
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An orthogonal O,S-CKA monomer for the introduction of thioester and/or thionoester functionalities by radical polymerization†
Marlena
Pięta
 a,
Vishal B.
Purohit
a,
Piotr
Paneth
a,
Joanna
Pietrasik
b,
Le
Li
cd and
Christopher M.
Plummer
*a
a,
Vishal B.
Purohit
a,
Piotr
Paneth
a,
Joanna
Pietrasik
b,
Le
Li
cd and
Christopher M.
Plummer
*a
aInternational Centre for Research on Innovative Biobased Materials (ICRI-BioM)— International Research Agenda, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland. E-mail: christopher.plummerp@p.lodz.pl
bInstitute of Polymer and Dye Technology, Faculty of Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
cKey Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, Sun Yat-sen University, Guangzhou 510275, P. R. China
dSchool of Chemistry, Sun Yat-sen University, Guangzhou, 510275, P. R. China
First published on 4th August 2023
Abstract
The introduction of thioester linkages into the backbone of free-radical polymerization-produced polymers has received recently renewed interest due to their use in preparing (bio)degradable polymers. To access monomers capable of introducing the thioester functionality, a novel monothiolated analogue of the cyclic ketene acetal (CKA) class of radical ring-opening polymerization (rROP) monomers was synthesized and examined, specifically O,S-BMDO. Interestingly, O,S-BMDO offers an example of an orthogonal rROP monomer, being capable of introducing thioesters, thionoesters, or a mixture of both functional groups into the polymer backbone, with the ratio of these functional groups affected by the comonomer and the reaction conditions. This unusual effect was explored both experimentally and computationally. This report constitutes the first time a thionoester-containing repeating unit has been introduced directly by radical polymerization. In addition, the polymerization of O,S-BMDO can produce a rare poly(thionoester) homopolymer.
Introduction
The low-cost and durability of plastics have resulted in an annual worldwide production exceeding 335 million tonnes. Single-use items constitute the major proportion of waste found in both marine and non-marine environments.1,2 The European Union (EU) has recently imposed a ban on single-use plastics within its member states, focusing on items including plastic bags, wrappers, cutlery, plates and straws.3 It should be noted that ca. 50% of all man-made polymers are synthesized by free-radical polymerization.4 Due to the plastic pollution problem, microplastic waste is also now becoming pervasive throughout the natural environment with long-term consequences yet to be established.5,6 Recycling is the most recognizable response to such waste production, but plastics are frequently soiled by biological matter which makes recycling impractical. In addition, each time a polymeric feedstock is recycled, the molecular chains within it are chemically degraded, and thus most polymers are actually ‘downcycled’ or mixed with virgin feedstock.7An emerging technique for the production of (bio)degradable polymers is the copolymerization of cyclic monomers with conventional monomer feedstock, named radical ring-opening polymerization (rROP) (Fig. 1). When such radical ring-opening processes are applied to copolymerization they allow for the installation of degradable linkages into the backbone of free-radical polymerization-produced polymers, thereby imparting (bio)degradability to already widely used commercial polymers. Such degradation processes can transform polymer chains into oligomers that can be readily consumed and mineralized by microbial action. To impart (bio)degradability to plastics, only a relatively minor number of rROP-induced linkages need to be introduced into the polymer backbone, pending homogeneous incorporation. An additional use of rROP monomers is their application in reversible-deactivation radical polymerization (RDRP) techniques which enable the preparation of complex macromolecular architectures (e.g., block, star, grafted copolymers, etc.) that can self-assemble at the nanoscale level to create degradable materials for applications such as in targeted drug delivery8–11 and biomedical applications,12–15 among others.
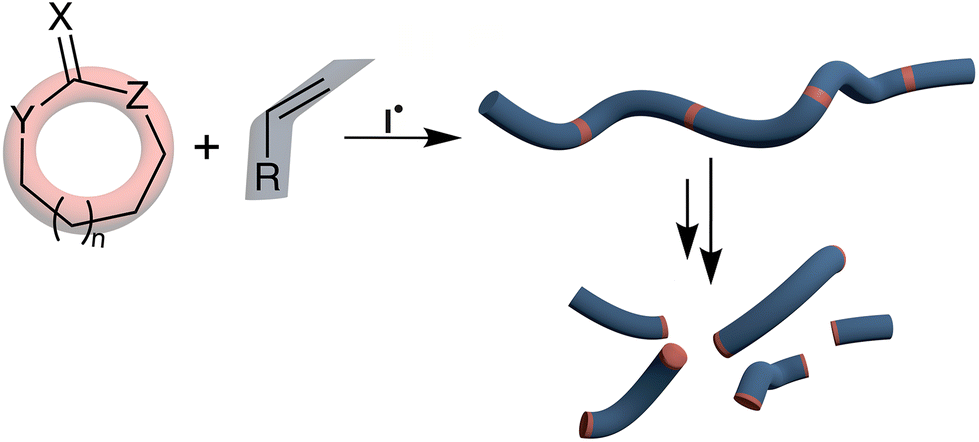 | ||
| Fig. 1 Copolymerization of cyclic rROP monomers with conventional vinyl monomers leading to incorporation of degradable functional groups. | ||
Among the various rROP monomer classes, cyclic ketene acetals (CKAs) have become the prototypical rROP monomer class, capable of introducing ester linkages into the backbone of radically produced polymers (Fig. 2, BMDO, compound 1).16 A relatively new class of rROP monomers is the cyclic thionoesters which, after the radical ring-opening process, can introduce thioester linkages which can be degraded by additional mechanisms including aminolysis, trans-thioesterification and oxidation (Fig. 2, DOT, compound 2).17–24 These additional susceptibilities have been envisioned to be able to assist in their environmental degradation as nitrogen and sulfur containing molecules (i.e., amino acids) are ubiquitous throughout the natural environment, as are natural oxidation processes.
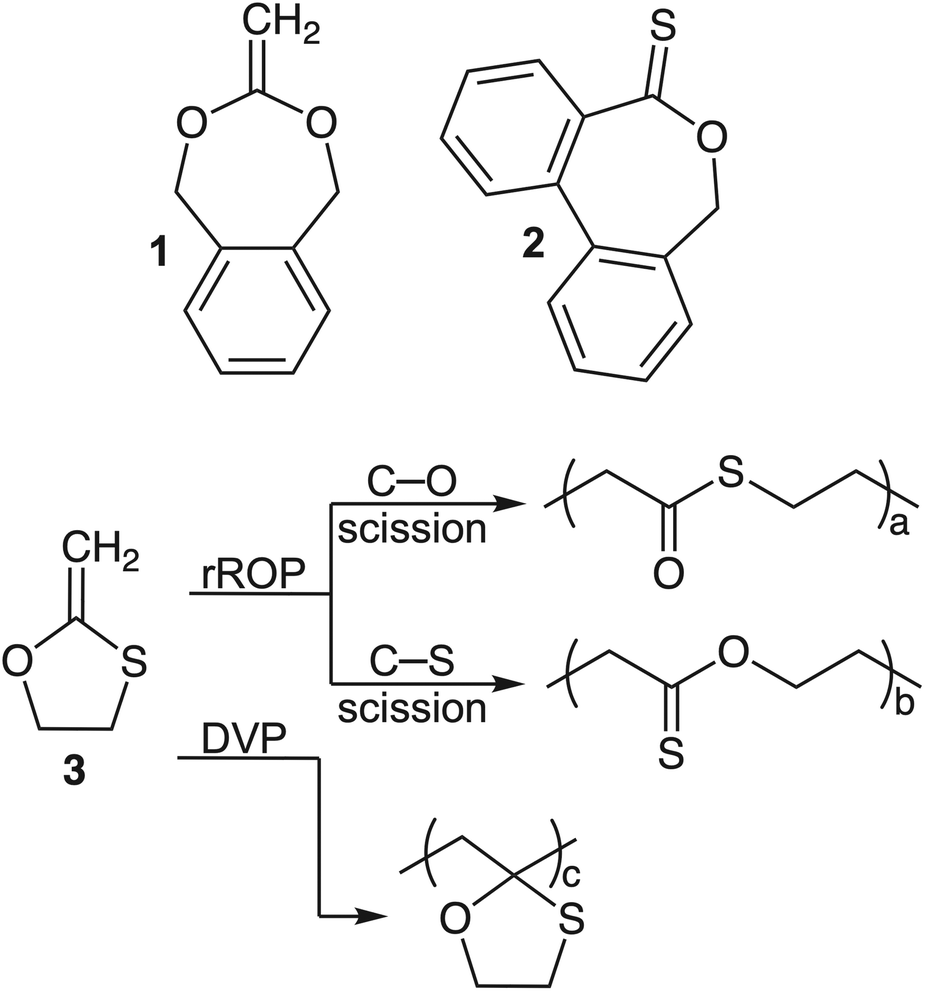 | ||
| Fig. 2 Monomers reported for rROP. Top: CKA monomer, BMDO (1), and thionolactone monomer, DOT (2); bottom: O,S-CKA analogue 3 reported by Bailey and coworkers displaying three possible reaction pathways during radical polymerization. rROP = radical ring-opening polymerization; DVP = direct vinylic propagation. | ||
Owing to the additional degradation susceptibilities of thioester functional groups, we were motivated to explore monothiolated analogues of the CKA class of rROP monomers. Such O,S-CKA monomers could be envisioned to perform radical polymerization to introduce thioester or thionoester functional groups into the polymer backbone, or even mixtures, depending on the chemical bond involved in the radical ring-opening process. Upon careful examination of the literature, it was found that this class of monomers was first reported by Bailey and co-workers in 1984.25,26 They described that the 5-membered O,S-CKA monomer could be subjected to homopolymerization at 120 °C to form a polymer containing 45% ring-opened units (Fig. 2, compound 3). It was never unambiguously established if the ring-opening process produced thioester or thionoester functional groups alongside the ring-retained pendant heterocycle. Thus, we turned our attention towards the synthesis of an O,S-CKA analogue of the prototypical CKA monomer BMDO (1) with the desire that the ring-opening process could be harnessed to introduce highly degradable thioester linkages.
Results and discussion
The targeted monothiolated BMDO analogue, O,S-BMDO (8), was obtained in a six-step synthetic pathway commencing from 2-methylbenzoic acid (Fig. 3). The synthesis of bromide 4 was performed by esterification and subsequent radical benzylic bromination of 2-methylbenzoic acid, as previously described in the literature.27–29 Intermediate 5 was then synthesized by nucleophilic substitution of bromide 4 with potassium thioacetate in ethanol. Subsequent reduction of both the ester and thioester moieties with lithium aluminum hydride furnished mercaptoalcohol 6.30 This intermediate was then cyclized using chloroacetaldehyde dimethyl acetal in a transacetalization reaction, followed by dehydrohalogenation with potassium tert-butoxide to furnish the target compound 5,6-benzo-2-methylene-1,3-oxthiepane (O,S-BMDO, 8) as a brown oil which was stored in a freezer before use.31,32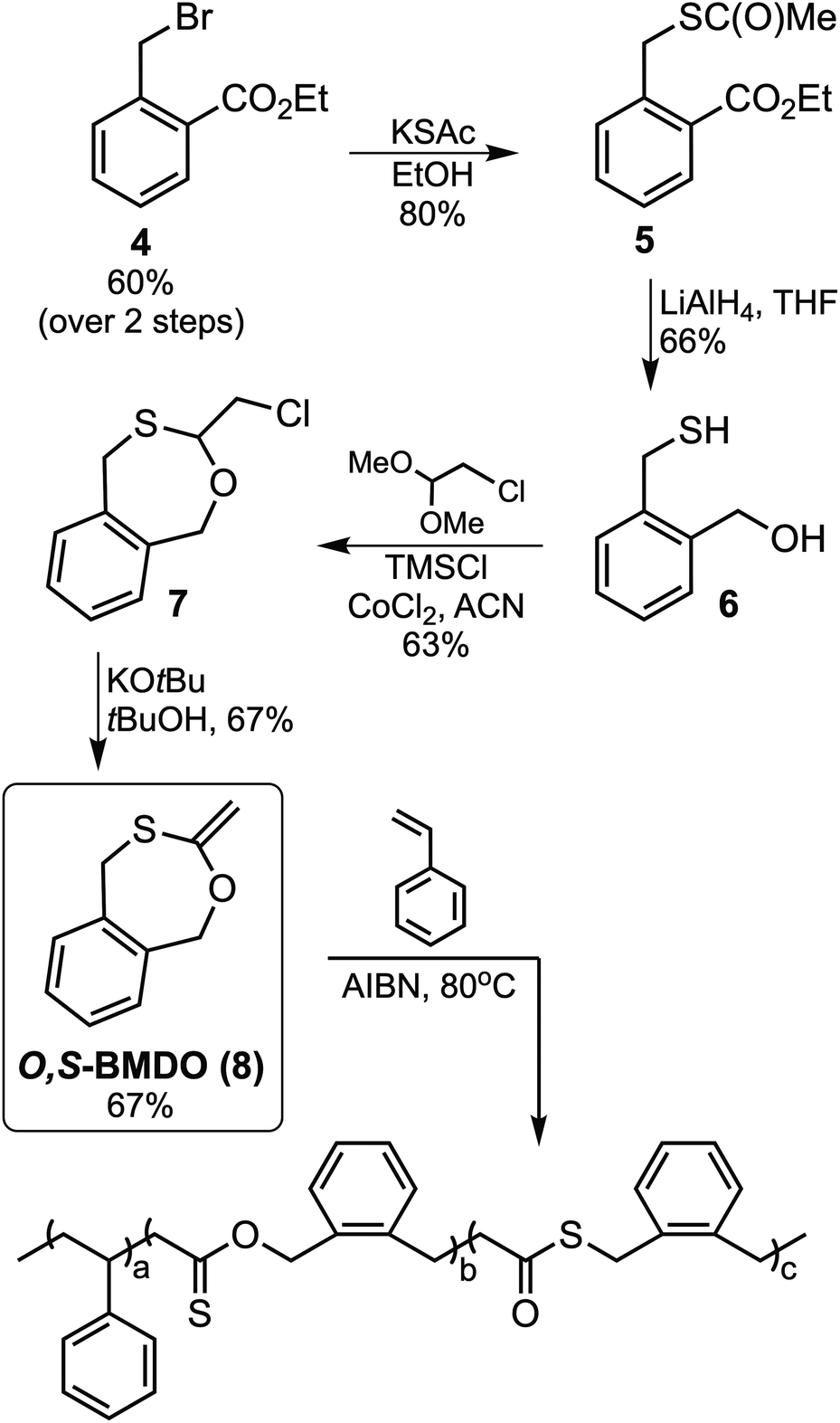 | ||
| Fig. 3 Synthesis of O,S-BMDO (8) and its radical copolymerization with styrene resulting in ring-opening polymerization. | ||
To examine the radical polymerization capabilities of O,S-BMDO, a series of copolymerizations of styrene (Sty) and O,S-BMDO were performed by applying 3 mol% AIBN at 80 °C with a feed ratio of Sty![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O,S-BMDO = 9:1, both in solution and under bulk conditions (Table 1, entries 2–5). The structures of the obtained polymers were examined in-depth by 1H NMR, 13C NMR, and FT-IR spectroscopy. Analogous to monomer 3, O,S-BMDO (8) can theoretically perform radical ring-opening polymerization via two distinct reaction pathways: (a) via O–C bond scission leading to the formation of a thioester-containing repeating unit (labelled O-rROP in Tables 1 and 2) and (b) via C–S bond scission leading to the formation of a thionoester-containing repeating unit (labelled S-rROP in Tables 1 and 2). Additionally, there exists the possibility of direct vinyl polymerization leading to ring-retained pendants within the polymer backbone which are not degradable (DVP, Fig. 2). Analysis of the spectroscopic results revealed that there was no detectable direct vinylic propagation and that exclusively S-rROP derived residues were obtained (Table 1, entries 2–5).
:O,S-BMDO = 9:1, both in solution and under bulk conditions (Table 1, entries 2–5). The structures of the obtained polymers were examined in-depth by 1H NMR, 13C NMR, and FT-IR spectroscopy. Analogous to monomer 3, O,S-BMDO (8) can theoretically perform radical ring-opening polymerization via two distinct reaction pathways: (a) via O–C bond scission leading to the formation of a thioester-containing repeating unit (labelled O-rROP in Tables 1 and 2) and (b) via C–S bond scission leading to the formation of a thionoester-containing repeating unit (labelled S-rROP in Tables 1 and 2). Additionally, there exists the possibility of direct vinyl polymerization leading to ring-retained pendants within the polymer backbone which are not degradable (DVP, Fig. 2). Analysis of the spectroscopic results revealed that there was no detectable direct vinylic propagation and that exclusively S-rROP derived residues were obtained (Table 1, entries 2–5).
| Entry | Feed ratio, Sty:O,S-BMDO |
Solvent | Copolymer ratio, Sty:O,S-BMDOb |
O-rROP:S-rROPb |
M
n
(kg mol−1) |
Đ |
|---|---|---|---|---|---|---|
| a 2.5 M solution. b Ring-opening ratio determined by 1H NMR. c Determined by GPC. | ||||||
| 1 | 10:0 |
Bulk | 1:0 |
— | 13.0 | 3.0 |
| 2 | 9:1 |
Toluenea | 99.3:0.7 |
0:1 |
5.77 | 1.4 |
| 3 | 9:1 |
THFa | 97.4:2.6 |
0:1 |
5.05 | 1.4 |
| 4 | 9:1 |
DMFa | 99.0:1.0 |
0:1 |
6.52 | 1.4 |
| 5 | 9:1 |
Bulk | 95.6:4.4 |
0:1 |
12.2 | 2.4 |
| 6 | 8:2 |
Bulk | 91.2:8.8 |
0:1 |
8.54 | 2.1 |
| 7 | 7:3 |
Bulk | 82.7:17.3 |
0:1 |
8.76 | 2.1 |
| 8 | 5:5 |
Bulk | 59.5:40.5 |
0.01:1 |
6.53 | 2.5 |
| 9 | 3:7 |
Bulk | 39.9:60.1 |
0.02:1 |
5.97 | 2.5 |
| 10 | 0:1 |
Bulk | 0:1 |
0.03:1 |
3.62 | 2.6 |
| Method | r O,S–BMDO 8 | r Sty | r O,S–BMDO 8 ·rSty |
|---|---|---|---|
| FR | 0.96 | 2.52 | 2.42 |
| KT | 1.09 | 2.67 | 2.91 |
| ML | 0.99 | 2.50 | 2.48 |
Interestingly, it was revealed that O,S-BMDO was more effectively incorporated into the copolymer when the polymerization was performed in THF versus toluene or DMF, but bulk polymerization resulted in the highest incorporation (Table 1, entry 5). In addition, bulk polymerization was found to provide superior molecular weights (Mn) and lower dispersities (Đ) and was therefore selected as the standard for further experimentation. Polymerizations were then conducted with a higher content of O,S-BMDO in the feed (Table 1, entries 6–9). These copolymerizations provided copolymers with reduced Mn values, from 12.2 kg mol−1 for Sty:O,S-BMDO = 9:1 (Table 1, entry 5) to 5.97 kg mol−1 for Sty:O,S-BMDO = 3:7 (Table 1, entry 9), with all samples having unimodal GPC traces. Unexpectedly, O,S-BMDO (8) even underwent homopolymerization with complete ring-opening to provide a poly(thionoester) with Mn = 3.62 kg mol−1 and Đ = 2.6 (Table 1, entry 10).
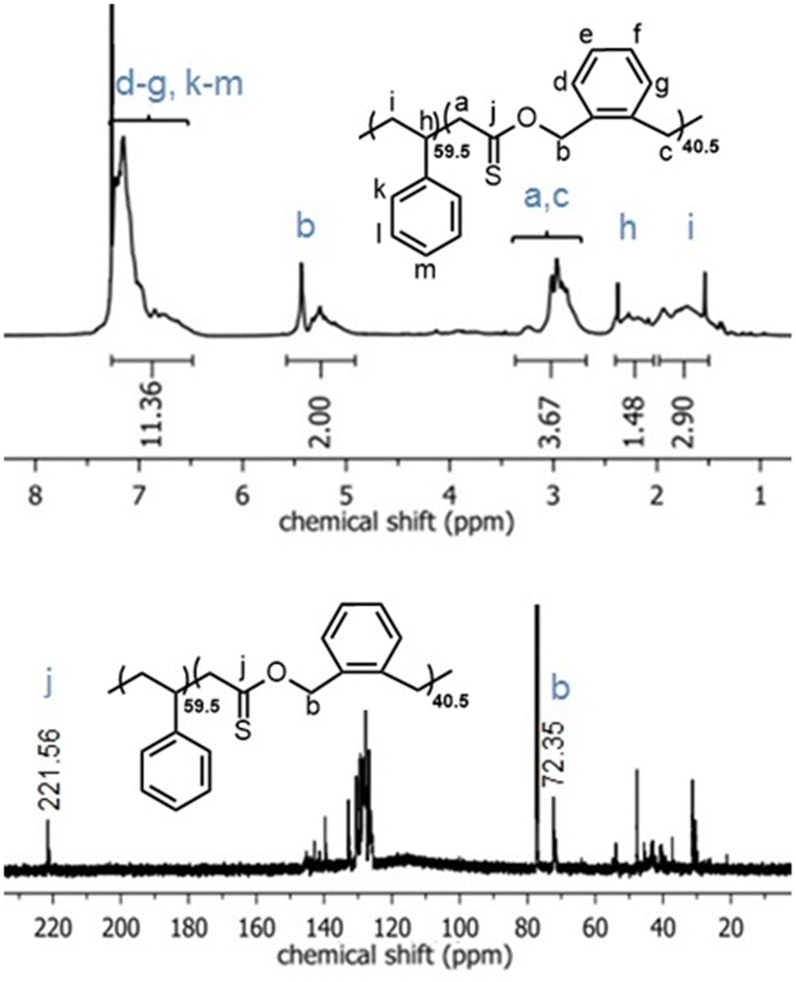
In the 1H NMR spectrum of poly(O,S-BMDO), the broad singlet at 5.39–5.43 ppm was assigned to the methylene protons adjacent to the thionoester –C(S)–O– moiety (Fig. 4, top) (Table 1, entry 10). Only a minor signal (ca. 3% relative to the signal at 5.39–5.43 ppm) which could be assigned to methylene protons adjacent to the thioester –C(O)–S– moiety was observed at 4.10 ppm.
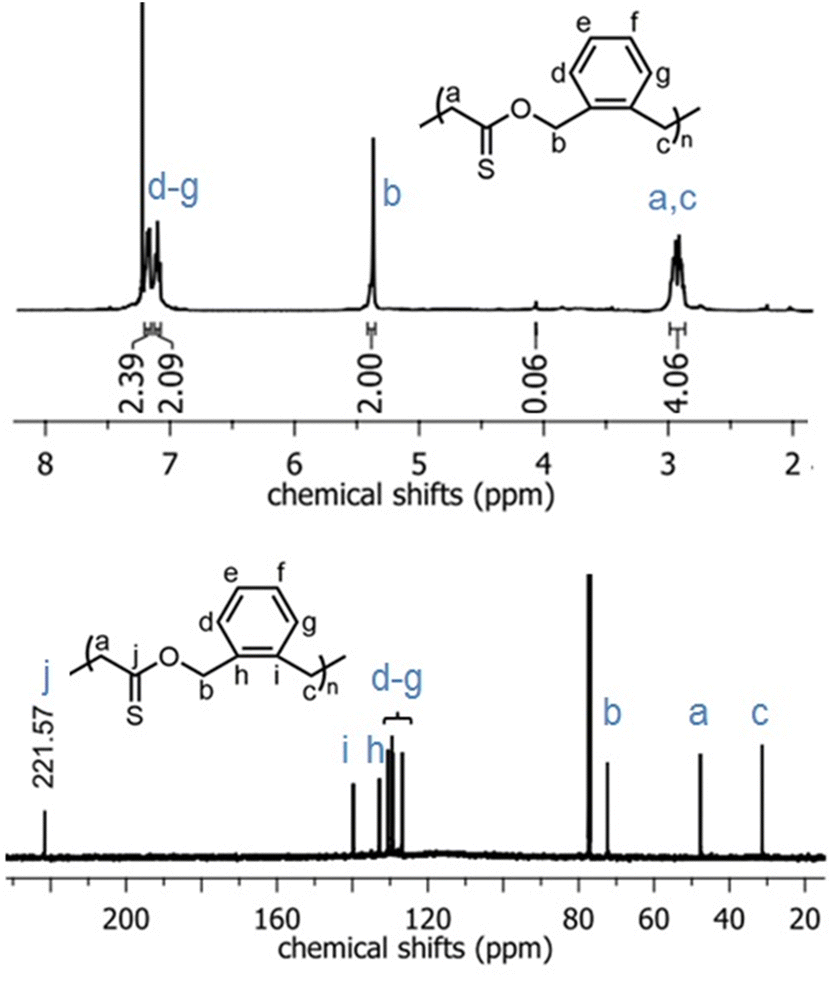 | ||
| Fig. 4 1H NMR (CDCl3, 700 MHz) and 13C NMR (CDCl3, 176 MHz) spectra of the poly(O,S-BMDO) homopolymer displaying virtually exclusively S-rROP derived signals. | ||
In addition, the signal at 221 ppm in the 13C NMR spectrum provided evidence for the selective formation of a quaternary thiocarbonyl-bearing carbon, while no signal could be detected in the characteristic region for quaternary (oxo)carbonyl-bearing carbons (ca. 200 ± 5 ppm) or quaternary monothioacetal carbons (ca. 95 ± 15 ppm) (Fig. 4, bottom). Moreover, the 1H and 13C NMR spectra of poly(O,S-BMDO) and poly(BMDO)33 correlated well but contained distinct differences for signals relating to the CH2 group adjacent to the (thio)carbonyl carbon. Based upon these analyses it was concluded that the molecular structure of poly(O,S-BMDO) was indeed that of a poly(thionoester). This can be considered significant as it represents the first report of a thionocarbonyl-containing functional group being introduced into a polymer backbone by radical polymerization, while also constituting a relatively rare example of the isolation of a poly(thionoester).
Similarly, the 1H NMR spectra of the poly(Sty-co-O,S-BMDO) copolymers contained signals for the methylene protons adjacent to the thionoester group –C(S)–O– present at 5.0–5.5 ppm, with only trace signals at 4.10–4.15 ppm observable in samples containing a higher incorporation of O,S-BMDO (Fig. 5, top). In the 13C NMR spectrum, the chemical shift at 221 ppm was assigned to a thionoester moiety, with no signals associated with thioester moieties being detected (Fig. 5, bottom). In addition, no signals could be detected to indicate direct vinylic propagation. The formation of thionoester functional groups was additionally confirmed by FT-IR analysis (Fig. 6). Increasing incorporation of O,S-BMDO derived residues in the copolymers resulted in increased absorption in the region of 1100–1350 cm−1 which is typical for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S (1000–1150 cm−1) and C–O (1050–1310 cm−1) stretching. The spectra also had a minor reinforcement in the region around 1650–1750 cm−1 which could be attributed to stretching modes of C–H and CC groups in the aromatic rings derived from O,S-BMDO, as well as small quantities of CO (≤3% in comparison with CS).
S (1000–1150 cm−1) and C–O (1050–1310 cm−1) stretching. The spectra also had a minor reinforcement in the region around 1650–1750 cm−1 which could be attributed to stretching modes of C–H and CC groups in the aromatic rings derived from O,S-BMDO, as well as small quantities of CO (≤3% in comparison with CS).
 | ||
| Fig. 5 1H NMR (CDCl3, 700 MHz) and 13C NMR (CDCl3, 176 MHz) spectra of poly(Sty-co-O,S-BMDO) copolymers displaying virtually exclusively S-rROP derived signals. | ||
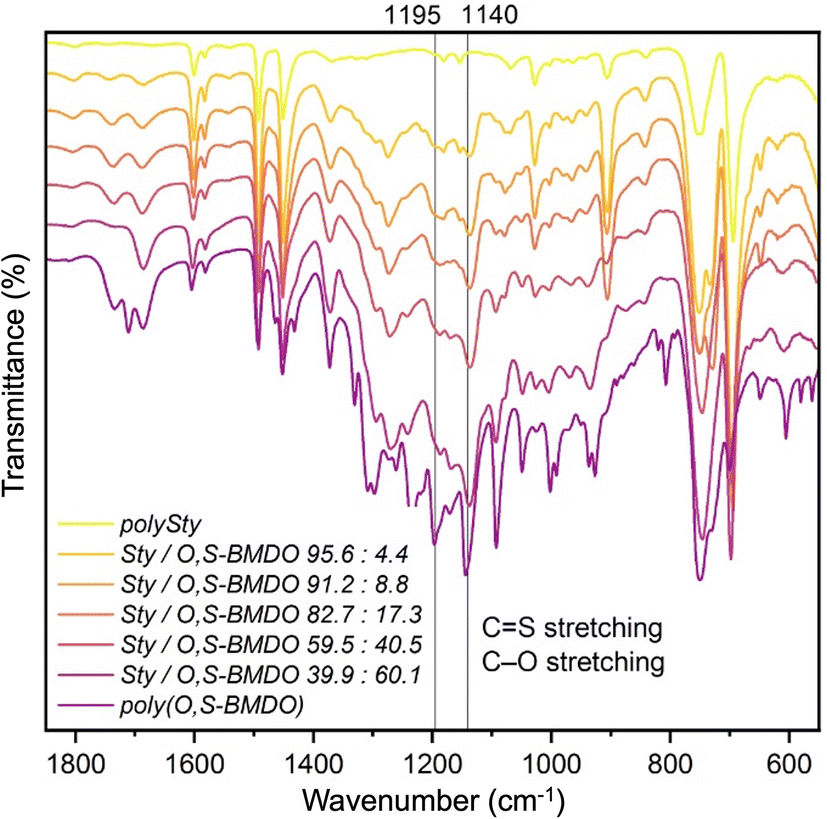 | ||
| Fig. 6 FT-IR of polystyrene, poly(O,S-BMDO) and various poly(Sty-co-O,S-BMDO) copolymers. | ||
To examine the kinetics of the polymerization system, a series of copolymerizations between styrene and O,S-BMDO (8) in a feed ratio of 8:2, respectively, were performed in bulk using AIBN (3 mol%), with the copolymerizations being stopped at various time intervals to determine the monomer conversion by 1H NMR (Fig. 7). The results indicated that O,S-BMDO copolymerizes at a slower rate than styrene, but the incorporation of O,S-BMDO begins at the onset and its incorporation rate increases with a reduction in styrene concentration. This phenomenon results in copolymers containing thionoester moieties located in a gradient-like distribution. Additionally, it was confirmed that the presence of O,S-BMDO does not produce a inhibition effect on the polymerization of styrene, which reached ca. 90% conversion after 90 min (with only 32% conversion of O,S-BMDO at this stage). This is in line with previous reports that statistical polymerization of CKA monomers with more-activated conventional monomers is challenging.34 In this case, after 120 minutes, radical copolymerization with styrene leads to ca. 43% consumption of the cyclic monomer via a radical ring-opening process, alongside ca. 93% consumption of styrene.
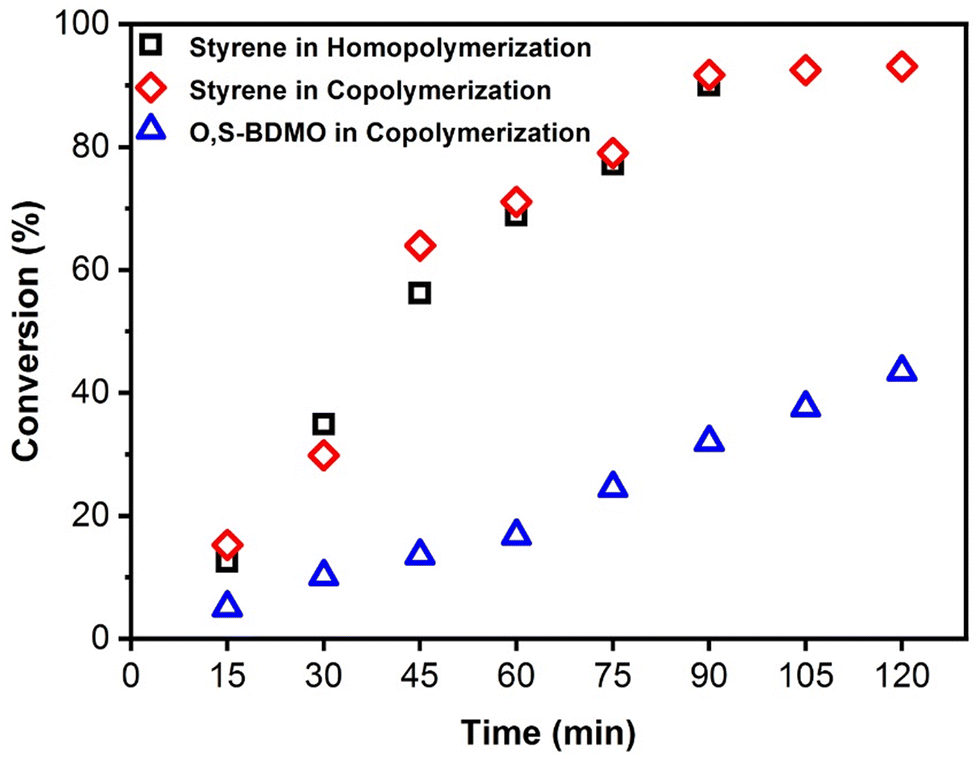 | ||
| Fig. 7 Kinetic plot displaying conversion of styrene in homopolymerization versus the conversion of styrene and O,S-BMDO (8) in copolymerization (feed ratio = 8:2, respectively). Conditions: 3 mol% AIBN at 80 °C in bulk. Details for the calculation of monomer conversion are given in the ESI.† | ||
It was observed that the molar fractions of O,S-BMDO were consistently lower in the obtained copolymers than in the initial feed, in line with previous reports for CKA monomers (Table 1). Thus, reactivity ratios were calculated using linearization methods including Fineman–Ross (FR)35 and Kelen–Tüdõs (KT)36 methods, which are derived from the Mayo–Lewis (ML) model37 (see the ESI† for methodology and calculations). The obtained results were similar in each case, and all results confirmed that styrene is incorporated into the copolymer preferentially over O,S-BMDO, and that some drift in composition can be expected to occur as the copolymerization proceeds (Table 2). Interestingly, the difference in the reactivity between styrene and O,S-BMDO was substantially smaller than that reported for BMDO (1) and styrene (rBMDO1 = 1.08 and rSty = 8.53; performed by ATRP and finished at lower conversions).38 However, the general consensus regarding sulfurated CKA monomers being less reactive than CKA monomers34 can likely be revised, and newer cyclic ketene O,S-acetal monomers with surprising aptitude for rROP can be explored.
To examine the copolymer degradation, as thionoester groups should be hydrolyzable under alkaline conditions,39 copolymers poly(Sty95.6-co-O,S-BMDO4.4) and poly(Sty82.7-co-O,S-BMDO17.3) were subjected to basic hydrolysis using KOH in EtOH/H2O at 80 °C. This process greatly reduced the Mn values of both copolymers, thereby demonstrating successful incorporation of thionoester linkages into the copolymer backbone in a relatively randomized manner (Fig. 8). Moreover, the reduction in Mn was significantly higher for the copolymer containing a higher quantity of O,S-BMDO-derived residues. Similarly, the homopolymer poly(O,S-BMDO) was hydrolyzed under basic conditions to produce a completely degraded mixture of small-molecule products, in which 3-(2-hydroxymethylphenyl) propionic acid and traces of 3-(2-hydroxymethylphenyl) propanethiocarboxylic acid could be examined by NMR (see the ESI, Fig. S31 and S32†).
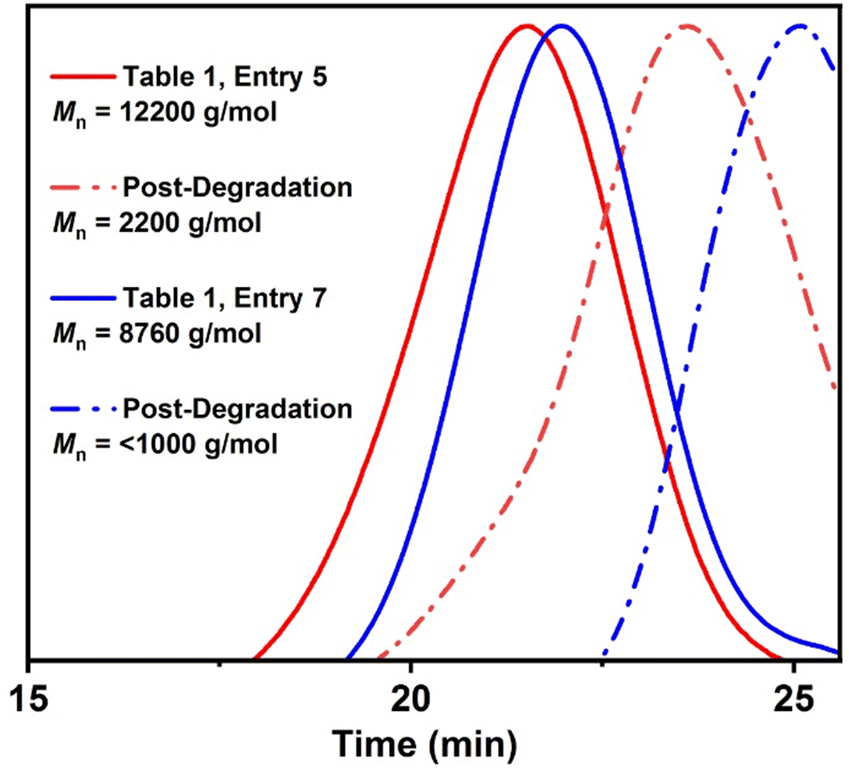 | ||
| Fig. 8 Comparison of GPC (DMF) chromatograms of poly(Sty95.6-co-O,S-BMDO4.4) (red) and poly(Sty82.7-co-O,S-BMDO17.3) (blue) before and after treatment with a KOH solution (2.0 M KOH, EtOH/H2O 4/1, 80 °C, 16 h). | ||
The unintended isomerization of the thionolactone moiety of monomer 2 to provide a thiolactone moiety was reported after heating to 150 °C in DMSO; however, under these conditions poly(O,S-BMDO) remained intact after 16 hours.17 It was additionally reported that dialkyl xanthates can rearrange to dithiocarbonates in the presence of trifluoroacetic acid (TFA)40 or aluminium chloride41 and thus the behavior of poly(O,S-BMDO) and poly(Sty82.7-co-O,S-BMDO17.3) was examined in their presence. However, in all cases the rearrangement of the thionoester moieties was accompanied by at least partial degradation of the polymer, and therefore no formal conversion of poly(thionoester) to poly(thioester) could be achieved (see the ESI, Fig. S33†).
To further examine the polymerization of O,S-BMDO (8), a series of bulk copolymerizations with various comonomers including methyl acrylate (MA), methyl methacrylate (MMA), N,N-dimethylacrylamide (DMAA), vinyl acetate (VAc), allyl acetate (AAc) and acrylamide (AM) with AIBN (3 mol%) at 80 °C were performed (Table 3). In all cases O,S-BMDO was incorporated into the polymer product; however, different reactivities were observed for different comonomer pairs. For DMAA, the molar fraction of O,S-BMDO was always lower in the obtained copolymer than in the feed (Table 3, entries 10–13). Unusually, this phenomenon was also true for MA at low feed ratios, e.g., for 10% feed of O,S-BMDO the copolymer had 5.2% incorporation (Table 3, entry 1), but at higher feeds the quantity of O,S-BMDO in the copolymer became enriched (Table 3, entry 4). Conversely, copolymerizations with MMA gave proportionate molar fractions of O,S-BMDO in the copolymer and the feed (Table 3, entries 5–9). Less-activated monomers AAc and VAc proved to be poorly reactive in copolymerizations with O,S-BMDO, yielding polymers highly enriched in O,S-BMDO (Table 3, entries 15–18). Analogous to the styrene copolymerization system, the copolymers generally displayed a decrease in their Mn values with an increase in O,S-BMDO feed.
| Entry | Comonomer (M) | Feed ratio, M:O,S-BMDO |
Copolymer ratio, M:O,S-BMDOc |
O-rROP:S-rROPc |
M
n
(kg mol−1) |
Đ |
|---|---|---|---|---|---|---|
| a 2.5 M in THF. b 2.5 M in toluene. c Determined by 1H NMR. d Determined by GPC. | ||||||
| 1 | MA | 9:1 |
94.8:5.2 |
0.03:1 |
8.84 | 2.5 |
| 2 | MA | 8:2 |
78.7:21.3 |
0.08:1 |
6.33 | 2.5 |
| 3 | MA | 7:3 |
55.7:44.3 |
0.06:1 |
7.84 | 2.2 |
| 4 | MA | 5:5 |
30.0:70.0 |
0.06:1 |
6.32 | 2.5 |
| 5 | MMA | 9:1 |
90.7:9.3 |
0.04:1 |
14.8 | 2.5 |
| 6 | MMA | 8:2 |
75.3:24.7 |
0.22:1 |
7.70 | 2.3 |
| 7 | MMAa | 8:2 |
81.2:18.8 |
0.05:1 |
5.37 | 2.7 |
| 8 | MMAb | 8:2 |
81.1:18.9 |
0.04:1 |
5.55 | 2.8 |
| 9 | MMA | 5:5 |
48.5:51.5 |
0.25:1 |
5.23 | 2.6 |
| 10 | DMAA | 9:1 |
93.0:7.0 |
1:0 |
9.38 | 2.5 |
| 11 | DMAA | 8:2 |
84.5:15.5 |
1:0 |
5.30 | 2.0 |
| 12 | DMAA | 7:3 |
78.4:21.6 |
1:0.01 |
3.98 | 2.6 |
| 13 | DMAA | 5:5 |
57.4:42.6 |
1: 0.03 | 2.18 | 2.4 |
| 14 | AM | 8:2 |
88.6:11.4 |
0.17:1 |
1.47 | 1.5 |
| 15 | AAc | 8:2 |
36.7:63.3 |
1:0.49 |
1.77 | 2.0 |
| 16 | AAc | 5:5 |
0:100 |
0.12:1 |
4.80 | 2.0 |
| 17 | VAc | 8:2 |
22.7:77.7 |
0.17:1 |
3.01 | 1.9 |
| 18 | VAc | 5:5 |
0:100 |
0.05:1 |
3.41 | 1.7 |
Interestingly, in contrast to the copolymerizations involving styrene, copolymerizations of O,S-BDMSO and DMAA underwent virtually exclusively O-side ring-opening (Table 3, entries 10–13). Thus, the obtained copolymers contained thioester linkages in the polymer backbone, as opposed to the thionoester linkages previously found (Fig. 9). Concerning the copolymerization system involving MA, regardless of the feed ratio it was found that ca. 3–8% of O,S-BDMSO was incorporated as thioester functionalities (Table 3, entries 1–4). However, proneness to ring-opening on the O-side was higher for MMA (Table 3, entries 6–9). Unusually, for the copolymerization system involving AAc there was a change from predominantly O-side ring-opening to predominantly S-side ring-opening when O,S-BMDO was applied in a higher feed ratio (Table 3, entries 15/16).
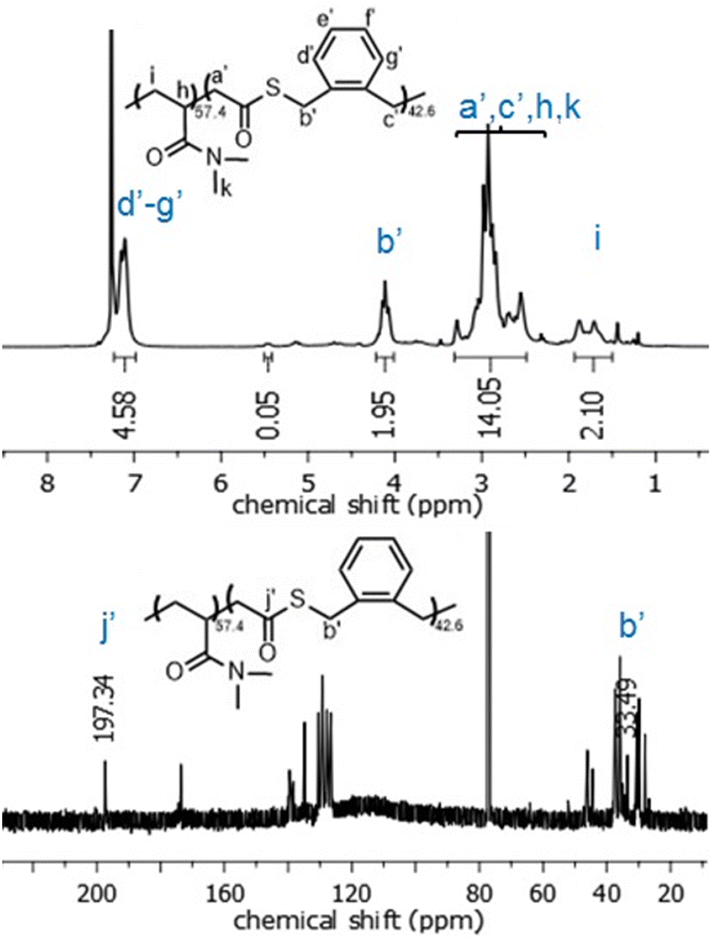 | ||
| Fig. 9 1H NMR (CDCl3, 700 MHz) and 13C NMR (CDCl3, 176 MHz) spectra of poly(DMAA-co-O,S-BMDO) copolymers displaying virtually exclusively O-rROP derived signals. | ||
It could be considered that the reaction of radical species with in-chain thionoester groups may be occurring. However, radical addition to the thionocarbonyl is expected to be an equilibrium process as the subsequent radical is non-stabilized. Therefore, if a radical species were to add onto an in-chain thionoester, unless there is a driving force that could facilitate further reactivity, the radical species should be released and the thionoester group regenerated. Ring-strain is one of the possible driving forces, this force being harnessed by thionolactone rROP monomers to introduce thioester groups during radical polymerization,17–24 but this phenomenon can be excluded in this case. If analogous processes are indeed occurring, they do not occur to a significant extent that could explain the formation of substantial levels of thioester moieties.
To examine the cause for the unusual reactivity profile of O,S-BMDO, a series of experiments were performed. As it had been examined that copolymerization with the non-polar monomer styrene occurred almost exclusively by S-side ring-opening, whereas copolymerization with the relatively polar monomer DMMA occurred almost exclusively by O-side ring-opening, it was speculated that monomer polarity may be the cause. Thus, a copolymerization with the highly polar monomer acrylamide (AM) was performed; however, this resulted in a predominantly S-side ring-opening process therefore ruling out this theory (Table 3, entry 14). To examine if the cause was the polarity of the reaction medium, copolymerizations of MMA and O,S-BMDO were performed in bulk, THF and toluene (Table 3, entries 6–8). Unusually, the outcome of the two copolymerizations in solvent was extremely similar despite the vastly different polarity of the solvents, with both resulting in copolymers with a higher ratio of thionoester to thioester moieties compared to the bulk experiment. These results were in disagreement with the styrene copolymerization system in which the solvent made no difference (Table 1, entries 2–5). It should be noted that for bulk polymerizations, the monomers themselves act as the solvent. These results point to the fact that polarity does play some role in the ring-opening process, but other effects are also in play.
Yet another theory that could explain the reactivity of O,S-BMDO relates to the penultimate effect. In this theory, the preceding monomer residues in the actively propagating polymer chain can affect the outcome of the ring-opening process of the ensuing reaction with a monomer. The copolymerization results offer support that this theory is likely in effect, but it is highly monomer specific, with some comonomers forcing virtually only S-side ring-opening in O,S-BMDO (i.e., styrene) and some monomers forcing virtually only O-side ring-opening (i.e., DMAA). It could even be considered that the penultimate O,S-BMDO monomer leads to primarily S-rROP polymerization (as per the homopolymerization results), and that, for example, O,S-BMDO monomers must be rarely incorporated sequentially during their copolymerization with DMAA. This effect would lead to the conclusion that the distribution of thiono- and thioester functionalities is highly dependent on the reactivity ratio of the two comonomers. Other copolymerization systems also appear temperamental to reaction conditions such as solvent polarity and feed ratio (i.e., MMA). In such cases it could be considered that S-rROP occurs more frequently at the end of the polymerization, where compositional drift causes higher sequential O,S-BMDO monomer incorporation.
To examine if any of these effects could be better rationalized computationally, a series of DFT calculations were performed. Although it is known that the theoretical modeling of radical reactions in general,42–44 and the ring-opening step in polymerization in particular,45,46 poses problems, we decided to examine if recent advances in computational chemistry could offer some clarity on our experimental results. Following literature data on the applicability of DFT calculations to radical ring-opening polymerization,45,46 alongside benchmark studies using radical dioxolanyl ring-opening47 as a model reaction, we selected the MN15-L/def2-TZVP theory level48,49 with a PCM continuum solvent model as implemented in the Gaussian 16 package50 for our calculations. Scaling51 of vibrational frequencies was applied, and all stationary points were characterized by vibrational analysis. All transition states exhibit exactly one imaginary frequency. Their correspondence to the transition of reactants to products for the studied reactions was confirmed by animation in GaussView.52
The computing of the initialization step (AIBN initiator) and radical site transfer to a monomer in the initial steps of the polymerization was straightforward. However, for the following reaction with O,S-BMDO, the reactivity competition between the ring-opening step at the C–O or C–S bond posed problems as indicated by the results in Table 4. The second column of Table 4 lists the differences between the Gibbs free energies of the corresponding transition states. Since the reactants for both competing ring-opening processes are the same, the difference in the energies of the corresponding transition states corresponds directly to the preference for the O-side or S-side ring-opening process. The third column in Table 4 reports the expected ratio of the respective products. The comparison of these results with the data reported in Tables 1 and 3 indicates that the theoretical predictions are not in systematic agreement with the experimental findings. It should be noted, however, that the energy differences between the transition states are very small, almost at the edge of the precision of theoretical calculations, and that due to exponential dependence small variations in these values lead to large changes in the predicted O-side or S-side selectivity of the ring-opening step. Thus, our results indicate that even using current theory levels it is not trivial to predict the selectivity of the radical ring-opening step for O,S-BMDO (8).
At the same time, it is apparent that there is a very delicate balance between the two competing reaction pathways as the energy differences are small and therefore likely sensitive to minor changes in substituents and reaction conditions. This conclusion is also supported by the lack of any correlation between the bond distances of breaking bonds (which reports geometrical changes on-going from reactants to the transition state) and the imaginary frequency of crossing the energy barrier (which reports the shape of the barrier and its location on the reaction path). In Table 5 these basic parameters of the calculated transition states are collected. For stable O,S-BMDO the corresponding bond lengths of C–O and C–S are 1.437 and 1.821 Å, respectively. Fig. 10 illustrates the structures of both transition states for the example of the reaction of a radical species composed of styrene and 2-cyano-2-propyl radical (AIBN derived) with O,S-BMDO, and the displacement vectors corresponding to the imaginary frequencies. Finally, it should be kept in mind that calculations reported here consider only the very initial steps of the polymerization process and do not include higher-level penultimate effects which could influence the energetics of the ring-opening process. Their inclusion, however, would require much larger models for which computationally performing at a sufficient level of theory is not practical. The possibility of using QM/MM techniques to address these problems is presently being explored by examining the suitability of the semiempirical methods (PM7 and xTB) for the description of the parts of the polymer chain not directly involved in the ring-opening step.
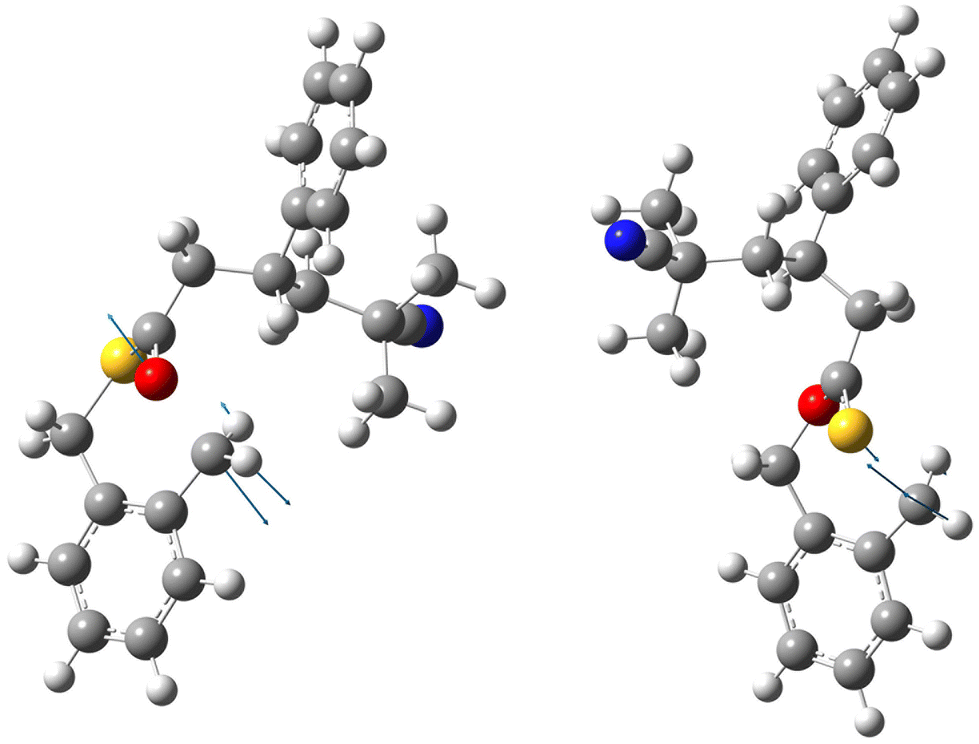 | ||
| Fig. 10 Calculated structures of the transition states for the O-side (left) and S-side (right) ring-opening step in the copolymerization of O,S-BMDO and a radical species involving styrene which had previously reacted with a 2-cyano-2-propyl radical. | ||
| Monomer | O-side ring-opening | S-side ring-opening | ||||
|---|---|---|---|---|---|---|
| d C–O [Å] | d C–S [Å] | ωi [cm−1] | d C–O [Å] | d C–S [Å] | ωi [cm−1] | |
| O,S-BMDO | 1.877 | 1.813 | 673.1 | 1.441 | 2.416 | 389.0 |
| Sty | 1.859 | 1.814 | 710.9 | 1.439 | 2.398 | 389.7 |
| MA | 1.860 | 1.815 | 690.2 | 1.438 | 2.408 | 378.6 |
| DMAA | 1.855 | 1.815 | 694.0 | 1.439 | 2.408 | 379.5 |
Conclusions
In summary, a novel monothiolated analogue of the prototypical CKA monomer BMDO, coined O,S-BMDO, was synthesized and evaluated for its polymerization ability. This monomer readily performed radical polymerization via either an O-side or S-side ring-opening process to install thioester or thionoester linkages, respectively, or a mixture of both into the polymer backbone. This monomer exhibited no detectable direct vinylic propagation. Subtle effects such as the substitution of the comonomer or the polarity of the reaction medium were found to affect this phenomenon practically on a case-by-case basis. Computational DFT calculations were performed which provided evidence that the energy barriers between the two competing reaction pathways were practically identical, offering an explanation as to why relatively minor variances can have a significant influence on the outcome. Homopolymerizations of O,S-BMDO provide the first example of a poly(thionoester) produced by radical polymerization, and copolymerizations provide the first example of the introduction of a thiocarbonyl-containing functional group into a polymer backbone by radical polymerization. Moreover, copolymers of styrene with installed thionoester linkages were proven to readily degrade under basic conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was conducted as part of the International Research Agendas PLUS Programme of the Foundation for Polish Science, co-financed by the European Union under the European Regional Development Fund (MAB PLUS/2019/11).References
- S. A. Miller, ACS Macro Lett., 2013, 2, 550–554 CrossRef CAS PubMed.
- I. E. Napper and R. C. Thompson, Environ. Sci. Technol., 2019, 53, 4775–4783 CrossRef CAS PubMed.
- Directive (EU) 2019/904 of the European Parliament and of the Council of 5 June 2019 on the Reduction of the Impact of Certain Plastic Products on the Environment, European Parliament, 2019.
- D. Braun, Int. J. Polym. Sci., 2009, 2009, 893234 Search PubMed.
- A. A. de Souza Machado, W. Kloas, C. Zarfl, S. Hempel and M. C. Rillig, Global Change Biol., 2018, 24, 1405–1416 CrossRef PubMed.
- K. D. Cox, G. A. Covernton, H. L. Davies, J. F. Dower, F. Juanes and S. E. Dudas, Environ. Sci. Technol., 2019, 53, 7068–7074 CrossRef CAS PubMed.
- Z. O. G. Schyns, M. P. Shaver, Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- E. Guégain, J. Tran, Q. Deguettes and J. Nicolas, Chem. Sci., 2018, 9, 8291–8306 RSC.
- M. Andrei, P. O. Stǎnescu, C. Drǎghici and M. Teodorescu, Colloid Polym. Sci., 2017, 295, 1805–1816 CrossRef CAS.
- Y. Gao, V. I. Böhmer, D. Zhou, T. Zhao, W. Wang and J. M. J. Paulusse, J. Controlled Release, 2016, 244, 375–383 CrossRef CAS PubMed.
- T. Cai, Y. Chen, Y. Wang, H. Wang, X. Liu, Q. Jin, S. Agarwal, J. Ji, T. Cai, Y. Chen, Y. Wang, H. Wang, X. Liu, Q. Jin, J. Ji and S. Agarwal, DOI:10.1002/macp.201400311.
- S. Louguet, V. Verret, L. Bédouet, E. Servais, F. Pascale, M. Wassef, D. Labarre, A. Laurent and L. Moine, Acta Biomater., 2014, 10, 1194–1205 CrossRef CAS PubMed.
- A. Galperin, T. J. Long and B. D. Ratner, Biomacromolecules, 2010, 11, 2583–2592 CrossRef CAS PubMed.
- J. Liu, J. Wang, Y.-F. Xue, T.-T. Chen, D.-N. Huang, Y.-X. Wang, K.-F. Ren, Y.-B. Wang, G.-S. Fu and J. Ji, J. Mater. Chem. B, 2020, 8, 5361 RSC.
- K. Zhao, B. Pi, L. Zhao, S. Tian, J. Ge, H. Yang, W. Sha and L. Wang, RSC Adv., 2019, 9, 11833–11841 RSC.
- W. J. Bailey, Z. Ni and S. R. Wu, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3021–3030 CrossRef CAS.
- N. M. Bingham and P. J. Roth, Chem. Commun., 2019, 55, 55–58 RSC.
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, J. Am. Chem. Soc., 2019, 141, 1446–1451 CrossRef CAS PubMed.
- C. M. Plummer, N. Gil, P.-E. Dufils, D. James Wilson, C. Lefay, D. Gigmes and Y. Guillaneuf, ACS Appl. Polym. Mater., 2021, 3, 3264–3271 CrossRef CAS.
- O. Ivanchenko, U. Authesserre, G. Coste, S. Mazières, M. Destarac and S. Harrisson, Polym. Chem., 2021, 12, 1931–1938 RSC.
- M. F. I. Rix, S. J. Higgs, E. M. Dodd, S. J. Coles, N. M. Bingham and P. J. Roth, DOI:10.26434/CHEMRXIV-2022-CDT52.
- O. Ivanchenko, S. Mazières, S. Harrisson and M. Destarac, Polym. Chem., 2022, 13, 5525 RSC.
- R. Kamiki, T. Kubo and K. Satoh, Macromol. Rapid Commun., 2023, 44, 2200537 CrossRef CAS PubMed.
- E. A. Prebihalo, A. M. Luke, Y. Reddi, C. J. Lasalle, V. M. Shah, C. J. Cramer and T. M. Reineke, DOI:10.26434/CHEMRXIV-2022-RGL2D.
- W. J. Bailey, T. Endo, B. Gapud, Y. N. Lin, Z. Ni, C. Y. Pan, S. E. Shaffer, S. R. Wu, N. Yamazaki and K. Yonezawa, J. Macromol. Sci., Part A: Pure Appl.Chem., 1984, 21, 979–995 CrossRef.
- V. B. Purohit, M. Pięta, J. Pietrasik and C. M. Plummer, Polym. Chem., 2022, 13, 4858–4878 RSC.
- L. Anzalone and J. A. Hirsch, J. Org. Chem., 1985, 50, 2128–2133 CrossRef CAS.
- M. J. Ahsan, J. G. Samy, H. Khalilullah, M. S. Nomani, P. Saraswat, R. Gaur and A. Singh, Bioorg. Med. Chem. Lett., 2011, 21, 7246–7250 CrossRef CAS PubMed.
- P. Heitel, L. Gellrich, L. Kalinowsky, J. Heering, A. Kaiser, J. Ohrndorf, E. Proschak and D. Merk, ACS Med. Chem. Lett., 2019, 10, 203–208 CrossRef CAS PubMed.
- K. E. Giesler and D. C. Liotta, J. Med. Chem., 2016, 59, 10244–10252 CrossRef CAS PubMed.
- J. Gaitzsch, P. C. Welsch, J. Folini, C. A. Schoenenberger, J. C. Anderson and W. P. Meier, Eur. Polym. J., 2018, 101, 113–119 CrossRef CAS.
- J. Folini, W. Murad, F. Mehner, W. Meier and J. Gaitzsch, Eur. Polym. J., 2020, 134, 109851 CrossRef CAS.
- H. Wickel, S. Agarwal and A. Greiner, Macromolecules, 2003, 36, 2397–2403 CrossRef CAS.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- N. Fineman and S. D. Ross, J. Polym. Sci., 1950, 259–262 CrossRef.
- T. Kelen and F. Tüdõs, J. Macromol. Sci., Part A: Pure Appl.Chem., 1975, 9, 1–27 CrossRef.
- F. R. Mayo and F. M. Lewis, J. Am. Chem. Soc., 1944, 66, 1594–1601 CrossRef CAS.
- H. Wickel and S. Agarwal, Macromolecules, 2003, 36, 6152–6159 CrossRef CAS.
- A. S. Narmon, C. A. M. R. van Slagmaat, S. M. A. De Wildeman and M. Dusselier, ChemSusChem, 2023, 5, e202202276 CrossRef PubMed.
- M. W. Fichtner and N. F. Haley, J. Org. Chem., 1981, 46, 3141–3143 CrossRef CAS.
- T. Kawata, K. Harano and T. Taguchi, Chem. Pharm. Bull., 1973, 604–608 CrossRef CAS.
- N. Latelli, N. Ouddai, M. Arotçaréna, P. Chaumont, P. Mignon and H. Chermette, Comput. Theor. Chem., 2014, 1027, 39–45 CrossRef CAS.
- E. I. Izgorodina, M. L. Coote and L. Radom, J. Phys. Chem. A, 2005, 109, 7558–7566 CrossRef CAS PubMed.
- I. Ying Zhang, J. Wu, Y. Luo and X. Xu, J. Chem. Theory Comput., 2010, 6, 1462–1469 CrossRef PubMed.
- B. Ochiai and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2827–2834 CrossRef CAS.
- A. Tardy, V. Delplace, D. Siri, C. Lefay, S. Harrisson, B. De Fatima Albergaria Pereira, L. Charles, D. Gigmes, J. Nicolas and Y. Guillaneuf, Polym. Chem., 2013, 4, 4776–4787 RSC.
- L. R. C. Barclay, D. Griller and K. U. Ingold, J. Am. Chem. Soc., 2002, 104, 4399–4403 CrossRef.
- H. S. Yu, X. He and D. G. Truhlar, J. Chem. Theory Comput., 2016, 12, 1280–1293 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, et al., Gaussian, Version 16, Revision A.03, Gaussian, Inc., Wallingford, CT, USA, 2016 Search PubMed.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00502j |
| This journal is © The Royal Society of Chemistry 2023 |