
Isomeric effect of naphthyl spacers on structures and properties of isostructural porous crystalline frameworks†
Yuto
Suzuki
,
Mao
Yamaguchi
,
Ryusei
Oketani
 and
Ichiro
Hisaki
*
and
Ichiro
Hisaki
*
Division of Chemistry, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: i.hisaki.es@osaka-u.ac.jp
First published on 28th November 2022
Abstract
In reticular chemistry, many studies have attempted to design a series of isostructural frameworks with specific structures and properties by manipulating the constituent molecules. Frameworks constructed through non-covalent interactions, such as hydrogen-bonded organic frameworks (HOFs), are highly sensitive to subtle molecular structural differences, but a systematic study of them has not been done. The present study describes the construction of three types of isostructural, dia-topological HOFs (C1N4DBC-1, C1N5DBC-1, and C2N6DBC-1) from dibenzo[g,p]chrysene (DBC) derivatives (C1N4DBC, C1N5DBC, and C2N6DBC) that possess 1,4-, 1,5-, and 2,6-substituted carboxynaphthyl groups, respectively. The frameworks were formed using directional H-bonds between carboxy groups and the shape-fitted docking of twisted DBC cores. However, despite the isostructural frameworks, they exhibited different dynamic behaviours toward guest removal, with C1N4DBC showing pore shrinkage, C1N5DBC retaining its structure, and C2N6DBC collapsing to an amorphous state. Furthermore, in situ PXRD measurements with benzene vapour adsorption/desorption showed that C1N4DBC underwent pore-breathing behaviour through crystal-to-crystal transition. These differences are attributed to the pore geometry and hydrogen bonding style due to the isomeric effect, which is an essential principle for constructing the desired HOFs.
Introduction
Many interesting porous structures based on organic molecules have been explored through reticular chemistry.1–4 Metal–organic frameworks (MOFs) and covalent–organic frameworks (COFs) are representative crystalline organic materials with permanent porosity, and the resulting diverse structures have been applied to a wide range of functional materials.5–9 To obtain frameworks with a finely tuned structure and properties, various isostructural or isoreticular10 frameworks have been constructed by designing network topology11–14 and an assembling of the network (e.g., interpenetration, stacking).15–19Porous organic frameworks constructed by self-assembly of molecules through hydrogen bonds are often called hydrogen-bonded organic frameworks (HOFs).20–24 Because of reversible, non-covalent bonds forming the framework, HOFs can be obtained as highly crystalline materials with a large domain by facile methods such as recrystallization and mechanochemistry.25 They can be also re-used and re-generated by solution processes via re-solubilization. In contrast, the weakness of the bonds results in fragile frameworks compared with other porous frameworks such as COFs. Furthermore, the resulting framework is not always the same as the one originally designed, and an unexpected structure sometimes forms. Recently, these shortcomings have been overcome by networking through charge-assisted hydrogen bonding20,26 and assembling via π–π stacking27–35 to give stable HOFs. However, systematically constructing a series of isostructural HOFs with different pore sizes is challenging because even molecules with the same geometry and H-bonding groups do not always give isostructural HOFs.36–39 Several isostructural HOFs were constructed successfully from dihydroimidazolone-annelated triptycene derivatives,40,41 pyrene-based tetratopic carboxylic acids,28,31 fluorinated trispyrazole derivatives,27,32 and tetrathiafulvalene-based tetratopic carboxylic acids.29,30 In these systems, the following three structural factors play a critical role in forming the isostructural frameworks: (1) well-defined supramolecular synthons formed through directional H-bonds, (2) a core skeleton with robust second interactions working orthogonally to the H-bonding, and (3) a suitable peripheral spacer bonded to the core. Hexaazatriphenylene (HAT) derivatives were also shown to form as many as four isostructural HOFs, in which robust shape-fitted docking among the twisted HAT cores significantly contributed to formation of the isostructural HOFs.33–35
However, isostructural construction via tuning peripheral spacers to change the dynamic properties has not been investigated, although this approach can provide a library of diverse porous functional materials. The present report demonstrates, for the first time, that three dibenzo[g,p]chrysene (DBC)-based isostructural HOFs exhibit different dynamic behaviours depending on the substitution positions of the isomeric naphthyl spacers during solvent desorption and re-adsorption (Fig. 1).
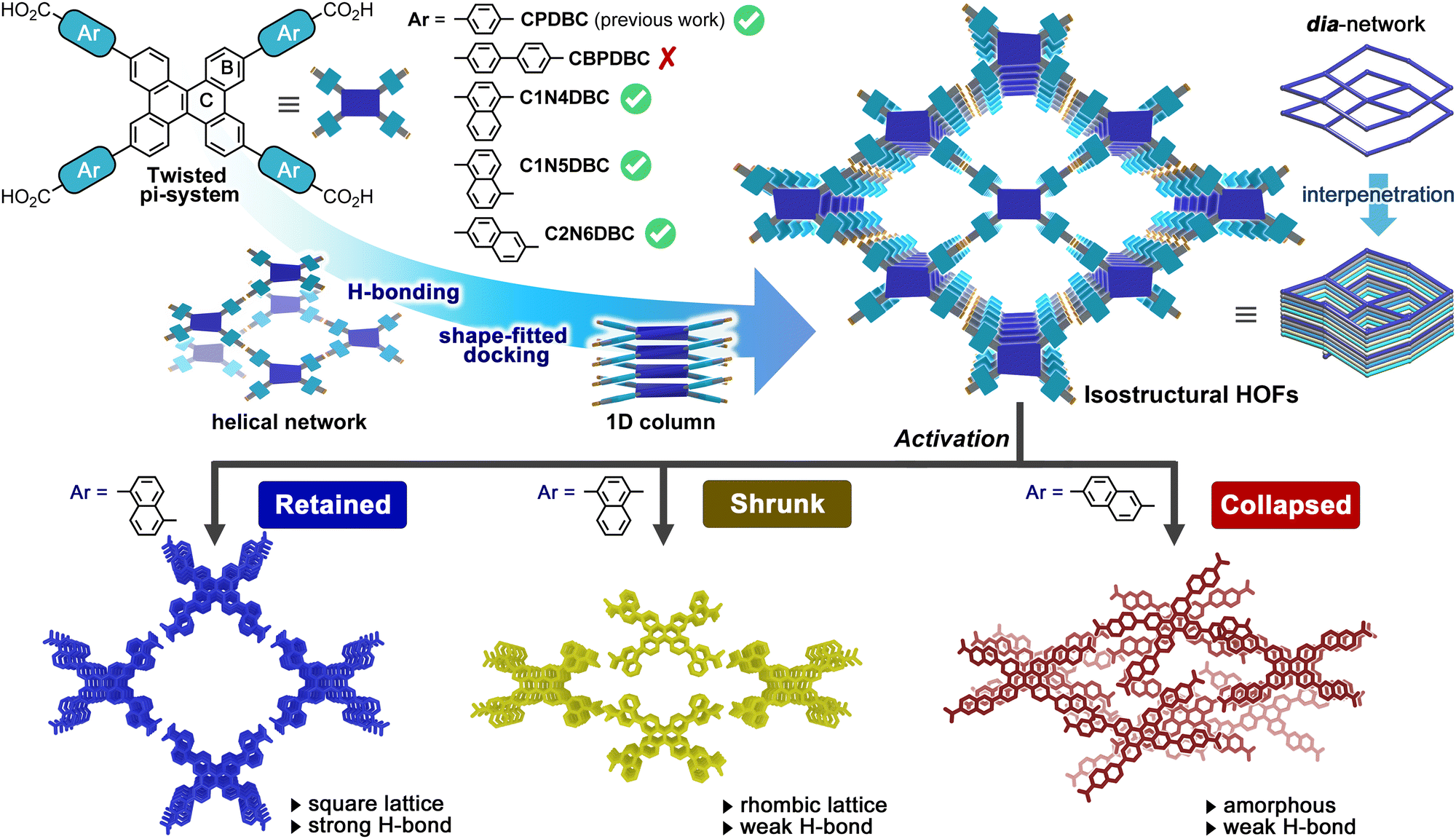 | ||
| Fig. 1 Construction of isostructural HOFs through shape-fitted docking of a dibenzo[g,p]chrysene (DBC) core and isomeric effects of the naphthyl spaces on the structures and properties. (Top) Molecular structures of DBC derivatives: CPDBC, C1N4DBC, C1N5DBC, and C2N6DBC that form isostructural HOFs via H-bonded helical networking and shape-fitted docking of a DBC core. (Bottom) Three activated isostructural HOFs with different substituted naphthyl groups. An isomeric effect led to differences in structural transition behaviour during solvent desorption and re-adsorption. | ||
DBC forms a twisted conformation due to steric hindrance at the fjord hydrogen atoms.42 A previous report stated that its derivative possessing a carboxyphenyl group, CPDBC, yielded a HOF with permanent porosity through a H-bonded dia network and the shape-fitted docking of DBC cores (Fig. 1). Based on this information, an attempt was made to introduce larger peripheral groups, a biphenyl group and naphthyl groups with different substitution positions, into the DBC system to investigate the isostructural construction of HOFs and the effects on their properties. A derivative with a carboxybiphenyl group, CBPDBC, underwent aggregation and had poor solubility, and produced only low crystalline structures. This is an example of the limitations of simple phenylene elongation of the peripheral group as in the previous HAT example.35 The DBC derivatives with 1,4-, 1,5-, and 2,6-substituted naphthyl groups (C1N4DBC, C1N5DBC, and C2N6DBC), however, yielded isostructural HOFs (C1N4DBC-1, C1N5DBC-1, and C2N6DBC-1, respectively), comparable to CPDBC.
Importantly, during the activation processes, C1N5DBC-1 maintained its porous structure, while C1N4DBC-1 showed reversible changes in aperture size upon guest desorption and adsorption, and C2N6DBC-1 experienced shrinkage of the pores followed by collapse to the amorphous phase. These structures were determined precisely by single crystal X-ray diffraction (SCXRD), single crystal electron diffraction (ED), or powder X-ray diffraction (PXRD).
These different behaviours resulted from differences in pore geometry and the H-bonding manner of the isostructural HOFs. Their physical properties are affected significantly by the isomeric effect52,53 of the peripheral groups, while DBC cores are useful for isostructural construction by shape-fitted docking. These results suggest the importance of peripheral groups and provide molecular design insights for the systematic construction of porous molecular crystalline materials.
Results and discussion
Synthesis and crystallization
The DBC tetracarboxylic acids, CBPDBC, C1N4DBC, C1N5DBC, and C2N6DBC, were synthesized as shown in Scheme 1. Biphenyl- and 2,6-naphthyl-substituted precursors 3a and 3b were obtained by Suzuki–Miyaura cross-coupling reaction with 2,7,10,15-tetrabromodibenzo[g,p]chrysene (1) and pinacol boronates with butyl 4′-bromobiphenyl-4-carboxylate and butyl 6-bromo-2-naphthoate moieties, respectively. 1,4- and 1,5-Naphthyl-substituted precursors 3c and 3d also were synthesized by the coupling reaction of 2,7,10,15-tetra(pinacolboryl)dibenzo[g,p]chrysene (2), which was synthesized by Miyaura borylation from 1, and the corresponding arylbromides; butyl 4-bromo-1-naphthoate and butyl 5-bromo-1-naphthoate, respectively. Precursors 3a–d were hydrolysed by KOH in a mixed solution of THF, methanol, and water to afford the building block molecules CBPDBC, C2N6DBC, C1N4DBC, and C1N5DBC, respectively. The synthesized DBC derivatives were dissolved in a mixture of a polar solvent (DMF or 1,4-dioxane) and an aromatic guest solvent (o-xylene: Xy, mesitylene: Mes, methyl benzoate: MeBz, 1,2,4-trichlorobenzene: TCB, or 5-tert-butyl-m-xylene: tBuXy), followed by recrystallization. For C1N4DBC, a vapour diffusion method was applied using a DMF/Xy solvent system. For the others, a solvent volatilization method was applied (for details, see Table S1, ESI†). As a result, CBPDBC and C1N4DBC yielded microcrystals CBPDBC-1 and C1N4DBC-1, respectively, while C1N5DBC and C2N6DBC yielded needle crystals C1N5DBC-1 and C2N6DBC-1, respectively (Fig. S1, ESI†). In solid states, naphthyl DBCs exhibited non or slight fluorescence (for details, see Fig. S25, ESI†).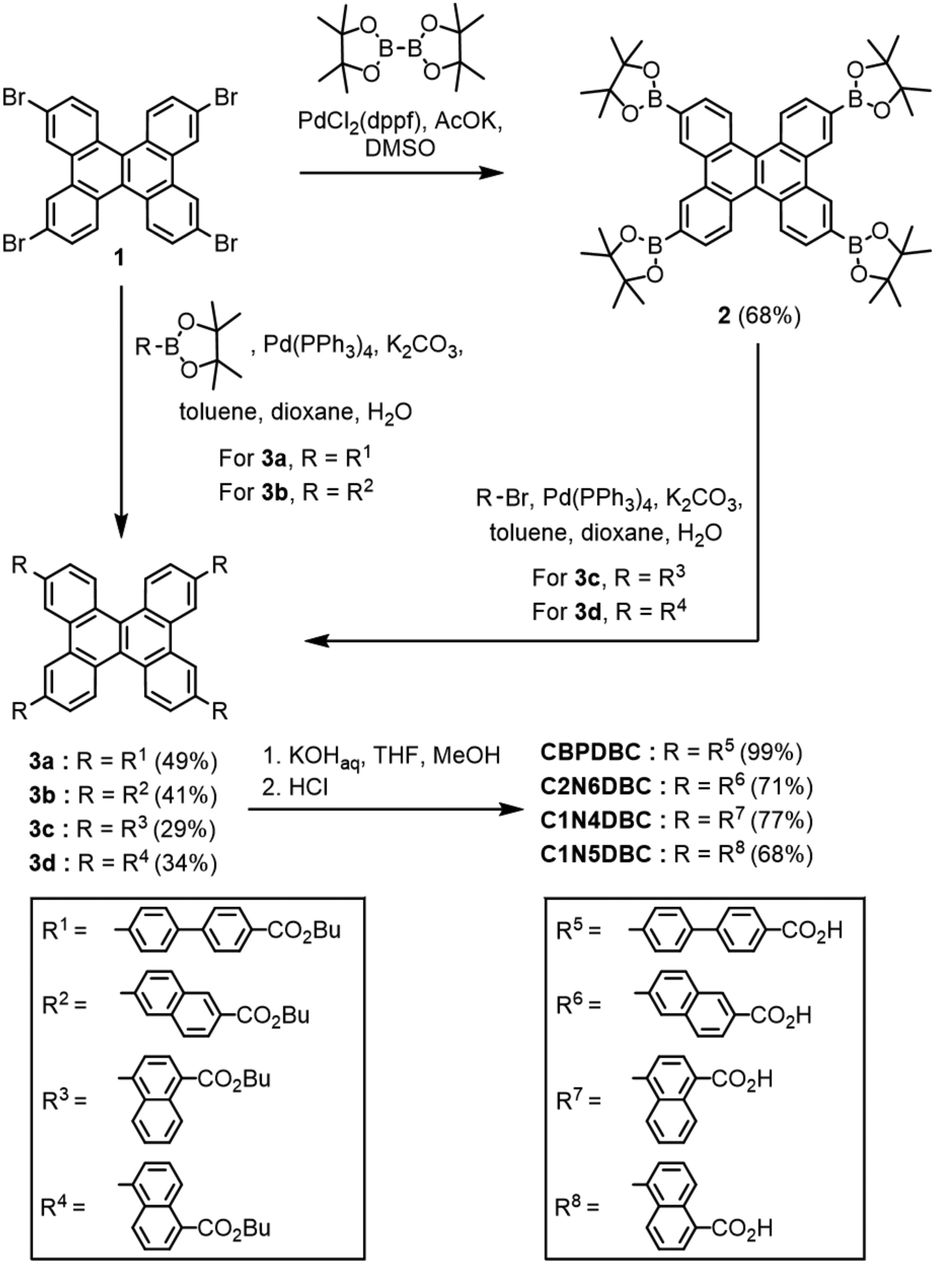 | ||
| Scheme 1 Synthesis of DBC derivatives CBPDBC, C1N4DBC, C1N5DBC, and C2N6DBC. | ||
Crystallography
To examine the crystals obtained, preliminary PXRD patterns were recorded on as-formed crystalline precipitates and compared with those of the previously reported HOF CPDBC-1 as shown in Fig. 2 and Fig. S7 (ESI†). The CBPDBC-1 showed weak diffraction peaks at 2θ = 1.54°, 3.08°, 4.74°, and 6.28°, which correspond to the (001), (002), (003), and (004) planes, respectively (Table S5, ESI†), of the estimated framework of CBPDBC, assuming that CBPDBC would form an isostructural network with CPDBC-1 (Fig. S9, ESI†). This indicates that CBPDBC-1 formed a larger isostructural network than did CPDBC-1. However, its low crystallinity precluded further investigation. The microcrystalline C1N4DBC-1 showed sharp diffraction peaks at 2θ = 4.30°, 7.45°, 8.55°, 11.34°, 12.89°, and 14.78°, which are similar to those of CPDBC-1 at 2θ = 4.21°, 7.27°, 8.43°, 11.11°, 12.67°, and 14.55°. This similarity is reasonable because CPDBC is a substructure of C1N4DBC and, therefore, periodicity of the resultant crystals is nearly the same as that of the isostructural HOF construction. The diffraction peaks of C1N4DBC-1 corresponded to the (011) (020), (022), (031), (033), and (040) of CPDBC-1 (Table S6, ESI†), suggesting that C1N4DBC-1 also is isostructural with CPDBC-1. The C1N5DBC-1 and C2N6DBC-1 had diffraction profiles similar to that of CPDBC-1, although they shifted slightly into a smaller angle region (for details, see Tables S7 and S8, ESI†).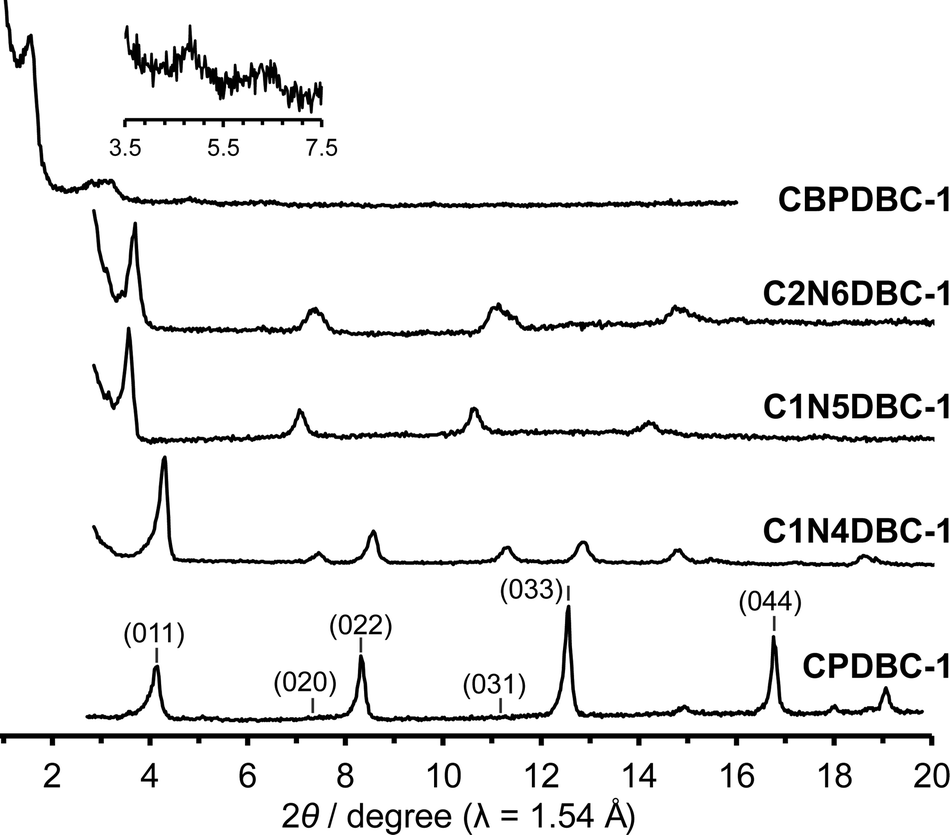 | ||
| Fig. 2 PXRD patterns of as-formed crystalline bulks of CBPDBC-1, C2N6DBC-1, C1N5DBC-1, and C1N4DBC-1, along with CPDBC-1 as the reference. Inset: PXRD pattern of CBPDBC-1 from 3.5° to 7.5°. | ||
The crystal structures of C1N5DBC-1 and C2N6DBC-1 were revealed by SCXRD measurements (Fig. 3, Table 2 and Table S2, ESI†). Both belong to the space group Pban and have isostructural porous frameworks with CPDBC-1 possessing quadrangular one-dimensional (1D) channels. The molecules were connected by intermolecular H-bonds between the peripheral carboxy groups to form three-dimensional (3D) dia-topological networks. Their peripheral groups were disordered in two parts, resulting in different H-bonding environments. For simplicity, further structural descriptions focus on the major moieties until describing the details (vide infra). The networks were interpenetrated 4- and 8-fold to yield HOFs C1N5DBC-1 and C2N6DBC-1,44 respectively (Fig. S11, ESI†). In both frameworks, the molecules were stacked one dimensionally via the shape-fitted docking of the DBC cores. Stacking distances between the DBC cores were 3.86 Å and 3.84 Å and those between the naphthyl groups were 3.35 Å and 3.54 Å for C1N5DBC-1 and C2N6DBC-1, respectively. The root mean square derivatives (RMSD) of the DBC cores in C1N5DBC-1 and C2N6DBC-1 were 0.669 and 0.716, respectively, indicating that the former distorted less and the latter distorted more than CPDBC-1 (RMSD: 0.680) (Table 2). The observed distortion of the core was smaller than that of theoretical structures optimized by the density functional theory (DFT) method at the B3LYP/6-311G** level: 0.764 for C1N5DBC and 0.761 for C2N6DBC (for details, see Table S9, ESI†). The dihedral angle between aromatic rings Ar–B (Fig. 1) also were smaller (C1N5DBC-1: calc. 56.2°, obs. 41.5°, C2N6DBC-1: calc. 37.8°, obs. 31.9°). The observed less-distorted conformations were due to shape-fitted docking, and this phenomenon also was observed in the CPDBC system.43 The C1N5DBC-1 had a rippled square aperture with dimensions of 26.8 Å × 23.3 Å, and that of C2N6DBC-1 had a rhombic aperture with dimensions of 19.8 Å × 30.6 Å. The void ratios of C1N5DBC-1 and C2N6DBC-1 were calculated to be 58.4% and 58.3%, respectively, by PLATON with a probe radius of 1.2 Å (Fig. S2 and S4, ESI†).45,46 Solvent molecules included in the channel were not solved crystallographically due to severe disorder.
 | ||
| Fig. 3 Crystal structures of (a) C1N5DBC-1, (b) C2N6DBC-1, and (c) CPDBC-1 as a ref. 43 (top) packing diagrams. Colour code: gray (major site), cyan (minor site). (Bottom) Single dia-network (coloured grey) and 1D stacked columnar structures, for which the number of colours in the stacked molecules correspond to the number of the frameworks interpenetrated. | ||
The terminal carboxy group in C1N5DBC-1 and the peripheral carboxynaphthyl group in C2N6DBC-1 are disordered in two positions (Fig. 4), which is significant because this is not observed in CPDBC-1. Particularly, the two disordered conformations in C2N6DBC-1 were caused by rotation of the low-symmetry naphthyl groups. One conformer has naphthyl groups far apart, and the other has them close together (Fig. 5c and d). In this way, rotation of the less symmetrical naphthyl portion causes the molecular conformation to be drastically different.
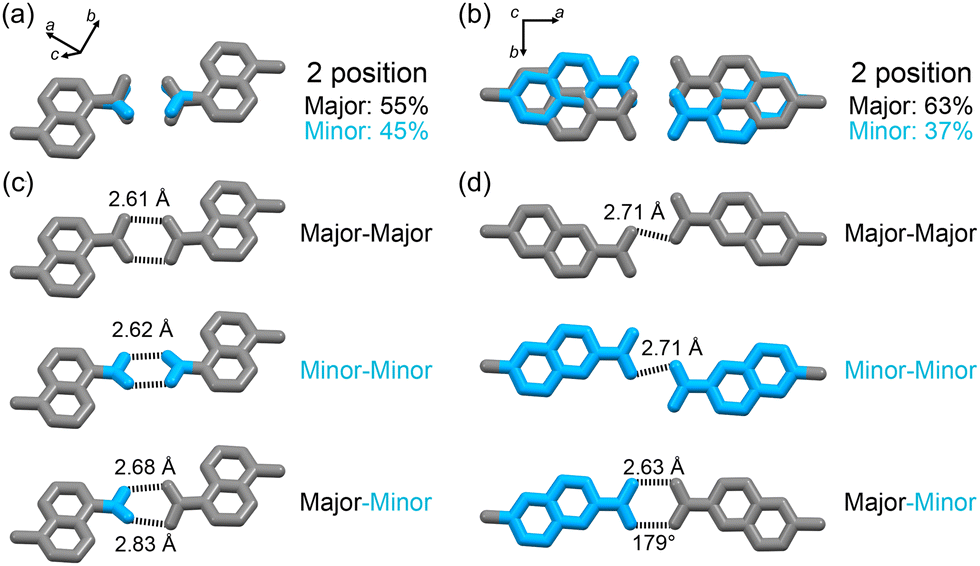 | ||
| Fig. 4 Disordered peripheral structures. (a) Carboxylic acid in C1N5DBC-1. Major: 55%, Minor: 45%. (b) 2-Carboxynaphthyl groups in C2N6DBC-1. Major: 63%, Minor: 37%. H-bonding networks in (c) C1N5DBC-1 and (d) C2N6DBC-1 between major–major, minor–minor, major–minor. Major and minor showed grey and cyan, respectively. | ||
 | ||
| Fig. 5 Predicted molecular structures of two representative conformations. C1N5DBC with naphthyl groups located (a) far apart and (b) close together. C2N6DBC with naphthyl groups located (c) far apart and (d) close together. | ||
In C1N5DBC-1, the conformation with naphthyl groups close together was too hindered for shape-fitted docking (Fig. 5b) and did not exist in crystalline form. Only the carboxylic acid moiety was disordered at major and minor positions with a site occupancy of 55% and 45%, respectively (Fig. 4a), and they are tilted from the naphthyl plane by +33.4° and −12.5°, respectively (Fig. S3a, ESI†). The O⋯O distances in the complementary H-bonded dimer between the same sites (i.e., the major–major and minor–minor) were 2.61 Å and 2.62 Å, respectively. The dimer between the major and minor sites was twisted by 45°, and the O⋯O distances were 2.68 Å and 2.81 Å (Fig. 4a and Fig. S3b, ESI†). However, both conformers of C2N6DBC-1 could form because their carboxynaphthyl groups did not prevent shape-fitted docking (Fig. 5c and d). All of the peripheral groups were disordered in two positions (Fig. 4b and d), suggesting that multiple conformations co-existed in the framework. The major and minor sites were occupied with ratios of 0.63 and 0.37, respectively (Fig. 4b). This disorder also made the H-bonding more complex (Fig. 4d). A truncated H-bond with a length of 2.71 Å existed between major–major and minor–minor, and complementary hydrogen bonds with a length of a 2.64 Å between major–minor, which can exist randomly in the crystal.
Thermogravimetric analysis of as-formed HOFs.
To estimate the stoichiometry of the molecules, as-formed crystalline bulks of C1N4DBC-1, C1N5DBC-1, and C2N6DBC-1 were subjected to thermogravimetric (TG) analysis (Fig. 6a, d, g and Fig. S12, ESI†). The TG curve of C1N4DBC-1 reached a plateau at ca. 180 °C with a weight loss of 32% in two steps, which indicated that the framework contained Xy molecules with a host/guest molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. The 1H-NMR spectrum of the as-formed crystals dissolved in DMSO-d6 also indicated a ratio of 1:3.8 (Fig. S13, ESI†). The C1N5DBC-1 showed a 55% weight loss in two steps, indicating a host/guest ratio of 1:7.6. This agreed well with the ratio of 1:7.1 obtained from the 1H-NMR analysis (Fig. S14, ESI†). The TG curve of C2N6DBC-1 did not plateau, but showed a first weight loss of 33% followed by continuous weight loss up to 47%, which was attributed to both release of guest solvent (tBuXy) and partial decomposition of the compound. The first weight loss corresponded to the host/guest ratios of 1:3, while the latter cannot be determined due to the unclear boundary between the removal of guest solvent and the decomposition of the carboxylic acid. The host/guest ratio was calculated from 1H-NMR to be 1:7.4 (Fig. S15, ESI†).
:4. The 1H-NMR spectrum of the as-formed crystals dissolved in DMSO-d6 also indicated a ratio of 1:3.8 (Fig. S13, ESI†). The C1N5DBC-1 showed a 55% weight loss in two steps, indicating a host/guest ratio of 1:7.6. This agreed well with the ratio of 1:7.1 obtained from the 1H-NMR analysis (Fig. S14, ESI†). The TG curve of C2N6DBC-1 did not plateau, but showed a first weight loss of 33% followed by continuous weight loss up to 47%, which was attributed to both release of guest solvent (tBuXy) and partial decomposition of the compound. The first weight loss corresponded to the host/guest ratios of 1:3, while the latter cannot be determined due to the unclear boundary between the removal of guest solvent and the decomposition of the carboxylic acid. The host/guest ratio was calculated from 1H-NMR to be 1:7.4 (Fig. S15, ESI†).
 | ||
| Fig. 6 (a, d and g) TG curves, (b, e and h) PXRD patterns after activation, and (c, f and i) VT-PXRD patterns of as-formed HOFs of (a–c) C1N4DBC-1, (d–f) C1N5DBC-1, and (g–i) C2N6DBC-1. Heating rate and radiation of VT-PXRD were 10 °C min−1 and synchrotron radiation (λ = 1.0800 Å) for C1N5DBC-1, and c.a. 6 °C min−1 and Cu Kα for others. TV and RV in PXRD patterns (b, e and h) indicate activation by thermal vacuum and replace vacuum methods, respectively. TG heating rate was 5 °C min−1. | ||
VT-PXRD measurements
As-formed crystalline bulks of C1N4DBC-1, C1N5DBC-1, and C2N6DBC-1 were subjected to variable temperature PXRD (VT-PXRD) under ambient atmosphere to investigate their structural transition behaviour and thermal stability. Patterns of C1N4DBC-1 were recorded every 10 °C increase using synchrotron radiation (λ = 1.0800 Å), and those of C1N5DBC-1 and C2N6DBC-1 were recorded every ca. 6 °C using Cu-Kα radiation (λ = 1.5418 Å) (Fig. 6c, f and i).A broad peak around 5–25° in C1N4DBC-1 was due to an excessive amount of mother liquor that remained and disappeared after the solvent volatilized. The initial phase started to decay at around 100 °C then disappeared entirely at 140 °C. The TG and PXRD results indicated that retaining the original porous structure was difficult after removing half of the guests. Interestingly, C1N5DBC-1 maintained its initial structure up to 220 °C, where its intensity began to shift. The TG analysis indicated guest removal terminated at 180 °C, showing that this porous structure was stable with no structural transition during guest removal. In contrast, the initial pattern for C2N6DBC-1 shifted to the lower angle region at 70 °C, and its intensity decayed and disappeared at 100 °C, resulting in an amorphous structure. Thus, the structure collapses until the first step of guest removal. These results indicate that frameworks C1N4DBC-1 and C2N6DBC-1 readily underwent a structural transition, and, therefore, should need attention when activation is conducted.
Activation condition
Based on the results of VT-PXRD measurements, activation (i.e., removing guest molecules from the pores) of the as-formed crystals was attempted using the following two methods. The first was a thermal vacuum (TV) method, in which as-formed crystalline powder was heated at 90–150 °C under vacuum. The other was a replaced vacuum (RV) method, in which as-formed crystalline powder was immersed in benzene to replace the included solvent with benzene, followed by heating at 60–100 °C under vacuum to remove benzene under milder conditions (for details, see Fig. S16, ESI†). The complete removal of the solvent was confirmed by 1H-NMR (Fig. S17–S19, ESI†).Although VT-PXRD indicated collapse of the framework by heating under ambient atmosphere, C1N4DBC-1 showed a good PXRD profile after activation by the TV method, but very low crystallinity by the RV method. Neither pattern after activation matched the initial one, indicating a structural transition occurred to provide the activated framework of C1N4DBC-1a. Therefore, C1N4DBC-1 is a flexible HOF. The C1N5DBC-1 showed no peak shift upon activation by either method, resulting in the structure-retained, activated framework of C1N5DBC-1a. This activated framework had chemical durability against several solvents (Fig. S24, ESI†). Therefore, C1N5DBC-1 is a stable HOF. The C2N6DBC-1 lost crystallinity after activation by both the TV and RV methods and became amorphous C2N6DBC-1a. Therefore, C2N6DBC-1 is a fragile HOF.
Structure of activated framework of C1N4DBC-1
Results showed that microcrystals of C1N4DBC-147 obtained from a Mes solution retained their single-crystallinity after heating under vacuum (Fig. S10 and S16b, ESI†). The microcrystals were subjected to ED analysis, which produced a preliminary structure for C1N4DBC-1x (for details, see Fig. S8, ESI†). The constituent molecule exhibited a conformation with the naphthyl groups far apart, (Fig. 5c) and stacked through shape-fitted docking of the DBC cores. The network created by intermolecular H-bonds among the carboxy groups gave an isostructural framework possessing 1D channels (Fig. 7a). Additionally, the analysis implied that some molecules of Mes remained.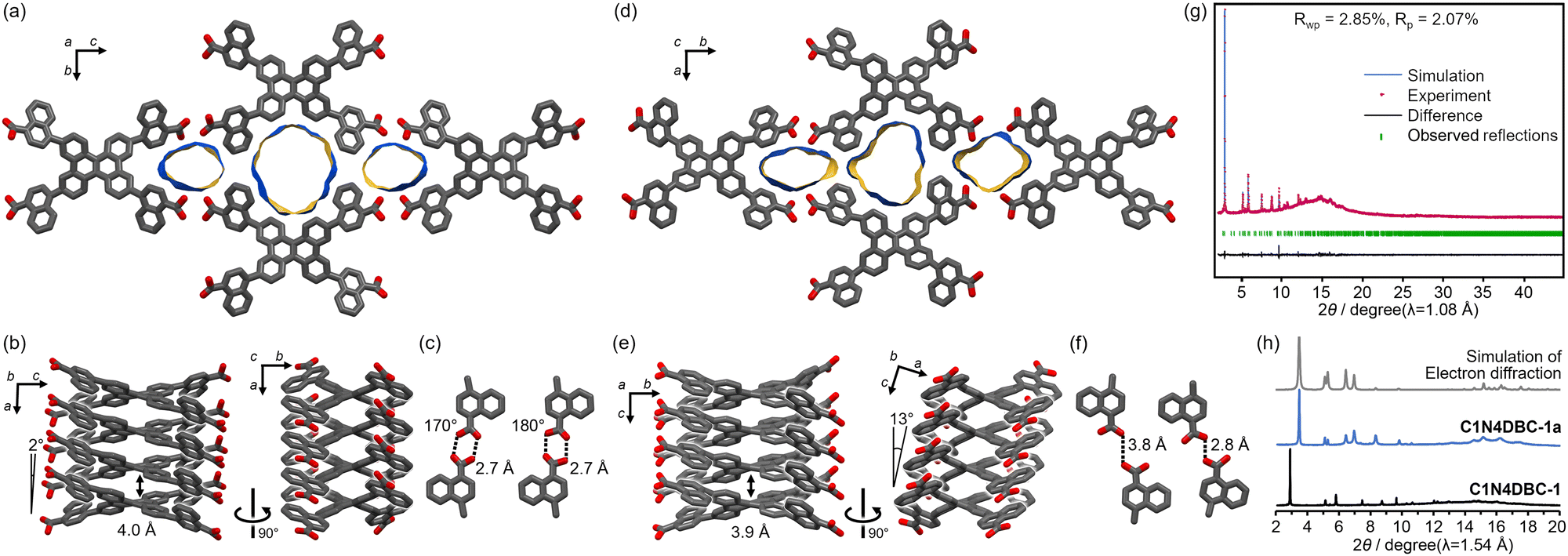 | ||
| Fig. 7 (a) Crystal structure of C1N4DBC-1x obtained from electron diffraction analysis. Void surfaces were visualized with blue (outside) and yellow (inside). (b) 1D columnar structure formed in C1N4DBC-1x through shape-fitted docking of the DBC core. (c) Complementary H-bonded dimers with a distorted manner. (d) Crystal structure of C1N4DBC-1a obtained from powder structure analysis. (e) 1D columns formed by shape-fitted docking of the DBC derivatives with a tilted angle of 13°. (f) Two different dimers with truncated H-bonding manner. (g) Rietveld refinement of C1N4DBC-1a. (h) PXRD pattern comparison among crystalline bulk of (bottom) C1N4DBC-1, (middle) -1a, and (top) simulation of the electron diffraction structure. | ||
Notably, compared with CPDBC-1 (Fig. 3c), the framework of C1N4DBC-1x shrank in the direction of the b axis, and the facing naphthyl groups divided the pores into three parts. This framework structure was affected by distortion caused by both the molecular conformation and H-bonding manner. The bond angle of the peripheral groups Ar–B (Fig. 1) in C1N4DBC-1x was 10°, which is larger than that of the optimized structure (0.3°) (Table S9, ESI†). The complementary H-bonded dimer also was distorted (Fig. 7c).
The crystal structure of the completely activated HOF C1N4DBC-1a47 (Fig. 7d), was obtained by Rietveld refinement using synchrotron PXRD patterns (Fig. 7g), where the molecular structure was constructed by optimization of the preliminary structure obtained in the ED while maintaining the molecular conformation. Crystal data are also listed in Tables S2 and S4 (ESI†). Although C1N4DBC-1a shows a very similar diffraction profile with C1N4DBC-1x (Fig. 7h), two structural differences between them were found. The first was the H-bonding manner. The molecules in C1N4DBC-1a were networked by truncated H-bonds with an O⋯O distance of 2.77 Å and 3.76 Å, instead of distorted complementary H-bonds (Fig. 7c and f). The second was the stacking direction. The molecules were stacked with an inclination of 13° (Fig. 7b and e). A 1D channel along the c axis was divided into three parts by the naphthyl group, with a larger central part (9.07 Å × 8.74 Å) and smaller parts on the left and right (4.53 Å × 9.07 Å, and 5.83 Å × 8.42 Å, respectively).
Gas sorption measurements
To evaluate porosity of the activated frameworks of DBC derivatives, N2 and CO2 absorption experiments were performed at 77 K and 195 K, respectively. The C1N4DBC-1a showed almost no adsorption of N2 in the low-pressure region, but showed type-I CO2 adsorption isotherms with an uptake of 3.60 mmol(STP) g−1 at a relative pressure of 0.99 and 195 K (Fig. 8a). These results indicate the existence of micropores. The C2N6DBC-1a showed similar CO2 sorption behaviour with an uptake of 3.22 mmol(STP) g−1, although the isotherm exhibited larger hysteresis (Fig. 8c). The Brunauer–Emmett–Teller (BET) specific surface area (SA(BET)) was calculated to be 281 m2 g−1 for C1N4DBC-1a and 113 m2 g−1 for C2N6DBC-1a. The smaller value of the latter system probably was due to the amorphous nature of C2N6DBC-1a without clear channels. The stable C1N5DBC-1a exhibited excellent porosity. Both N2 and CO2 were adsorbed, with type IV and type V adsorption isotherms, respectively, suggesting the presence of mesopores (Fig. 8b). Uptake was 16.8 mmol(STP) g−1 for N2 and 27.5 mmol(STP) g−1 for CO2. The SA(BET) was calculated from the N2 adsorption isotherm to be 1300 m2 g−1, indicating that large pores were present. During adsorption of CO2, a sharp rise was observed at a relative pressure of approximately 1.0. In contrast, desorption did not show a sharp change but hysteresis with a gradual decrease to the plateau until a relative pressure of 0.7, due to CO2 aggregation in the pores.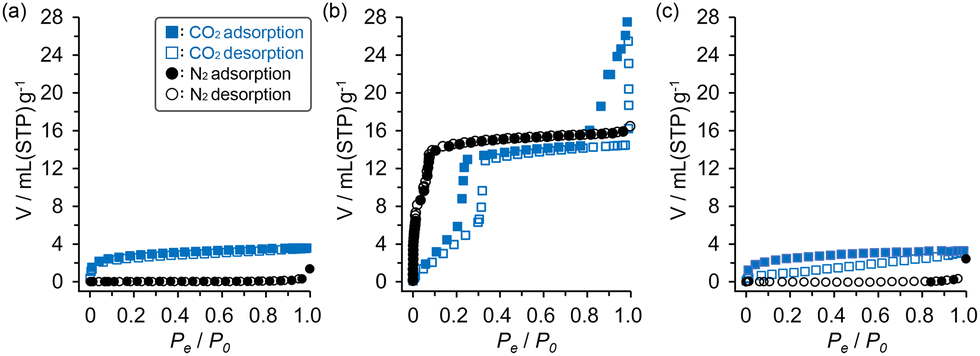 | ||
| Fig. 8 CO2 (blue) and N2 (black) absorption isotherms of (a) C1N4DBC-1a, (b) C1N5DBC-1a, and (c) C2N6DBC-1a. Solid and open symbols correspond to adsorption and desorption processes, respectively. | ||
In situ PXRD measurements with loaded molecules
The C1N4DBC-1a that underwent a structural transition upon activation was expected to undergo a structural change induced by loading guest molecules. Rigid HOF C1N5DBC-1a showed no structural changes upon adsorption of benzene vapour (Fig. S22a, ESI†). However, C1N4DBC-1a showed obvious structural changes that were confirmed by in situ PXRD experiments during adsorption and desorption of benzene vapour (Fig. 9). The patterns were recorded under a benzene atmosphere from 0 kPa to 10 kPa with 1 kPa steps at 298 K, followed by a decrease to 0 kPa. The structural transition began at 2 kPa, where the characteristic peak of the initial structure at 2θ = 3.48° (d = 16.5 Å) decayed and a new peak appeared at 2θ = 2.91° (d = 19.7 Å). At 5 kPa, the initial structural pattern completely disappeared and was replaced by that of the benzene-filled HOF C1N4DBC-1(benzene). During the desorption process, the structural transition began between 3 and 0 kPa. At 0 kPa, the pattern of C1N4DBC-1a was recovered accompanied by a broad pattern. The broad peak at around 2θ = 3.0–3.3° was attributed to an intermediate state of the activated structure, in which the number of benzenes in the pores decreased (for details see Fig. S22d and e, ESI†). Heating at 60 °C under vacuum finally gave a pure phase of C1N4DBC-1a. The C1N4DBC-1a also was subjected to the in situ experiment using N2 and CO2 at 298 K, but no transition occurred (Fig. S20, ESI†). | ||
| Fig. 9 In situ PXRD patterns of C1N4DBC-1a upon introduction of benzene vapor. | ||
Proposed lattice change in C1N4DBC-1
Since the PXRD patterns of C1N4DBC-1 maintained a good signal/noise ratio during the in situ experiment, the mechanism of the structural changes could be proposed based on PXRD patterns. The patterns of C1N4DBC-1, C1N4DBC-1(benzene), and C1N4DBC-1a are shown in Fig. 10c. The broad signals around 10–20° was a background from a glass capillary. Cell parameters and space group were determined using the patterns (Fig. 10b and Fig. S23, ESI†); C1N4DBC-1 belongs to the orthorhombic space group Pnnn, while C1N4DBC-1a and C1N4DBC-1(benzene) belong to the less symmetric monoclinic space group Pn (Table S5, ESI†). The lattices and proposed changes are shown in Fig. 10a. The C1N4DBC-1 lattice was similar to the single crystal structure of CPDBC-1. The a, b, and c axes of C1N4DBC-1 reflect the width and height of the aperture and stacking distance of the rhombic framework, respectively. Axes lengths were −1.04 Å, +0.343 Å, and +0.337 Å compared with the corresponding axes of CPDBC-1. Therefore, it formed a slightly wider H-bonding network, and the stacking distance was longer due to the bulky naphthyl groups. Upon activation to C1N4DBC-1a, a significant shrinkage by 5.47 Å was observed along the a axis direction. Simultaneously, the b axis extended by 0.6 Å to form three divided pores. The cell volume decreased by ca. 900 Å3 from C1N4DBC-1 (3985.5 Å3). This anisotropic shrinkage was expected to be accompanied by a decrease in the number of guest molecules. | ||
| Fig. 10 (a) Proposed lattice changes in C1N4DBC-1, (b) table of predicted lattice, and (c) PXRD patterns of C1N4DBC-1, C1N4DBC-1a, and C1N4DBC-1(benzene). | ||
Removal of guest molecules from the pores of HOFs results in pore shrinkage and/or structural transitions of the framework associated with recombination or dissociation.48–51 The molecular conformation, stacking manner, and hydrogen bonds of C1N4DBC-1 were perturbed, resulting in shrinkage of the pore aperture along the shorter direction. However, structurally hindered 1-naphthyl spacers as well as the shape-fitted docking of DBC cores prevented a drastic transition that would have led to collapse of the framework into an amorphous state.
Upon loading benzene to C1N4DBC-1a, the divided channel was restored and C1N4DBC-1(benzene) was formed. The a axis elongated by 4.5 Å and the b axis contracted by 1.2 Å compared to the activated structure, indicating that the rhombic channel opened along the height direction. This HOF was not identical to C1N4DBC-1 but had a similar structure with all peaks shifted slightly to the wide angle, and large values of the c axis (4.68 Å) and β angle (95.9°). The cell volume increased by ca. 1316 Å3 from C1N4DBC-1a (3080.1 Å3).
Thus, results showed that C1N4DBC-1 exhibits breathing behaviour in which the loading/removal of the guests causes expansion and shrinkage of the pores.
Computational evaluation of stability
DFT calculations of the intermolecular interactions between two peripheral group molecules were done to investigate the stability of the HOFs (for details, see Table S16, ESI†). Dispersion-corrected complexation energies were calculated for each stack of carboxynaphthyl groups (major–major, major–minor, minor–minor stacking) and each type of H-bond (major–major, major–minor, minor–minor bonding) (Table 1). No significant differences were observed in the complexation energy of stacking. The energy ranged from −7.40 kcal mol−1 to −8.31 kcal mol−1. The complexation energy of H-bonding was significantly different depending on whether the H-bonding was dimeric or monomeric. The major–major and minor–minor bonds of C2N6DBC, which were truncated monomers, show small stabilization values of −9.42 kcal mol−1 and −3.14 kcal mol−1, respectively. In addition, the self-complementary dimer, C1N5DBC, and the major–minor bond of C2N6DBC were strongly stabilized at approximately −20 kcal mol−1.| CPDBC-1 | C1N4DBC-1x | C1N5DBC-1 | C2N6DBC-1 | |
|---|---|---|---|---|
| a “Ar” and “B” means aromatic moieties shown in Fig. 1. This table focused on the major moieties of the disorder for simplicity. | ||||
| RMSD of the DBC core plane | 0.680 | 0.73 | 0.669 | 0.716 |
| Bond angle of peripheral groups Ar–B/° | 121.5 | 122 | 123.7 | 122.9 |
| Dihedral angle of peripheral groups Ar–B/° | 36.7 | 29 | 41.5 | 31.4 |
| Stacking distance between DBC cores/Å | 3.76 | 4.0 | 3.86 | 3.84 |
| Aperture size of the void/Å × Å | 29.1 × 15.3 | 9.8 × 8.4 and 4.9 × 6.4 | 26.8 × 23.3 | 22.7 × 17.9 |
| Void ratio | 0.56 | 0.32 | 0.58 | 0.58 |
| Structure after guest removal | Retained the framework | Shrunk the pore | Retained the framework | Collapsed into an amorphous solid |
| BET surface area/m2 g−1 | 1548 | 281 | 1300 | 113 |
| Reference | Ref. 43 | This work | This work | This work |
The C1N5DBC-1 with square pores did not experience an anisotropic shrinkage as C1N4DBC-1, and the strong complementary dimers appeared to successfully maintain the initial structure. In C2N6DBC-1, while the dissociation of fragile bonds was more likely to occur, it was less likely to occur with strong bonds. While complementary H-bonded dimers firmly networked in C1N5DBC-1, C2N6DBC-1 consisted of a mixture of less stabilized monomers and stable dimers. This coexistence of H-bonds resulted in disorderly structural collapse, leading to crystalline-to-amorphous transition.
Conclusions
The isomeric effect of the peripheral groups in DBC-based building block molecules on structures and properties of isostructural HOFs were investigated. The HOFs were composed of DBC derivatives with 4-carboxybiphenyl, 1-carboxy-4-naphthyl, 1-carboxy-5-naphthyl, and 2-carboxy-6-naphthyl substituents (CBPDBC, C1N4DBC, C1N5DBC, and C2N6DBC, respectively). Results indicated that CBPDBC formed a homomorphic network with previously reported CPDBC, but only a low crystalline precipitate was obtained. In contrast, HOFs composed of naphthyl-substituted DBC derivatives successfully gave three isostructural HOFs that exhibited different transition behaviours depending on the naphthyl substituents (Table 2).The C1N5DBC-1 formed a square pore through the complementary H-bonded dimer, which resulted in a stable HOF without a structural transition during guest removal. The activated HOF, C1N5DBC-1a, retained large pores with a SA(BET) of 1300 m2 g−1 and was stable up to 220 °C. The C1N4DBC-1, which is a HOF with a structure similar to that of CPDBC-1, showed pore shrinkage along the b axis during guest removal, giving activated HOF, C1N4DBC-1a, with SA(BET) of 281 m2 g−1. In situ PXRD measurements revealed that C1N4DBC-1a underwent reversible crystal-to-crystal structural transition upon loading and removal of benzene. These dynamic behaviours were due to the shape-fitted docking and hindered 1-naphthyl groups: i.e., they prevented structural collapse against activation, and alternately, provided the framework with shrunken and divided channels. The C2N6DBC-1 showed a crystalline-to-amorphous structural change because of large rhombic pores and the coexistence of weak and strong hydrogen bonds caused by disordering of the peripheral groups.
The results implied that the DBC core is fundamental to isostructural constructed HOFs, but that the isomeric effect at the peripheral groups significantly affected structural properties, ranging from stable to flexible to fragile. The behaviours observed were attributed to differences in pore geometry and H-bonding manner, due to the peripheral structure or its disorder. Investigation of the molecular design must include the study of peripheral groups as well what is needed to control the systematic construction of HOFs with the desired structure and function.
Author contributions
Y. S. and M. Y. designed, synthesized, and characterized all materials, evaluated properties of the materials, and co-wrote the paper. R. O. contributed to the theoretical calculations. I. H. planned and supervised the research, analysed the data, and co-wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by KAKENHI (JP21H05485, JP21H01919, JP21K18961, JP22H05461) from MEXT and JSPS, Japan. I.H. thanks Hoansha Foundation and Nagase Science Technology Foundation. I.H. also thanks MRL, Graduate School of Engineering Science, Osaka University. Y.S. is thankful for a Grant-in-Aid for JSPS Research Fellow (20J20301). The authors thank Prof. N. Tohnai at Osaka University for VT-PXRD and gas sorption experiments, Dr H. Sato at Rigaku Corporation for electron diffraction crystallographic analyses, and Ms R. Miyake at Osaka University for the HR-MS analysis. Theoretical calculations were conducted at the Cybermedia Center, Osaka University, using the Supercomputer for Quest to Unsolved Interdisciplinary Datascience (SQUID). X-ray diffraction data, including preliminary datasets, were collected at BL40XU in SPring-8 with approval of JASRI (proposal No. 2021A1080, 2021A1567, 2019B1134, and 2019A1192).Notes and references
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Reticular synthesis and the design of new materials, Nature, 2003, 423, 705–814 CrossRef PubMed.
- W. Xu, B. Tu, Q. Liu, Y. Shu, C. C. Liang, C. S. Diercks, O. M. Yaghi, Y. B. Zhang, H. Deng and Q. Li, Anisotropic reticular chemistry, Nat. Rev. Mater., 2020, 5, 764–779 CrossRef.
- E. Ploetz, H. Engelke, U. Lächelt and S. Wuttke, The Chemistry of Reticular Framework Nanoparticles: MOF, ZIF, and COF Materials, Adv. Funct. Mater., 2020, 30, 1909062 CrossRef.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, 25 Years of Reticular Chemistry, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef PubMed.
- C. Wang, D. Liu and W. Lin, Metal–Organic Frameworks as A Tunable Platform for Designing Functional Molecular Materials, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. I. Llabrés Xamena, V. Van Speybroeck and J. Gascon, Metal–organic and covalent organic frameworks as single-site catalysts, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- M. Wang, H. Guo, R. Xue, Q. Li, H. Liu, N. Wu, W. Yao and W. Yang, Covalent Organic Frameworks: A New Class of Porous Organic Frameworks for Supercapacitor Electrodes, ChemElectroChem, 2019, 6, 2984–2997 CrossRef.
- Z. Li, T. He, Y. Gong and D. Jiang, Covalent Organic Frameworks: Pore Design and Interface Engineering, Acc. Chem. Res., 2020, 53, 1672–1685 CrossRef PubMed.
- J. Lü and R. Cao, Porous Organic Molecular Frameworks with Extrinsic Porosity: A Platform for Carbon Storage and Separation, Angew. Chem., Int. Ed., 2016, 55, 9474–9480 CrossRef PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage, Science, 2002, 295, 469–472 CrossRef.
- N. Huang, L. Zhai, D. E. Coupry, M. A. Addicoat, K. Okushita, K. Nishimura, T. Heine and D. Jiang, Multiple-component covalent organic frameworks, Nat. Commun., 2016, 7, 12325 CrossRef.
- W. Lu, Z. Wei, Z. Y. Gu, T. F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle, M. Bosch and H. C. Zhou, Tuning the structure and function of metal–organic frameworks via linker design, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Secondary building units as the turning point in the development of the reticular chemistry of MOFs, Sci. Adv., 2018, 4, eaat9180 CrossRef.
- F. Beuerle and B. Gole, Covalent Organic Frameworks and Cage Compounds: Design and Applications of Polymeric and Discrete Organic Scaffolds, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef.
- Z. Chen, H. Jiang, M. Li, M. O’Keeffe and M. Eddaoudi, Reticular Chemistry 3.2: Typical Minimal Edge-Transitive Derived and Related Nets for the Design and Synthesis of Metal–Organic Frameworks, Chem. Rev., 2020, 120, 8039–8065 CrossRef PubMed.
- A. J. Howarth, P. Li, O. K. Farha and M. O’Keeffe, Bottom-Up Design and Generation of Complex Structures: A New Twist in Reticular Chemistry, Cryst. Growth Des., 2018, 18, 449–455 CrossRef.
- Z. Chen, K. O. Kirlikovali, P. Li and O. K. Farha, Reticular Chemistry for Highly Porous Metal–Organic Frameworks: The Chemistry and Applications, Acc. Chem. Res., 2022, 55, 579–591 CrossRef PubMed.
- V. Guillerm and D. Maspoch, Geometry Mismatch and Reticular Chemistry: Strategies To Assemble Metal–Organic Frameworks with Non-default Topologies, J. Am. Chem. Soc., 2019, 141, 16517–16538 CrossRef PubMed.
- S. Y. Ding and W. Wang, Covalent organic frameworks (COFs): from design to applications, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- S. Yu, G. Xing, L. Chen, T. Ben and B. Su, Crystalline Porous Organic Salts: From Micropore to Hierarchical Pores, Adv. Mater., 2020, 32, 2003270 CrossRef PubMed.
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Multifunctional porous hydrogen-bonded organic framework materials, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Designing Hydrogen-Bonded Organic Frameworks (HOFs) with Permanent Porosity, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, Hydrogen-Bonded Organic Frameworks as a Tunable Platform for Functional Materials, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef PubMed.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, Design Rules of Hydrogen-Bonded Organic Frameworks with High Chemical and Thermal Stabilities, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef.
- W. Qin, D. Si, Q. Yin, X. Gao, Q. Huang, Y. Feng, L. Xie, S. Zhang, X. Huang, T. Liu and R. Cao, Reticular Synthesis of Hydrogen-Bonded Organic Frameworks and Their Derivatives via Mechanochemistry, Angew. Chem., Int. Ed., 2022, 61, e202202089 Search PubMed.
- T. Ami, K. Oka, K. Tsuchiya and N. Tohnai, Porous Organic Salts: Diversifying Void Structures and Environments, Angew. Chem., Int. Ed., 2022, 61, e202202597 Search PubMed.
- T.-H. Chen, I. Popov, W. Kaveevivitchai, Y.-C. Chuang, Y.-S. Chen, O. Daugulis, A. J. Jacobson and O. Š. Miljanić, Thermally robust and porous noncovalent organic framework with high affinity for fluorocarbons and CFCs, Nat. Commun., 2014, 5, 5131 CrossRef PubMed.
- Y. Wang, K. Ma, J. Bai, T. Xu, W. Han, C. Wang, Z. Chen, K. O. Kirlikovali, P. Li, J. Xiao and O. K. Farha, Chemically Engineered Porous Molecular Coatings as Reactive Oxygen Species Generators and Reservoirs for Long-Lasting Self-Cleaning Textiles, Angew. Chem., Int. Ed., 2022, 61, e202115956 Search PubMed.
- I. Hisaki, N. Q. Emilya Affendy and N. Tohnai, Precise elucidations of stacking manners of hydrogen-bonded two-dimensional organic frameworks composed of X-shaped π-conjugated systems, CrystEngComm, 2017, 19, 4892–4898 RSC.
- K. O. Kirlikovali, S. Goswami, M. R. Mian, M. D. Krzyaniak, M. R. Wasielewski, J. T. Hupp, P. Li and O. K. Farha, An Electrically Conductive Tetrathiafulvalene-Based Hydrogen-Bonded Organic Framework, ACS Mater. Lett., 2022, 4, 128–135 CrossRef.
- K. Ma, P. Li, J. H. Xin, Y. Chen, Z. Chen, S. Goswami, X. Liu, S. Kato, H. Chen, X. Zhang, J. Bai, M. C. Wasson, R. R. Maldonado, R. Q. Snurr and O. K. Farha, Ultrastable Mesoporous Hydrogen-Bonded Organic Framework-Based Fiber Composites toward Mustard Gas Detoxification, Cell Rep. Phys. Sci., 2020, 1, 100024 CrossRef.
- M. I. Hashim, H. T. M. Le, T. H. Chen, Y. S. Chen, O. Daugulis, C. W. Hsu, A. J. Jacobson, W. Kaveevivitchai, X. Liang, T. Makarenko, O. Miljanić, I. Popovs, H. V. Tran, X. Wang, C. H. Wu and J. I. Wu, Dissecting Porosity in Molecular Crystals: Influence of Geometry, Hydrogen Bonding, and [π···π] Stacking on the Solid-State Packing of Fluorinated Aromatics, J. Am. Chem. Soc., 2018, 140, 6014–6026 CrossRef PubMed.
- I. Hisaki, Y. Suzuki, E. Gomez, B. Cohen, N. Tohnai and A. Douhal, Docking Strategy to Construct Thermostable, Single-Crystalline, Hydrogen-Bonded Organic Framework with High Surface Area, Angew. Chem., Int. Ed., 2018, 57, 12650–12655 CrossRef.
- I. Hisaki, N. Ikenaka, E. Gomez, B. Cohen, N. Tohnai and A. Douhal, Hexaazatriphenylene-Based Hydrogen-Bonded Organic Framework with Permanent Porosity and Single-Crystallinity, Chem. – A Eur. J., 2017, 23, 11611–11619 CrossRef PubMed.
- Y. Suzuki, M. Gutiérrez, S. Tanaka, E. Gomez, N. Tohnai, N. Yasuda, N. Matubayasi, A. Douhal and I. Hisaki, Construction of isostructural hydrogen-bonded organic frameworks: limitations and possibilities of pore expansion, Chem. Sci., 2021, 12, 9607–9618 RSC.
- C. A. Zentner, H. W. H. Lai, J. T. Greenfield, R. A. Wiscons, M. Zeller, C. F. Campana, O. Talu, S. A. Fitzgerald and J. L. C. Rowsell, High surface area and Z′ in a thermally stable 8-fold polycatenated hydrogen-bonded framework, Chem. Commun., 2015, 51, 11642–11645 RSC.
- H. W. H. Lai, R. A. Wiscons, C. A. Zentner, M. Zeller and J. L. C. Rowsell, Supramolecular Assembly of Tris(4-carboxyphenyl)arenes: Relationship between Molecular Structure and Solid-State Catenation Motifs, Cryst. Growth Des., 2016, 16, 821–833 CrossRef.
- W. Yang, W. Zhou and B. Chen, A Flexible Microporous Hydrogen-Bonded Organic Framework, Cryst. Growth Des., 2019, 19, 5184–5188 CrossRef.
- Y. L. Li, E. V. Alexandrov, Q. Yin, L. Li, Z. Bin Fang, W. Yuan, D. M. Proserpio and T. F. Liu, Record Complexity in the Polycatenation of Three Porous Hydrogen-Bonded Organic Frameworks with Stepwise Adsorption Behaviors, J. Am. Chem. Soc., 2020, 142, 7218–7224 CrossRef PubMed.
- M. Mastalerz and I. M. Oppel, Rational Construction of an Extrinsic Porous Molecular Crystal with an Extraordinary High Specific Surface Area, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef PubMed.
- A. Pulido, L. Chen, T. Kaczorowski, D. Holden, M. A. Little, S. Y. Chong, B. J. Slater, D. P. McMahon, B. Bonillo, C. J. Stackhouse, A. Stephenson, C. M. Kane, R. Clowes, T. Hasell, A. I. Cooper and G. M. Day, Functional materials discovery using energy-structure-function maps, Nature, 2017, 543, 657–664 CrossRef PubMed.
- Y. Fujii, T. Maruyama, R. Akasaka, K. Sakao, S. Tokai, Y. Taguchi, Y. Matsumoto, S. Kamiguchi, N. Yoshida and T. Iwasawa, Regio-defined syntheses of tetra-brominated dibenzo[g,p]chrysene scaffolds with high solubility, Tetrahedron Lett., 2021, 65, 152758 CrossRef.
- Y. Suzuki, N. Tohnai, A. Saeki and I. Hisaki, Hydrogen-bonded organic frameworks of twisted polycyclic aromatic hydrocarbon, Chem. Commun., 2020, 56, 13369–13372 RSC.
- The crystal included tBuXy C2N6DBC-1 showed 8-fold interpenetration, but the one included MeBz C2N6DBC-2 showed 9-fold (for details, see Fig. S10, ESI†).
- P. V. D. Sluis and A. L. Spek, BYPASS: an effective method for the refinement of crystal structures containing disordered solvent regions, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- A. L. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef PubMed.
- The single microcrystal for electron diffraction was obtained by the activation of mesitylene-included crystal through VT method. The crystalline balk was obtained by the activation of xylene-included crystals through VT method (for details, see Fig. S16 and S17, ESI†).
- X. Y. Gao, Y. L. Li, T. F. Liu, X. S. Huang and R. Cao, Single-crystal-to-single-crystal transformation of tetrathiafulvalene-based hydrogen-bonded organic frameworks, CrystEngComm, 2021, 23, 4743–4747 RSC.
- H. Kubo, R. Oketani and I. Hisaki, Quasi single-crystalline transformation of porous frameworks accompanied by interlayer rearrangements of hydrogen bonds, Chem. Commun., 2021, 57, 8568–8571 RSC.
- B. Yu, S. Geng, H. Wang, W. Zhou, Z. Zhang, B. Chen and J. Jiang, A Solid Transformation into Carboxyl Dimers Based on a Robust Hydrogen-Bonded Organic Framework for Propyne/Propylene Separation, Angew. Chem., Int. Ed., 2021, 60, 25942–25948 CrossRef.
- Q. Ji, K. Takahashi, S. I. Noro, Y. Ishigaki, K. Kokado, T. Nakamura and I. Hisaki, A Hydrogen-Bonded Organic Framework Based on Pyrazinopyrazine, Cryst. Growth Des., 2021, 21, 4656–4664 CrossRef.
- Y. Li, L. Guo, Y. Lv, Z. Zhao, Y. Ma, W. Chen, G. Xing, D. Jiang and L. Chen, Polymorphism of 2D Imine Covalent Organic Frameworks, Angew. Chem., Int. Ed., 2021, 60, 5363–5369 CrossRef PubMed.
- R.-R. Liang, F.-Z. Cui, R.-H. A, Q.-Y. Qi and X. Zhao, A Study on Constitutional Isomerism in Covalent Organic Frameworks: Controllable Synthesis, Transformation, and Distinct Difference in Properties, CCS Chem., 2020, 2, 139–145 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, characterization, thermal analysis, spectroscopic data, theoretical calculations, and crystallographic data. CCDC 2210179–2210181. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qm01091g |
| This journal is © the Partner Organisations 2023 |