
Supramolecular squares of Sn(IV)porphyrins with Re(I)-corners for the fabrication of self-assembled nanostructures performing photocatalytic degradation of Eriochrome Black T dye†
Nirmal Kumar
Shee
and
Hee-Joon
Kim
 *
*
Department of Chemistry and Bioscience, Kumoh National Institute of Technology, Gumi 39177, Republic of Korea. E-mail: hjk@kumoh.ac.kr
First published on 16th November 2022
Abstract
Two Sn(IV)porphyrin-based supramolecular squares (1 and 2) were synthesized via the reaction of Re(CO)5Cl with two [trans-bis(4-pyridyl)(porphyrinato)]Sn(IV) complexes (SnP1 and SnP2). Although the structural framework of these architectures was fixed with a closed 2D tetrameric array, altering the substituent on the Sn(IV)porphyrin units not only produced conformationally different forms but also led to distinctly different nanostructures. These nanostructured materials efficiently performed photocatalytic degradation of Eriochrome Black T (EBT) dye, thereby exhibiting degradation efficiencies between 88 and 95% within 90 min in aqueous media under the irradiation of visible-light. It was also found that the pseudo-first-order degradation rate constant of 2 was higher than that of 1.
Introduction
Our Mother Earth suffers from water pollution due to growing industrial and agricultural activities.1 Every year, huge amounts of toxic materials such as dyes, pesticides, pigments, herbicides, phenol, and nitro compounds are extensively discharged into water bodies and cause serious problems for the aquatic life by damaging the water quality. In particular, because of their extensive use in textiles and dyeing industries, reactive azo dyes represent more than half of the world's dye production. Azo dyes are not only carcinogenic, but also hazardous enough that trace amounts of azo dyes present in water bodies cause serious problems to aquatic life. Hence, azo dyes are of serious concern worldwide for the active treatment of wastewater. A number of scientific efforts have been applied to eradicate these hazardous chemicals from wastewater.2,3 Various physicochemical techniques such as filtration,4 adsorption,5 precipitation,6 and advanced oxidation processes (AOPs) have been reported.7–9 AOPs are a set of chemical treatments designed to remove organic materials from wastewater by oxidation through reactions with reactive oxygen species. Undoubtedly, AOPs are the most appropriate methods owing to their simple operation, low cost, high degradation rate, and the degradation of the entire chemical compound to less toxic CO2 and H2O without generating secondary pollution. In AOPs, photocatalysts absorbed the light from the sunlight and generate reactive oxygen species (ROS) in situ, thereby facilitating the degradation of chemical pollutants in water.On the other hand, porphyrin-based self-organizing nano- or micro-materials have drawn considerable attention for photocatalytic application.10,11 Porphyrin-based materials exhibit extensive features, including a high surface area, high physiochemical stability, and versatile geometry compared to their precursor compounds. Porphyrinoids (free-base porphyrins or metalloporphyrins) self-assemble and form large-scale aggregates in solid as well as in solution. Various intermolecular non-covalent interactions, such as π–π stacking, hydrogen bonding, van der Waals force, metal–ligand coordination, electrostatic interaction, and hydrophobic and hydrophilic effects are responsible for the self-assembly of porphyrinoids.12–15 Various methods, including ionic self-assembly,16 surfactant-assistance,17 sonication,18 re-precipitation,19 and metal–ligand coordination20 have been used for the construction of self-organized porphyrin nanoaggregates. Because the morphology of self-organizing nano- or micro-materials is not always regular and thus unsuitable for many applications, the design, selection of an appropriate building block, and fabrication of these materials with well-defined dimensions are challenging tasks. In this context, supramolecular assemblies constructed by the directional coordination between porphyrin building blocks and metal complexes are very attractive for the fabrication of nanostructured porphyrin materials. Metal-coordinated directional bonding has successfully provided various multiporphyrin architectures, such as rectangles,21 squares,22 ladders,23 cube cages,24 double-deckers,25 and wheel type nano-rings.26
Sn(IV)porphyrins among metalloporphyrins especially form stable six-coordinated complexes with two trans-oxyanions of carboxylates or alkoxides because of the oxophilic nature of the Sn(IV) center, and the structural information can be easily obtained by NMR spectroscopy due to the diamagnetic nature of Sn(IV)porphyrins. Therefore, Sn(IV)porphyrin-based building blocks can be ideal scaffolds for the creation of supramolecular assemblies.27–30 Besides, attractive photophysical properties of Sn(IV)porphyrins with distinct absorption and emission have led to develop interesting optoelectronic supramolecular systems.31–35 We have been devoting ourselves to studying the nanostructures based on the supramolecular Sn(IV)porphyrin assembly and their photocatalytic performances for water-splitting H2 generation36,37 and degradation of pollutant dyes.38–43 Specifically, the complementary Zn(II)–Sn(IV)–Zn(II) porphyrin triads,38–41 the coordination frameworks of Sn(IV)porphyrin with Ag(I) ions,42 and the ionic assembly of cationic Sn(IV)porphyrin43 were investigated, where we intensively explored important aspects such as the fabrication of nanostructures, the morphology control, and photocatalytic performances. With these studies, our interest has been now focused on the nanomaterials based on supramolecular assemblies featuring well-defined dimensions and innate porosity, such as porphyrin squares, for the application of photocatalytic degradation of pollutant dyes.44
Here, we report two porphyrin-based supramolecular architectures (1 and 2) constructed by the coordination of the Re(I) complex to the pyridyl groups of Sn(IV)porphyrin (Scheme 1). The reaction of trans-dihydroxo-[5,15-bis(4-pyridyl)-10,20-bis(phenyl)porphyrinato]tin(IV) (SnP1) and trans-dihydroxo-[5,15-bis(4-pyridyl)-10,20-bis(4-tert-butylphenyl)porphyrinato]tin(IV) (SnP2) with Re(CO)5Cl (pentacarbonylchlororhenium(I)) leads to the formation of closed 2D tetrameric supramolecular square 1 and 2, respectively. We also explored the thermodynamic stability, photophysical properties, surface morphology, and photocatalytic performances toward the degradation of an azo dye, Eriochrome Black T (EBT), in an aqueous solution. EBT dye has been widely used in textile industries and for estimating the amount of Zn2+, Mg2+, and Ca2+ ions by complexometric titrations. The EBT dye is carcinogenic, non-biodegradable, and long-lasting in aqueous solutions. Hence, efficient treatments are required to eliminate this potentially toxic pollutant from wastewater.45–49
 | ||
| Scheme 1 Chemical structures of supramolecular squares used in this study. | ||
Results and discussion
Syntheses and spectroscopic characterization
To synthesize porphyrin-based self-assembled supramolecular squares, we used two Sn(IV)porphyrin-based building block units, SnP1 and SnP2 (Scheme 2). The supramolecular squares (1 and 2) featuring fac-Re(CO)3Cl corners were synthesized using the same procedures as reported previously by Hupp and co-workers.50,51 Multi-porphyrin supramolecular squares 1 and 2 were constructed from the reaction of Re(CO)5Cl with SnP1 and SnP2, respectively, under an Ar atmosphere in a mixed solvent mixture (ratio THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene = 1:1). Initially, the trans-labilizing effect of CO assisted the activation of two carbonyl groups for substitution by THF. The THF molecules were then replaced by pyridylporphyrins. The yields were higher, when a mixed solvent (THF and toluene) instead of pure THF was used. The full details of all experiments are described in the Experimental sections. Average yield was found to be more than 74% in all cases. All synthesized compounds were fully characterized by various spectroscopic techniques, including elemental analysis, 1H NMR, ESI-MS, FT-IR, UV-vis spectroscopy, and fluorescence spectroscopy.
:toluene = 1:1). Initially, the trans-labilizing effect of CO assisted the activation of two carbonyl groups for substitution by THF. The THF molecules were then replaced by pyridylporphyrins. The yields were higher, when a mixed solvent (THF and toluene) instead of pure THF was used. The full details of all experiments are described in the Experimental sections. Average yield was found to be more than 74% in all cases. All synthesized compounds were fully characterized by various spectroscopic techniques, including elemental analysis, 1H NMR, ESI-MS, FT-IR, UV-vis spectroscopy, and fluorescence spectroscopy.
 | ||
| Scheme 2 Synthesis of 1 and 2. | ||
1H NMR spectra of H2P2, SnP2, 1, and 2 are shown in the ESI (Fig. S1–S4†). Fig. S3† shows that the 1H NMR spectrum of 1 are more complex than that of SnP1 and shows a downfield shift for the pyridyl protons, which is consistent with Re(I)–pyridine complexation (Fig. S3†).51 Initially, the 1H NMR spectrum of SnP1 exhibits a doublet for the α-pyridyl (H3,5) protons at 9.26 ppm and for the β-pyridyl (H2,6) protons at 8.27 ppm. After Re(I) binds with the pyridyl groups, the (H3,5) protons significantly shift downfield at 9.54 ppm, while the resonance shift and splitting of the β-pyridyl (H2,6) protons in 1 are less affected (8.45 ppm). However, the proton resonances for both the phenyl and the β-pyrrole split in 1 (due to the difference in the outer and inner square environments) exhibit a smaller downfield shift as compared to SnP1. The β-pyrrole protons in 1 shift from 9.1–9.15 ppm in SnP1 to 9.15–9.24 ppm. Similarly, the α-pyridyl (H3,5) protons in SnP2 appear as a doublet at 9.22 ppm. After coordination with Re(I), these protons exhibit significant downfield shifts, and appearing at 9.41 ppm in 2. The β-pyrrole protons also shift downfield (9.05–9.10 ppm to 9.21–9.27 ppm) after the coordination with Re(I) in 2. On the other hand, the 1H NMR spectrum of phenyl and the β-pyridyl (H2,6) protons shift downfield in 2 compared to SnP2 and are slightly more pronounced.
By comparing the 1H NMR spectra of 1 and 2, as shown in Fig. 1, it was found that the resonances for 1 were significantly broader than those for 2. In the case of 1, the porphyrin rings could freely rotate along the Re(I)–N bond axis. However, as can be expected, the rotation in 2 was almost forbidden owing to the large steric hindrance among the tertiary butyl groups. Consequently, the resonances for 2 appeared as sharp peaks. In this context, a supramolecular square can adopt two different conformations. Either, the porphyrin rings are positioned horizontally with the plane defined by the four Re(I) centres, or the porphyrin rings face each other in an upright form (Scheme 3). Notably, 2 favours the upright form with no horizontal form, whereas there is little barrier between the two forms of 1.
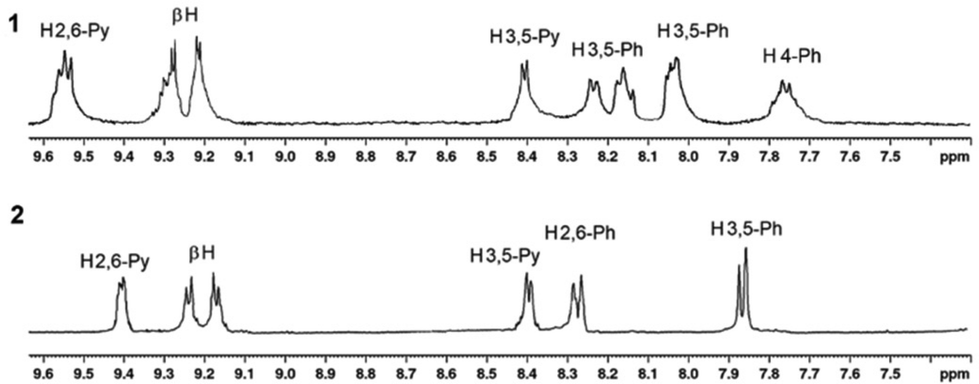 | ||
| Fig. 1 Comparison of 1H NMR spectra of 1 and 2. | ||
 | ||
| Scheme 3 Possible conformations in 1 and 2. | ||
The electrospray ionization mass spectra (ESI-MS) of SnP2, 1, and 2 are provided in the ESI (Fig. S5–S7†). The molecular ion peak for [SnP2 + H]+ appeared at 881.20 (Fig. S5†). As shown in Fig. S6,† a peak for fragment [SnP1 + H]+ appeared at 769.10 and a peak indicating protonated species of 1, [1 + 4H]4+ appeared at 1075.02 with low intensity. Similarly, in the case of 2, a peak for [SnP2 + H]+ appeared at 881.20 and a peak for the protonated species of 2, [2 + 4H]4+ appeared at 1187.12 (Fig. S7†). The appearance of the protonated species of 1 and 2 in ESI-MS supports the existence of supramolecular squares.
The FT-IR spectra of 1 and 2 along with their porphyrin units (SnP1 and SnP2) are shown in Fig. 2. The presence of a characteristic peak for the carbonyl ligand from the fac-Re(CO)3Cl corners at 2022 and 1905 cm−1 for 1, and at 2015 and 1890 cm−1 for 2, proved the formation of coordinative linkages between Re(I) complex and Sn(IV)porphyrins in the supramolecular squares of 1 and 2.51
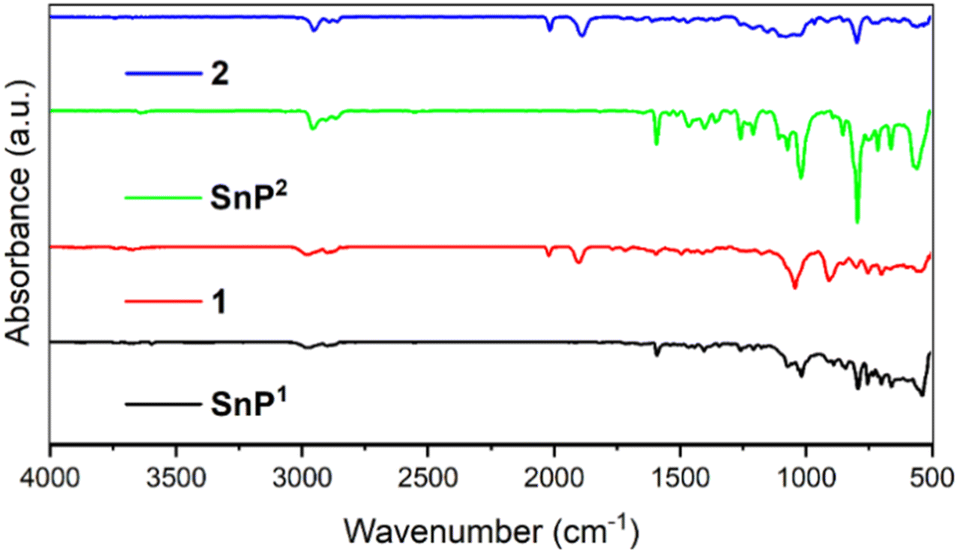 | ||
| Fig. 2 Fourier transform infrared (FT-IR) spectra of SnP1, SnP2, 1, and 2. | ||
The optical properties of SnP1, SnP2, 1, and 2 were investigated using UV-vis spectroscopy in chloroform (Fig. 3). Initially, the Soret band in SnP1 displayed a sharp peak at 423 nm along with the Q-band at 518, 558, and 598 nm. Upon coordination with Re(I), these peaks were red-shifted in 1, and the Soret band appeared at 428 nm. A peak shift of 5 nm confirmed the coordination of the pyridyl N with the Re(I) complex in 1. The Q-bands were also red-shifted and appeared at 518, 561, and 604 nm. By contrast, the Soret band in SnP2 appeared at 424 nm along with the Q-bands at 517, 558, and 598 nm. All these bands were red-shifted in 2 compared to the bands in the starting SnP2. Supramolecular square 2 exhibited a strong Soret band at 428 nm, while the Q-bands appeared at 518, 560, and 603 nm. Therefore, bathochromic shifts and peak broadening are proof of the formation of Re(I) that is coordinated to the pyridyl N atoms. The above results are consistent with those reported previously.51
 | ||
| Fig. 3 UV-vis absorption spectra of SnP1, SnP2, 1, and 2 in chloroform. | ||
The fluorescence spectra of 1 and 2 in chloroform along with their respective porphyrin building units (SnP1 and SnP2), are shown in Fig. 4. The fluorescence spectrum of SnP1 showed two typical emission bands at 603 and 655 nm. Upon complexation with Re(I) in 1, these bands were red-shifted to 606 and 655 nm, respectively, with a relatively lower intensity compared to SnP1. Similarly, the emission spectra of SnP2 displayed two-band spectra at 604 and 655 nm (Fig. 4): upon complexation with Re(I) in 2, the bands were red-shifted to 608 and 656 nm, respectively. From Fig. 4, it is inferred that fluorescence is largely quenched upon ligation and changes the peak-to-peak ratio. This is because the heavy metal atom (Re) accelerates the singlet-to-triplet excited-state intersystem crossing and energy transfer between two porphyrins via π–π stacking interactions.22
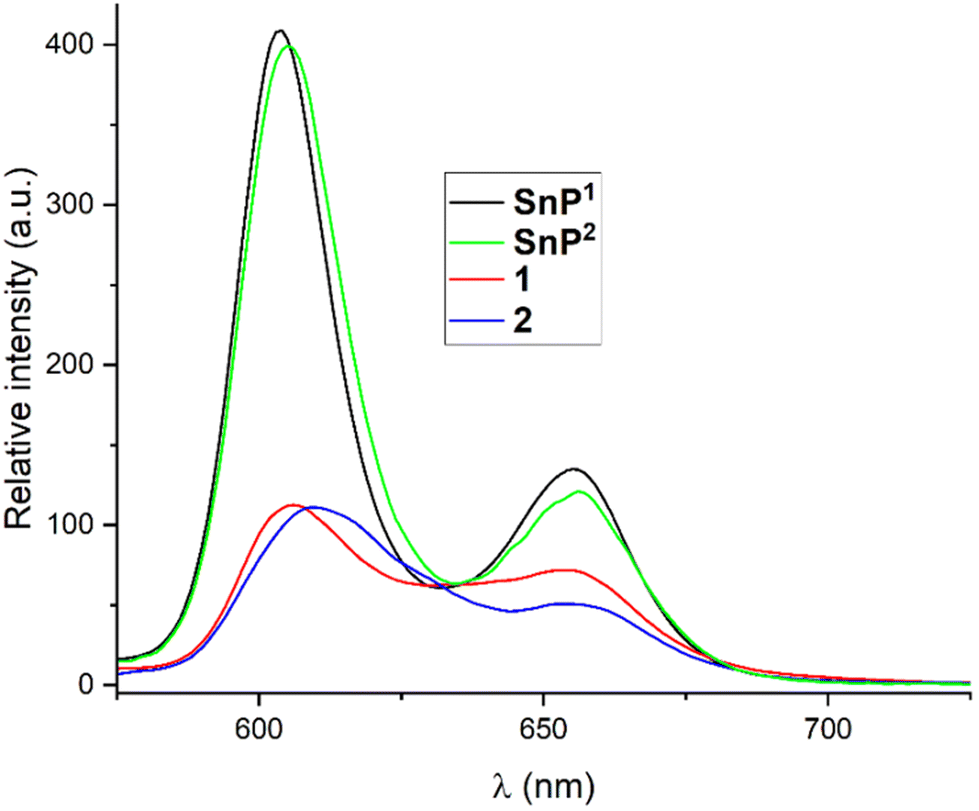 | ||
| Fig. 4 Fluorescence spectra of SnP1, SnP2, 1, and 2 in chloroform (λex = 560 nm). The optical density (OD) of each sample solution was fixed at 0.25. | ||
The thermal stabilities of both supramolecular squares (1 and 2) were measured by thermogravimetric analysis (TGA) under helium flow (Fig. S8†). In case of 1, the only major weight loss occurred in the range of 400–600 °C. This is because of the decomposition of the framework (loss of Re(I) linkage with Sn(IV)porphyrin). Similarly, in case of 2, small weight losses happened as temperature increased to approximately 100 °C. This was due to the removal of some volatile molecules from 2. Major weight loss due to the decomposition of the framework occurred in the range of 400–600 °C. These results clearly indicate that both supramolecular squares are stable up to 400 °C. We also examined the crystallinity of the supramolecular squares by analyzing powder X-ray diffraction (PXRD) patterns (Fig. S9†). From Fig. S9,† it is confirmed that 1 is more amorphous than 2 (or 2 is more crystalline than 1).
The surface morphologies of 1 and 2 were observed using field emission scanning electron microscopy (FE-SEM) (Fig. 5). Spherical shape nanoparticles were observed in the case of 1, and the average diameter of the largest sphere was 310 nm. Small particle diameters of 180 and 130 nm were also observed for 1. This size distribution was also confirmed by dynamic light scattering (DLS) studies, as shown in Fig. S10.† By contrast, for 2, the average particle sizes ranged from 60 to 170 nm. In addition, the nanoparticles were interconnected. The distinct difference in the morphology of the nanoaggregates for the two supramolecular squares was caused by the difference in the conformational structures of 1 and 2. As confirmed by 1H NMR spectral analysis, two conformational isomers in 1, namely, horizontal and upright structures, could co-exist. By contrast, 2 favoured the upright structure and did not exhibit a horizontal structure due to its rotational restrictions, which was attributed to the large steric hindrance among the bulky tert-butyl substituents. In addition, the surface morphologies of the starting porphyrin units SnP1 and SnP2 (Fig. S11†) were compared. In SnP1 and SnP2, the large particles with no specific shapes were fused and interconnected to each other. These observations support that the formation of supramolecular square through the coordination with the Re(I) center tremendously affected the morphology of 1 and 2.
 | ||
| Fig. 5 FE-SEM images for 1 (a), 2 (b). | ||
N2 sorption isotherms were recorded at 77 K in a liquid nitrogen bath to determine the surface area and permanent porosity of 1 and 2 (Fig. S12†). The estimated Brunauer–Emmett–Teller (BET) surface areas were 42 m2 g−1 for 1 and 54 m2 g−1 for 2. The difference in surface areas comes mainly from the conformational difference of 1 and 2. Preferring the upright over horizontal conformation in 2 results in a larger surface area of 2 than 1. Moreover, the presence of permanent porosity within these supramolecular squares could enable access to the metalloporphyrin sites in the pores for small molecules and thus provide a higher catalytic performance toward pollutant dyes.
Photocatalytic degradation of the EBT dye
The photocatalytic performance of these nanostructured materials from supramolecular squares (1 and 2) was examined by the degradation of a pollutant dye in aqueous solution under visible-light irradiation. Eriochrome Black T (EBT) dye was selected as the target for the photocatalytic degradation reaction. As shown in Fig. S13,† approximately 30 min are required to reach the adsorption–desorption equilibrium, under which 11 and 17% of EBT are adsorbed by 1 and 2, respectively. This presumably indicates that the supramolecular squares 1 and 2 provided a suitable porous environment for adsorbing pollutant dyes. The time-dependent absorption spectra of EBT in the presence of photocatalyst 2 under visible-light irradiation are shown in Fig. S14.† Negligible decay of the EBT dye was observed either in the absence of a photocatalyst or visible-light (Fig. 6). Therefore, the photocatalysts, as well as visible-light, were essential for the degradation of the EBT dye. As depicted in Fig. S14,† the absorbance of the EBT dye at 534 nm decreased upon prolongation of the visible-light irradiation time. This indicates that the EBT dye degraded photochemically. Both supramolecular squares (1 and 2) showed significant progress in the photodegradation of the EBT dye in aqueous solution (Fig. 6). The degradation of the EBT dye in the presence of a photocatalyst can be described by its degradation efficiency expressed as follows: (C0 − C)/C0, where C0 is the initial concentration of EBT and C is the concentration at time t. The observed degradation rates of the EBT dye were 88% for 1 and 95% for 2 (Fig. 6). Therefore, photocatalyst 2 showed better performance in the degradation of EBT dye than 1. | ||
| Fig. 6 Photocatalytic degradation of the EBT dye in an aqueous solution (pH 7, T = 298 K) under visible-light irradiation in the presence of photocatalysts 1 and 2. | ||
To further interpret the reaction kinetics for the decay of EBT dye, we applied a pseudo-first-order rate kinetic as expressed by the equation ln(C0/C) = kt. This equation is generally used for photocatalytic degradation experiments if C0 of the dye is low, and k is the pseudo-first-order rate constant. The reaction kinetics of the EBT dye degradation as shown in Fig. S15† were obtained based on the data plotted in Fig. 6. The first-order rate constant for the degradation of EBT dye for 1 and for 2 were 0.023 and 0.032 min−1, respectively. These results were notable compared to the other reported values for the degradation of EBT dye (Table 1).
The reusability of photocatalysts 1 and 2 is important for practical application, and was determined by the recycling tests of 2 with respect to the EBT dye degradation under visible-light (Fig. S16 and Table S1†). In the first cycle, the EBT dye was degraded by 95%, and 90% degradation was achieved in the tenth cycle. Therefore, after 10 consecutive cycles, 2 still maintained 95% of the initial performance regarding the degradation of EBT dye, thereby indicating the spectacular stability of photocatalyst 2. The rate constant also decreased slightly from 0.032 min−1 (for 1st cycle) to 0.029 min−1 (for 10th cycle) (Table S1†). Moreover, the surface morphologies of 1 and 2 were examined after the degradation reaction to confirm the stability of these catalysts. By comparison, the FE-SEM images (Fig. S17†) of 1 and 2 were similar to their initial images, indicating that the morphology of these photocatalysts was intact during the photocatalytic reaction.
We attempted to optimize the reaction conditions in terms of temperature, pH of the dye solution, and EBT dye/catalyst ratio. Degradation experiments were performed at different temperatures to evaluate the effect of temperatures on the degradation of EBT dye by 2. As the temperature increased, the degradation efficiency first increased up to a temperature of 308 K and then decreased (Fig. S18†). As shown in Fig. S19,† the degradation performance was maximized at pH 7, and decreased under acidic and basic conditions. It was likely that in a strongly acidic or basic medium, the molecular square framework in 2 was broken. In addition, it was noticed that the degradation performance was affected more in an acidic medium than in a basic medium. The absorption of EBT dye was changed by altering the pH of solution. The peak at 534 nm (pH 7) changed to 515 nm (pH 2) or to 529 (pH 10). However, the degradation pattern was the same in all conditions. Additionally, the effect of the EBT dye/catalyst ratio on the degradation of the EBT dye was examined using various concentrations of EBT solution (10, 20, 30, 40, 50, 60, 70, and 80 mg L−1) with a constant amount of photocatalyst 2 (20 mg was used in every experiment). Most of the EBT dye was degraded at concentrations of 10–50 mg L−1, and approximately 70% of the dye was degraded even at 80 mg L−1 (Fig. S20†).
Previously, we established a possible mechanism for the photodegradation of pollutant dyes by Sn(IV)porphyrin-based nanomaterials in aqueous solutions.38,39 Generally, the mechanism consists of five steps. In step (I), the photocatalyst absorbs light when exposed to visible-light irradiation along with the EBT dye in an aqueous solution. The valence band (VB) electrons are promoted to the conduction band (CB) after crossing the bandgap. This leads to the formation of electron–hole (e−/h+) pairs at the surface of the photocatalyst. Strong intermolecular π–π interactions among the porphyrin molecules intensified electronic delocalization within the photocatalysts. This can minimize the recombination energy of the excited electrons. Subsequently, the photogenerated hole (h+) reacts with H2O to generate a highly reactive hydroxyl radical (˙OH) (step (II)). In step (III), excited electrons react with dissolved O2 molecules to produce reactive superoxide radical anions (O2˙−). These photogenerated superoxide radical anions and hydroxyl radicals react with EBT dye and thus degrade it into smaller molecules, CO2, and H2O (steps (IV) and (V)).
| SnP + hν → SnP (e− + h+) | (I) |
| H2O + h+ → ˙OH + H+ | (II) |
| O2 + 2e− → O2˙− | (III) |
| ˙OH + EBT → degraded products | (IV) |
| O2˙− + EBT → degraded products | (V) |
Radical trapping experiments were performed to detect the photogenerated reactive species during the photocatalytic degradation of the EBT dye.52,53 We used tert-butanol (tBuOH) to capture hydroxyl radicals (˙OH), para-benzoquinone (p-BQ) for superoxide radical anions (O2˙−), and ethylenediaminetetraacetic acid disodium (Na2-EDTA) was used to capture holes during the photodegradation experiments in the presence of 2, (Fig. S21†). Fig. S21† reveals that the degradation rate is critically affected in the presence of tBuOH and p-BQ. Superoxide radical anions (O2˙−) are the major reactive species compared to hydroxyl radicals (˙OH), which are responsible for the catalytic oxidation of the EBT dye. However, the degradation of EBT dye was not affected by the presence of Na2–EDTA or photogenerated holes. It should be noted that the starting porphyrin building blocks such as SnP1 and SnP2 show very low to negligible catalytic efficiency in the degradation of EBT dye (Fig. S21†).
Additionally, the photocatalytic activity of 2 in EBT degradation under various monochromatic light wavelengths (Fig. S22†) was investigated. The variation trend in the wavelength-dependent photodegradation of the EBT dye demonstrated that optical absorption makes a significant contribution toward solar energy conversion and its photocatalytic performance. In addition, 2 still showed a small degradation ability even at λ > 700 nm.
We also focused on revealing the details of the degradation products of the EBT dye after irradiation with visible-light in the presence of photocatalyst 2. For this purpose, the reaction mixture obtained after 45 min was analyzed by ESI-MS (Fig. S23†). New peaks emerged in the mass spectra, which confirmed the degradation of EBT dye to small molecules.54 Possible intermediates for the degradation of EBT dye based on the mass spectra in Fig. S23† are shown in Fig. 7.
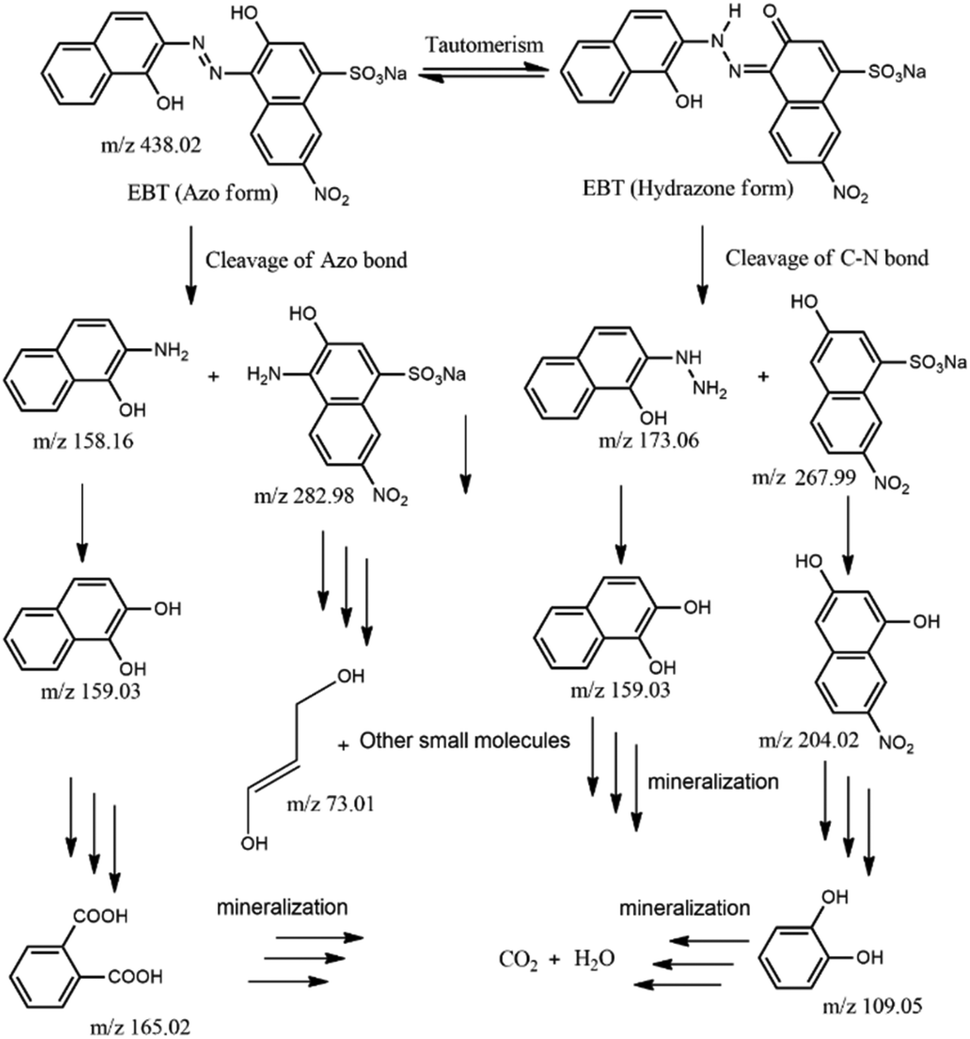 | ||
| Fig. 7 Possible intermediates for the degradation of EBT dye in presence of 2. | ||
Initially, the base peak (m/z = 438.02; EBT–Na+) belonged to the anionic EBT dye and existed in solution as keto–enol tautomerism. The EBT dyes can be fragmented in two ways. First, the cleavage of the azo bond by the hydroxyl radical (˙OH) and the formation of two low molecular weight fragments with 2-amino-1-naphthol (m/z 158) and 4-amino-3-hydroxy-7-nitro-napthalenesulphonic acid (m/z 283) occurred. Further, 2-amino-1-naphthol could undergo hydrolysis to generate either 1,2-dihydroxynapthalen (m/z 159) or phthalic acid (m/z 165). However, successive oxidation of 4-amino-3-hydroxy-7-nitro-napthalenesulphonic acid led to the formation of lower molecular fragments (m/z 73). In the second process, the cleavage of the C–N bond leads to the formation of lower fragments of 2-hydrazino-1-naphthol (m/z 173) and 3-hydroxy-7-nitro-napthalenesulphonic acid (m/z 268). Then, 2-hydrazino-1-naphthol could undergo hydrolysis leading to the formation of 1,2-dihydroxynapthalen. On the other hand, successive oxidation, hydrolysis, and ring opening of 3-hydroxy-7-nitro-napthalenesulphonic acid led to the formation of lower-weight catechol molecules (m/z 109). Finally, all lower molecular weight intermediate molecules were further fragmented and mineralized into H2O and CO2. Additionally, the total organic carbon (TOC) value was calculated to quantify the removal of the EBT dye by the photocatalysts.55 Using the photocatalyst from 1 and 2, TOC removal percentages of only 71 and 74%, respectively, were achieved.
Experimental
Materials and methods
All chemicals were purchased from Sigma-Aldrich and used without further purification, unless stated otherwise. Further, 5-(4-pyridyl)dipyrromethane and trans-dihydroxo-[5,15-bis(4-pyridyl)-10,20-bis(phenyl)porphyrinato]tin(IV) (SnP1) were synthesized according to a previously reported procedure.56,57 THF and toluene were purified by distillation from a sodium/benzophenone ketyl solution, whereas pyrrole was obtained from a solution of calcium hydride. Elemental analysis was performed using an EA 1110 Fisons analyzer (Used Lab Machines Limited, London, England). 1H NMR spectra were obtained on a Bruker BIOSPIN/AVANCE III 400 spectrometer at 293 K (Bruker BioSpin GmbH, Silberstreifen, Rheinstetten, Germany). Electrospray ionization mass (ESI-MS) spectra were recorded using a Thermo Finnigan linear ion trap quadrupole mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). The FT-IR spectra (KBr) were obtained using a Shimadzu FTIR-8400S spectrophotometer (Shimadzu, Tokyo, Japan). Steady-state UV-vis spectra were recorded on a Shimadzu UV-3600 spectrophotometer (Shimadzu, Tokyo, Japan). Fluorescence spectra were recorded using a Shimadzu RF-5301PC fluorescence spectrophotometer (Shimadzu, Tokyo, Japan). Thermogravimetric analysis (TGA) was performed using an Auto-TGA Q500 instrument (TA Instruments, New Castle, DE, USA). Powder X-ray diffraction (PXRD) patterns were obtained using a Bruker AXS D8 Advance powder X-ray diffractometer (Bruker, Billerica, MA, USA). The Brunauer–Emmett–Teller (BET) surface area was determined with an analyzer (BELSORP-mini volumetric adsorption equipment) using N2 adsorption isotherms at 77 K. Field emission scanning electron microscope (FE-SEM) images were obtained from MAIA III (TESCAN, Brno, Czech Republic). Dynamic light scattering (DLS) experiments were performed using a NanoBrook 90Plus DLS size analyzer (Brookhaven, NY, USA).Syntheses
ε) 417 (5.20), 518 (4.18), 549 (3.90), 592 (3.66), 647 (3.52). Emission (CHCl3, nm): λmax 650, 718.
ε) 404(4.1), 424(5.51), 517(3.6), 558(4.15), 598(3.86). Emission (CHCl3, nm): λmax 604, 654.
:toluene = 1:1) under Ar atmosphere. Subsequently, the solution was allowed to reflux under Ar in the dark for 48 h. n-Hexane (20 mL) was added to precipitate the desired product. The wine-red solid was filtered, washed with cold toluene (5 mL), and dried in air. Yield: 42 mg (78%). Anal calcd for C180H112Cl4N24O20Re4Sn4: C, 50.37; H, 2.63; N, 7.83; R, 39.17. Found: C, 49.98; H, 2.95; N, 7.76; R, 39.31. 1H NMR (400 MHz, CDCl3, ppm): δ 7.75 (d, J = 7.2 Hz, 8H, H4-phenyl), 8.16 (m, 16H, H3,5-phenyl), 8.23 (d, J = 7.6 Hz, 8H, meso-O-phenyl), 8.29 (d, J = 7.6 Hz, 8H, meso-O-phenyl), 8.45 (d, J = 6.4 Hz, 16H, meso-O-Py), 9.15 (d, J = 4.8 Hz, 16H, β-pyrrole), 9.24 (d, J = 4.8 Hz, 16H, β-pyrrole), 9.54 (d, J = 6.8 Hz, 16H, H3,5-Py). FT-IR (KBr pellet, ν/cm−1): 2930 (br), 2022 (s), 1905 (br), 1594 (w), 1490 (w), 1415 (w), 1179 (w), 1040 (vs), 906 (vs), 082 (w), 755 (s), 698 (s), 555 (br). UV-vis (CHCl3, nm): λmax(logε); 428(5.70), 518(4.17), 561(4.62), 604(4.48). Emission (CHCl3, λnm): 606, 655.
:toluene = 1:1) while stirring under Ar atmosphere. The reaction mixture was refluxed for 48 h in the dark. n-Hexane (20 mL) was added to the above reaction mixture and stirred for 1 h. Then, the brown-red precipitate was filtered, washed with 5 mL of cold toluene, and dried in air. Yield: 44 mg (74%). Anal calcd for C212H176Cl4N24O20Re4Sn4: C, 53.70; H, 3.74; N, 7.09; R, 35.47. Found: C, 53.36; H, 4.02; N, 6.89; R, 35.73. 1H NMR (400 MHz, CDCl3, ppm): δ 1.61 (s, 72H, tBu), 7.87 (d, J = 8.2 Hz, 16H, H3,5-Ar), 8.27 (d, J = 7.8 Hz, 16H, meso-O-Ar), 8.53 (d, J = 6.4 Hz, 16H, meso-O-Py), 9.21 (d, J = 6.0 Hz 16H, β-pyrrole), 9.27 (d, J = 6.0 Hz, 16H, β-pyrrole), 9.41 (d, J = 5.4 Hz, 16H, H3,5-Py). FT-IR (KBr pellet, ν/cm−1): 2958 (br), 2015 (s), 1890 (br), 1610 (w), 1470 (w), 1070 (br), 915 (w), 798 (vs), 720 (br), 560 (br). UV-vis (CHCl3, nm): λmax(logε) 428(5.63), 518(4.11), 560(4.55), 603(4.40). Emission (CHCl3, nm): λmax 608, 656.
Photocatalytic degradation experiment
The photocatalytic efficiencies of supramolecular squares (1 and 2) were evaluated by the degradation of Eriochrome Black T (EBT) dye in an aqueous solution. The photodegradation of EBT was carried out under the irradiation of a 150 W xenon arc lamp with a UV cut-off filter (ABET technologies, Old gate lane Milford, Connecticut, USA) at 298 K. In a typical procedure, 20 mg of the photocatalyst was added to a 250 mL aqueous solution of EBT (50 mg L−1, distilled water with a pH of 7.0) while stirring at 298 K. The reaction mixture was then kept in the dark for 30 min to reach an adsorption–desorption equilibrium. After irradiation with visible light, a 2 mL suspension was collected at regular intervals. The photocatalyst was removed from the solution by centrifugation and collected after filtration through a filter paper. The concentration of EBT was measured by determining the absorbance at 534 nm by a Vis spectrophotometer. Recovery of the photocatalyst was readily handled by a continuous procedure. First, the catalyst was separated from the reaction mixture by filtration and washed with water. After that, it was dried in the air and ready for further use.Conclusions
We synthesized Sn(IV)porphyrin-based supramolecular squares (1 and 2) by the reaction of Re(CO)5Cl with two porphyrin units (SnP1 and SnP2). Although the structural framework of these architectures was fixed with a closed 2D tetrameric array, altering the substituent on the Sn(IV)porphyrin units produced not only conformationally different squares but also led to distinctly different nanostructures. Using these nanostructures as a photocatalyst, the photocatalytic degradation of Eriochrome Black T (EBT) dye was efficiently performed in an aqueous solution under visible-light irradiation. Therefore, the strategy in this report is of considerable value as a supramolecular engineering approach for controlling various structural modifications, including a large porous surface area, high thermodynamic stability, and specific morphology. As a future prospect, the Sn(IV)porphyrin unit can adopt functional ligands at the axial position, leading to further structural and chemical modification of the supramolecular squares. Our study holds great importance in tackling the treatment of dyeing wastewater because our nanomaterials exhibit congenital porosity, efficient degradation performance, low catalyst loading, and reusability.Author contributions
Investigation, methodology, data curation, visualization, formal analysis, validation, software, and writing, N. K. S.; conceptualization, review and editing, supervision, project administration, and funding acquisition, H.-J. K.Conflicts of interest
There are no conflicts to be declared.Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) (grant no. 2022R1F1A1074420) funded by the Korean government (MSIT).References
- Z. Wang, G. W. Walker and D. C. G, Muir and K. Nagatani-Yoshida, Toward a Global Understanding of Chemical Pollution: A First Comprehensive Analysis of National and Regional Chemical Inventories, Environ. Sci. Technol., 2020, 54, 2575–2584 CrossRef CAS PubMed.
- R. P. Schwarzenbach, T. Egli, T. Hofstetter, U. Von Gunten and B. Wehrli, Global Water Pollution and Human Health, Annu. Rev. Environ. Resour., 2010, 35, 109–136 CrossRef.
- R. Naidu, B. Biswas, I. R. Willett, J. Cribb, B. Kumar Singh, C. Paul Nathanail, F. Coulon, K. T. Semple, K. C. Jones, A. Barclay and R. J. Aitken, Chemical pollution: A growing peril and potential catastrophic risk to humanity, Environ. Int., 2021, 156, 106616 CrossRef CAS.
- L. P. Glugoski, P. de Jesus Cubas and S. T. Fujiwara, Reactive Black 5 dye degradation using filters of smuggled cigarette modified with Fe3+, Environ. Sci. Pollut. Res., 2017, 24, 6143–6150 CrossRef CAS PubMed.
- Q. U. Ain, U. Rasheed, M. Yaseen, H. Zhang and Z. Tong, Superior dye degradation and adsorption capability of polydopamine modified Fe3O4-pillared bentonite composite, J. Hazard. Mater., 2020, 397, 122758 CrossRef CAS.
- A. Balcha, O. P. Yadav and T. Dey, Photocatalytic degradation of methylene blue dye by zinc oxide nanoparticles obtained from precipitation and sol-gel methods, Environ. Sci. Pollut. Res., 2016, 23, 25485–25493 CrossRef CAS PubMed.
- W. Kim, J. Park, H. J. Jo, H.-J. Kim and W. Choi, Visible Light Photocatalysts Based on Homogeneous and Heterogenized Tin Porphyrins, J. Phys. Chem. C, 2008, 112, 491–499 CrossRef CAS.
- H. Kim, W. Kim, Y. Mackeyev, G.-S. Lee, H.-J. Kim, T. Tachikawa, S. Hong, S. Lee, J. Kim, L. J. Wilson, T. Majima, P. J. J. Alvarez, W. Choi and J. Lee, Selective Oxidative Degradation of Organic Pollutants by Singlet Oxygen Photosensitizing Systems: Tin Porphyrin versus C60 Aminofullerene Systems, Environ. Sci. Technol., 2012, 46, 9606–9613 CrossRef CAS.
- J. Xu, Q. Z. Gao, Z. P. Wang and Y. Zhu, An all-organic 0D/2D supramolecular porphyrin/g-C3N4 heterojunction assembled via π-π interaction for efficient visible photocatalytic oxidation, Appl. Catal., B, 2021, 291, 120059 CrossRef CAS.
- C. M. Drain, A. Varotto and I. Radivojevic, Self-Organized Porphyrinic Materials, Chem. Rev., 2009, 109, 1630–1658 CrossRef CAS.
- Y. Chen, A. Li, Z.-H. Huang, L.-N. Wang and F. Kang, Porphyrin-Based Nanostructures for Photocatalytic Applications, Nanomaterials, 2016, 6, 51–67 CrossRef.
- J. A. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Molecular Materials by Self–Assembly of Porphyrins, Phthalocyanines, and Perylenes, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS.
- I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Supramolecular chemistry of metalloporphyrins, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS PubMed.
- S. Durot, J. Taesch and V. Heitz, Multiporphyrinic Cages: Architectures and Functions, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS PubMed.
- S. Shao, V. Rajendiran and J. F. Lovell, Metalloporphyrin nanoparticles: Coordinating diverse theranostic functions, Coord. Chem. Rev., 2019, 379, 99–120 CrossRef CAS.
- Z. Wang, C. J. Medforth and J. A. Shelnutt, Porphyrin Nanotubes by Ionic Self-Assembly, J. Am. Chem. Soc., 2004, 126, 15954–15955 CrossRef CAS PubMed.
- C. Zhang, P. Chen, H. Dong, Y. Zhen, M. Liu and W. Hu, Porphyrin Supramolecular 1D Structures via Surfactant-Assisted Self-Assembly, Adv. Mater., 2015, 27, 5379–5387 CrossRef CAS PubMed.
- T. Hasobe, H. Oki, A. S. D. Sandanayakaa and H. Murata, Sonication-assisted supramolecular nanorods of meso-diaryl-substituted porphyrins, Chem. Commun., 2008, 724–726 RSC.
- X. Gong, T. Milic, C. Xu, J. D. Batteas and C. M. Drain, Preparation and Characterization of Porphyrin Nanoparticles, J. Am. Chem. Soc., 2002, 124, 14290–14291 CrossRef CAS PubMed.
- N. K. Shee, C.-J. Lee and H.-J. Kim, Hexacoordinated Sn(IV) porphyrin-based square-grid frameworks exhibiting selective uptake of CO2 over N2, Bull. Korean Chem. Soc., 2022, 43, 103–109 CrossRef CAS.
- K. D. Benkstein, C. L. Stern, K. E. Splan, R. C. Johnson, K. A. Walters, F. W. M. Vanhelmont and J. T. Hupp, Collapsed Molecular Rectangles Based on Rhenium(I) Coordination of Ethynylpyridyl Porphyrins - Synthesis, Structure, and Bending-Induced Charge-Transfer Behavior, Eur. J. Inorg. Chem., 2002, 2002, 2818–2822 CrossRef.
- E. Iengo, P. Cavigli, D. Milano and P. Tecilla, Metal mediated self-assembled porphyrin metallacycles: Synthesis and multipurpose applications, Inorg. Chim. Acta, 2014, 417, 59–78 CrossRef CAS.
- H. L. Anderson, Conjugated porphyrin ladders, Inorg. Chem., 1994, 33, 972–981 CrossRef CAS.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, A Self-Assembled M8L6 Cubic Cage That Selectively Encapsulates Large Aromatic Guests, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- N. K. Shee, J.-W. Seo and H.-J. Kim, Spectrophotometric Study of Bridging N-Donor Ligand-Induced Supramolecular Assembly of Conjugated Zn-Trisporphyrin with a Triphenylamine Core, Molecules, 2021, 26, 4771 CrossRef CAS PubMed.
- M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. L. Anderson, Vernier templating and synthesis of a 12-porphyrin nano-ring, Nature, 2011, 469, 72–75 CrossRef.
- J. K. M. Sanders, N. Bampos, Z.-C. Watson, S. L. Darling, J. C. Hawley, H.-J. Kim, C. C. Mak and S. J. Webb, Axial Coordination Chemistry of Metalloporphyrins, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, 2000, vol. 3, pp. 1–48 Search PubMed.
- H.-J. Kim, N. Bampos and J. K. M. Sanders, Assembly of Dynamic Heterometallic Oligoporphyrins Using Cooperative Zinc-Nitrogen, Ru-Nitrogen and Tin-Oxygen Coordination, J. Am. Chem. Soc., 1999, 121, 8120–8121 CrossRef CAS.
- H. J. Kim, N. K. Shee, K. M. Park and H.-J. Kim, Assembly and X-ray crystal structures of heterometallic multiporphyrins with complementary coordination between ruthenium(II) and tin(IV) porphyrins, Inorg. Chim. Acta, 2019, 488, 1–7 CrossRef CAS.
- H.-J. Kim, H. J. Jo, J. Kim, S.-Y. Kim, D. Kim and K. Kim, Supramolecular self-assembly of tin(IV) porphyrin channels stabilizing single-file chains of water molecules, CrystEngComm, 2005, 7, 417–420 RSC.
- H. J. Kim, K.-M. Park, T. K. Ahn, S. K. Kim, K. S. Kim, D. Kim and H.-J. Kim, Novel fullerene–porphyrin–fullerene triad linked by metal axial coordination: Synthesis, X-ray crystal structure, and spectroscopic characterizations of trans-bis([60]fullerenoacetato)tin(iv) porphyrin, Chem. Commun., 2004, 2594–2595 RSC.
- H. J. Kim, J. H. Jang, H. Choi, T. Lee, J. Ko, M. Yoon and H.-J. Kim, Photoregulated Fluorescence Switching in Axially-Coordinated Tin(IV) Porphyrinic Dithienylethene, Inorg. Chem., 2008, 47, 2411–2415 CrossRef CAS PubMed.
- M. T. Indelli, C. Chiorboli, M. Ghirotti, M. Orlandi, F. Scandola, H. J. Kim and H.-J. Kim, Photoinduced Electron Transfer in Ruthenium(II)/Tin(IV) Multiporphyrin Arrays, J. Phys. Chem. B, 2010, 114, 14273–14282 CrossRef CAS PubMed.
- N. K. Shee, M. K. Kim and H.-J. Kim, Fluorescent chemosensing for aromatic compounds by supramolecular complex composed of tin(IV) porphyrin, viologen, and cucurbit[8]uril, Chem. Commun., 2019, 55, 10575–10578 RSC.
- M. K. Kim, N. K. Shee, J. Lee, M. Yoon and H.-J. Kim, Photoinduced Electron Transfer upon Supramolecular Complexation of (Porphyrinato) Sn-Viologen with Cucurbit [7] uril, Photochem. Photobiol. Sci., 2019, 18, 1996–2002 CrossRef CAS.
- C. Li, K.-M. Park and H.-J. Kim, Ionic assembled hybrid nanoparticle consisting of tin(IV) porphyrin cations and polyoxomolybdate anions, and photocatalytic hydrogen production by its visible light sensitization, Inorg. Chem. Commun., 2015, 60, 8–11 CrossRef CAS.
- S. H. Kim and H.-J. Kim, Photocatalytic Hydrogen Production by the Sensitization of Sn(IV)-Porphyrin Embedded in a Nafion Matrix Coated on TiO2, Molecules, 2022, 27, 3770 CrossRef CAS PubMed.
- N. K. Shee, M. K. Kim and H.-J. Kim, Supramolecular Porphyrin Nanostructures Based on Coordination-Driven Self-Assembly and Their Visible Light Catalytic Degradation of Methylene Blue Dye, Nanomaterials, 2020, 10, 2314 CrossRef CAS PubMed.
- N. K. Shee and H.-J. Kim, Self-Assembled Nanomaterials Based on Complementary Sn(IV) and Zn(II)-Porphyrins, and Their Photocatalytic Degradation for Rhodamine B Dye, Molecules, 2021, 26, 3598 CrossRef CAS.
- N. K. Shee and H.-J. Kim, Three Isomeric Zn(II)-Sn(IV)-Zn(II) Porphyrin-Triad-Based Supramolecular Nanoarchitectures for the Morphology-Dependent Photocatalytic Degradation of Methyl Orange, ACS Omega, 2022, 7, 9775–9784 CrossRef CAS PubMed.
- N. K. Shee and H.-J. Kim, Morphology-controlled self-assembled nanostructures of complementary metalloporphyrin triads through intermolecular coordination tuning and their photocatalytic degradation for Orange II, Inorg. Chem. Front., 2022, 9, 5538–5548 RSC.
- N. K. Shee, H. J. Jo and H.-J. Kim, Coordination framework materials fabricated by the self-assembly of Sn(IV) porphyrins with Ag(I) ions for the photocatalytic degradation of organic dyes in wastewater, Inorg. Chem. Front., 2022, 9, 1270–1280 RSC.
- N. K. Shee and H.-J. Kim, Sn(IV) Porphyrin-Based Ionic Self-Assembled Nanostructures and Their Application in Visible Light Photo-Degradation of Malachite Green, Catalysts, 2022, 12, 799 CrossRef CAS.
- N. K. Shee and H.-J. Kim, Sn(IV)-Porphyrin-Based Nanostructures Featuring Pd(II)-Mediated Supramolecular Arrays and Their Photocatalytic Degradation of Acid Orange 7 Dye, Int. J. Mol. Sci., 2022, 23, 13702 CrossRef.
- S. K. Kansal, S. Sood, A. Umar and S. K. Mehta, Photocatalytic degradation of Eriochrome Black T dye using well-crystalline anatase TiO2 nanoparticles, J. Alloys Compd., 2013, 581, 392–397 CrossRef CAS.
- H. M. Gobara, R. A. Elsalamony and S. A. Hassan, Sonophotocatalytic degradation of eriochrome black-T dye in water using Ti grafted SBA-15, J. Porous Mater., 2016, 23, 1311–1318 CrossRef CAS.
- A. A. Oladipo, A. O. Ifebajo and M. Gazi, Magnetic LDH-based CoO–NiFe2O4 catalyst with enhanced performance and recyclability for efficient decolorization of azo dye via Fenton-like reactions, Appl. Catal., B, 2018, 243, 243–252 CrossRef.
- M. F. Lanjwani, M. Y. Khuhawar, T. M. J. Khuhawar, A. H. Lanjwani, S. Q. Memon, W. A. Soomro and I. K. Rind, Photocatalytic Degradation of Eriochrome Black T Dye by ZnO Nanoparticles Using Multivariate Factorial, Kinetics and Isotherm Models, J. Cluster Sci., 2022 DOI:10.1007/s10876-022-02293-8.
- X.-H. Chen, Y.-S. Zhang, W.-B. Li, X.-W. Guan, J.-W. Ye, L. Chen, H.-P. Wang, J. Bai, Z.-W. Mo and X.-M. Chen, A porphyrin-based metal–organic framework with highly efficient adsorption and photocatalytic degradation performances for organic dyes, Inorg. Chem. Front., 2022, 9, 2328–2335 RSC.
- S. J. Lee and J. T. Hupp, Porphyrin-containing molecular squares: design and applications, Coord. Chem. Rev., 2006, 250, 1710–1723 CrossRef CAS.
- R. V. Slone and J. T. Hupp, Synthesis, Characterization, and Preliminary Host–Guest Binding Studies of Porphyrinic Molecular Squares Featuring fac-Tricarbonylrhenium(I) Chloro Corners, Inorg. Chem., 1997, 36, 5422–5423 CrossRef CAS.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Environmental Implications of Hydroxyl Radicals (˙OH), Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- Z. Zhang, J. Fan, J. Du and X. Peng, Two-channel responsive luminescent chemosensors for dioxygen species: Molecular oxygen, singlet oxygen and superoxide anion, Coord. Chem. Rev., 2021, 427, 213575 CrossRef CAS.
- B. Pal, R. Kaur and I. S. Grover, Superior adsorption and photodegradation of eriochrome black-t dye by Fe3+ and Pt4+ impregnated TiO2 nanostructures of different shapes, J. Ind. Eng. Chem., 2016, 33, 178–184 CrossRef CAS.
- Y. Hong, J. Oh, I. Lee, C. Fan, S. Y. Pan, M. Jang, Y. K. Park and H. Kim, Total-organic-carbon-based quantitative estimation of microplastics in sewage, Chem. Eng. J., 2021, 423, 130182 CrossRef CAS.
- M. Boccalon, E. Iengo and P. Tecilla, Metal-Organic Transmembrane Nanopores, J. Am. Chem. Soc., 2012, 134, 20310–20313 CrossRef CAS PubMed.
- N. K. Shee, C.-J. Lee and H.-J. Kim, Crystal structure of bis (benzoato-κO)[5,15-di-phenyl-10,20-bis(pyridin-4-yl)porphyrinato-κ4N,N′,N′′,N′′′]tin(IV), IUCrData, 2019, 4, x190787.A CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental results, together with additional figures. See DOI: https://doi.org/10.1039/d2qi02194c |
| This journal is © the Partner Organisations 2023 |