
Tuning intramolecular electron transfer of cyanide bridged [Co2Fe2] squares through chemical modifications in solid and solution†
Pei-Ye
You
 a,
Rong-Jia
Wei
*a,
Mo
Xie
a,
Guo-Hong
Ning
a and
Hiroki
Oshio
*bc
a,
Rong-Jia
Wei
*a,
Mo
Xie
a,
Guo-Hong
Ning
a and
Hiroki
Oshio
*bc
aCollege of Chemistry and Materials Science, Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangzhou, Guangdong 510632, P. R. China. E-mail: rjwei@jnu.edu.cn
bFaculty of Pure and Applied Sciences, Department of Chemistry, University of Tsukuba, Tennodai 1-1-1, Tsukuba 305-8571, Japan. E-mail: oshio@chem.tsukuba.ac.jp
cState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, China
First published on 9th November 2022
Abstract
Cyanide bridged heterometallic complexes exhibiting intramolecular electron transfer associated with a spin conversion (so-called electron-transfer-coupled spin transition, ETCST) have attracted considerable attention for future applications in new generation molecular switching devices. Yet, the rational control of intermetallic electron transfer, especially in bistable molecules in solid and solution, remains challenging. In this work, we report three structurally related [Co2Fe2] molecular square complexes, {[Co(bpy)2]2[Fe(tp′)(CN)3]2}(PF6)2 (1), {[Co(dmbpy)2]2[Fe(tp′)(CN)3]2}(PF6)(OTf)·2MeOH (2), and {[Co(dtbbpy)2]2[Fe(tp′)(CN)3]2}(PF6)·2MeOH·3EtOH (3), (tp′ = hydrotris(3-methylpyrazol-1-yl)borate, bpy = 2,2′-bipyridine, dmbpy = 4,4′-dimethyl-2,2′-bipyridine, and dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, OTf = trifluoromethanesulfonate), which showed different ETCST behavior with electronic configurations in the high-spin (HS: [(CoIIHS)2(FeIIILS)2]) and low-spin (LS: [(CoIIILS)2(FeIILS)2]) states in solid. Solution experiments revealed that the substituents of the bipyridyl ligand lead to different ETCST behavior, which was caused by the changes in CoII/III redox potentials. Moreover, hydrogen bonding interactions of the terminal cyanide group on the Fe sites with solvent molecules of MeOH, EtOH, and n-PrOH shifted the equilibrium temperature between the HS and LS states in the square molecules.
Introduction
Prussian blue analogues (PBAs) are a class of three-dimensional (3D) coordination networking materials with homo- or hetero-metal ions bridged by cyanide ions.1,2 Due to the outstanding electronic exchange interactions between the neighboring metal ions, remarkable chemical and physical properties, such as molecular magnets with high critical temperatures, ferroelectricity, spin transition, and tunable optical, electric, and magnetic properties, have been explored based on the cyanide bridged networks which may afford future applications such as data storage elements, molecule-based sensors, and miniature display devices.3–12 In particular, mixed valence [CoFe] PBA systems have been reported to show an intramolecular electron transfer between the cyanide bridged Co and Fe ions, associated with the spin conversion of the Co ions.13,14 This so-called electron-transfer-coupled spin transition (ETCST) phenomenon was first discovered by Hashimoto et al. in bulk PBA of K0.2Co1.4[Fe(CN)6]3·6.9H2O upon photoinduced magnetization in 1996.15 In a thermally induced ETCST system, the PBA consisted of two thermodynamically stable phases, in which the high-temperature phase (HT) of paramagnetic [CoIIHSFeIIILS] and the low-temperature (LT) phase of diamagnetic [CoIIILSFeIILS] are reversibly switched by external stimuli such as temperature and light.16,17Compared with the well-studied coordination networks, cyanide bridged discrete [CoFe] complexes can be viewed as the functional units of bulk [CoFe] PBAs and would have a variety of electronic structures, allowing systematically tunable physical and chemical properties via chemical substitution and external stimuli.18 In 2004, K. Dunbar et al. reported a molecular trigonal pyramidal [Co3Fe2] complex and its thermally induced ETCST at the molecular level for the first time.19 Up to now, cyanide-bridged molecular complexes with [CoFe],20 [CoFe2],21–23 [Co2Fe2],24–32 [Co3Fe2],19,33,34 [Co4Fe4],35 [Co5Fe4],36 and [Co6Fe4]37 cores were reported to show ETCST upon temperature changes and light irradiation in solids. Since thermal ETCST is an intramolecular redox process, the redox potentials of the donor and acceptor sites are important for designing molecular ETCST complexes. Oshio et al. reported a serial of cyanide bridged tetranuclear square complexes with the general formula of {[Co(bpyR)2]2[Fe(tpR)(CN)3]2[X]2}·solvent, where tpR and bpyR are functional capping ligands coordinated to the Fe and Co sites, respectively, and X is the counter anion. In this system, the redox potentials of the Co and Fe sites were controlled by chemical modifications of the capping ligands, resulting in tunable spin and oxidation states of the metal ions.24 Since then, some [Co2Fe2] square complexes were reported to show different ETCST behavior upon ligand modifications.29–31 Holmes et al. suggested that the donor strength of the ancillary TpR ligand, coordinated with the iron ion, appears to be the dominant factor for tuning the electron transfer properties of these [Co2Fe2] complexes.29 Mondal and co-workers reported that in their [Co2Fe2] squares, changing the capping ligands on the Co sites caused different crystal packings and bistable behavior.30 The ETCST behavior in solid is also influenced by intermolecular interactions. For example, Liu and co-workers reported a series of {[Fe(PzTp)(CN)3]2[Co(dpq)2]2}X2 (PzTp− = tetrakis(pyrazolyl)borate, dpq = dipyrido[3,2-d:2′,3′-f]quinoxaline, and X = BF4−, PF6−, and OTf−) that have different intermolecular interactions, leading to quite different ETCST behavior and T1/2 values.38 It should be noted that ETCST molecules in solution evade the crystal packing effects, and detailed solution studies might give crucial information on how to tune the ETCSTs by chemical modifications. However, the studies on ETCST in solution are still limited because of the accessible temperature range for ETCST equilibrium and solubility in organic solvents.39
We presented herein the syntheses of a series of structurally related cyanide bridged [Co2Fe2] square complexes: {[Co(bpy)2]2[Fe(tp′)(CN)3]2}(PF6)2 (1), {[Co(dmbpy)2]2[Fe(tp′)(CN)3]2}(PF6)(OTf)·2MeOH (2), and {[Co(dtbbpy)2]2[Fe(tp′)(CN)3]2}(PF6)(OTf)·2MeOH·3EtOH (3), (tp′ = hydrotris(3-methylpyrazol-1-yl)borate, bpy = 2,2′-bipyridine, dmbpy = 4,4′-dimethyl-2,2′-bipyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, and OTf = trifluoromethanesulfonate). In 1–3, systematic substitutions were made on the capping ligand of the cobalt ion. The rest of the molecule has the same cyanide building block, [Fe(tp′)(CN)3]−, constructing the [Co2Fe2] core units (Scheme 1). Note that the carefully selected substituent groups lead to the ETCST properties in the room temperature range and increase the solubility, allowing for the investigation of the ETCST in solid and solution. In solid, the complexes showed different magnetic behaviors: 1 remained paramagnetic and ETCST was not observed in the temperature range measured. 2 exhibited thermal ETCST with a transition temperature (T1/2) value of 274 K. 3 was in the LS state up to 300 K. Still, the desolvated sample showed a gradual and incomplete ETCST. Interestingly, the substitution effects on ETCST were clear in solution, showing the transition temperatures (T1/2) in the order of 1 (231 K) < 2 (283 K) < 3 (329 K) in MeOH. Moreover, the ETCST was further tuned by selecting alcohol solvents with different pKa values, understood by different hydrogen bonding interactions with the terminal cyanide group on the Fe sites, and by shifting the transition temperature.
 | ||
| Scheme 1 (Left) Controlling thermal ETCST by ligand substitution in {[Co(L)2]2[Fe(tp′)(CN)3]2}2+ squares. (Right) Representative bidentate capping ligands for 1–3. | ||
Results and discussion
Syntheses and crystallography
The square-type tetranuclear complexes 1–3 were obtained as red (for 1 and 2) and green (for 3) block crystals (Fig. S1–S3†) by the reactions of (Bu4N)[Fe(CN)3(tp′)] and Co(BF4)2·6H2O or Co(OTf)2·6H2O with the bidentate capping ligand L (bpy, dmbpy and dtbbpy for 1–3, respectively) and the structures were confirmed by single crystal X-ray diffractions (SCXRD). The crystal colors indicate the electronic states in solid, and the red and green colors suggest the HS and LS states, respectively.24 The phase purities of the complexes were confirmed by powder X-ray diffraction (PXRD) (Fig. S4–S6†). 1–3 contained a tetranuclear cyclic core with the formula of [Co2Fe2(CN)2(μ-CN)4(tp′)2(L)4]2+, comprised of alternately bridged Co and Fe ions linked by CN− anions, and the core resides on a crystallographic center of inversion (Fig. 1). The Co ion is coordinated by two bidentate capping ligands, L (bpy, dmbpy and dtbbpy for 1–3, respectively), and two cyanide N atoms, forming a distorted [CoN6] octahedron. The six-coordination sites of the Fe ions are occupied by three N atoms of the tp′ tridentate ligand and three carbon atoms from the CN− ions, two of which bridge the neighboring Co ions, and the remainder is a monodentate terminal ligand. Note that the terminal cyanide group shows hydrogen bond interactions with solvent molecules.40 The square molecules are weakly connected by the counterions through hydrogen bonding interactions (Fig. S12–S14†). Note that the tetranuclear square cores are either in the high-spin (HS: [(CoIIHS)2(FeIIILS)2]) or the low-spin (LS: [(CoIIILS)2(FeIILS)2]) state and their structures are significantly dependent upon their electronic states. | ||
| Fig. 1 Molecular structures of square cations in complex 1–3 (a–c, respectively) at 100 K. Hydrogen atoms, counter anions, and solvent molecules have been omitted for clarity. | ||
1 crystallized in the monoclinic space group P21/c at 100 K. The [Co2Fe2] core showed a distorted square shape with bent Co–N–C linkages of 163.09(18)° and 160.36(18)° (Fig. 1a). The Co–N bond lengths range from 2.099(2) to 2.168(2) Å with the average value (d〈Co–N〉) of 2.129 Å, which is close to the typical value for CoIIHS–N (2.1–2.2 Å) bonds.34 The average bond length for the Fe ions is 1.968 Å. Note that the coordination bond lengths of FeIILS and FeIIILS ions are close. By considering the Mössbauer spectral data (see below), however, the electronic configuration of 1 can be regarded as [(CoIIHS)2(FeIIILS)2] at 100 K. The core structure of 3 is markedly different from that of 1. 3 adopted the monoclinic space group P21/c at 100 K. The [Co2Fe2] core of 3 has a regular square shape with almost linear Co–N–C linkages of 177.3(3)° and 170.1(4)° (Fig. 1c). The average bond lengths for the Co and Fe ions were 1.918 and 1.964 Å, respectively. The short Co–N bonds featured characteristic CoIIILS ions,34 and the Mössbauer spectral analysis (see below) indicates the Fe ions are FeIILS. Thus, the square cation in 3 is in the LS phase with the electronic configuration of [(CoIIILS)2(FeIILS)2] at 100 K. In 3, the substitution of the strong electron-donating tert-butyl group on the bpy moiety destabilizes the cobalt d-orbitals, leading to the extra valence electron being localized on the FeII ions.
In contrast, the cobalt ions in 2 are coordinated by a moderate electron-donating ligand, dmbpy; it showed thermally induced ETCST with dramatic structural changes upon temperature variations. 2 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) at both 100 and 300 K. Similar to 3, the Co–N–C linkages in 2 were almost linear, resulting a relatively regular [Co2Fe2] square cation (Fig. 1b). When the temperature was increased from 100 to 300 K, the d〈Co–N〉 for 2 showed a remarked increase from 1.938 to 2.107 Å, suggesting intramolecular electron transfer. It should be noted that the average Co–N bond lengths at 300 K are slightly shorter than 1 in the HS phase, suggesting incomplete ETCST. Unfortunately, the SCXRD data of 2 were not collected at higher temperatures (320 K) because of solvent loss upon heating. Nevertheless, the Mössbauer spectra of 2 revealed that the Fe ions were partially in the FeIIILS state upon the temperature increase from 200 to 300 K. A thermally induced phase transition from [(CoIIILS)2(FeIILS)2] (LS) to [(CoIIHS)2(FeIIILS)2] (HS) was thus suggested.
at both 100 and 300 K. Similar to 3, the Co–N–C linkages in 2 were almost linear, resulting a relatively regular [Co2Fe2] square cation (Fig. 1b). When the temperature was increased from 100 to 300 K, the d〈Co–N〉 for 2 showed a remarked increase from 1.938 to 2.107 Å, suggesting intramolecular electron transfer. It should be noted that the average Co–N bond lengths at 300 K are slightly shorter than 1 in the HS phase, suggesting incomplete ETCST. Unfortunately, the SCXRD data of 2 were not collected at higher temperatures (320 K) because of solvent loss upon heating. Nevertheless, the Mössbauer spectra of 2 revealed that the Fe ions were partially in the FeIIILS state upon the temperature increase from 200 to 300 K. A thermally induced phase transition from [(CoIIILS)2(FeIILS)2] (LS) to [(CoIIHS)2(FeIIILS)2] (HS) was thus suggested.
Magnetic studies
Temperature-dependent magnetic susceptibility data were collected to elucidate the electronic states and the possible thermal-induced ETCST behavior of 1–3. The corresponding χMT versus T plots are shown in Fig. 2. Upon temperature variations, the χMT values of 2 showed an S-shaped curve centered at 270 K. The χMT value of 2 at 221 K was 0.26 emu mol−1 K and was nearly constant down to 2 K. The magnetic data are indicative of the diamagnetic [(CoIIILS)2(FeIILS)2] (LS) phase of 2 at 221 K, which was consistent with the SCXRD and 57Fe Mössbauer data. When the temperature was increased from 221 to 320 K, the χMT value raised to 6.40 emu mol−1 K, which was close to the value expected for two CoIIHS and two FeIIILS ions (assuming S = 3/2, g = 2.5 and S = 1/2, g = 2.3 for CoIIHS and FeIIILS, respectively).36 The results implied that 2 turned to the HS phase at 320 K. Interestingly, upon cooling, 2 showed a χMT versus T curve similar to that in the warming process but with a lower T1/2 value of 263 K (Fig. S15†). This could be due to the desolvation of solvent molecules at 320 K, which was confirmed by the TG analysis (Fig. S17†). In sharp contrast, the χMT value of 1 at 300 K was 6.97 emu mol−1 K, and the values remained nearly constant down to 25 K, suggesting the HS phase with the [(CoIIHS)2(FeIIILS)2] configuration over the whole temperature range measured and no ETCST occurred. It is noted that the sudden increase followed by the decrease in the χMT value below 25 K is due to the intramolecular ferromagnetic and intermolecular antiferromagnetic interactions, respectively. 3 is in the diamagnetic LS phase below 300 K with a constant χMT value of 0.33 emu mol−1 K. Upon heating above 300 K, the χMT exhibited a sharp increase centered at 352 K and reached a value of 6.97 cm3 mol−1 K at 400 K. The corresponding cooling process showed a gradual and incomplete ETCST process with a T1/2 value of 257 K and a constant χMT value of 3.12 cm3 mol−1 K from 100 to 45 K (Fig. S16†). The incomplete ETCST behavior in the cooling process might be caused by the desolvation and loss of crystallinity upon heating (Fig. S8 and S18†).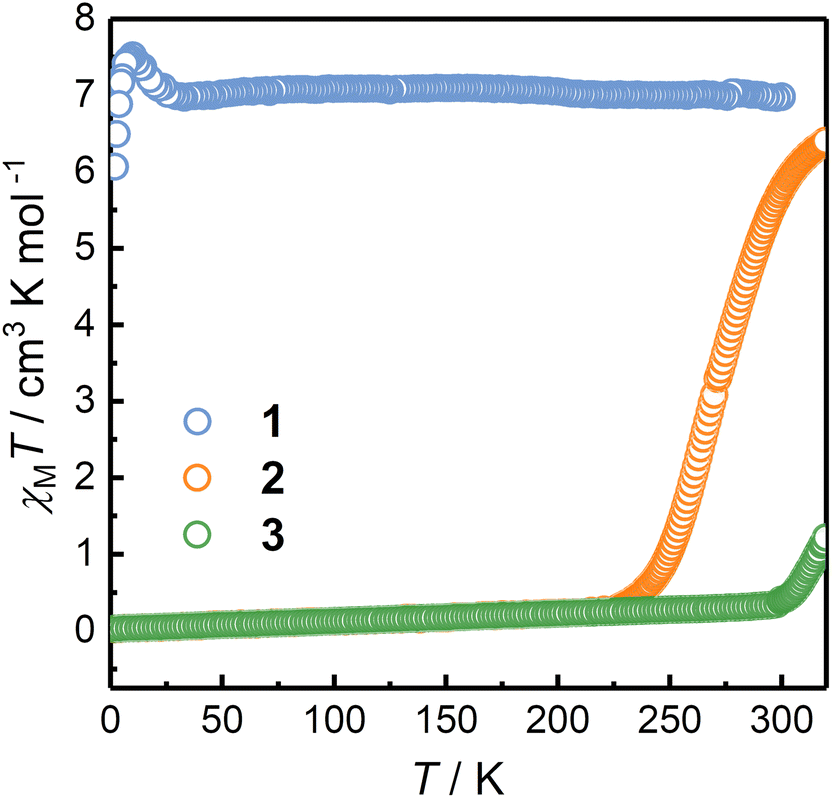 | ||
| Fig. 2 χ M T versus T plots of 1–3 in solid. | ||
The significantly different magnetic behavior of 1–3 can be explained by the modified redox potentials of the metal ions caused by the substituent groups on the capping ligands. However, the bistable behavior is also affected by crystal packing and intermolecular interactions in solid.38,41–43 Holmes et al. reported the cyanide bridged complexes {[Co(bpy)2]2[Fe(tp′)(CN)3]2}[Fe(tp′)(CN)3]2·12H2O and {[Co(bpy)2]2[Fe(tp′)(CN)3]2}[BPh4]2·6MeCN, of which the [Co2Fe2] core cations are isostructural to that in 1, showing gradual ETCST with the T1/2 values of 230 and 244 K, respectively. It should be noted that solution experiments are necessary to elucidate the substituent effects on ETCST behavior.24,31
57Fe Mössbauer spectra
57Fe Mössbauer spectra were recorded to investigate the oxidation and spin states of the Fe ions in the squares. The Mössbauer spectrum of 1 at 300 K showed one quadrupole doublet with an isomer shift (δIS) of 0.03 mm s−1 and a quadrupole splitting (ΔEQ) of 0.77 mm s−1, relative to the α-Fe foil at 300 K (Fig. S22†). The obtained parameters agree with the typical FeIIILS spices,13 suggesting the square complex in the HS phase. The Mössbauer spectrum of 3 at 300 K showed a single quadrupole doublet with the Mössbauer parameters of δIS = 0.06 mm s−1 and ΔEQ = 0.51 mm s−1, suggesting the LS phase of [(CoIIILS)2(FeIILS)2].13 This was supported by the magnetic susceptibility data (Fig. S23†). The Mössbauer spectra of 2 were recorded at various temperatures to confirm the thermally induced ETCST behavior (Fig. 3). At 200 K, 2 exhibited one quadrupole doublet with Mössbauer parameters of δIS = 0.12 mm s−1 and ΔEQ = 0.44 mm s−1 similar to 3, confirming that 2 was in the LS phase at 200 K. When the temperature was increased to 265 K, an additional doublet (δIS = −0.06 and ΔEQ = 0.75 mm s−1) corresponding to FeIIILS ions, appeared. The peak area ratio (FeIIILS/FeIILS) was 0.39 at 265 K and 0.91 at 300 K, which agreed with the magnetic data of 0.37 and 0.90 at 265 and 300 K, respectively. The Mössbauer and magnetic data of 2 revealed the occurrence of a substantial intramolecular electron transfer between Co and Fe ions.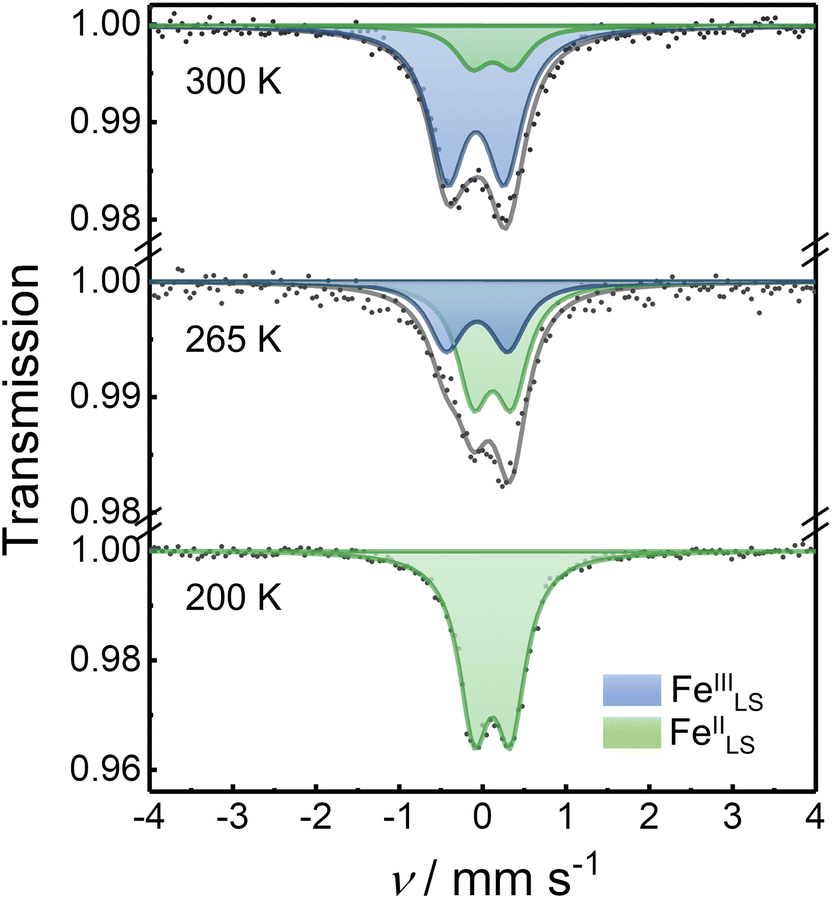 | ||
| Fig. 3 Mössbauer spectra of 2 collected at various temperatures. The experimental data points are shown in black spots while the fitting curves are displayed as solid lines. The fitting parameters are given in Table S3, ESI.† | ||
Infrared absorption spectra
Since stretching frequencies of the cyanide groups are sensitive to the electronic states of the linked metal ions,24 the infrared (IR) absorption spectra were recorded to further elucidate the electronic states of 1–3. 1 showed a νCN stretching band at 2154 cm−1, corresponding to the cyanide ions of the CoIIHS-(μ-CN)-FeIIILS linkages in the HS [(CoIIHS)2(FeIIILS)2] phase (Fig. 4a).26 A relatively weak band at 2136 cm−1 was also found in the spectrum, characteristic of the stretching absorption band of the terminal cyanide group. 3 exhibited the νCN bands at 2107 and 2089 cm−1, assigned to the bridging cyanides in CoIIILS-(μ-CN)-FeIILS species in the LS phase (Fig. 4b).33 Note that the absorption peak of the terminal cyanide ions was found at 2065 cm−1. The IR spectra of 2 were recorded at various temperatures to investigate the variations in the CN− stretching modes upon ETCST (Fig. 4c–e). At 225 K, 2 exhibited νCN absorption bands at 2104 and 2085 cm−1 similar to those of 3, suggesting the LS [(CoIIILS)2(FeIILS)2] phase at this temperature. When the temperature was increased to 270 K, the νCN absorption band corresponding to the CoIIILS-(μ-CN)-FeIILS species decreased in intensity, associated with the increase in the absorption band that is assignable to the cyanide ions in CoIIHS-(μ-CN)-FeIIILS linkages. Upon further heating to 320 K, the νCN absorption bands associated with the CoIIILS-(μ-CN)-FeIILS species vanished, and 2 exhibited an IR spectrum similar to 1, indicative of the complete conversion from the LS to HS phase.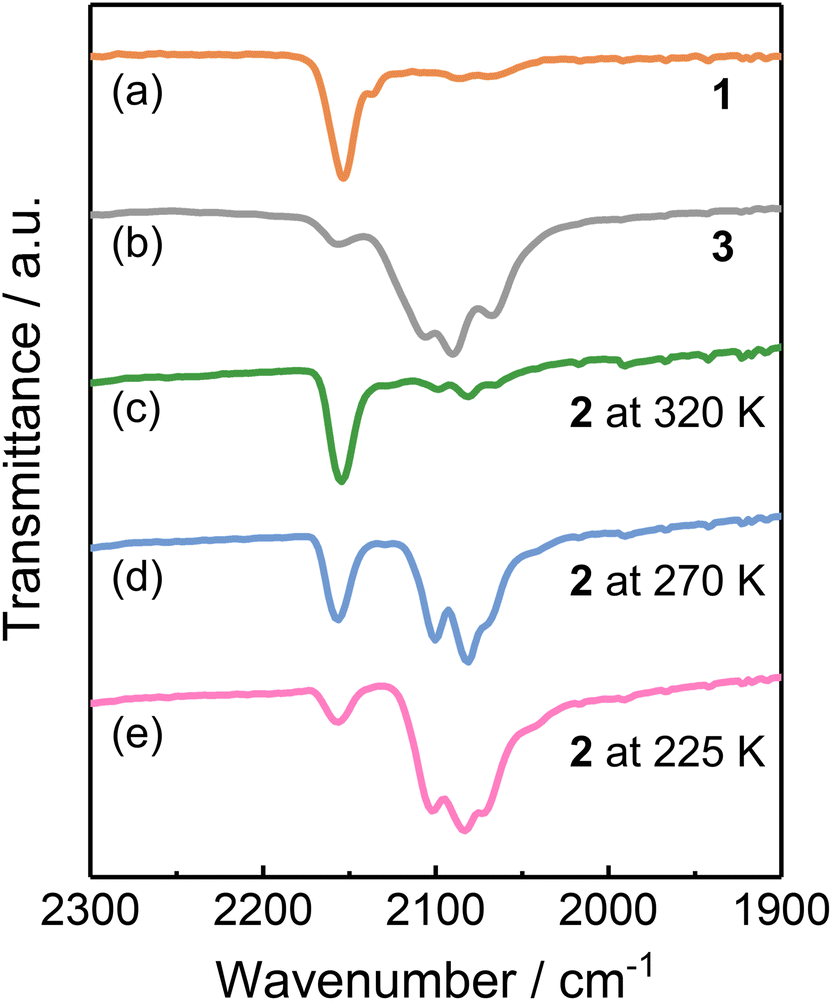 | ||
| Fig. 4 IR spectra of complex 1 (a) and 3 (b) collected at room temperature and 2 (c–e) recorded at various temperatures. | ||
UV-vis-NIR absorption spectra
The UV-vis-NIR absorption spectra of 1–3 in MeOH were recorded to investigate the electronic structures of the complexes (Fig. 5a). 1 showed an absorption band with a peak maximum at 420 nm at room temperature, characteristic of the ligand-to-metal charge transfer (LMCT) and metal-to-ligand charge transfer (MLCT) bands in the FeIII and CoII ions, respectively. A shoulder band at 460 nm corresponds to the intervalence charge transfer (IVCT) band of CoII → FeIII,24,31 suggesting that 1 is in the HS [(CoIIHS)2(FeIIILS)2] state. 3 showed an electronic spectrum consisting of two absorption maxima at 420 and 740 nm, assignable to the MLCT band of the low-spin FeII ion and the IVCT band of FeII → CoIII, respectively.24,31 The results suggest that 3 is in the LS [(CoIIILS)2(FeIILS)2] state in MeOH at room temperature. The electronic spectrum of 2 exhibited two absorption bands at 420 and 740 nm. Note that the absorption intensity for the lower energy peak is relatively weak compared to that of 3 and that the electronic state of 2 in MeOH is a mixture of the HS and LS states. | ||
| Fig. 5 (a) UV-vis-NIR spectra of 1–3 at room temperature. (b) The UV-vis-NIR spectral change of 2 from 335–245 K. (c) [(CoIIHS)2(FeIIILS)2] fractions versus temperature plots of 1–3 in MeOH. (d) [(CoIIHS)2(FeIIILS)2] fractions versus temperature plots of 2 in various solvents. For (c) and (d), the [(CoIIILS)2(FeIILS)2] fractions are shown as open cycles which were estimated by the intensity changes in absorption at 740 nm upon temperature change, while the solid lines are fitting curves estimated using first-order thermodynamic equilibrium. | ||
ETCST behavior in solution
State conversions of 1, 2, and 3 were investigated by UV-vis-NIR and electrochemistry to understand the substitution effects. The UV-vis-NIR absorption spectra were recorded at various temperatures and in various solvents. At 335 K, 2 in MeOH showed a very similar absorption spectrum to that of 1 in the same solvent, suggesting that 2 was in the HS state in this condition. Upon cooling, the CoII MLCT band at 420 nm decreased in intensity and a new band at 740 nm corresponding to the FeII → CoIII IVCT band appeared. The results confirmed that 2 underwent an electronic state conversion from the HS [(CoIIHS)2(FeIIILS)2] to the LS [(CoIIILS)2(FeIILS)2] states as the temperature was decreased in MeOH (Fig. 5b). Moreover, an isosbestic point at 554 nm was observed in the spectra upon temperature change, suggesting that ETCST in solution was following the first order equilibrium. Similar temperature-dependent spectral variations were also found in the methanolic solutions of 1 and 3 at different temperature ranges, thus confirming the occurrence of thermally induced ETCST. The temperature dependence of the HS [(CoIIHS)2(FeIIILS)2] fractions (γHS) was plotted by the absorption intensities of the FeII → CoIII IVCT band at 740 nm (Fig. 5c). Assuming the first-order thermodynamic ETCST equilibrium (ln[(1 − γHS)/γHS] = ΔH/RT − ΔS/R),44 a systematical change in the T1/2 values was obtained in the order of electron donation of the substituent groups on the Co sites, 1 (T1/2 = 231 K) < 2 (283 K) < 3 (329 K), which was due to the destabilization of the cobalt d-orbitals upon the substitutions.On the other hand, the Fe ions in the square molecules are coordinated by the non-bridging (terminal) cyanide groups, which may serve as proton acceptors for solvent alcohol. Such hydrogen bonding interactions should affect the redox potentials of the Fe ions. It is, therefore, suggested that alcoholic solvents with different pKa values would change the ETCST equilibrium.40,45 The temperature-dependent UV-vis-NIR spectra of 1–3 were recorded in MeOH, EtOH, and n-PrOH (Fig. 5d and S24–S30†). The results showed that the three complexes showed drastic solvent effects with spectral changes in the MLCT and IVCT bands. The T1/2versus solvent pKa plots showed that the T1/2 values are inversely proportional to the pKa values (Fig. 6). Note that the pKa values of MeOH, EtOH, and n-PrOH are 15.3, 15.9, and 16.1, respectively. The alcohol with the larger pKa value forms weaker hydrogen bonds with the terminal cyanide nitrogen atoms coordinated with the iron ions. Therefore, n-PrOH mostly destabilizes the Fe d-orbital among the alcohols, leading to the lowest T1/2 value.
 | ||
| Fig. 6 T 1/2 versus pKa for the ETCST equilibrium of 1 (green), 2 (blue), and 3 (red) in various solvents. The T1/2 shows a positive correlation with the electron donating strength of capping ligands and a reverse correlation with the pKa value of the solvent. | ||
The electrochemical properties of 1, 2, and 3 were investigated in acetonitrile, and their cyclic voltammograms are shown in Fig. S35–S37.† The complexes showed two redox waves at (0.608 and 0.479 V), (0.570 and 0.403 V), and (0.075 and −0.129 V) versus SCE in 1, 2, and 3, respectively, which were assigned to the CoII/CoIII redox waves in the square molecules. Moreover, DFT calculations were performed to estimate the redox potentials of [Co(L)3]2+ ions, which showed negative shifts in the order of electron-donating groups substituted on the capping ligand: [Co(bpy)3]2+ > [Co(dmbpy)3]2+ > [Co(dtbbpy)3]2+ (Fig. S41†). The cobalt d-orbitals are destabilized by ligand substitutions with stronger electron-donating groups, such as in the order of proton, methyl, and tert-butyl groups, resulting in the delocalized Co d-orbitals and leading to the stabilization of the LS state and higher T1/2 values (Fig. 6). Note that the potential scans from +0.8 to −0.4 V gave irreversible waves for the complexes (Fig. S38–S40†).
Conclusions
In summary, we synthesized three cyanide-bridged [Co2Fe2] molecular squares by systematic ligand substitution, and tunable intramolecular electron transfer behavior was explored both in solid and solution. 1 is in the HS phase with an electronic configuration of [(CoIIHS)2(FeIIILS)2] over the whole temperature range in solid. 2 shows thermal ETCST with a T1/2 value of 274 K, while 3 exhibits different ETCST behaviors associated with the desolvation. Solution experiments, which exclude the crystal packing effects on ETCST behavior, were performed to obtain a deeper understanding of the substitution effects and to prove tunable ETCST equilibrium using solvents. Transition temperatures (T1/2) in solution showed a positive correlation with the electron donating strength of the substituent groups on the bpy ligands coordinated with the Co ions. In contrast, a reverse correlation was observed between the T1/2 and pKa values of the selected alcohol solvents, exhibiting different hydrogen bonding interactions with the terminal cyanide groups on the Fe sites. The present study offers vivid examples for tuning intramolecular electron transfer behavior in molecular complexes both in solid and in solution by chemical modifications. The deeper understanding obtained in this work helps construct molecular materials with predictable and controllable switching properties.Author contributions
R.-J. W., and H. O. designed the research. P.-Y. Y. and R.-J. W. conducted the experiments and data analysis. M. X. contributed to theoretical calculations. P.-Y. Y., R.-J. W., G.-H. N., and H. O. co-wrote the manuscript. All authors read and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported financially by the National Natural Science Foundation of China (no. 21975104, 22150004 and 21901085) and the Guangdong Major Project of Basic and Applied Research (no. 2019B030302009). G. H. N. is thankful for the financial support from the Guangdong Basic and Applied Basic Research Foundation (no. 2019B151502024 and 2021A0505030037) and the Guangdong Province Pearl River Scholar Funded Scheme (2019).Notes and references
- T. Mallah, S. Thiébaut, M. Verdaguer and P. Veillet, High-Tc Molecular-Based Magnets: Ferrimagnetic Mixed-Valence Chromium(III)-Chromium(II) Cyanides with Tc at 240 and 190 Kelvin, Science, 1993, 262, 1554–1557 CrossRef CAS PubMed.
- G. N. Newton, M. Nihei and H. Oshio, Cyanide-Bridged Molecular Squares-the Building Units of Prussian Blue, Eur. J. Inorg. Chem., 2011, 3031–3042 CrossRef CAS.
- O. Sato, J. Tao and Y. Z. Zhang, Control of Magnetic Properties through External Stimuli, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS PubMed.
- O. Sato, Dynamic Molecular Crystals with Switchable Physical Properties, Nat. Chem., 2016, 8, 644–656 CrossRef CAS.
- W. Kosaka, K. Nomura, K. Hashimoto and S.-i. Ohkoshi, Observation of an Fe(II) Spin-Crossover in a Cesium Iron Hexacyanochromate, J. Am. Chem. Soc., 2005, 127, 8590–8591 CrossRef CAS.
- S. Ferlay, T. Mallah, R. Ouahes, P. Veillet and M. Verdaguer, A Room-Temperature Organometallic Magnet Based on Prussian Blue, Nature, 1995, 378, 701–703 CrossRef CAS.
- K.-T. Lian, W.-W. Wu, G.-Z. Huang, Y. Liu, S.-G. Wu, Z.-P. Ni and M.-L. Tong, Reversible Step Spin Crossover Modulation Via Water Absorption and Dehydration in a 3D Hofmann-Type Framework, Inorg. Chem. Front., 2021, 8, 4334–4340 RSC.
- K.-P. Xie, Z.-Y. Ruan, X.-X. Chen, J. Yang, S.-G. Wu, Z.-P. Ni and M.-L. Tong, Light-Induced Hidden Multistability in a Spin Crossover Metal-Organic Framework, Inorg. Chem. Front., 2022, 9, 1770–1776 RSC.
- C.-J. Zhang, K.-T. Lian, S.-G. Wu, Y. Liu, G.-Z. Huang, Z.-P. Ni and M.-L. Tong, The Substituent Guest Effect on Four-Step Spin-Crossover Behavior, Inorg. Chem. Front., 2020, 7, 911–917 RSC.
- Y.-Y. Peng, S.-G. Wu, Y.-C. Chen, W. Liu, G.-Z. Huang, Z.-P. Ni and M.-L. Tong, Asymmetric Seven-/Eight-Step Spin-Crossover in a Three-Dimensional Hofmann-Type Metal-Organic Framework, Inorg. Chem. Front., 2020, 7, 1685–1690 RSC.
- A. Sulaiman, Y.-Z. Jiang, M. K. Javed, S.-Q. Wu, Z.-Y. Li and X.-H. Bu, Tuning of Spin-Crossover Behavior in Two Cyano-Bridged Mixed-Valence FeIII2FeII Trinuclear Complexes Based on a TpR Ligand, Inorg. Chem. Front., 2022, 9, 241–248 RSC.
- Q. Liu, N.-T. Yao, H.-Y. Sun, J.-X. Hu, Y.-S. Meng and T. Liu, Light Actuated Single-Chain Magnet with Magnetic Coercivity, Inorg. Chem. Front., 2022, 9, 5093–5104 RSC.
- N. Hoshino, F. Iijima, G. N. Newton, N. Yoshida, T. Shiga, H. Nojiri, A. Nakao, R. Kumai, Y. Murakami and H. Oshio, Three-Way Switching in a Cyanide-Bridged [CoFe] Chain, Nat. Chem., 2012, 4, 921–926 CrossRef CAS PubMed.
- Y.-S. Meng, O. Sato and T. Liu, Manipulating Metal-to-Metal Charge Transfer for Materials with Switchable Functionality, Angew. Chem., Int. Ed., 2018, 57, 12216–12226 CrossRef CAS.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Photoinduced Magnetization of a Cobalt-Iron Cyanide, Science, 1996, 272, 704–705 CrossRef CAS PubMed.
- D. Aguilà, Y. Prado, E. S. Koumousi, C. Mathonière and R. Clérac, Switchable Fe/Co Prussian Blue Networks and Molecular Analogues, Chem. Soc. Rev., 2016, 45, 203–224 RSC.
- Y.-S. Meng and T. Liu, Manipulating Spin Transition to Achieve Switchable Multifunctions, Acc. Chem. Res., 2019, 52, 1369–1379 CrossRef CAS PubMed.
- M. Nihei, Molecular Prussian Blue Analogues: From Bulk to Molecules and Low-Dimensional Aggregates, Chem. Lett., 2020, 49, 1206–1215 CrossRef CAS.
- C. P. Berlinguette, A. Dragulescu-Andrasi, A. Sieber, J. R. Galán-Mascarós, H.-U. Güdel, C. Achim and K. R. Dunbar, A Charge-Transfer-Induced Spin Transition in the Discrete Cyanide-Bridged Complex {[Co(Tmphen)2]3[Fe(CN)6]2}, J. Am. Chem. Soc., 2004, 126, 6222–6223 CrossRef CAS PubMed.
- E. S. Koumousi, I.-R. Jeon, Q. Gao, P. Dechambenoit, D. N. Woodruff, P. Merzeau, L. Buisson, X. Jia, D. Li and F. Volatron, Metal-to-Metal Electron Transfer in Co/Fe Prussian Blue Molecular Analogues: The Ultimate Miniaturization, J. Am. Chem. Soc., 2014, 136, 15461–15464 CrossRef CAS PubMed.
- T. Liu, D.-P. Dong, S. Kanegawa, S. Kang, O. Sato, Y. Shiota, K. Yoshizawa, S. Hayami, S. Wu, C. He and C.-Y. Duan, Reversible Electron Transfer in a Linear {Fe2Co} Trinuclear Complex Induced by Thermal Treatment and Photoirraditaion, Angew. Chem., Int. Ed., 2012, 51, 4367–4370 CrossRef CAS PubMed.
- J.-X. Hu, L. Luo, X.-J. Lv, L. Liu, Q. Liu, Y.-K. Yang, C.-Y. Duan, Y. Luo and T. Liu, Light-Induced Bidirectional Metal-to-Metal Charge Transfer in a Linear Fe2Co Complex, Angew. Chem., Int. Ed., 2017, 56, 7663–7668 CrossRef CAS.
- N.-T. Yao, L. Zhao, C. Yi, Q. Liu, Y. M. Li, Y.-S. Meng, H. Oshio and T. Liu, Manipulating Selective Metal-to-Metal Electron Transfer to Achieve Multi-Phase Transitions in an Asymmetric [Fe2Co]-Assembled Mixed-Valence Chain, Angew. Chem., Int. Ed., 2022, 61, e202115367 CAS.
- M. Nihei, Y. Sekine, N. Suganami, K. Nakazawa, A. Nakao, H. Nakao, Y. Murakami and H. Oshio, Controlled Intramolecular Electron Transfers in Cyanide-Bridged Molecular Squares by Chemical Modifications and External Stimuli, J. Am. Chem. Soc., 2011, 133, 3592–3600 CrossRef CAS.
- Y.-Z. Zhang, D. Li, R. Clérac, M. Kalisz, C. Mathonière and S. M. Holmes, Reversible Thermally and Photoinduced Electron Transfer in a Cyano-Bridged {Fe2Co2} Square Complex, Angew. Chem., Int. Ed., 2010, 49, 3752–3756 CrossRef CAS.
- M. Nihei, Y. Sekine, N. Suganami and H. Oshio, Thermally Two-Stepped Spin Transitions Induced by Intramolecular Electron Transfers in a Cyanide-Bridged Molecular Square, Chem. Lett., 2010, 39, 978–979 CrossRef CAS.
- L. Meng, Y.-F. Deng, S. Liu, Z. Zheng and Y.-Z. Zhang, A Smart Post-Synthetic Route Towards [Fe2Co2] Molecular Capsules with Electron Transfer and Bidirectional Switching Behaviors, Sci. China: Chem., 2021, 64, 1340–1348 CrossRef CAS.
- L. Meng, Y.-F. Deng and Y.-Z. Zhang, Anion-Dependent Electron Transfer in the Cyanide-Bridged [Fe2Co2] Capsules, Inorg. Chem., 2021, 60, 14330–14335 CrossRef CAS PubMed.
- Y.-Z. Zhang, P. Ferko, D. Siretanu, R. Ababei, N. P. Rath, M. J. Shaw, R. Clérac, C. Mathonière and S. M. Holmes, Thermochromic and Photoresponsive Cyanometalate Fe/Co Squares: Toward Control of the Electron Transfer Temperature, J. Am. Chem. Soc., 2014, 136, 16854–16864 CrossRef CAS PubMed.
- S. Kamilya, S. Ghosh, S. Mehta and A. Mondal, Effect of Ligand Modulation on Metal-to-Metal Electron Transfer in a Series of [Fe2Co2] Molecular Square Complexes, J. Phys. Chem. A, 2021, 125, 4775–4783 CrossRef CAS PubMed.
- M. Nihei, K. Shiroyanagi, M. Kato, R. Takayama, H. Murakami, Y. Kera, Y. Sekine and H. Oshio, Intramolecular Electron Transfers in a Series of [Co2Fe2] Tetranuclear Complexes, Inorg. Chem., 2019, 58, 11912–11919 CrossRef CAS PubMed.
- Y. Sekine, M. Nihei, R. Kumai, H. Nakao, Y. Murakami and H. Oshio, Investigation of the Light-Induced Electron-Transfer-Coupled Spin Transition in a Cyanide-Bridged [Co2Fe2] Complex by X-Ray Diffraction and Absorption Measurements, Inorg. Chem. Front., 2014, 1, 540–543 RSC.
- C. P. Berlinguette, A. Dragulescu-Andrasi, A. Sieber, H.-U. Güdel, C. Achim and K. R. Dunbar, A Charge-Transfer-Induced Spin Transition in a Discrete Complex: The Role of Extrinsic Factors in Stabilizing Three Electronic Isomeric Forms of a Cyanide-Bridged Co/Fe Cluster, J. Am. Chem. Soc., 2005, 127, 6766–6779 CrossRef CAS PubMed.
- R.-J. Wei, R. Nakahara, J. M. Cameron, G. N. Newton, T. Shiga, H. Sagayama, R. Kumai, Y. Murakami and H. Oshio, Solvent-Induced On/Off Switching of Intramolecular Electron Transfer in a Cyanide-Bridged Trigonal Bipyramidal Complex, Dalton Trans., 2016, 45, 17104–17107 RSC.
- D. Li, R. Clérac, O. Roubeau, E. Harté, C. Mathonière, R. Le Bris and S. M. Holmes, Magnetic and Optical Bistability Driven by Thermally and Photoinduced Intramolecular Electron Transfer in a Molecular Cobalt-Iron Prussian Blue Analogue, J. Am. Chem. Soc., 2008, 130, 252–258 CrossRef CAS PubMed.
- R.-J. Wei, T. Shiga, G. N. Newton, D. Robinson, S. Takeda and H. Oshio, A Cyanide-Bridged Magnetically Switchable Cage with Encapsulated Water Molecules, Inorg. Chem., 2016, 55, 12114–12117 CrossRef CAS.
- T. Shiga, T. Tetsuka, K. Sakai, Y. Sekine, M. Nihei, G. N. Newton and H. Oshio, Cyanide-Bridged Decanuclear Cobalt–Iron Cage, Inorg. Chem., 2014, 53, 5899–5901 CrossRef CAS.
- C.-Q. Jiao, Y.-S. Meng, Y. Yu, W. J. Jiang, W. Wen, H. Oshio, Y. Luo, C.-Y. Duan and T. Liu, Effect of Intermolecular Interactions on Metal-to-Metal Charge Transfer: A Combined Experimental and Theoretical Investigation, Angew. Chem., Int. Ed., 2019, 58, 17009–17015 CrossRef CAS.
- D. Siretanu, D. Li, L. Buisson, D. M. Bassani, S. M. Holmes, C. Mathonière and R. Clérac, Controlling Thermally Induced Electron Transfer in Cyano-Bridged Molecular Squares: From Solid State to Solution, Chem. – Eur. J., 2011, 17, 11704–11708 CrossRef CAS PubMed.
- M. Nihei, Y. Yanai, I.-J. Hsu, Y. Sekine and H. Oshio, A Hydrogen-Bonded Cyanide-Bridged [Co2Fe2] Square Complex Exhibiting a Three-Step Spin Transition, Angew. Chem., Int. Ed., 2017, 56, 591–594 CrossRef CAS PubMed.
- C.-Q. Jiao, W.-J. Jiang, Y.-S. Meng, W. Wen, L. Zhao, J.-L. Wang, J.-X. Hu, G. G. Gurzadyan, C.-Y. Duan and T. Liu, Manipulation of Successive Crystalline Transformations to Control Electron Transfer and Switchable Functions, Natl. Sci. Rev., 2018, 5, 507–515 CrossRef CAS.
- T.-T. Ma, X.-P. Sun, Z.-S. Yao and J. Tao, Homochiral Versus Racemic Polymorphs of Spin-Crossover Iron(II) Complexes with Reversible Liesst Effect, Inorg. Chem. Front., 2020, 7, 1196–1204 RSC.
- Y.-Y. Zhu, H.-Q. Li, Z.-Y. Ding, X.-J. Lü, L. Zhao, Y.-S. Meng, T. Liu and S. Gao, Spin Transitions in a Series of [Fe(Pybox)2]2+ Complexes Modulated by Ligand Structures, Counter Anions, and Solvents, Inorg. Chem. Front., 2016, 3, 1624–1636 RSC.
- S. De, S. Tewary, D. Garnier, Y. Li, G. Gontard, L. Lisnard, A. Flambard, F. Breher, M. L. Boillot and G. Rajaraman, Solution and Solid-State Study of the Spin-Crossover [FeII(R-BIK)3](BF4)2 Complexes (R = Me, Et, Vinyl), Eur. J. Inorg. Chem., 2018, 414–428 CrossRef CAS.
- Y. Sekine, M. Nihei and H. Oshio, Dimensionally Controlled Assembly of an External Stimuli-Responsive [Co2Fe2] Complex into Supramolecular Hydrogen-Bonded Networks, Chem. – Eur. J., 2017, 23, 5193–5197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystal data, additional crystallographic figures, magnetic measurements, TGA, 57Fe Mössbauer spectra, IR spectra, UV-vis-NIR spectra, and theoretical calculations. CCDC 2189899–2189902. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi02108k |
| This journal is © the Partner Organisations 2023 |