 124.0, 122.6, 120.2, 119.1, 113.4, 96.2, 44.5, 25.6, 25.2, 22.6; HRMS (ESI) exact mass calcd for C19H16ClNOS + H (M + Na), 364.0533; found: 364.0541.
11-((3-Bromophenyl)thio)-6,7,8,9-tetrahydro-10H-cyclohepta[b] indolizin-10-one (3bf).
The title compound was prepared by following the general procedure for Table 2 using 6,7,8,9-tetrahydro-10H-cyclohepta[b]indolizin-10-one 1b (0.5 mmol, 0.100 g) and bis(3-bromophenyl) disulfide 2f (0.5 mmol, 0.188 g), and after column chromatography (12–15% EtOAc/hexanes), 3bf was obtained as a green solid; yield: 0.149 g, 77%; M.P.: 106 °C; 1H NMR (400 MHz, CDCl3) δ 7.85 (dt, J = 7.2 and 1.2 Hz, 1H), 7.62 (dt, J = 9.2 and 1.2 Hz, 1H), 7.16–7.13 (m, 1H), 7.11–7.10 (m, 1H), 7.03–6.98 (m, 2H), 6.87 (ddd, J = 9.2, 6.8 and 1.2 Hz, 1H), 6.75 (ddd, J = 7.2, 6.4 and 1.2 Hz, 1H), 3.13 (t, J = 6.4 Hz, 2H), 2.82 (t, J = 6.4 Hz, 2H), 2.14–2.08 (m, 2H), 2.01–1.95 (m, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 198.8, 142.9, 136.2, 129.6, 129.2, 128.2, 128.1, 127.6, 124.4, 122.7, 122.6, 120.2, 119.1, 113.4, 96.2, 44.5, 25.6, 25.2, 22.6; HRMS (ESI) exact mass calcd for C19H16BrNOS + H (M + H), 386.0209; found: 386.0216.
11-((2-Bromophenyl)thio)-6,7,8,9-tetrahydro-10H-cyclohepta[b] indolizin-10-one (3bg).
The title compound was prepared by following the general procedure for Table 2 using 6,7,8,9-tetrahydro-10H-cyclohepta[b]indolizin-10-one 1b (0.5 mmol, 0.100 g) and bis(2-bromophenyl) disulfide 2g (0.5 mmol, 0.188 g), and after column chromatography (12–15% EtOAc/hexanes), 3bg was obtained as a green solid; yield: 0.129 g, 67%; M.P.: 138 °C; 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 7.2 Hz, 1H), 7.51 (dt, J = 9.2 and 1.2 Hz, 1H), 7.36 (dd, J = 8.0 and 1.2 Hz, 1H), 6.89 (td, J = 8.0 and 1.6 Hz, 1H), 6.80–6.73 (m, 2H), 6.64 (td, J = 6.8 and 1.6 Hz, 1H), 6.41 (td, J = 8.0 and 1.6 Hz, 1H), 3.04 (t, J = 6.0 Hz, 2H), 2.72 (t, J = 6.4 Hz, 2H), 2.05–1.98 (m, 2H), 1.91–1.85 (m, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 198.5, 141.4, 136.2, 132.2, 129.3, 128.3, 127.2, 126.4, 125.3, 122.7, 120.2, 119.5, 119.0, 96.0, 44.4, 25.6, 25.2, 22.5; HRMS (ESI) exact mass calcd for C19H16BrNOS + H (M + H), 386.0209; found: 386.0218.
11-((4-Bromophenyl)thio)-6,7,8,9-tetrahydro-10H-cyclohepta[b] indolizin-10-one (3bh).
The title compound was prepared by following the general procedure for Table 2 using 6,7,8,9-tetrahydro-10H-cyclohepta[b]indolizin-10-one 1b (0.5 mmol, 0.100 g) and bis(2–4romophenyl) disulfide 2h (0.5 mmol, 0.188 g), and after column chromatography (12–15% EtOAc/hexanes), 3bh was obtained as a green solid; yield: 0.163 g, 84%; M.P.: 119 °C; 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.2 Hz, 1H), 7.51 (d, J = 8.8 Hz, 1H), 7.14 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 6.77–6.73 (m, 1H), 6.63 (t, J = 6.8 Hz, 1H), 3.01 (t, J = 6.8 Hz, 2H), 2.71 (t, J = 7.2 Hz, 2H), 1.99 (quint, J = 6.0 Hz, 2H), 1.86 (quint, J = 6.4 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 198.7, 139.7, 136.0, 131.3, 129.1, 128.0, 127.5, 122.6, 120.0, 118.9, 118.0, 113.3, 96.6, 44.4, 25.5, 25.1, 22.5; HRMS (ESI) exact mass calcd for C19H16BrNOS + H (M + H), 386.0209; found: 386.0215.
10-(Phenylselanyl)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (5aa).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and diphenyl diselenide 4a (0.5 mmol, 0.156 g), and after column chromatography (18–22% EtOAc/hexanes), 5aa was obtained as a black solid; yield: 0.111 g, 65%; M.P.: 78 °C; 1H NMR (400 MHz, CDCl3) δ 7.71 (dq, J = 7.2 and 1.2 Hz, 1H), 7.60 (dq, J = 9.2 and 1.2 Hz, 1H), 7.29–7.27 (m, 2H), 7.14–7.06 (m, 3H), 6.79 (ddd, J = 9.2, 6.4 and 1.2 Hz, 1H), 6.68 (tt, J = 6.4 and 1.2 Hz, 1H), 3.01 (t, J = 6.4 Hz, 2H), 2.63 (t, J = 6.4 Hz, 2H), 2.30 (quint, J = 6.4 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 195.1, 136.4, 134.1, 133.5, 129.7, 128.8, 125.8, 123.3, 122.7, 120.4, 120.1, 113.2, 91.2, 39.4, 23.1, 21.4; HRMS (ESI) exact mass calcd for C18H15NOSe + H (M + H), 342.0392; found: 342.0405.
10-(p-Tolylselanyl)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (5ab).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(p-tolyl) diselenide 4b (0.5 mmol, 0.170 g), and after column chromatography (12–15% EtOAc/hexanes), 5ab was obtained as a dark green solid; yield: 0.119 g, 67%; M.P.: 94 °C; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 6.8 Hz, 1H), 7.50 (dt, J = 9.2 and 1.2 Hz, 1H), 7.15 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 7.6 Hz, 2H), 6.68 (dd, J = 9.2 and 6.4 Hz, 1H), 6.56 (td, J = 6.8 and 1.2 Hz, 1H), 2.89 (t, J = 6.4 Hz, 2H), 2.53 (t, J = 6.4 Hz, 2H), 2.19 (quint, J = 6.4 Hz, 2H), 2.14 (s, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 195.0, 136.2, 135.7, 133.3, 130.3, 130.0, 129.6, 123.2, 122.6, 120.4, 119.8, 113.0, 90.8, 39.3, 23.1, 21.3, 21.0; HRMS (ESI) exact mass calcd for C19H17NOSe + H (M + H), 356.0548; found: 356.0559.
10-((4-Methoxyphenyl)selanyl)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (5ac).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(4-methoxyphenyl) diselenide 4c (0.5 mmol, 0.186 g), and after column chromatography (18–20% EtOAc/hexanes), 5ac was obtained as a dark green solid; yield: 0.134 g, 72%; M.P.: 101 °C; 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 7.2 Hz, 1H), 7.53 (d, J = 9.2 Hz, 1H), 7.31 (d, J = 8.8 Hz, 2H), 6.70–6.54 (m, 4H), 3.63 (s, 3H), 2.88 (t, J = 6.4 Hz, 2H), 2.55–2.52 (m, 2H), 2.20 (quint, J = 6.4 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 195.1, 158.6, 135.9, 133.2, 133.0, 123.7, 123.1, 122.62, 120.4, 119.7, 114.5, 113.0, 93.0, 55.2, 39.4, 23.1, 21.3; HRMS (ESI) exact mass calcd for C19H17NO2Se + H (M + H), 372.0498; found: 372.0505.
10-((4-Bromophenyl)selanyl)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (5ad).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(4-bromophenyl) diselenide 4d (0.5 mmol, 0.235 g), and after column chromatography (18–20% EtOAc/hexanes), 5ad was obtained as a green solid; yield: 0.147 g, 70%; M.P.: 92 °C; 1H NMR (400 MHz, CDCl3) δ 7.73 (dt, J = 7.2 and 1.2 Hz, 1H), 7.59 (dt, J = 9.2 and 1.2 Hz, 1H), 7.22 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 6.85–6.81 (m, 1H), 6.70 (td, J = 6.8 and 1.2 Hz, 1H), 3.02 (t, J = 6.4 Hz, 2H), 2.63 (d, J = 6.4 Hz, 2H), 2.30 (quint, J = 6.0 Hz, 2H); 13C{H} NMR (100 MHz, CDCl3) δ 195.0, 136.5, 133.7, 133.2, 131.8, 131.2, 123.2, 122.8, 120.4, 120.2, 119.7, 113.3, 90.6, 39.3, 23.1, 21.4; HRMS (ESI) exact mass calcd for C18H14BrNOSe + H (M + H), 419.9497; found: 419.9504.
10-(Butylselanyl)-3,4-dihydropyrido[1,2-a]indol-1(2H)-one (5ae).
The title compound was prepared by following the general procedure for Table 2 using 3,4-dihydropyrido[1,2-a]indol-1(2H)-one 1a (0.5 mmol, 0.093 g) and bis(butyl) diselenide 4e (0.5 mmol, 0.136 g), and after column chromatography (10–12% EtOAc/hexanes), 5ae was obtained as a yellow liquid; yield: 0.077 g, 48%; 1H NMR (400 MHz, CDCl3) δ 7.61–7.57 (m, 2H), 6.71–6.68 (m, 1H), 6.56 (t, J = 5.6 Hz, 1H), 2.91 (t, J = 5.2 Hz, 2H), 2.78 (t, J = 6.0 Hz, 2H), 2.57 (t, J = 5.2 Hz, 2H), 2.22 (quint, J = 5.2 Hz, 2H), 1.45 (quint, J = 6.0 Hz, 2H), 1.30 (sext, J = 6.0 Hz, 2H), 0.76 (t, J = 5.6 Hz, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 195.3, 136.0, 133.2, 123.7, 122.4, 120.8, 119.0, 112.9, 92.5, 39.5, 32.2, 29.0, 23.2, 22.8, 21.4, 13.6; HRMS (ESI) exact mass calcd for C16H19NOSe + H (M + H), 322.0705; found: 322.0712
General procedure for Table 3
An undivided ElectraSyn 2.0 cell equipped with a graphite anode and a nickel cathode was charged with indolizines 6 (0.5 mmol), disulfides 2 (0.75 mmol)/diselenides 4 (0.75 mmol), KI (100 mol%), and n-Bu4NPF6 (50 mol%) in acetonitrile (4 mL) solvent. The reaction mixtures were stirred and electrolyzed at a constant current of 10 mA at room temperature for 12 h via the manual programming of the IKA ElectraSyn 2.0 instrument. After the completion of the reaction, the acetonitrile solvents were evaporated under reduced pressure to obtain crude products, which were further purified through column chromatography using EtOAc:hexanes as an eluent.
2-Phenyl-3-(phenylthio)indolizine (7aa′)8.
The title compound was prepared by following the general procedure for Table 3 using 2-phenylindolizine 6a (0.5 mmol, 0.097 g) and diphenyl disulfide 2a (0.5 mmol, 0.109 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7aa′ was obtained as a yellow liquid; yield: 0.046 g, 30%; 1H NMR (400 MHz, CDCl3) δ 8.14 (dt, J = 7.2 and 1.2 Hz, 1H), 7.64–7.61 (m, 2H), 7.36 (dt, J = 8.8 and 1.2 Hz, 1H), 7.31–7.27 (m, 2H), 7.23–7.18 (m, 1H), 7.10–7.06 (m, 2H), 7.01–6.97 (m, 1H), 6.82–6.74 (m, 3H), 6.71 (br s, 1H), 6.48 (td, J = 6.8 and 1.3 Hz, 1H); 13C{H} NMR (100 MHz, CDCl3) δ 137.2, 137.0, 135.9, 135.2, 129.2, 128.9, 128.4, 127.2, 125.4, 125.1, 124.0, 119.9, 118.8, 111.3, 104.6, 100.2.
2-Phenyl-1,3-bis(phenylthio)indolizine (7aa)8.
The title compound was prepared by following the general procedure for Table 3 using 2-phenylindolizine 6a (0.5 mmol, 0.097 g) and diphenyl disulfide 2a (0.75 mmol, 0.164 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7aa was obtained as a white solid; yield: 0.187 g, 91%; M.P.: 144 °C; 1H NMR (400 MHz, CDCl3) δ 8.33 (dt, J = 6.8 and 1.2 Hz, 1H), 7.69 (dt, J = 9.2 and 1.2 Hz, 1H), 7.43–7.41 (m, 2H), 7.33–7.29 (m, 3H), 7.23–7.15 (m, 4H), 7.14–7.09 (m, 1H), 7.07–6.99 (m, 4H), 6.92–6.89 (m, 2H), 6.75 (td, J = 6.8 and 1.2 Hz, 1H); 13C{H} NMR (100 MHz, CDCl3) δ 141.7, 140.2, 139.5, 136.7, 133.2, 130.5, 129.3, 128.8, 127.8, 127.7, 125.6, 125.37, 125.32, 124.7, 124.6, 122.3, 117.9, 112.8, 107.7, 97.5.
2-Phenyl-1,3-bis(p-tolylthio)indolizine (7ab)7.
The title compound was prepared by following the general procedure for Table 3 using 2-phenylindolizine 6a (0.5 mmol, 0.097 g) and bis(p-tolyl) disulfide 2b (0.75 mmol, 0.185 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7ab was obtained as a white solid; yield: 0.211 g, 96%; M.P.: 136 °C; 1H NMR (400 MHz, CDCl3) δ 8.25 (dt, J = 7.2 and 1.2 Hz, 1H), 7.61 (dt, J = 8.8 and 1.2 Hz, 1H), 7.37–7.35 (m, 2H), 7.27–7.22 (m, 3H), 6.95–6.90 (m, 5H), 6.82 (d, J = 8.4 Hz, 2H), 6.73 (d, J = 8.4 Hz, 2H), 6.65 (td, J = 6.8 and 1.6 Hz, 1H), 2.19 (s, 3H), 2.18 (s, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 141.5, 139.3, 136.7, 135.5, 134.4, 133.3, 133.1, 130.6, 130.1, 129.6, 127.8, 127.6, 125.5, 125.4, 124.6, 122.1, 117.9, 112.7, 108.1, 97.9, 20.97, 20.94.
2-(4-Methoxyphenyl)-1,3-bis(phenylthio)indolizine (7ba).
The title compound was prepared by following the general procedure for Table 3 using 2-(4-methoxyphenyl)indolizine 6b (0.5 mmol, 0.112 g) and diphenyl disulfide 2a (0.75 mmol, 0.164 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7ba was obtained as a white solid; yield: 0.148 g, 67%; M.P.: 83 °C; 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 7.2 Hz, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 2.0 Hz, 1H), 7.25 (dd, J = 8.4 and 2.0 Hz, 1H), 7.13–6.90 (m, 10H), 6.80 (d, J = 7.6 Hz, 2H), 6.72 (d, J = 8.4 Hz, 1H), 6.65 (td, J = 6.8 and 1.2 Hz, 1H), 3.73 (s, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 155.3, 139.9, 139.8, 139.5, 136.5, 135.3, 131.6, 130.6, 129.4, 128.9, 126.9, 125.8, 125.4, 125.3, 124.8, 124.5, 122.4, 117.8, 113.4, 112.9, 111.2, 110.8, 107.8, 97.5, 56.1; HRMS (ESI) exact mass calcd for C27H21NOS2 + H (M + H), 440.1138; found: 440.1142.
1,3-Bis((2-bromophenyl)thio)-2-(4-methoxyphenyl)indolizine (7bg).
The title compound was prepared by following the general procedure for Table 3 using 2-(4-methoxyphenyl)indolizine 6b (0.5 mmol, 0.112 g) and bis(2-bromophenyl) disulfide 2g (0.75 mmol, 0.282 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7bg was obtained as a white solid; yield: 0.168 g, 56%; M.P.: 106 °C; 1H NMR (400 MHz, CDCl3) δ 8.21 (dt, J = 6.8 and 1.2 Hz, 1H), 7.59 (dt, J = 9.2 and 1.2 Hz, 1H), 7.48 (d, J = 2.0 Hz, 1H), 7.46 (dd, J = 8.0 and 1.6 Hz, 1H), 7.41 (dd, J = 8.0 and 1.6 Hz, 1H), 7.23 (dd, J = 8.4 and 2.0 Hz, 1H), 7.02–6.89 (m, 5H), 6.84 (dt J = 7.6 and 1.6 Hz, 1H), 6.76–6.70 (m, 2H), 6.44 (dd, J = 8.0 and 1.6 Hz, 1H), 6.18 (dd, J = 7.6 and 1.6 Hz, 1H), 3.76 (s, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 155.5, 140.7, 140.5, 139.8, 137.3, 135.0, 133.4, 132.8, 130.4, 128.2, 127.7, 126.9, 126.2, 125.95, 125.91, 125.5, 124.7, 123.0, 120.2, 119.8, 117.9, 113.3, 111.4, 111.0, 107.3, 97.0, 56.1; HRMS (ESI) exact mass calcd for C27H19Br2NOS2 + H (M + H), 595.9348; found: 595.9357.
2-(3-Bromophenyl)-1,3-bis((3-bromophenyl)thio)indolizine (7cf).
The title compound was prepared by following the general procedure for Table 3 using 2-(3-bromophenyl)indolizine 6c (0.5 mmol, 0.136 g) and bis(3-bromophenyl) disulfide 2f (0.75 mmol, 0.282 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7cf was obtained as a white solid; yield: 0.201 g, 62%; M.P.: 124 °C; 1H NMR (400 MHz, CDCl3) δ 8.27 (dt, J = 6.8 and 1.2 Hz, 1H), 7.63 (dt, J = 9.2 and 1.2 Hz, 1H), 7.45–7.44 (m, 1H), 7.38–7.35 (m, 1H), 7.22 (dt, J = 7.6 and 1.6 Hz, 1H), 7.19–7.16 (m, 1H), 7.13 (d, J = 7.6 Hz, 1H), 7.12–7.09 (m, 1H), 7.06–7.02 (m, 1H), 7.01–6.93 (m, 4H), 6.82–6.79 (m, 1H), 6.75 (td, J = 6.8 and 1.2 Hz, 1H), 6.65–6.63 (m, 1H); 13C{H} NMR (100 MHz, CDCl3) δ 142.2, 140.4, 139.7, 138.8, 134.8, 133.4, 130.9, 130.8, 130.2, 129.5, 129.1, 129.0, 128.1, 128.0, 124.5, 124.0, 123.7, 123.4, 123.2, 123.1, 121.9, 117.9, 113.5, 107.5, 97.1; HRMS (ESI) exact mass calcd for C26H16Br3NS2 + H (M + H), 643.8347; found: 643.8358.
2-(4-Chlorophenyl)-1,3-bis((4-chlorophenyl)thio)indolizine (7dd).
The title compound was prepared by following the general procedure for Table 3 using 2-(4-chlorophenyl)indolizine 6d (0.5 mmol, 0.114 g) and bis(4-chlorophenyl) disulfide 2d (0.75 mmol, 0.215 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7dd was obtained as a green solid; yield: 0.172 g, 67%; M.P.: 199 °C; 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 6.8 Hz, 1H), 7.61 (d, J = 8.8 Hz, 1H), 7.32–7.19 (m, 5H), 7.11 (d, J = 8.0 Hz, 2H), 7.06 (d, J = 8.0 Hz, 2H), 6.81 (d, J = 8.0 Hz, 2H), 6.76–6.69 (m, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 140.6, 139.6, 138.4, 134.9, 134.1, 131.9, 131.7, 131.3, 130.7, 129.6, 129.0, 128.3, 126.6, 126.5, 124.5, 122.9, 117.9, 113.4, 107.5, 97.3; HRMS (ESI) exact mass calcd for C26H16Cl3NS2 + H (M + H), 511.9863 found: 511.9869.
2-(4-Fluorophenyl)-1,3-bis(phenylthio)indolizine (7ea).
The title compound was prepared by following the general procedure for Table 3 using 2-(4-fluorophenyl)indolizine 6e (0.5 mmol, 0.106 g) and diphenyl disulfide 2a (0.75 mmol, 0.164 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7ea was obtained as a white solid; yield: 0.150 g, 70%; M.P.: 166 °C; 1H NMR (400 MHz, CDCl3) δ 8.26 (dt, J = 6.8 and 1.2 Hz, 1H), 7.62 (dt, J = 9.2 and 1.2 Hz, 1H), 7.31 (dd, J = 8.8 and 5.2 Hz, 2H), 7.18–7.09 (m, 4H), 7.08–7.03 (m, 2H), 7.00–6.94 (m, 3H), 6.92–6.90 (m, 2H), 6.83–6.81 (m, 2H), 6.69 (td, J = 6.8 and 1.6 Hz, 1H); 13C{H} NMR (100 MHz, CDCl3) δ 162.5 (JC–F = 245.0 Hz), 140.7, 140.0, 139.5, 136.6, 132.2 (JC–F = 8.0 Hz), 129.4, 129.1 (JC–F = 3.0 Hz), 128.9, 125.8, 125.3, 125.2, 124.8, 124.6, 122.4, 117.9, 114.9 (JC–F = 21.0 Hz), 113.0, 107.7, 97.5; 19F NMR (376 MHz, CDCl3) δ −114.4; HRMS (ESI) exact mass calcd for C26H18FNS2 + H (M + H), 428.0938; found: 428.0950
2-(Naphthalen-2-yl)-1,3-bis(phenylthio)indolizine (7fa).
The title compound was prepared by following the general procedure for Table 3 using 2-(naphthalen-2-yl)indolizine 6f (0.5 mmol, 0.122 g) and diphenyl disulfide 2a (0.75 mmol, 0.164 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7fa was obtained as a green solid; yield: 0.166 g, 72%; M.P.: 140 °C; 1H NMR (400 MHz, CDCl3) δ 8.40 (dt, J = 6.8 and 1.2 Hz, 1H), 7.87 (br s, 1H), 7.82–7.79 (m, 2H), 7.76 (dt, J = 9.2 and 1.2 Hz, 1H), 7.67–7.64 (m, 1H), 7.60 (dd, J = 8.4 and 1.6 Hz, 1H), 7.46–7.39 (m, 2H), 7.23–7.18 (m, 4H), 7.16–7.12 (m, 1H), 7.09–7.05 (m, 4H), 6.97–6.94 (m, 2H), 6.78 (td, J = 6.8 and 1.2 Hz, 1H); 13C{H} NMR (100 MHz, CDCl3) δ 141.6, 140.3, 139.6, 136.7, 133.0, 132.7, 130.7, 130.0, 129.4, 128.9, 128.5, 128.4, 127.6, 127.2, 126.0, 125.8, 125.7, 125.5, 125.4, 124.8, 124.6, 122.3, 117.9, 112.9, 108.1, 97.9; HRMS (ESI) exact mass calcd for C30H21NS2 + H (M + H), 460.1188; found: 460.1197.
2-(Naphthalen-2-yl)-1,3-bis(p-tolylthio)indolizine (7fb).
The title compound was prepared by following the general procedure for Table 3 using 2-(naphthalen-2-yl)indolizine 6f (0.5 mmol, 0.122 g) and bis(p-tolyl) disulfide 2b (0.75 mmol, 0.185 g), and after column chromatography (0–0.5% EtOAc/hexanes), 7fb was obtained as a white solid; yield: 0.181 g, 74%; M.P.: 126 °C; 1H NMR (400 MHz, CDCl3) δ 8. 30 (dt, J = 7.2 and 1.2 Hz, 1H), 7.79 (d, J = 1.6 Hz, 1H), 7.73–7.69 (m, 2H), 7.66 (dt, J = 9.2 and 1.2 Hz, 1H), 7.60–7.58 (m, 1H), 7.51 (dd, J = 8.4 and 1.6 Hz, 1H), 7.37–7.30 (m, 2H), 6.98–6.85 (m, 7H), 6.76 (d, J = 8.4 Hz, 2H), 6.67 (td, J = 6.8 and 1.2 Hz, 1H), 2.19 (s, 3H), 2.18 (s, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 141.4, 139.5, 136.7, 135.6, 134.5, 133.1, 133.0, 132.7, 130.9, 130.1, 130.0, 129.6, 128.6, 128.4, 127.6, 127.2, 126.0, 125.7, 125.6, 124.5, 122.1, 118.0, 112.8, 108.5, 98.3, 20.98, 20.96; HRMS (ESI) exact mass calcd for C32H25NS2 + H (M + H), 488.1501; found: 488.1514.
Methyl 1,3-bis(p-tolylthio)indolizine-2-carboxylate (7gb).
The title compound was prepared by following the general procedure for Table 3 using 1-(indolizin-2-yl)ethan-1-one 6g (0.5 mmol, 0.080 g) and bis(p-tolyl) disulfide 2b (0.75 mmol, 0.185 g), and after column chromatography (2–5% EtOAc/hexanes), 7gb was obtained as a white solid; yield: 0.093 g, 46%; M.P.: 102 °C; 1H NMR (400 MHz, CDCl3) δ 8.26 (dt, J = 7.2 and 1.2 Hz, 1H), 7.63 (dt, J = 8.8 and 1.2 Hz, 1H), 6.97–6.92 (m, 5H), 6.88 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 8.0 Hz, 2H), 6.69 (td, J = 6.8 and 1.2 Hz, 1H), 2.61 (s, 3H), 2.19 (s, 3H), 2.18 (s, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 197.5, 138.8, 138.4, 136.2, 135.5, 135.0, 131.6, 130.2, 129.7, 126.6, 126.1, 124.5, 122.6, 118.8, 114.0, 111.2, 98.8, 31.9, 20.99, 20.95; HRMS (ESI) exact mass calcd for C24H21NOS2 + H (M + Na), 426.0957; found: 426.0964.
1-(1,3-Bis(p-tolylthio)indolizin-2-yl)propan-1-one (7hb).
The title compound was prepared by following the general procedure for Table 3 using 1-(indolizin-2-yl)propan-1-one 6h (0.5 mmol, 0.087 g) and bis(p-tolyl) disulfide 2b (0.75 mmol, 0.185 g), and after column chromatography (2–5% EtOAc/hexanes), 7hb was obtained as a green solid; yield: 0.132 g, 63%; M.P.: 118 °C; 1H NMR (400 MHz, CDCl3) δ 8.24 (dt, J = 7.2 and 1.2 Hz, 1H), 7.61 (dt, J = 9.2 and 1.2 Hz, 1H), 6.97–6.91 (m, 5H), 6.89–6.82 (m, 4H), 6.68 (td, J = 6.8 and 1.6 Hz, 1H), 3.01 (q, J = 7.2 Hz, 2H), 2.19 (s, 3H), 2.18 (s, 3H), 1.03 (t, J = 7.2 Hz, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 201.1, 139.1, 138.8, 136.2, 135.5, 135.0, 131.7, 130.1, 129.7, 126.6, 126.2, 124.5, 122.5, 118.7, 113.8, 110.4, 98.3, 37.4, 21.0, 20.9, 8.0; HRMS (ESI) exact mass calcd for C25H23NOS2 + H (M + H), 418.5923; found: 418.5930.
Methyl 1,3-bis(phenylthio)indolizine-2-carboxylate (7ia).
The title compound was prepared by following the general procedure for Table 3 using methyl indolizine-2-carboxylate 6i (0.5 mmol, 0.088 g) and diphenyl disulfide 2a (0.75 mmol, 0.164 g), and after column chromatography (2–5% EtOAc/hexanes), 7ia was obtained as a green solid; yield: 0.079 g, 40%; M.P.: 115 °C; 1H NMR (400 MHz, CDCl3) δ 8.28 (dt, J = 7.2 and 1.2 Hz, 1H), 7.65 (dt, J = 9.2 and 1.2 Hz, 1H), 7.15–7.06 (m, 5H), 7.03–6.93 (m, 6H), 6.71 (td, J = 7.2 and 1.2 Hz, 1H), 3.75 (s, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 164.5, 139.2, 138.8, 135.3, 130.4, 129.3, 128.8, 126.6, 126.2, 125.1, 124.5, 122.6, 118.9, 114.1, 112.6, 100.2, 52.1; HRMS (ESI) exact mass calcd for C22H17NO2S2 + H (M + H), 392.0774; found: 392.0789.
2-Phenyl-1,3-bis(phenylselanyl)indolizine (8aa)6.
The title compound was prepared following the general procedure for Table 3 using 2-phenylindolizine 6a (0.5 mmol, 0.097 g) and diphenyl diselenide 4a (0.75 mmol, 0.234 g), and after column chromatography (0–0.5% EtOAc/hexanes), 8aa was obtained as a brown solid; yield: 0.182 g, 72%; M.P.: 122 °C; 1H NMR (400 MHz, CDCl3) δ 8.31 (dt, J = 7.2 and 1.2 Hz, 1H), 7.62 (dt, J = 8.8 and 1.2 Hz, 1H), 7.29–7.27 (m, 2H), 7.26–7.23 (m, 3H), 7.10–7.05 (m, 4H), 7.04–7.00 (m, 4H), 6.94–6.92 (m, 2H), 6.91–6.89 (m, 1H), 6.63 (td, J = 6.8 and 1.6 Hz, 1H); 13C{H} NMR (100 MHz, CDCl3) δ 142.6, 140.1, 135.0, 134.7, 132.3, 131.5, 130.9, 129.5, 129.2, 129.0, 128.2, 128.0, 127.6, 127.5, 126.3, 125.9, 125.5, 121.8, 118.9, 112.6, 105.6, 94.8.
1,3-Bis(butylselanyl)-2-phenylindolizine (8ae).
The title compound was prepared by following the general procedure for Table 3 using 2-phenylindolizine 6a (0.5 mmol, 0.097 g) and bis(butyl) diselenide 4e (0.75 mmol, 0.204 g), and after column chromatography (0–0.5% EtOAc/hexanes), 8ae was obtained as a yellow solid; yield: 0.197 g, 85%; M.P.: 46 °C; 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 6.8 Hz, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.49–7.41 (m, 4H), 7.39–7.35 (m, 1H), 6.93–6.87 (m, 1H), 6.69 (t, J = 6.8 Hz, 1H), 2.50 (t, J = 7.2 Hz, 2H), 2.41 (t, J = 7.2 Hz, 2H), 1.33 (quint, J = 7.2 Hz, 4H), 1.22–1.15 (m, 4H), 0.73 (t, J = 7.2 Hz, 6H); 13C{H} NMR (100 MHz, CDCl3) δ 141.3, 138.8, 135.9, 131.3, 127.4, 127.0, 125.7, 119.9, 119.0, 111.5, 106.6, 95.7, 32.1, 32.0, 29.4, 28.8, 22.64, 22.5, 13.57, 13.51; HRMS (ESI) exact mass calcd for C22H27NSe2 + H (M + H), 466.0547; found: 466.0554.
2-(3-Bromophenyl)-1,3-bis((4-bromophenyl)selanyl)indolizine (8cd).
The title compound was prepared by following the general procedure for Table 3 using 2-(3-bromophenyl)indolizine 6c (0.5 mmol, 0.136 g) and bis(4-bromophenyl) diselenide 4d (0.75 mmol, 0.352 g), and after column chromatography (0–0.5% EtOAc/hexanes), 8cd was obtained as a white solid; yield: 0.237 g, 64%; M.P.: 150 °C; 1H NMR (400 MHz, CDCl3) δ 8.30 (dt, J = 6.8 and 1.2 Hz, 1H), 7.62 (dt, J = 8.8, and 1.2 Hz, 1H), 7.39–7.35 (m, 2H), 7.21 (d, J = 8.8 Hz, 2H), 7.17 (d, J = 8.8 Hz, 2H), 7.14–7.11 (m, 2H), 6.97 (ddd, J = 9.2, 6.8 and 1.2 Hz, 1H), 6.88 (d, J = 8.4 Hz, 2H), 6.77 (d, J = 8.4 Hz, 2H), 6.70 (td, J = 6.8 and 1.2 Hz, 1H); 13C{H} NMR (100 MHz, CDCl3) δ 141.0, 140.1, 136.5, 133.8, 133.5, 132.6, 132.1, 130.9, 130.6, 130.2, 129.9, 129.4, 129.2, 125.8, 122.5, 121.6, 120.6, 119.8, 118.9, 113.1, 105.7, 94.9; HRMS (ESI) exact mass calcd for C26H16Br3NSe2 + H (M + H), 739.7236; found: 739.7248.
2-(Naphthalen-2-yl)-1,3-bis(phenylselanyl)indolizine (8fa).
The title compound was prepared by following the general procedure for Table 3 using 2-(naphthalen-2-yl)indolizine 6f (0.5 mmol, 0.122 g) and diphenyl diselenide 4a (0.75 mmol, 0.234 g), and after column chromatography (0–0.5% EtOAc/hexanes), 8fa was obtained as a brown solid; yield: 0.219 g, 79%; M.P.: 116 °C; 1H NMR (400 MHz, CDCl3) δ 8.35 (dt, J = 7.2 and 1.2 Hz, 1H), 7.74–7.65 (m, 4H), 7.58–7.55 (m, 1H), 7.44 (dd, J = 8.4 and 2.0 Hz, 1H), 7.37–7.30 (m, 2H), 7.10–7.02 (m, 8H), 6.97–6.90 (m, 3H), 6.65 (td, J = 6.4 and 1.6 Hz, 1H); 13C{H} NMR (100 MHz, CDCl3) δ 142.5, 140.3, 135.1, 132.9, 132.7, 132.4, 132.3, 130.3, 129.6, 129.1, 128.9, 128.4, 128.3, 128.2, 127.7, 126.9, 126.4, 125.9, 125.9, 125.8, 125.6, 121.9, 119.0, 112.6, 106.0, 95.2; HRMS (ESI) exact mass calcd for C30H21NSe2 + H (M + H), 556.0077; found: 556.0085.
1-(1,3-Bis(phenylselanyl)indolizin-2-yl)propan-1-one (8ha).
The title compound was prepared by following the general procedure for Table 3 using 1-(indolizin-2-yl)propan-1-one 6h (0.5 mmol, 0.087 g) and diphenyl diselenide 4a (0.75 mmol, 0.234 g), and after column chromatography (2–5% EtOAc/hexanes), 8ha was obtained as a yellow solid; yield: 0.097 g, 40%; M.P.: 75 °C; 1H NMR (400 MHz, CDCl3) δ 8.26 (dt, J = 7.2 and 1.2 Hz, 1H), 7.59 (dt, J = 7.2 and 1.2 Hz, 1H), 7.13–7.04 (m, 10H), 6.90 (ddd, J = 9.2, 6.8 and 1.2 Hz, 1H), 6.66 (td, J = 6.8 and 1.2 Hz, 1H), 3.00 (quint, J = 7.2 Hz, 2H), 1.04 (t, J = 7.2 Hz, 3H); 13C{H} NMR (100 MHz, CDCl3) δ 202.2, 140.9, 139.6, 133.9, 131.5, 131.0, 129.6, 129.2, 129.0, 128.7, 126.8, 126.0, 125.8, 122.2, 119.6, 113.6, 106.4, 93.4, 37.7, 8.1; HRMS (ESI) exact mass calcd for C23H19NOSe2 + H (M + H), 485.9870; found: 485.9881.
Conclusions
In summary, we have developed electrochemical direct site-selective C–H mono and bis-sulfenylation and selenylation of indolizine compounds in 40–96% yields. A wide variety of electron-rich and electron-poor disulfides and diselenides were successfully employed to functionalize cyclohexenone/cycloheptenone-fused indolizines and aryl/alkyl-substituted indolizines under the standard conditions of the developed protocol. A radical reaction pathway was also validated with control experiments and cyclic voltammetry data. The simple operating module, avoidance of unpleasant odor, and photocatalytic-free, catalyst-free and oxidant-free protocol with high functional group tolerance are the significant features of this electrochemical protocol.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The SERB-DST, New Delhi (CRG/2020/003634) is gratefully acknowledged for financial support. PKJ thanks CSIR-India for her fellowship. LY thanks SERB-DST for his fellowship. We thank MRC-MNIT Jaipur for NMR data collection and the Department of Chemistry, BITS-Pilani, Pilani Campus, India for HRMS data collection.
References
-
(a) E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538 CrossRef CAS PubMed;
(b) G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 5237 CrossRef CAS PubMed.
- V. Sharma and V. Kumar, Med. Chem. Res., 2014, 23, 3593 CrossRef CAS.
-
(a) C. Bailly, Pharmcol. Res., 2019, 148, 104398 CrossRef CAS PubMed;
(b) G. J. Creemers, B. Lund and J. Verweij, Cancer Treat. Rev., 1994, 20, 73 CrossRef CAS PubMed.
-
(a) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed;
(b) C. Zhao, K. P. Rakesh, L. Ravidar, W. Y. Fang and H. L. Qin, Eur. J. Med. Chem., 2019, 162, 679 CrossRef CAS PubMed;
(c) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2019, 1 Search PubMed;
(d) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed;
(e) Z. W. Chen, R. Bai, P. Annamalai, S. S. Badsara and C. F. Lee, New J. Chem., 2022, 46, 15 RSC;
(f) P. Devendar and G. F. Yang, Sulfur Chem., 2019, 35 Search PubMed;
(g) D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486 CrossRef CAS PubMed;
(h) H. Liu and X. Jiang, Chem. – Asian J., 2013, 8, 2546 CrossRef CAS PubMed;
(i) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS PubMed;
(j) G. Mugesh, W. W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125 CrossRef CAS PubMed.
-
(a)
G. Hynd, J. G. Montana, H. Finch, M. C. Cramp, J. Gold and G. Carnevale, WO2009044134, 2009;
(b) R. Pettipher and M. Whittaker, J. Med. Chem., 2012, 55, 2915 CrossRef CAS PubMed;
(c)
J. Gubin, P. Chatelain, M. Descamps, D. Nisato, H. Inion, J. Lucchetti, J. M. Mahaux and J. N. Vallat, U.S. Patent, 4957925A, 1990 Search PubMed;
(d) J. Gubin, J. Lucchetti, J. Mahaux, D. Nisato, G. Rosseels, M. Clinet, P. Polster and P. Chatelain, J. Med. Chem., 1992, 35, 981 CrossRef CAS PubMed;
(e) J. Gubin, H. de Vogelaer, H. Inion, C. Houben, J. Lucchetti, J. Mahaux, G. Rosseels, M. Peiren and M. Clinet, J. Med. Chem., 1993, 36, 1425 CrossRef CAS PubMed.
-
(a) K. Nishino, S. Tsukahara, Y. Ogiwara and N. Sakai, Eur. J. Org. Chem., 2019, 2019, 1588 CrossRef CAS;
(b) F. Penteado, L. Bettanin, K. Machado, G. Perin, D. Alves and E. J. Lenardão, Asian J. Org. Chem., 2020, 9, 1631 CrossRef CAS.
-
(a) B. Li, Z. Chen, H. Cao and H. Zhao, Org. Lett., 2018, 20, 3291 CrossRef CAS PubMed;
(b) S. S. Badsara, C.-C. Chan and C.-F. Lee, Asian J. Org. Chem., 2014, 3, 1197 CrossRef CAS.
-
(a) F. Penteado, C. S. Gomes, L. I. Monzon, G. Perin, C. C. Silveira and E. J. Lenardão, Eur. J. Org. Chem., 2020, 2020, 2110 CrossRef CAS;
(b) Y. Zhu, L. Yang, L. Fang, X. Du, K. Chen, C. Yu and X. Jiang, Tetrahedron Lett., 2020, 61, 152368 CrossRef CAS;
(c) J.-C. Liou, S. S. Badsara, Y.-T. Huang and C.-F. Lee, RSC Adv., 2014, 4, 41237 RSC.
-
(a) G. M. Martins, G. C. Zimmer, S. R. Mendes and N. Ahmed, Green Chem., 2020, 22, 4849 RSC;
(b) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC;
(c) T. V. Münchow, S. Dana, Y. Xu, B. Yuan and L. Ackermann, Science, 2023, 379, 1036 CrossRef PubMed;
(d) Y. Wu, H. Ding, M. Zhao, Z. H. Ni and J. P. Cao, Green Chem., 2020, 22, 4906 RSC;
(e) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC.
-
(a) G. M. Martins, A. G. Meirinho, N. Ahmed, A. L. Braga and S. R. Mendes, ChemElectroChem, 2019, 6, 5928 CrossRef CAS;
(b) Z.-W. Hou, L. Li and L. Wang, Org. Chem. Front., 2022, 9, 2815 RSC;
(c) Q. Wan, Z. Zhang, Z.-W. Hou and L. Wang, Org. Chem. Front., 2023, 10, 2830 RSC.
-
(a) K. Ucheniya, A. Chouhan, L. Yadav, P. K. Jat and S. S. Badsara, J. Org. Chem., 2023, 88, 6096 CrossRef CAS PubMed;
(b) P. K. Jat, L. Yadav, A. Chouhan, K. Ucheniya and S. S. Badsara, Chem. Commun., 2023, 59, 5415 RSC;
(c) P. K. Jat, K. K. Dabaria, R. Bai, L. Yadav and S. S. Badsara, J. Org. Chem., 2022, 87, 12975 CrossRef CAS PubMed;
(d) L. Yadav, M. Maneesha, K. K. Dabaria, P. K. Jat, A. Gurjar and S. S. Badsara, Synthesis, 2022, 54, 5479 CrossRef CAS;
(e) B. K. Malviya, P. K. Jaiswal, V. P. Verma, S. S. Badsara and S. Sharma, Org. Lett., 2020, 22, 2323 CrossRef CAS PubMed.
-
(a) K. Liu, C. Song and A. Lei, Org. Biomol. Chem., 2018, 16, 2375 RSC;
(b) H. Zhang, H. Wang, Y. Jiang, F. Cao, W. Gao, L. Zhu, Y. Yang, X. Wang, Y. Wang, J. Chen and Y. Feng, Chem. – Eur. J., 2020, 26, 17289 CrossRef CAS PubMed.
- D. Basavaiah and A. J. Rao, Tetrahedron Lett., 2003, 44, 4365 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*