
Precision syntheses of poly(NIPAM-alt-HEMA) and effects of the alternating sequence on thermoresponsive behaviors in water†
Xiaoyan
Xu
,
Kentaro
Shibata
and
Makoto
Ouchi
 *
*
Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: ouchi.makoto.2v@kyoto-u.ac.jp
First published on 18th November 2022
Abstract
In this work, an alternating copolymer of 2-hydroxyethyl methacrylate (HEMA) and N-isopropylacrylamide (NIPAM) [poly(NIPAM-alt-HEMA)] was synthesized via ATRP-based cyclopolymerization of a divinyl monomer carrying a transformable linker and aminolysis transformation of the resulting cyclopolymer with isopropylamine. The selective propagation in the cyclopolymerization leading to the alternating sequence was supported by the reactivity ratios of the two model monomers cut out from the divinyl monomer. The quantitative transformation and the alternating sequence were confirmed by 1H NMR, FT-IR, and MALDI-TOF-MS. The thus-obtained poly(NIPAM-alt-HEMA) underwent an LCST-type temperature-induced phase transition in water, and the thermoresponsive behavior was different from that of the corresponding statistical copolymers [poly(NIPAM-sta-HEMA)] as well as the homopolymer of NIPAM. The thermoresponsiveness specific to the alternating sequence is likely derived from the structural character that can affect the dehydration behavior, such as no existence of the consecutive NIPAM sequence and the adjacent circumstance of NIPAM units and hydroxy pendants of HEMA.
Introduction
Poly(N-isopropylacrylamide) [poly(NIPAM)] exhibits a temperature-induced phase separation behavior in an aqueous solution, where the macromolecular chains are soluble in water below the lower critical solution temperature (LCST) but become immiscible with water molecules upon heating.1–3 These chains are hydrated via hydrogen bonds between polymer chains and water molecules at low temperature giving the transparent solution, whereas they become hydrophobic due to the destruction of hydrogen bonds in the heating process, making the solution turbid via chain aggregation. The thermal response features are of great interest from the viewpoint of fundamental polymer physics and material applications such as drug delivery and tissue engineering.4,5 The development of precision polymerization has allowed the synthesis of poly(NIPAM)s having well-defined structures, and it has been clarified that the thermal response behaviors are affected by the primary structures, such as end groups,6 tacticity7 and topology.8,9Several approaches for obtaining sequence-controlled copolymers composed of vinyl monomer units have been developed by some research groups in recent years,10–13 but research studies demonstrating sequence-oriented properties/functions are still limited. Our group has reported some examples of syntheses of alternating copolymers made of commodity repeating units, such as (meth)acrylate, styrene, and acrylamide, as well as their unique features different from the corresponding 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 statistical copolymers.14–19 One approach for synthesizing such alternating copolymers relies on two steps including radical cyclopolymerization of a divinyl monomer and post-polymerization modification of the resultant cyclo-repeating units,16–19 as generalized in Fig. 1. Herein, two types of selectivities in the propagation are crucial for realizing the alternating sequence. One is the control of the intramolecular cyclo-propagation of the radical species derived from one vinyl group, competing with intermolecular non-cyclo propagation. This selectivity could be achieved under diluted conditions and/or by spacer design leading to the adjacent circumstance of the two vinyl groups. Another kind of selectivity is required in the intramolecular propagation of the radical species for two vinyl groups of the divinyl monomer after intramolecular cyclo-propagation. The selectivity can be evaluated using the reactivity ratio (r2) of the cut-out model monomers (M1 and M2): the smaller the r2 is, the higher the selectivity is expected. Furthermore, the embedded spacer is designed to be transformable into commodity repeating units via post-polymerization modification.20,21
:1 statistical copolymers.14–19 One approach for synthesizing such alternating copolymers relies on two steps including radical cyclopolymerization of a divinyl monomer and post-polymerization modification of the resultant cyclo-repeating units,16–19 as generalized in Fig. 1. Herein, two types of selectivities in the propagation are crucial for realizing the alternating sequence. One is the control of the intramolecular cyclo-propagation of the radical species derived from one vinyl group, competing with intermolecular non-cyclo propagation. This selectivity could be achieved under diluted conditions and/or by spacer design leading to the adjacent circumstance of the two vinyl groups. Another kind of selectivity is required in the intramolecular propagation of the radical species for two vinyl groups of the divinyl monomer after intramolecular cyclo-propagation. The selectivity can be evaluated using the reactivity ratio (r2) of the cut-out model monomers (M1 and M2): the smaller the r2 is, the higher the selectivity is expected. Furthermore, the embedded spacer is designed to be transformable into commodity repeating units via post-polymerization modification.20,21
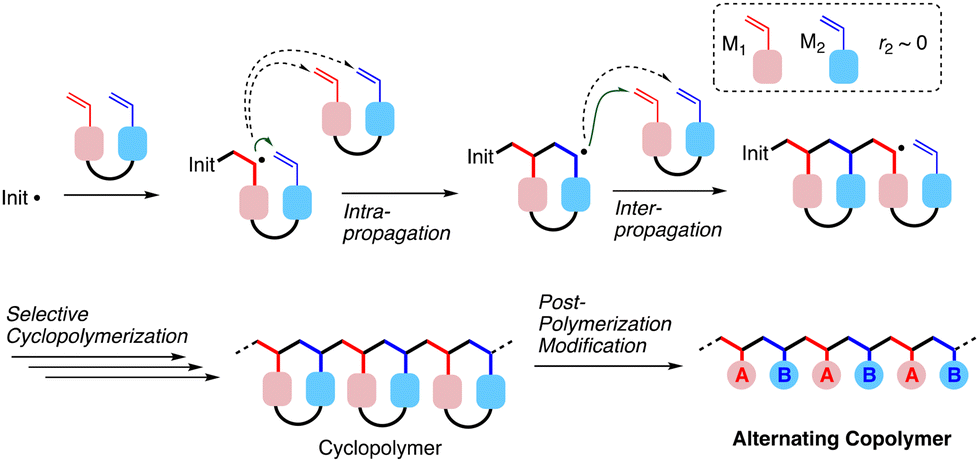 | ||
| Fig. 1 General scheme for syntheses of alternating copolymers via selective cyclopolymerization and post-polymerization modification. | ||
In this work, our interest was directed to the syntheses of alternating copolymers made of the NIPAM unit to study the sequence effects on the thermal response behaviors. We selected the 2-hydroxyethyl methacrylate (HEMA) unit as the partner. Poly(HEMA) is a commodity hydrophilic polymer carrying a hydroxy pendant group, but its solubility in water is not so high as that of poly(2-hydroxyethyl acrylate) due to the presence of a hydrophobic methyl group at the α-position. According to the paper by S. P. Armes et al.,22 the water solubility of poly(HEMA) is dependent on the molecular weight: it is soluble in water when the degree of polymerization (DP) is lower than 30 (DP < 30); it shows the LCST behavior in a range of 30 < DP < 45; and it becomes insoluble when DP is above 45. Considering the moderate hydrophilicity and the incomplete water solubility of poly(HEMA), so far, there have been plenty of research studies on the effects of composition23,24 and architectures25,26 on the thermal response behaviors of the copolymers of NIPAM and HEMA. However, the effects of the sequence, i.e., the arrangement of NIPAM and HEMA units, still remain understudied. It is of great interest to us whether the adjacent hydroxy group in the alternating sequence could affect the behaviors of water molecules in the thermal response process, and there is a possibility that the alternating copolymer of NIPAM with HEMA shows specific thermal response features. Thus, we designed a divinyl monomer (1) to synthesize the alternating copolymer of NIPAM with HEMA (Fig. 2). The OH group of HEMA is linked with the carboxylic acid of CF3-substituted salicylic acid via ester bond formation and an acrylate is connected to the phenol group of salicylic acid via another ester bond. Both ester bonds are activated by the electron-withdrawing CF3 group to be cleavable by the attack of a nucleophilic amine compound for aminolysis transformation.19 When isopropylamine is used for aminolysis transformation, the acrylate unit in the repeating cyclo-unit is converted to a NIPAM unit while the methacrylate one is converted to a HEMA unit. We first studied the validity of the divinyl monomer design for the synthesis of the alternating copolymer of NIPAM with HEMA by using the model monomers. Cyclopolymerization was controlled via ATRP for molecular weight control and poly(NIPAM-alt-HEMA) samples of different molecular weights were synthesized via post-polymerization modification by aminolysis of the resultant cyclopolymers with isopropylamine. Then, the effects of the sequence and molecular weight on the thermal response behaviors were examined through comparison with those of the corresponding statistical copolymer [poly(NIPAM-sta-HEMA)] samples with various composition ratios.
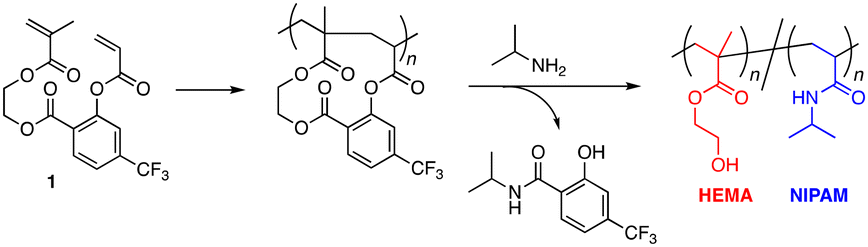 | ||
| Fig. 2 Divinyl monomer 1 for synthesis of poly(NIPAM-alt-HEMA). | ||
Results and discussion
The methacrylate-acrylate divinyl monomer 1 was synthesized according to the scheme shown in Fig. 3A. The electron densities of two vinyl groups were first evaluated by 1H NMR in comparison with HEMA and methyl acrylate (MA) (Fig. 3B). The positions of the peaks derived from the methacrylate vinyl group protons (A and B) are almost the same as those of HEMA (a and b). This implies that the vinyl group is little affected by the CF3 group on the salicylic acid spacer, which makes sense due to the presence of a non-conjugated ethylene spacer between the vinyl and salicylic acid groups. On the other hand, the peaks from the acrylate vinyl group protons (C–E) clearly shifted downfield relative to those of MA, indicating that the electron density of the vinyl group is decreased owing to the electron-withdrawing feature of the CF3 group on the salicylic acid spacer.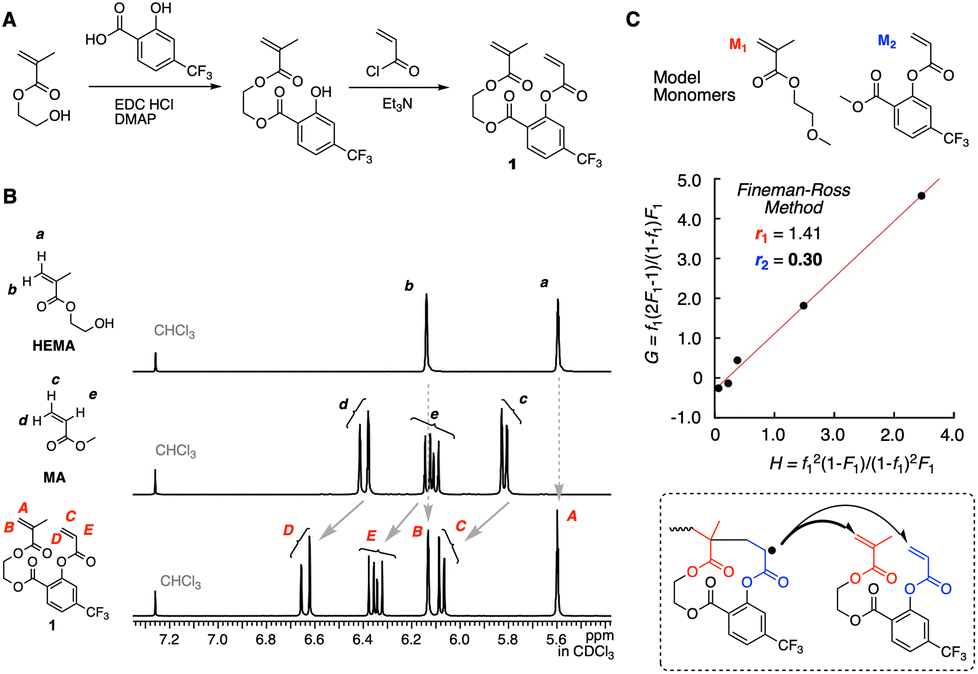 | ||
Fig. 3 Rational design of the methacrylate-acrylate divinyl monomer 1: (A) the synthesis scheme; (B) evaluation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond electron densities by 1H NMR; (C) monomer reactivity ratios of the model monomers calculated by the Fineman–Ross method. C bond electron densities by 1H NMR; (C) monomer reactivity ratios of the model monomers calculated by the Fineman–Ross method. | ||
Next, the cut-out model monomers (M1 and M2) of 1 were synthesized for evaluating the selectivity of cyclopolymerization (Fig. 3C). The peaks attributed to the vinyl groups of M1 and M2 in the 1H NMR spectrum were located at the same position as those of 1, supporting that they are the model monomers (Fig. S1†). Radical copolymerization of M1 and M2 was conducted at different injection ratios ([M1]0:[M2]0 = 84:16, 71:29, 47:53, 30:70, and 14:86) with AIBN as an initiator in dimethoxyethane (DME) at 60 °C. The conversions at the earlier stage were measured by 1H NMR (Table S1†) for the determination of the reactivity ratios by the Fineman–Ross method27 (Fig. S2†): r1 = 1.41 and r2 = 0.30. We also determined the reactivity ratios by a non-linear fitting method and obtained similar values (r1 = 1.66 and r2 = 0.40, Fig. S2†). The small value of r2 is appropriate for the control of the selective propagation where the cyclo-acrylate radical species favours the methacrylate monomer over acrylate in the divinyl monomer 1 (Fig. 3C). The higher value of r1 indicates that diluted conditions are required to control cyclopolymerization without cross-linking reactions as well as the alternating sequence.
We then conducted the radical cyclopolymerization of 1 at 60 °C via ATRP to control both the molecular weight and sequence. The initiator/catalyst system that is useful for controlling both methacrylate and acrylate was selected: ethyl 2-bromoisobutyrate (EMA-Br) as the initiator; CuBr/tris[2-(dimethylamino)ethyl] amine (Me6TREN) as the catalyst. Dimethylsulfoxide (DMSO) was used as the solvent, and each component was set at a relatively diluted concentration: [1]0 = 200 mM, [EMA-Br]0 = 4.0 mM, [CuBr]0 = 0.4 mM, and [Me6TREN]0 = 2.4 mM. Despite the lower concentration, the conversions of two vinyl groups (i.e., Conv.M and Conv.A for methacrylate and acrylate respectively) were smoothly increased at the same rate and eventually reached 90% in 3 hours (Fig. 4B). The smooth and parallel conversion despite the large difference in the reactivity ratios of two model monomers indicates the progress of cyclopolymerization as expected. The molecular weight distributions of the obtained cyclopolymers were broader (Mw/Mn = 1.6–1.9) than those of linear polymers synthesized by a general ATRP process. The higher dispersity is due to that cyclopolymerization requires diluted conditions for the suppression of the cross-linking reaction. Such diluted conditions are not favoured in ATRP for the promotion of the activation–deactivation equilibrium. In spite of the broad distribution, the SEC curves were basically unimodal and shifted to a higher molecular weight as the conversions were increased, indicating that the polymerization was basically controlled (Fig. 4C).
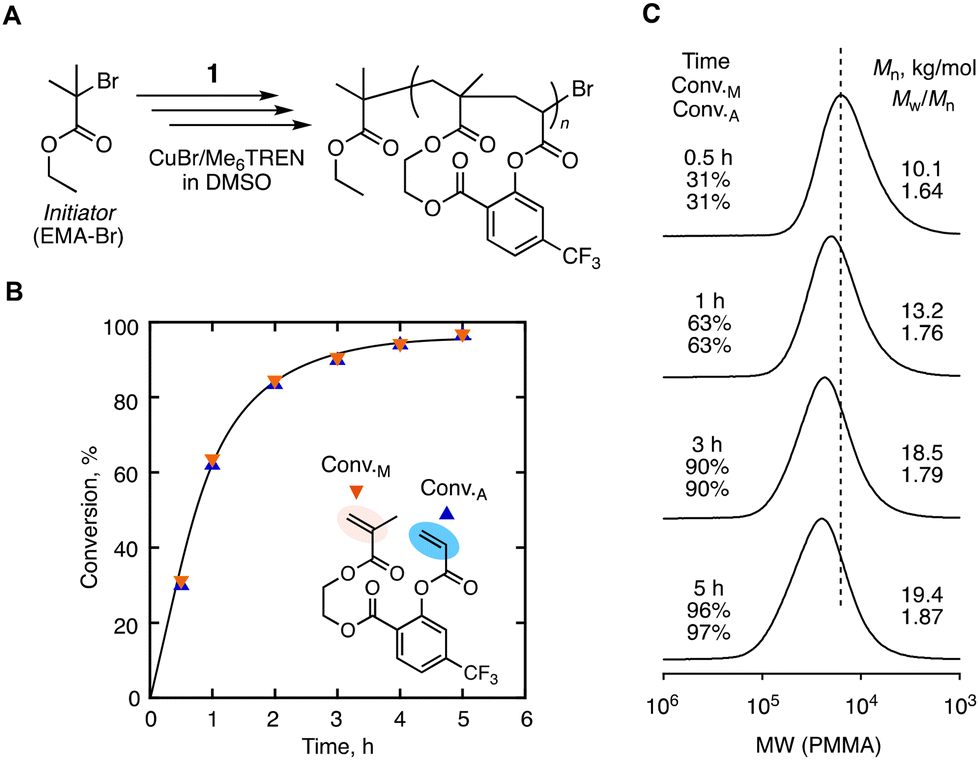 | ||
| Fig. 4 ATRP-based cyclopolymerization of 1: (A) the synthesis scheme; (B) time-conversion plots and (C) SEC curves of the obtained cyclopolymers. Polymerization conditions: [1]0/[EMA-Br]0/[CuBr]0/[Me6TREN]0 = 200:4.0:0.4:2.4 mM in DMSO at 60 °C. | ||
An excess of isopropylamine (i-PrNH2, ca. 10 equiv. for the cyclo-repeating units) was added to the solution of the obtained cyclopolymer (Mn = 19.4 kg mol−1, Mw/Mn = 1.87, and SEC with THF as an eluent), followed by heating at 60 °C to ensure the quantitative transformation into the copolymers of NIPAM and HEMA. The resultant solution was purified via dialysis over methanol and the structure of the obtained product was characterized by SEC, FT-IR and 1H NMR (Fig. 5A–C). The aminolysis treatment afforded the copolymer that was soluble in polar solvents rather than non-polar ones, and therefore the molecular weight was measured by SEC, using DMF as an eluent (Mn = 23.1 kg mol−1, Mw/Mn = 1.39). After the aminolysis reaction, the FT-IR spectrum (Fig. 5B) gave the characteristic peaks of the HEMA unit (O–H stretch at ∼3300 cm−1 and CO stretch of ester at 1720 cm−1) and the NIPAM unit (N–H stretch at ∼3250 cm−1, CO stretch of amide at 1635 cm−1, and N–H bend at ∼1525 cm−1), which were observed with the NIPAM and HEMA homopolymers, indicating the transformation into these units. Indeed, the IR spectrum was similar to that of the 1:1 NIPAM-HEMA statistical copolymer (Mn = 39.8 kg mol−1, Mw/Mn = 1.33, DPn,NIPAM:DPn,HEMA = 49:51, ESI†). In detail, however, slight differences from the statistical copolymer were observed, which might be derived from the controlled sequence. The transformation was further confirmed by 1H NMR (Fig. 5C): the characteristic peaks to the aromatic protons of the salicylic acid spacer (g) around 7.2–8.2 ppm disappeared, and the peak from the amide proton (n) appeared instead. The spectrum was almost similar to that of the 1:1 statistical copolymer, but only slight differences were observed, which is also attributed to the sequence difference. The peak integration ratio supported the equal polymerization degrees of both NIPAM and HEMA units.
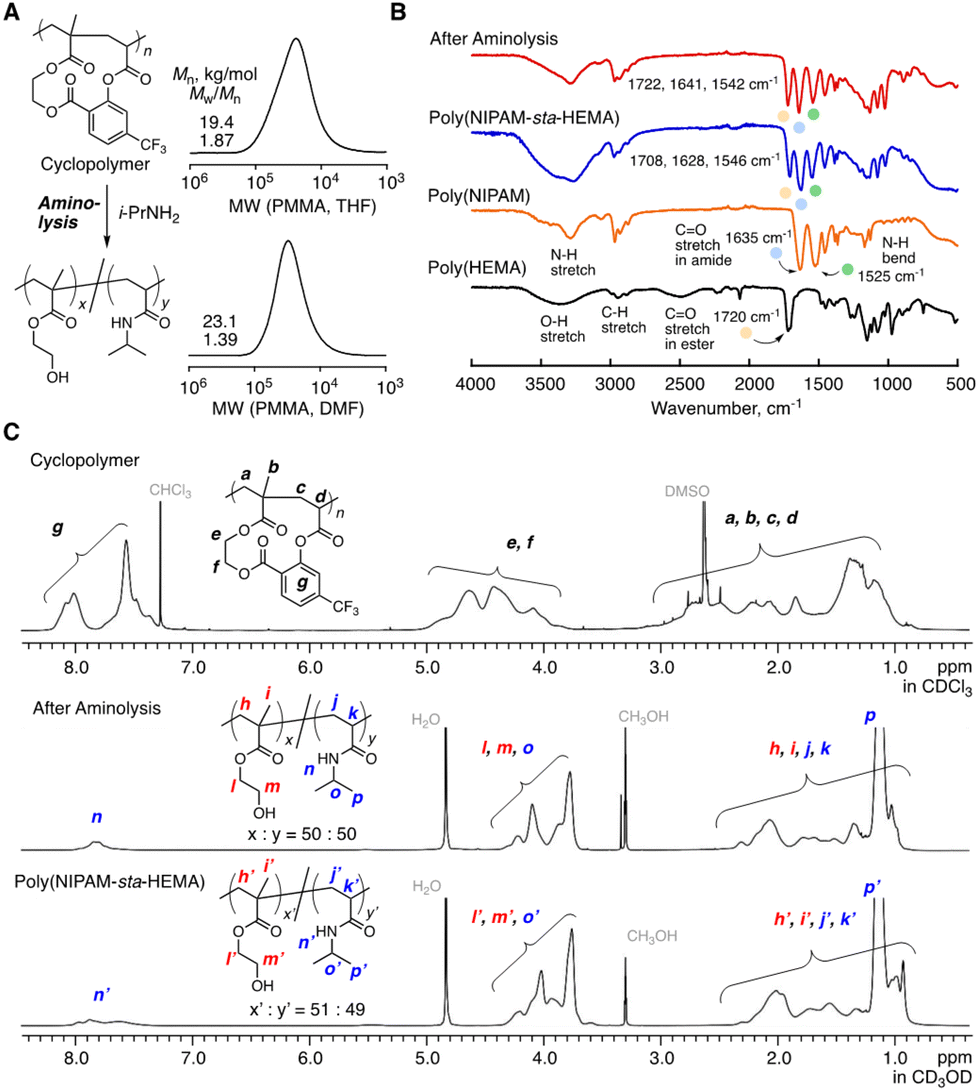 | ||
| Fig. 5 Post-polymerization modification of the cyclopolymer: (A) the synthesis scheme with SEC curves of the obtained cyclopolymer and the polymer obtained after modification; (B) FT-IR spectra of the polymer obtained after modification, poly(NIPAM-sta-HEMA), poly(HEMA) and poly(NIPAM); (C) 1H NMR spectra of the obtained cyclopolymer, the polymer obtained after modification and poly(NIPAM-sta-HEMA). | ||
The copolymer of a relatively low molecular weight (Mn = 6.6 kg mol−1 and Mw/Mn = 1.24) was synthesized via ATRP of 1 and subsequent aminolysis reaction with i-PrNH2 for further structural characterization by MALDI-TOF-MS. There were two types of main peak series (Fig. 6), and in both cases, the peak interval almost corresponded to the molecular weight for the sum of HEMA and NIPAM units (∼243.15). The larger peak series was likely attributed to the HEMA–NIPAM copolymer of the 1:1 composition ratio having the ethyl methacrylate moiety from the initiator at α-end and isopropylamine-substituted terminal at ω-end. The peak mass agreed with the molecular weight for the adduct of the 1:1 copolymer structure with 2,5-dihydroxybenzoic acid (2,5-DHB), which was used as the matrix. The isopropyl amine ω-terminal group was likely generated via the nucleophilic substitution for the carbon–bromine dormant species using isopropylamine during aminolysis transformation. On the other hand, the smaller series was derived from the 1:1 copolymer having an olefinated HEMA or NIPAM unit at ω-end, which was presumably derived from the elimination of HBr of the bromine-terminated copolymer by laser during MALDI-TOF-MS measurements. The MALDI-TOF-MS analysis does not strictly support the alternating sequence, but it is noteworthy that the main peak series corresponded to the copolymer having the 1:1 composition ratio of HEMA to NIPAM. Given the results of the cyclopolymerization behavior, reactivity ratios of the model monomers, and structural analyses by FT-IR, 1H NMR and MALDI-TOF-MS, we concluded that the alternating-rich copolymer of HEMA and NIPAM [poly(NIPAM-alt-HEMA)] was formed via the cyclopolymerization of 1 and quantitative aminolysis transformation with i-PrNH2.
 | ||
| Fig. 6 MALDI-TOF mass spectrum of poly(NIPAM-alt-HEMA) (Mn = 6.6 kg mol−1, Mw/Mn = 1.24 by SEC). The plausible structures are shown below with the calculated and observed values of m/z. | ||
We changed the ratios of the divinyl monomer 1 to the initiator (EMA-Br) in the ATRP-based cyclopolymerization to prepare four samples of poly(NIPAM-alt-HEMA)s having different molecular weights (Mn = 33.3, 23.1, 10.8, and 6.6 kg mol−1, Fig. 7A) via aminolysis transformation with i-PrNH2. (Table S2 and Fig. S3†) Ultrapure water was added to the resultant copolymers at an ambient temperature, but they were not perfectly dissolved. However, they were soluble after standing at 5 °C for 24 h giving transparent solutions, and the transparent solutions became turbid upon heating, i.e., the copolymers showed LCST-type thermoresponsive behaviors in aqueous solution. The thermoresponsive behavior of each copolymer solution at 0.5 wt% was further investigated by variable-temperature transmittance measurements with the light of 670 nm wavelength at a heating rate of 1 °C min−1. For all of the samples, the transmittance sharply dropped around 25–30 °C, as supported by the visual observation, but the detailed profiles varied depending on the molecular weights (Fig. 7B). The cloud point (TCP: the temperature giving 90% transmittance) decreased as the molecular weight increased: TCP = 25.1 °C (Mn = 33.3 kg mol−1), 25.9 °C (23.1 kg mol−1), 28.3 °C (10.8 kg mol−1), and 28.8 °C (6.6 kg mol−1). Given the fact that the water solubility of poly(HEMA) declines as molecular weight increases,22 the trend is likely reasonable. Additionally, the decrease of cloud points with the increasing molecular weights was found in many other LCST-type thermoresponsive polymers.28–32
 | ||
| Fig. 7 (A) SEC curves of poly(NIPAM-alt-HEMA)s with different molecular weights; (B) effects of molecular weights on thermoresponsive behaviors of poly(NIPAM-alt-HEMA)s in 0.5 wt% aqueous solutions; (C) concentration effects of poly(NIPAM-alt-HEMA) on thermoresponsive behaviors in comparison with poly(NIPAM); (D) DLS measurements of 0.1 wt% aqueous solution of poly(NIPAM-alt-HEMA) at different temperatures in comparison with poly(NIPAM). TCP is defined as the temperature at which the transmittance reached 90%. | ||
Furthermore, solutions of different concentrations (1.0, 0.5, 0.2, and 0.1 wt%) of poly(NIPAM-alt-HEMA) (Mn = 33.3 kg mol−1) were prepared and effects of the concentration on thermoresponsive behaviors were also examined in comparison with the NIPAM homopolymer [poly(NIPAM)] of similar molecular weight (Mn = 36.8 kg mol−1). The thermal response behaviors widely varied and the cloud point increased as the concentration decreased (Fig. 7C), whereas such a clear trend on the concentration dependency was not observed with poly(NIPAM). We thus measured dynamic light scattering (DLS) of 0.1 wt% solution at different temperatures to see the thermal response chain aggregation behaviors (Fig. 7D).33 Larger particles (∼100 nm) were observed in addition to the smaller ones derived from an unimer (∼10 nm) even at 20 °C, which was a lower temperature than TCP (28.0 °C) giving the transparent solution. The trend remained unchanged even at 25 °C, which was slightly below TCP. When it was heated to 30 °C above TCP, much larger particles were formed (∼570 nm), and the intensity was increased without any change in the size upon further heating. On the other hand, in the case of poly(NIPAM), such pre-aggregated particles were not observed even at 5 °C lower than TCP (34.8 °C). When the temperature reached 35 °C, the aggregation eventually took place, which likely corresponded to the increase in turbidity. Thus, the pre-aggregation behavior below TCP was peculiar to the alternating copolymer, and thus the interaction between polymer chains likely played an important role in the thermoresponsive behavior of the alternating copolymer unlike poly(NIPAM). The concentration effect became more remarkable as the molecular weight decreased (Fig. S4†).
Finally, we synthesized five samples of the HEMA–NIPAM statistical copolymers having different composition ratios via the direct copolymerization under ATRP conditions (Table S3†) and compared the thermal response behaviors with those of the alternating copolymer to clarify sequence-specific properties. The mole ratios of monomer units were calculated from the peak integrals of 1H NMR spectra: DPn,NIPAM/(DPn,NIPAM + DPn,HEMA) = 26%, 36%, 49%, 54% and 62% (Fig. S23–27†). Herein, the copolymers whose molecular weights are high enough (Mn > 30 kg mol−1) were prepared so that the effects of molecular weight on thermal response behaviors are negligible. The turbidity of 1.0 wt% solutions was measured by variable-temperature UV/Vis spectrophotometry, and all the statistical copolymers were found to exhibit LCST behaviors like alternating copolymers (Fig. S7†). Notably, the alternating copolymer (Mn = 33.3 kg mol−1) was found to exhibit 8 °C higher TCP than the 1:1 statistical copolymer having a 49% NIPAM ratio (Mn = 39.8 kg mol−1) (24.9 °C vs. 16.9 °C, Fig. 8A). In addition, the transition was much sharper than that of the statistical copolymer: the temperature range for the decrease in the transmittance from 95% to 5% was 1.4 °C, whereas that of the statistical copolymer was 4.9 °C. The DLS measurement was also performed at different temperatures for the solution of the 1:1 statistical copolymer at 0.1 wt%, and the data were collated with the variable-temperature transmittance measurement at the same concentration (Fig. S8 and S9†). The pre-aggerated particles were observed at a temperature below TCP similar to that of the alternating copolymers (Fig. 7D). However, in contrast to the alternating copolymer, the large aggregate of the size range from 102 to 103 nm was observed even at a temperature (i.e., 15 °C) lower than TCP. The HEMA-rich chains were present in the statistical copolymer with a certain probability and could aggregate even at a lower temperature, as supported by the trend in Fig. 8B as well as the previous research studies on poly(NIPAM-sta-HEMA).23,24 In this manner, the alternating copolymer showed higher Tcp than the statistical analogue, and the transition was sharper likely due to none of the compositional distribution among polymer chains.
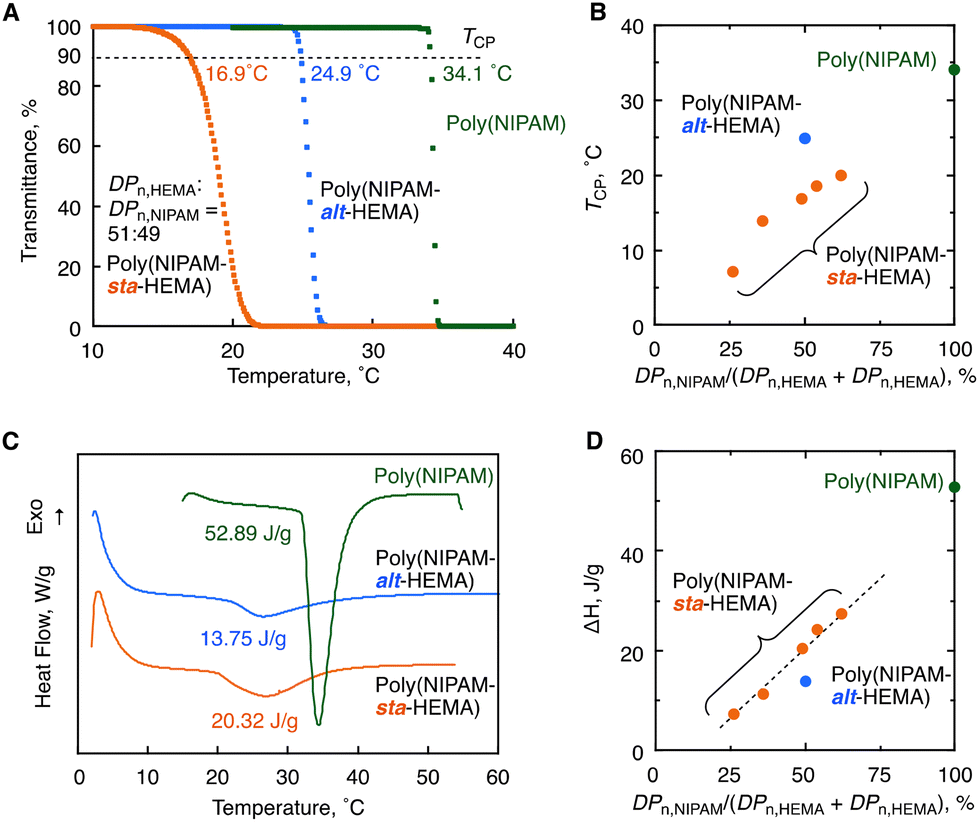 | ||
| Fig. 8 Comparison of thermoresponsive behaviors among poly(NIPAM-alt-HEMA) (Mn = 33.3 kg mol−1), poly(NIPAM-sta-HEMA)s (Mn: 30.0–50.0 kg mol−1) and poly(NIPAM) (Mn = 36.8 kg mol−1) in aqueous solutions: (A) transmittance versus temperature plots of 1.0 wt% aqueous solutions on the heating process at 1 °C min−1; (B) TCPversus the mole fraction of NIPAM of the polymers; (C) DSC thermograms of 10 wt% aqueous solutions on the heating process at 10 °C min−1; (D) ΔH versus the mole fraction of NIPAM of the polymers. | ||
We also measured differential scanning calorimetry (DSC) for the 10 wt% aqueous solution of the alternating copolymer (Mn = 33.3 kg mol−1) to study the enthalpy change on the thermal response process in comparison with the statistical copolymers (Mn = 39.8 kg mol−1) and poly(NIPAM) (Mn = 36.8 kg mol−1) (Fig. 8C). All the three samples exhibited endothermic peaks around the cloud points, which is absolutely attributed to dehydration from the polymer chains.34–36 In sharp contrast to poly(NIPAM) giving the large endothermic peak (ΔH = 52.89 J g−1), the alternating and statistical copolymers showed smaller peaks. The smaller peaks are reasonable because the relative number of NIPAM units being involved in dehydration is lower than that of the homopolymer, which is supported by the trend of the ΔH value for the NIPAM unit ratio in the series of statistical copolymers (Fig. 8D). Interestingly, the ΔH value of the alternating copolymer (13.75 J g−1) was much lower than that of the 1:1 statistical copolymer (20.32 J g−1) as well as the half value of poly(NIPAM) (26.45 J g−1). A similar trend was reported in the literature where ΔH was decreased as the number of OH pendant groups increased for copolymers of NIPAM with 2-hydroxyisopropylacrylamide.37 Probably, the dehydration upon heating probably occurs only from NIPAM units, and the number of water molecules involved in the dehydration increases with the number of NIPAM continuous sequences resulting in a higher enthalpy change at the phase transition. The relatively low enthalpy change of the alternating copolymer is likely due to the lack of the NIPAM continuous sequence.
Conclusions
We designed a methacrylate-acrylate divinyl monomer 1 to control the alternating sequence as well as the molecular weight of the HEMA–NIPAM copolymer via ATRP-based cyclopolymerization and subsequent aminolysis with i-PrNH2. The reactivity ratios of the cut-out model monomers indicated that the high degree of the alternating sequence was achieved via selective cyclopolymerization. The resultant poly(NIPAM-alt-HEMA) showed LCST-type thermal response behaviors in water. These behaviors were specific to the features derived from the alternating sequence, i.e., “none of the composition distribution” and “none of the continuous NIPAM sequence”, which was supported by DLS and DSC analyses in comparison with the corresponding statistical copolymers as well as poly(NIPAM).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Takaya Terashima and Dr Tsuyoshi Nishikawa (Kyoto University) for fruitful discussions. This work was supported by JSPS KAKENHI grants (17H06453, 19H00911, and 20K21222, for M. O.).References
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl.Chem., 1968, 2, 1441–1455 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- K. Nagase, M. Yamato, H. Kanazawa and T. Okano, Biomaterials, 2018, 153, 27–48 CrossRef CAS PubMed.
- L. Tang, L. Wang, X. Yang, Y. Fen, Y. Li and W. Feng, Prog. Mater. Sci., 2021, 115, 100702 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- B. Ray, Y. Okamoto, M. Kamigaito, M. Sawamoto, K. Seno, S. Kanaoka and S. Aoshima, Polym. J., 2005, 37, 234–237 CrossRef CAS.
- R. Plummer, D. J. T. Hill and A. K. Whittaker, Macromolecules, 2006, 39, 8379–8388 CrossRef CAS.
- J. Xu, J. Ye and S. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- M. Ouchi, Polym. J., 2021, 53, 239–248 CrossRef CAS.
- D. Oh, Y. Furuya and M. Ouchi, Macromolecules, 2019, 52, 8577–8586 CrossRef CAS.
- Y. Kametani and M. Ouchi, ACS Polym. Au, 2021, 1, 10–15 CrossRef CAS.
- M. Ouchi, M. Nakano, T. Nakanishi and M. Sawamoto, Angew. Chem., Int. Ed., 2016, 55, 14584–14589 CrossRef CAS PubMed.
- Y. Kametani, M. Sawamoto and M. Ouchi, Angew. Chem., Int. Ed., 2018, 57, 10905–10909 CrossRef CAS.
- Y. Kametani, F. Tournilhac, M. Sawamoto and M. Ouchi, Angew. Chem., Int. Ed., 2020, 59, 5193–5201 CrossRef CAS.
- K. Shibata, Y. Kametani, Y. Daito and M. Ouchi, J. Am. Chem. Soc., 2022, 144, 9959–9970 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS PubMed.
- J. V. M. Weaver, I. Bannister, K. L. Robinson, X. Bories-Azeau, S. P. Armes, M. Smallridge and P. McKenna, Macromolecules, 2004, 37, 2395–2403 CrossRef CAS.
- Z. Shen, K. Terao, Y. Maki, T. Dobashi, G. Ma and T. Yamamoto, Colloid Polym. Sci., 2006, 284, 1001–1007 CrossRef CAS.
- M. Kubo, T. Sone, M. Ohata and T. Tsukada, Ultrason. Sonochem., 2018, 49, 310–315 CrossRef CAS PubMed.
- X. Zhao, W. Liu, D. Chen, X. Lin and W. W. Lu, Macromol. Chem. Phys., 2007, 208, 1773–1781 CrossRef CAS.
- T. M. Quynh, M. Yoneyamab, Y. Maki and T. Dobashi, J. Appl. Polym. Sci., 2012, 123, 2368–2376 CrossRef CAS.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 5, 259–262 CrossRef CAS.
- V. Bütün, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794–799 RSC.
- M. A. Ward and T. K. Georgiou, Soft Matter, 2012, 8, 2737–2745 RSC.
- Q. Li, A. P. Constantinou and T. K. Georgiou, J. Polym. Sci., 2021, 59, 230–239 CrossRef CAS.
- We performed the DLS measurements for the solutions at higher concentrations (e.g., 1.0 or 0.5 wt%), but the measurements did not work resulting in error data likely due to less homogeneous scattering for the too-opaque solution. Thus, we carried out the DLS measurement for the diluted solution at 0.1 wt%.
- H. G. Schild and D. A. Tirrell, J. Phys. Chem., 1990, 94, 4352–4356 CrossRef CAS.
- M. Shibayama, M. Morimoto and S. Nomura, Macromolecules, 1994, 27, 5060–5066 CrossRef CAS.
- V. Aseyev, H. Tenhu and F. M. Winnik, in Self Organized Nanostructures of Amphiphilic Block Copolymers II, ed. A. H. E. Müller and O. Borisov, Springer, Berlin, Heidelberg, 2011, pp. 29–89 Search PubMed.
- T. Maeda, K. Yamamoto and T. Aoyagi, J. Colloid Interface Sci., 2006, 302, 467–474 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01196d |
| This journal is © The Royal Society of Chemistry 2023 |