
Synthesis and solution properties of telechelic poly(2-isopropyl-2-oxazoline) bearing perfluoro end groups†
Na
Xue
 a,
Xiaoming
Hou
b,
Xing-Ping
Qiu
c,
Xiaotao
Song
b,
Qingguo
Feng
*b and
Xiaozhi
Liu
*a
a,
Xiaoming
Hou
b,
Xing-Ping
Qiu
c,
Xiaotao
Song
b,
Qingguo
Feng
*b and
Xiaozhi
Liu
*a
aTianjin Key Laboratory of Epigenetics for Organ Development of Preterm Infants, Central Laboratory, Tianjin Fifth Central Hospital, Tianjin 300450, China. E-mail: lxz7997@126.com
bDepartment of Critical Care Medicine, Tianjin Fifth Central Hospital, Tianjin 300450, China. E-mail: anginagi@163.com
cDepartment of Chemistry, University of Montreal, CP6128 Succursale Centre Ville, Montreal, QC H3C 3J7, Canada
First published on 9th November 2022
Abstract
Amphiphilic telechelic poly(2-isopropyl-2-oxazoline) (Mn = 7.7 kg mol−1) end-capped with 1H,1H,2H,2H-perfluoro-1-decanyl end groups (FPIPOZ) was prepared through cationic ring-opening polymerization followed by the ligation of alkyne–azide cycloaddition. At room temperature, FPIPOZ self-assembled into flower-like micelles in water above cac (0.35 g L−1). In comparison with its precursor polymer N3PIPOZ, the FPIPOZ aqueous solution had a lower Tc, a reduced phase transition enthalpy, and a much lower degree of crystallization. When exposed to THF vapor, an initially smooth FPIPOZ film reassembled into discs comprised of multiple lamella layers on silicon wafers, according to a THF vapor annealing experiment. This is the first observation of such an assembly pattern for PIPOZ. The N3PIPOZ films, on the other hand, generated fibers on the surface upon exposure to THF vapor. The distinct surface assembly behaviors of FPIPOZ and its precursor N3PIPOZ during solvent annealing added to our knowledge of polyoxazoline phase behaviors.
Introduction
Poly(2-isopropyl-2-oxazoline) (PIPOZ) is a thermo-responsive polymer that undergoes phase separation at the cloud point temperature (Tc) of around 40 °C.1,2 A long time heating treatment of PIPOZ aqueous solutions results in the formation of micron-sized particles composed of nanofibers.3–5 TEM, optical microscopy, IR, NMR, light scattering, XRD, and other techniques have been used to investigate the phase separation process of PIPOZ in hot water.3,6–11 A liquid–liquid phase separation followed by the crystallization mechanism of this process has been broadly accepted, which involves the conformational change of the backbone and packing of the PIPOZ chains through dipolar and hydrophobic interactions.7,9,12–15 The effect of hydrophobic end group modification on the Tc of PIPOZ has been examined.16–19 However, the end groups used in most of these trials were so bulky that they significantly hampered the assembly and phase separation of PIPOZ by the steric effect and diverted people's attention. Therefore, more research on the phase behavior of PIPOZ is required, taking into account the influencing factors such as the hydrophobicity and structure of functional groups.Because of their chemical and biological inertness, thermal stability, and oleophobicity, fluorocarbon surfactants outperform their hydrocarbon counterparts in synthetic blood, liquid ventilation agents, imaging agents, cleaners, cosmetics, fire-fighting foams, adhesives, and antifogging and antistatic agents among other applications.20–22 To take advantage of their unique properties, fluorocarbon segments have been widely used to functionalize polymer chains.23–25 Fluorinated polymerizable hydrogels with a chitosan main chain and fluorinated side groups have been utilized for wound dressing by delivering dissolved oxygen or other small oxygenated molecules upon exposure to a lower-tension environment.22 Amphiphilic nanocomplexes were prepared by grafting fluorocarbons onto hyaluronic acid chains, which formed micelles in water and acted as O2 delivering vehicles.23 Different kinds of fluorinated 2-oxazolines have been used to fabricate block polymers with specific micellization properties.26–31 Jodan et al.26 studied the self-assembly of PMeOx-b-PnonOx and PmeOxb-2-(1H,1H′,2H,2H′-perfluorohexyl)-2-oxazoline, which formed spherical and slightly elongated shapes, respectively. Triblock polymers containing a 2-(2,6-difluorophenyl)-2-oxazoline block were reported to self-assemble into flat rolled-up cylindrical micelles and vesicles.29 To synthesize fluorinated PIPOZ while retaining its phase transition characteristics and investigate the effects of fluorocarbon modification on the phase behavior of PIPOZ must be enlightening.
In this work, we synthesized telechelic poly(2-isopropyl-2-oxazoline) (Mn = 7.7 kg mol−1) bearing two 1H,1H,2H,2H-perfluoro-1-decanyl end groups (FPIPOZ). The influence of fluorocarbon functionalization on the properties of PIPOZ was examined in solution and on the surface. The telechelic FPIPOZ assembled into flower-like micelles in water and featured a lower Tc and a reduced phase transition enthalpy in comparison with its precursor N3PIPOZ (PIPOZ bearing two azide end groups). In hot water, FPIPOZ exhibits almost no crystallization behavior. In contrast to N3PIPOZ, a THF vapor annealing investigation revealed a slower crystallization and distinct morphologies of FPIPOZ films via different approaches. Our study offers a viable way to modify the physiochemical characteristics of polyoxazoline. The different assembly approaches of telechelic FPIPOZ and its pristine analogue enrich our knowledge about the phase behaviors of polyoxazolines.
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich Chemicals Co. and used as received unless otherwise stated. 2-Isopropyl-2-oxazoline was prepared from isobutyric acid and 2-aminoethanol following a known procedure.32 Diethylene glycol di(p-toluenesulfonate) was synthesized according to a reported procedure and purified by recrystallization from methanol.33 Acetonitrile was dried under reflux over CaH2 under a dry nitrogen atmosphere and distilled prior to use. Water was deionized with a Millipore Milli-Q system. THF was purified using a solvent purification system. Silicon wafers were purchased from Hefei Kejing Materials and Technology Co. and cleaned with piranha acid (VH2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VH2O2 = 7:3) before use.
:VH2O2 = 7:3) before use.
Synthetic procedures
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2O–), 3.85 (t, –OCH2CH2CF2–), and 4.20 (s, HCCCH2O–).
CCH2O–), 3.85 (t, –OCH2CH2CF2–), and 4.20 (s, HCCCH2O–).
Methods
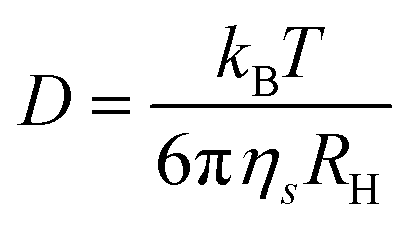 | (1) |
For SLS experiments, the apparent weight-average molar mass (Mw,app) and the z-average root-mean square radius of gyration (Rg) of the scattering objects in dilute solution are evaluated according to the angular dependence of the excess absolute scattering intensity known as the excess Rayleigh ratio R(q,c) given by eqn (2):
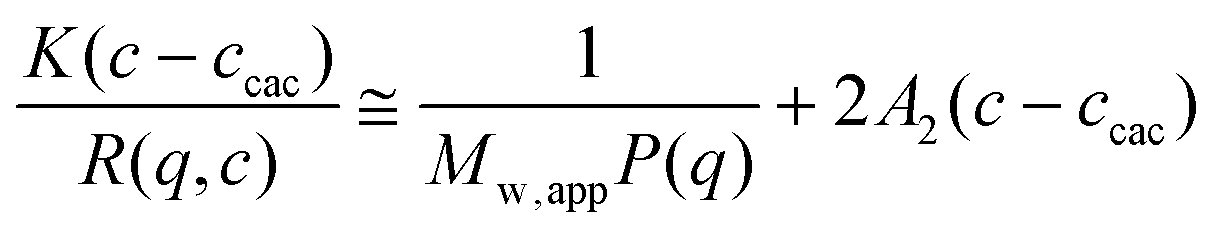 | (2) |
Results and discussion
Polymer synthesis and characterization
In previous studies of α,ω-di n-octadecyl-poly(2-isopropyl-2-oxazolines), the two octadecyl end groups were introduced onto a polymer chain consecutively; one group was attached to the initiator and the second group was inserted subsequently by post-modification.35 This approach resulted in pairs of semitelechelic and telechelic polymers of identical molar mass, which allowed us to compare and contrast the solution properties of star micelles and flower micelles. Because the two classes of polymers have comparable solubility characteristics, this synthetic strategy suffers from drawbacks related to the purification of telechelic polymers contaminated by residual semitelechelic polymers. The approach chosen here entails utilizing a difunctional initiator to synthesize α,ω-di-azido-poly(2-isopropyl-2-oxazoline) (N3PIPOZ), followed by “click” ligation of the end groups (Scheme 1). This route benefits from the high conversion yield characteristic of the azide–alkyne Huisgen cycloaddition and allows for the production of polymers with identical chain lengths but different end groups, as demonstrated here by the preparation of α,ω-di-(1H,1H,2H,2H-perfluoro-1-decanyl)-PIPOZ (FPIPOZ).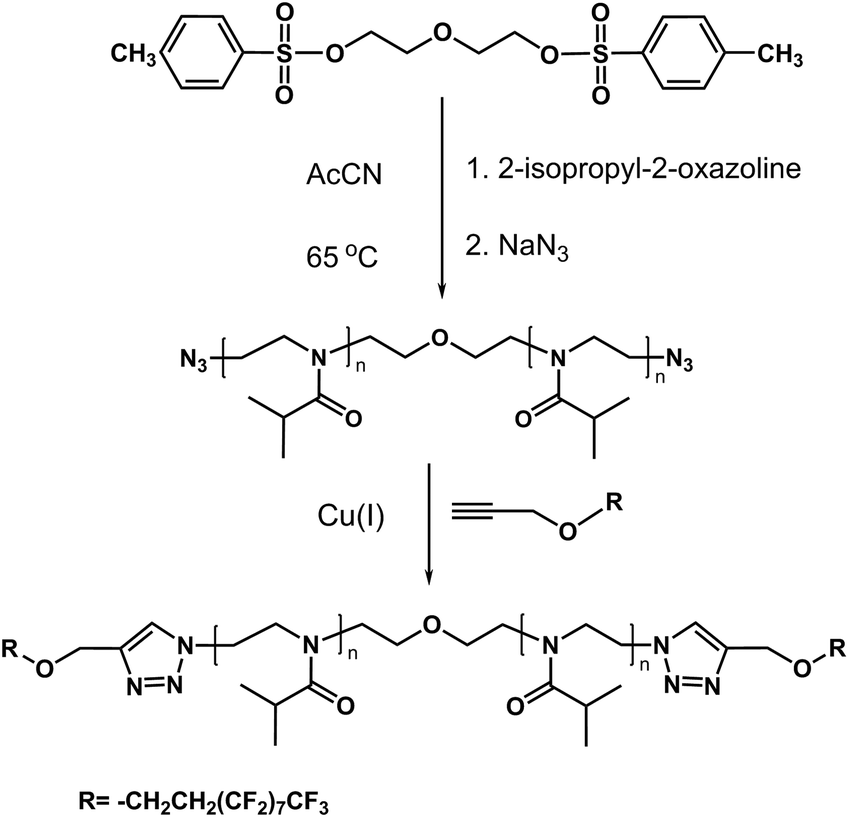 | ||
| Scheme 1 Synthetic procedure of α,ω-di-(1H,1H,2H,2H-perfluoro-1-decanyl) poly(2-isopropyl-2-oxazoline). | ||
The polymerization of 2-isopropyl-2-oxazoline was started with a bifunctional initiator, diethylene glycol di(p-toluenesulfonate), which resulted in a chain with propagating species on both ends.36 The polymerization was quenched by adding azide. The successful linkage of azide to the polymer chain was confirmed by the FT-IR spectrum of N3PIPOZ, which presents a moderately strong –N3 vibration at 2105 cm−1 (Fig. 1, bottom trace). The 1H NMR spectrum of N3PIPOZ (Fig. 2, bottom trace) confirmed the success of the polymerization. The molar mass Mw and dispersity (Đ) of N3PIPOZ were 7.6 kDa and 1.12, respectively, as determined by GPC. The Đ value is slightly larger than that expected for living polymerizations. Such a phenomenon is usually encountered for CROP polymerization initiated by alkyl (p-toluenesulfonates).38
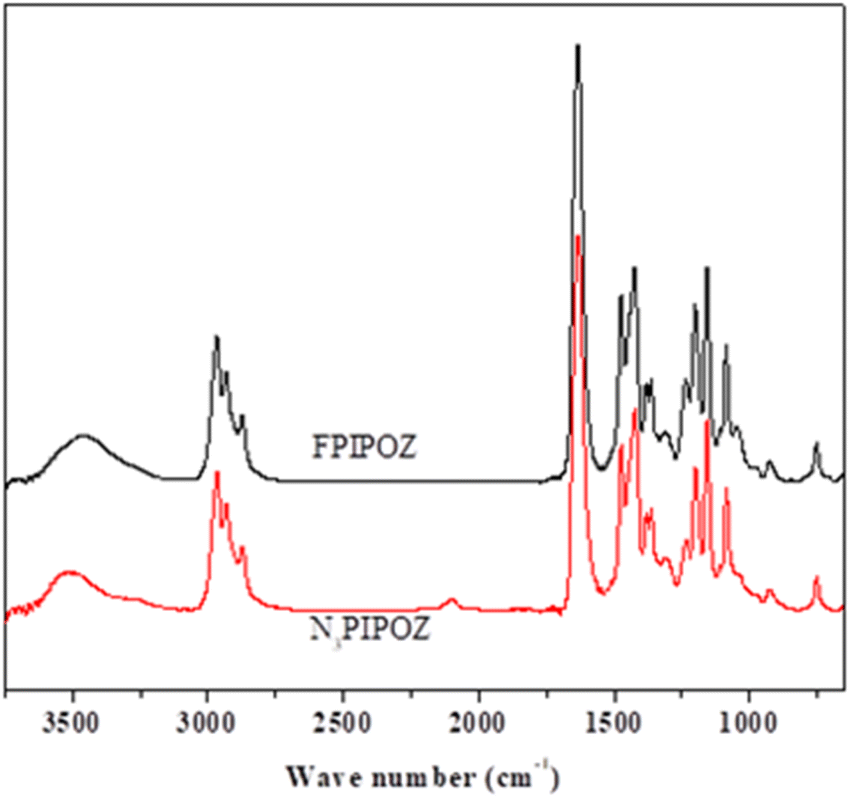 | ||
| Fig. 1 FT-IR spectra of telechelic FPIPOZ (upper trace) and N3PIPOZ (lower trace). | ||
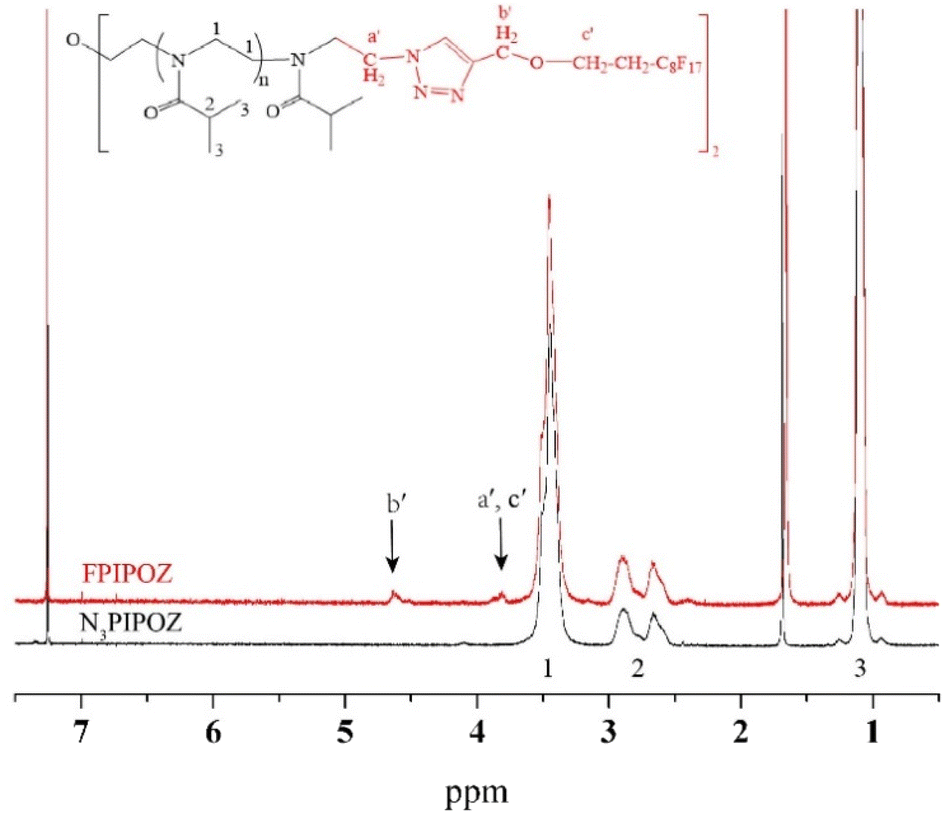 | ||
| Fig. 2 1H NMR spectra of N3PIPOZ (lower trace) and telechelic FPIPOZ (upper trace) in CDCl3. The arrows denote the signals used to calculate Mn. | ||
The ‘click’ ligation between the terminal azido groups of N3PIPOZ and 1H,1H,2H,2H-perfluoro-1-decanol propargyl ether was performed in DMF at room temperature using Cu(I) as the catalyst. The molar mass and dispersity of the resulting polymers, determined by GPC, were nearly identical to those of N3PIPOZ (Table 1 and Fig. S2†). The completion of this reaction was confirmed by the elimination of the band at 2105 cm−1 due to azide stretching in the FT-IR spectra of FPIPOZ (Fig. 1) and the appearance of resonances around 4.6 and 3.8 ppm originating from the methylene groups of the corresponding end groups in the 1H NMR spectra (Fig. 2). The aromatic triazole ring downfield shifts the adjacent methylene group from 4.21 ppm (original –OCH2CCH) to 4.66 ppm (–OCH2-triazole) in FPIPOZ (group b′). The nearby methylene group (–CH2-OCH2-triazole) was also downfield shifted to 3.85 ppm (signal a′) for FPIPOZ. The ratio of the integrated values of signal b′ with respect to that of signal 1 of the backbone methylene groups (Fig. S3†) yielded a polymerization degree close to that measured by GPC-MALLS, indicating that the hydrophobic end functionality of FPIPOZ was nearly quantitative. Furthermore, the near overlapping of the GPC traces of N3PIPOZ before and after the end capping reaction demonstrates that the ‘click’ ligation of the end groups did not affect the molecular weight and dispersity of the precursor.
FPIPOZ micellization and heat-induced phase transition
The telechelic FPIPOZ was easily soluble in water at room temperature, yielding a clear solution. Spherical or oval micelles were observed for FPIPOZ aqueous solutions as depicted in the inset of Fig. 3a. The average radius of the micelles in the image was 7.3 nm, which was smaller than the hydrodynamic radius (RH, 8.8 nm) of the micelles in water, as determined by dynamic light scattering. It makes sense because the hydrophilic corona of the micelles dehydrated during sample preparation for TEM measurement. In the studied concentration range (1.0 to 10.0 g L−1), a unimodal size distribution was detected for FPIPOZ micelles as shown in Fig. S4a.† The RH values of these micelles at different angles were almost identical, indicating that the flower micelles were isotropic, possibly flexible spherical shapes. SLS measurements provided more information about the flower-like micelles, including the apparent weight-average molar mass Mw,app of the micelles, the radius of gyration Rg, Rg/RH, and the aggregation number, Nagg, in terms of the number of polymer chains per micelles.37 An Nagg value of 19 was determined for the FPIPOZ micelle in a 1.0 g L−1 solution, which was close to the reported value (17) for a telechelic PEO (Mn = 10 kDa) end-capped with the same end group (C8F17C2H4–) using SANS.39 It is well known that the shape factor, Rg/RH, provides information on the shape of polymer assemblies in solution. The Rg/RH value of the FPIPOZ micelle in a 1.0 g L−1 solution was found to be 1.38, suggesting a disordered organization of the micelle loops.40 The critical micellization concentration (cmc) value of FPIPOZ in water was determined by recording their light scattering intensities as a function of polymer concentration (Fig. 3a). The scattering intensity remained constant as the polymer concentration grew up to cac, beyond which it increased steadily, indicating the emergence of massive assemblies. For FPIPOZ, the cac was 0.35 g L−1, as estimated by the intersection of two extrapolate plots. A study on telechelic PEO end-capped with C16H33 groups showed a comparable cac value of 0.2 g L−1 based on the concentration dependence of scattering intensities.41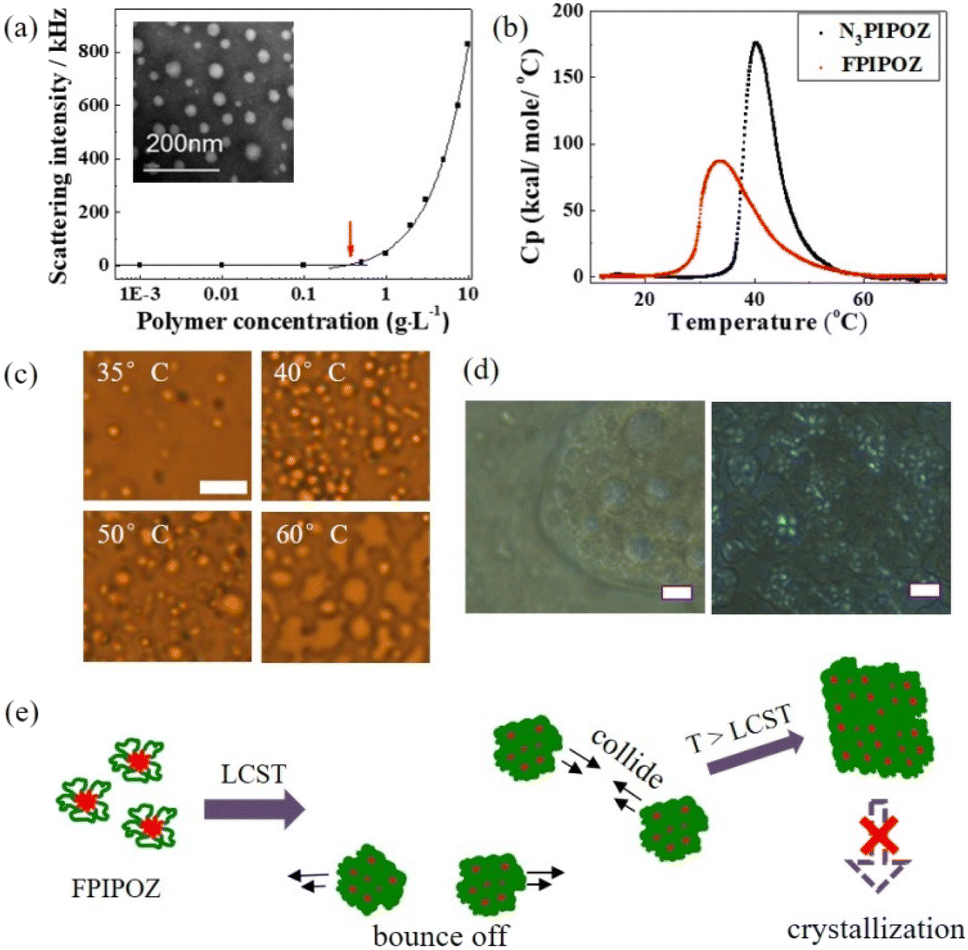 | ||
| Fig. 3 Solution properties of an FPIPOZ aqueous solution. (a) The cmc of FPIPOZ determined by the changes in the scattering intensity of the assemblies at 24 °C. The arrow indicates the cmc value. A TEM image of the solution is shown in the inset. (b) DSC results of an FPIPOZ aqueous solution at 1.0 g L−1. (c) Optical images of an FPIPOZ aqueous solution (0.8 g L−1) on a glass slide at 35 °C, 40 °C, 50 °C and 60 °C, respectively. The magnification on all panels is the same (scale bar, 10 μm). (d) Optical micrographs of FPIPOZ and N3PIPOZ aqueous solutions (5.0 g L−1) incubated for 1 day at 60 °C through crossed Nichols polarizers. The sporadic blue hues in the image of the FPIPOZ sample indicates a very low degree of crystallinity, whereas a large number of blue hues for the N3PIPOZ sample imply a high degree of crystallinity. The magnification of all panels is the same (scale bar, 10 μm). (e) Schematic presentation of FPIPOZ phase transition in water. There was no evidence of crystallization. | ||
The thermoresponses of the FPIPOZ micelles in water were studied by UV, light scattering, optical microscopy, and HS-DSC. For a 1.0 g L−1 FPIPOZ aqueous solution, the turbidity measurements revealed a phase transition temperature (Tc) of 30.3 °C. Fig. S5† depicts the turbidity curves of FPIPOZ and N3PIPOZ in water. FPIPOZ had a lower Tc than its unmodified precursor by 9 °C. Fig. S4b† shows the RH, Rg and Rg/RH values for FPIPOZ in water at a concentration of 0.8 g L−1 as the solution was gradually heated from 10 to 60 °C. Above Tc, the solution became milky, and micro-sized aggregates appeared, as shown by the shaded area in Fig. S4b,† which were also probed by optical microscopy (see Fig. 3c). The temperature dependency of the RH and Rg values of FPIPOZ in water was similar, with near constant values of 9 nm below Tc, micrometer-sized aggregates at Tc, and greater values above 40 °C. The corona of the telechelic FPIPOZ micelles collapsed during the dehydration process at Tc. The hydrophobic interaction between collapsed chains resulted in micrometer-sized aggregates with an abrupt increase in RH and Rg. As the temperature was gradually elevated above 50 °C, the telechelic FPIPOZ formed aggregates with constant RH values of 140 nm and a very narrow size distribution. The Rg/RH ratio for FPIPOZ fell from 1.36 below Tc to 0.81 at 60 °C, typical of block copolymer aggregates.
To investigate the disappearance of micrometer-sized particles detected by light scattering experiments above 40 °C, an FPIPOZ aqueous solution at 0.8 g L−1 was examined by optical microscopy upon heating. Fig. 3c shows the images of the FPIPOZ sample after equilibration for 1 h at a certain temperature. Droplets with diameters of ca. 1 μm emerged from the aqueous solution at 35 °C, which grew in size by coalescence and gradually settled down on the bottom glass slide, reaching an average of ca. 7 μm at 60 °C. Fig. 3d shows large droplets (2−50 μm) observed by optical microscopy from a 5.0 g L−1 FPIPOZ solution (left panel). The substantially larger droplets (20–50 μm) reported by Katsumoto et al. for unmodified PIPOZ at 50 °C must be related to the much higher polymer concentration adopted in their study.7 A recent SANS analysis of micelles formed by the PS-PNIPAM-PS (polystyrene-b-poly(N-isopropylacrylamide-b-polystyrene) copolymer demonstrated that micelle aggregation growth was governed by a diffusion-limited model for low concentration samples at shallow targeting temperatures (just above Tc).42 The small droplets that appeared just above Tc of FPIPOZ in our study were likely due to the limited effective collision of droplets to coalesce in the low concentration regime (0.8 g L−1). Fig. 3d shows the images of the FPIPOZ and N3PIPOZ aqueous solutions (5.0 g L−1) incubated at 60 °C for 1 day under polarized light. The sporadic blue hues in the image of the FPIPOZ sample indicated a quite low degree of crystallinity, whereas the large number of blue hues for the N3PIPOZ sample implied a high degree of crystallinity. The diffractogram (Fig. S6†) of the FPIPOZ sample recovered from the aqueous solution after heating at 65 °C for 1 day exhibited two weak peaks at 2θ = 8.08° and 18.1°. In contrast to the reported diffractogram of pristine PIPOZ,3 the weak peaks of FPIPOZ demonstrated a low degree of crystallinity. According to Katsumoto's7 study, the crystallization of pristine PIPOZ beyond 50 °C required all-trans conformation of the polymer chain, which started to occur at a lower temperature, rendering the phase transition of PIPOZ irreversible. In our study, however, at 60 °C, the constraint imposed by the FPIPOZ micelle structure precluded the all-trans configuration of the backbone and the alignment of polymer chains through hydrophobic and dipolar interactions,3 resulting in a very low degree of crystallinity. The phase separation process of the FPIPOZ aqueous solution after a long-term heating treatment is schematically shown in Fig. 3e, which included the micelle collapse and aggregation growth steps without crystallization. As demonstrated in Fig. 3b, the end-group constraint of FPIPOZ was also manifested by a lower phase transition enthalpy ΔH (4.61 kJ mol−1) and lower Tm (temperature of the absorption maximum, 33.9 °C) compared to its precursor N3PIPOZ (ΔH = 5.4 kJ mol−1, Tm = 40.2 °C). When compared to N3PIPOZ, the reduced accessibility of water molecules for the main chain segments adjacent to the hydrophobic end groups leads to the reduced hydration of the FPIPOZ backbone, resulting in a lower ΔH.
FPIPOZ and N3PIPOZ films annealed by THF vapor at 22 °C
Fig. 4 showed the morphologies of the FPIPOZ and N3PIPOZ films after annealing in THF vapor for 1/2, 3 and 7 h at 22 °C. The thicknesses of the spin-coated FPIPOZ and N3PIPOZ films were found to be 30.9 nm and 30.1 nm, respectively, by scratching experiments (Fig. S7†). Fibers sprouted from seeding centers were distributed randomly across the surface of the N3PIPOZ films and grew in number. The fibers had an average height of 21 nm and a width of 21–80 nm. In the all-trans configuration, the experimental periodicity along the polyoxazoline backbone is 6.4 Å.43 The average contour length of N3PIPOZ consisting of 68 repeating units is approximately 22 nm, which is in accordance with the height of the N3PIPOZ fibers, implying that the fibers are packed similarly to polyoxazoline fibers crystallized from hot water. The home-made annealing apparatus protected the films from exposure to the atmosphere during sample preparation. The shorter length of the N3PIPOZ fibers compared to those formed in hot water in the literature,3ca. 0.2–0.6 μm, was most likely due to the weak hydrophobic and dipolar interactions between the polymer chains as well as the diffusion limited aggregation during their assembly on the surface when exposed to THF vapors.44,45 In the case of FPIPOZ films, large aggregates of 10–30 nm in height and 0.2–0.6 μm in diameter were formed upon spin coating due to dewetting of the films. After being annealed in THF vapor for 2 hours, the peripherals of the aggregates became serrated as shown in Fig. 4b left panel. The serrated aggregates had a thickness of ca. 22 nm (Fig. S8†), implying the assembly of fibril structures. Increasing the THF vapor exposure time, the serrated aggregates developed into discs of 2–3 μm in diameter (see Fig. 4 middle and right panels). Fig. S8† shows the disc heights ranging from 20 to 80 nm, with 20 nm representing the most population. Independent crystallizations of both the fluorophilic end groups and hydrophilic backbones were most likely to be responsible for disc self-assembly. THF annealed both segments throughout the exposure process as a common solvent for the end groups and backbones of FPIPOZ, resulting in the crystallization of both segments. The fluorophilic characteristics of the end groups caused them to stick together during crystallization, suggesting the lamellar arrangement of the discs. Because of the interactions between the fluorophilic end groups and the polyoxazoline backbones, the discs have various heights (ca. 20, 40, 60 and 80 nm), signifying multiple lamella layers. Fig. 4c schematically illustrates the assembly process of FPIPOZ films during THF vapor annealing.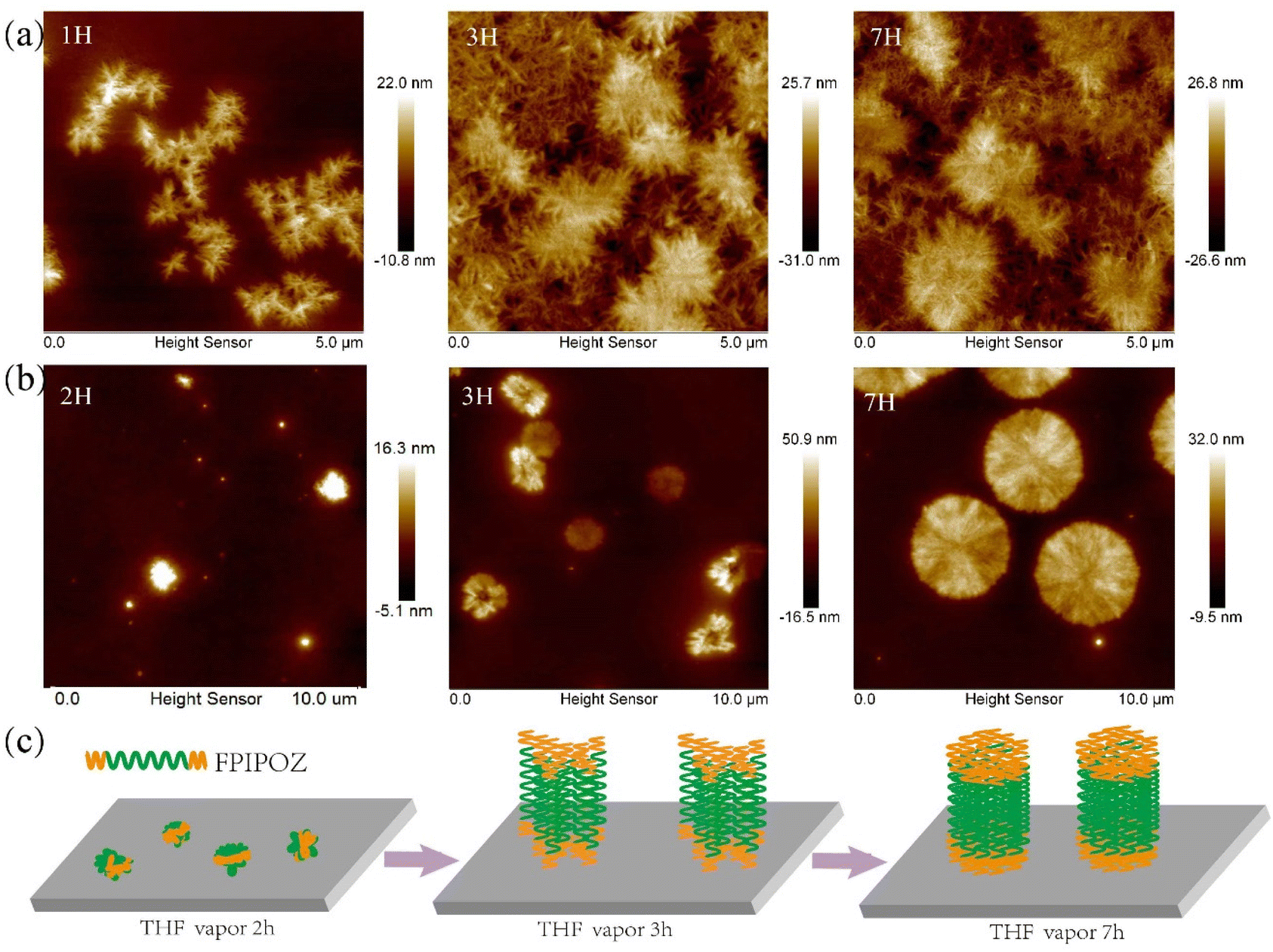 | ||
| Fig. 4 Morphologies of the (a) N3PIPOZ and (b) FPIPOZ films after annealing in THF vapor at 70% saturation for 1/2, 3 and 7 hours, respectively, at 22 °C. (c) Schematic presentation of the assembly process of FPIPOZ films during THF vapor annealing. | ||
Conclusions
We have demonstrated that telechelic α,ω-diazido-polyoxazoline with nearly quantitative end functionality and narrow distribution can be readily synthesized by CROP using diethylene glycol di(p-toluenesulfonate) as a bifunctional initiator and NaN3 as the terminating agent. The azido end groups of the polymers are viable for further functionalization via the efficient alkyne–azide ‘click’ cycloaddition. Perfluorodecanyl end-functionalized telechelic polyoxazoline (FPIPOZ) was thus prepared, which formed flower-like micelles in water. The perfluoro-end-group functionalization affected the phase transition behavior of polyoxazoline and inhibited its crystallization in hot water. A THF vapor annealing experiment revealed the distinct morphologies of the FPIPOZ films generated through different assembly approaches compared to their unmodified precursor, providing new insights into the functionalization and phase behaviors of polyoxazolines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was funded by the National Natural Science Foundation of China (NSFC) (No. 22002109) and the Tianjin Municipal Health Commission (No. KJ20163). The solvent annealing experiments were performed in Prof. Sabine Ludwigs’ Lab at the Institute of Polymer Chemistry, University of Stuttgart. The authors are grateful to Prof. Ludwigs for the constructive discussions and advices.References
- S. Hageneder, V. Jungbluth, R. Soldo, C. Petri, M. Pertiller, M. Kreivi, A. Weinhäusel, U. Jonas and J. Dostalek, ACS Appl. Mater. Interfaces, 2021, 13, 27645–27655 CrossRef CAS.
- T. Sezonenko, X.-P. Qiu, F. M. Winnik and T. Sato, Macromolecules, 2019, 52, 935–944 CrossRef.
- A. L. Demirel, M. Meyer and H. Schlaad, Angew. Chem., Int. Ed., 2007, 46, 8622–8624 CrossRef CAS PubMed.
- M. Meyer, M. Antonietti and H. Schlaad, Soft Matter, 2007, 3, 430–431 RSC.
- N. Oleszko-Torbus, A. Utrata-Wesołek, M. Bochenek, D. Lipowska-Kur, A. Dworak and W. Wałach, Polym. Chem., 2020, 11, 15–33 RSC.
- N. Oleszko, W. Wałach, A. Utrata-Wesołek, A. Kowalczuk, B. Trzebicka, A. Klama-Baryła, D. Hoff-Lenczewska, M. Kawecki, M. Lesiak, A. L. Sieroń and A. Dworak, Biomacromolecules, 2015, 16, 2805–2813 CrossRef CAS.
- Y. Katsumoto, A. Tsuchiizu, X. Qiu and F. M. Winnik, Macromolecules, 2012, 45, 3531–3541 CrossRef CAS.
- R. Konefał, P. Černoch, M. Konefał and J. Spěváček, Polymers, 2020, 12, 1879 CrossRef.
- T. Li, H. Tang and P. Wu, Langmuir, 2015, 31, 6870–6878 CrossRef CAS.
- N. Oleszko-Torbus, Polym. Rev., 2022, 62, 529–548 CrossRef CAS.
- C. Gourier, E. Beaudoin, M. Duval, D. Sarazin, S. Maître and J. François, J. Colloid Interface Sci., 2000, 230, 41–52 CrossRef CAS PubMed.
- M. M. Bloksma, R. M. Paulus, H. P. van Kuringen, F. van der Woerdt, H. M. Lambermont-Thijs, U. S. Schubert and R. Hoogenboom, Macromol. Rapid Commun., 2012, 33, 92–96 CrossRef CAS PubMed.
- J. Zhao, R. Hoogenboom, G. V. Assche and B. V. Mele, Macromolecules, 2010, 43, 6853–6860 CrossRef CAS.
- C. Diehl, P. Černoch, I. Zenke, H. Runge, R. Pitschke, J. Hartmann, B. Tiersch and H. Schlaad, Soft Matter, 2010, 6, 3784–3788 RSC.
- S. Sun and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 32232–32240 RSC.
- A. I. Amirova, T. Y. Kirile, A. V. Ten'kovtsev and A. P. Filippov, Fibre Chem., 2018, 50, 293–296 CrossRef CAS.
- J. Lee, J. M. Park and W.-D. Jang, Carbohydr. Polym., 2019, 221, 48–54 CrossRef CAS PubMed.
- J. M. Park, Y. J. Kim and W.-D. Jang, ACS Appl. Polym. Mater., 2020, 2, 3535–3542 CrossRef CAS.
- S. Huber, N. Hutter and R. Jordan, Colloid Polym. Sci., 2008, 286, 1653–1661 CrossRef CAS.
- J. Eastoe, A. Downer, A. Paul, D. C. Steytler, E. Rumsey, J. Penfold and R. K. Heenan, Phys. Chem. Chem. Phys., 2000, 2, 5235–5242 RSC.
- X. Wang, J. Zhang, X. Li, Y. Liu, H. Yang, X. Zhao, L. Xie and L. Yin, Clin. Respir. J., 2014, 8, 160–166 CrossRef CAS PubMed.
- J. K. Weigel, D. Steinmann, P. Emerich, C. A. Stahl, D. V. Elverfeldt and J. Guttmann, Physiol. Meas., 2011, 32, 251–262 CrossRef PubMed.
- N. Leipzig and A. Wijekoon, WO2013112863A1, 2013.
- J. Li, Y. Xue, J. Tian, Z. Liu, A. Zhuang, P. Gu, H. Zhou, W. Zhang and X. Fan, Carbohydr. Polym., 2020, 237, 116119 CrossRef CAS.
- C. Zhang, S. S. Moonshi, W. Wang, H. T. Ta, Y. Han, F. Y. Han, H. Peng, P. Král, B. E. Rolfe, J. J. Gooding, K. Gaus and A. K. Whittaker, ACS Nano, 2018, 12, 9162–9176 CrossRef CAS PubMed.
- R. Ivanova, T. Komenda, T. B. Bonné, K. Lüdtke, K. Mortensen, P. K. Pranzas, R. Jordan and C. M. Papadakis, Macromol. Chem. Phys., 2008, 209, 2248–2258 CrossRef CAS.
- M. Lobert, H. M. L. Thijs, T. Erdmenger, R. Eckardt, C. Ulbricht, R. Hoogenboom and U. S. Schubert, Chem. – Eur. J., 2008, 14, 10396–10407 CrossRef CAS PubMed.
- M. Lobert, R. Hoogenboom, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5859–5868 CrossRef CAS.
- K. Kempe, R. Hoogenboom, S. Hoeppener, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, Chem. Commun., 2010, 46, 6455–6457 RSC.
- K. Kempe, A. Baumgaertel, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5100–5108 CrossRef CAS.
- U. Mansfeld, S. Hoeppener, K. Kempe, J.-M. Schumers, J.-F. Gohy and U. S. Schubert, Soft Matter, 2013, 9, 5966–5974 RSC.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Thier and H. Hellmann, Angew. Chem., Int. Ed., 1966, 5, 875–888 CrossRef CAS.
- A. Schwabacher, J. W. Lane, M. Schiesher, K. M. Leigh and C. W. Johnson, J. Org. Chem., 1998, 63, 1727–1729 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960–4965 CrossRef CAS.
- R. Obeid, E. Maltseva, A. F. Thünemann, F. Tanaka and F. M. Winnik, Macromolecules, 2009, 42, 2204–2214 CrossRef CAS.
- R. Luxenhofer, M. Bezen and R. Jordan, Macromol. Rapid Commun., 2008, 29, 1509–1513 CrossRef CAS.
- M. W. M. Fijten, C. Haensch, B. M. van Lankvelt, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2008, 209, 1887–1895 CrossRef CAS.
- P. Kujawa, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 3048–3055 CrossRef CAS.
- G. Tae, J. A. Kornfield, J. A. Hubbell and J. Lal, Macromolecules, 2002, 35, 4448–4457 CrossRef CAS.
- M. Bhatt and A. M. Jamieson, Macromolecules, 1988, 21, 3015–3022 CrossRef CAS.
- S. Dai, S. T. Sio, K. C. Tam and R. D. Jenkins, Macromolecules, 2003, 36, 6260–6266 CrossRef CAS.
- J. Adelsberger, I. Grillo, A. Kulkarni, M. Sharp, A. M. Bivigou-Koumba, A. Laschewsky, P. Müller-Buschbaum and C. M. Papadakis, Soft Matter, 2013, 9, 1685–1699 RSC.
- M. Litt, F. Rahl and L. G. Roldan, J. Polym. Sci., Part A-2, 1969, 7, 463–473 CrossRef CAS.
- S. Shin, M. L. Gu, C. Y. Yu, J. Jeon, E. Lee and T. L. Choi, J. Am. Chem. Soc., 2018, 140, 475–482 CrossRef CAS PubMed.
- L. M. Sander, Contemp. Phys., 2000, 41, 203–218 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional measurements. See DOI: https://doi.org/10.1039/d2py00815g |
| This journal is © The Royal Society of Chemistry 2023 |