
C–N bond metathesis: mechanistic insight into palladium-catalyzed ring-closing using aminal species†
Song
Liu
 *abc,
Dianmin
Zhang
a,
Min
Xiao
a,
Chengling
Pu
a,
Xiaoqing
Zhang
a,
Xiuwen
Yang
a,
Tao
Zhang
*d and
Ruopeng
Bai
*b
*abc,
Dianmin
Zhang
a,
Min
Xiao
a,
Chengling
Pu
a,
Xiaoqing
Zhang
a,
Xiuwen
Yang
a,
Tao
Zhang
*d and
Ruopeng
Bai
*b
aChongqing Key Laboratory of Environmental Materials and Remediation Technologies, College of Chemistry and Environmental Engineering, Chongqing University of Arts and Sciences, Chongqing, 402160, P. R. China. E-mail: sliu@cqwu.edu.cn
bSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400030, China
cChongqing Precision Medical Industry Technology Research Institute, Chongqing 400084, P. R. China
dZhengZhou JiShu Institute of AI Science, Zhengzhou, Henan, China 450000
First published on 24th November 2022
Abstract
C–N bond metathesis is a straightforward and step-economical approach for C–N bond construction. Typically, the oxidation state of the transition metal remains unchanged during C–N bond metathesis. In this report, we present computational evidence that supports a mechanism in which a new type of reversible reductive elimination/oxidative addition mode is involved in the C–N bond metathesis reaction. This reversible reductive elimination/oxidative addition-induced C–N bond metathesis pathway was found to be lower in energy compared with transimination from a Huang-complex, which is important for understanding the mechanism of Pd-catalyzed C–N bond formation reactions. Non-covalent interaction analysis was conducted to elucidate the details of the diene 1,4-migratory insertion step process and to investigate the origin of stereoselectivity for this type of reaction. We anticipate that this novel C–N bond metathesis mode may extend to other cross-coupling processes and may provide a theoretical guide for further experimental investigations.
Introduction
Owing to the ubiquitous and necessary presence of amines in biologically active molecules, the development of methods for constructing amine-based organic frameworks is an important research topic in synthetic chemistry.1 C–N bond metathesis, which transfers an amine moiety between two nitrogen-containing substrates leading to the breaking and formation of C–N or N–H bonds (Scheme 1), is acknowledged as one of the most efficient and step-economical approaches for C–N bond construction.2 Regardless of its apparent simplicity, C–N bond metathesis is a convenient approach for amine synthesis.3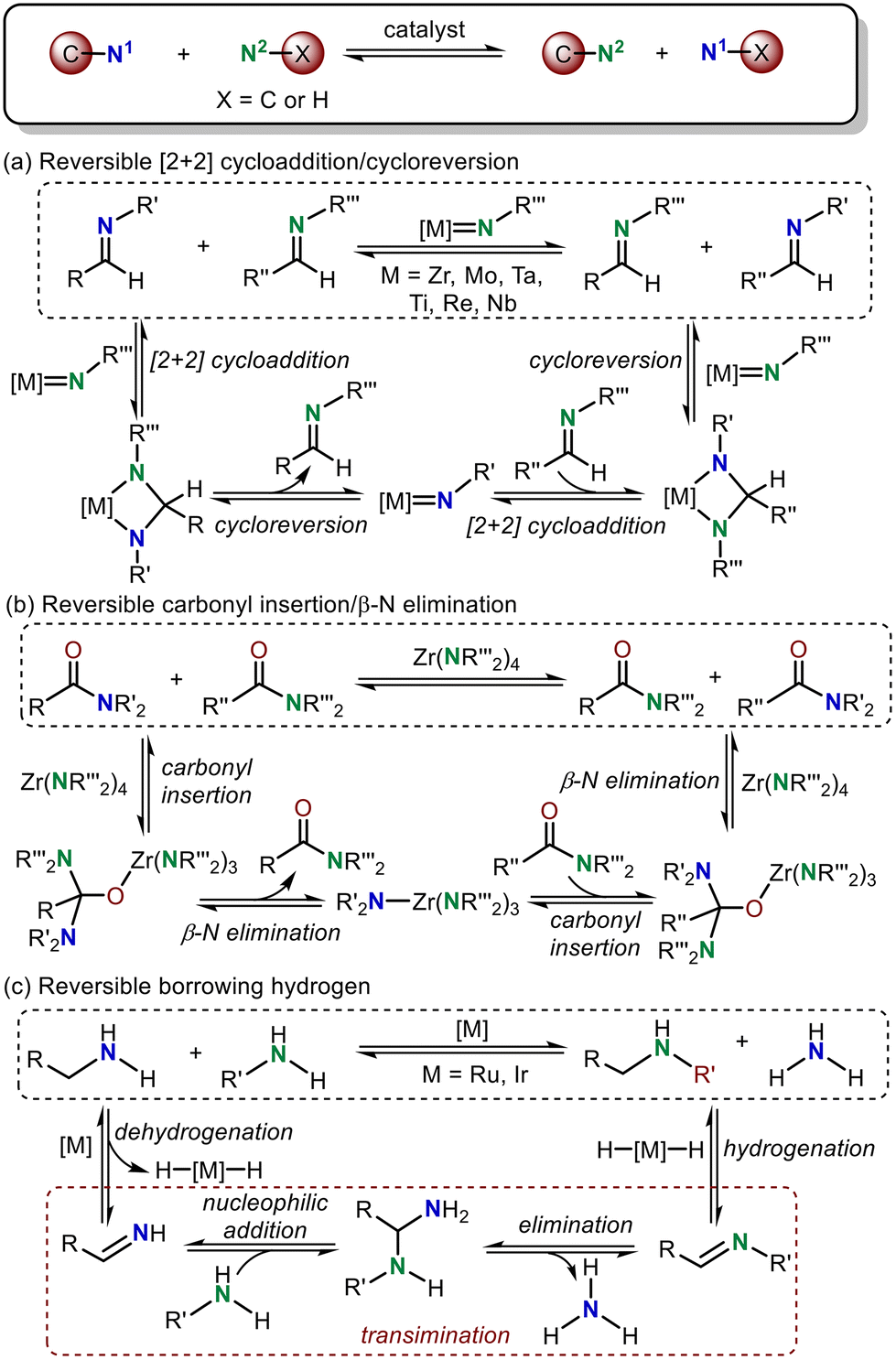 | ||
| Scheme 1 Three strategies of C–N bond metathesis. | ||
According to previous studies in this area, three possible reaction strategies were developed for the C–N bond metathesis (Scheme 1). In strategy A, the imine C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N metathesis transfers an amine moiety in a manner analogous to alkene metathesis, which proceeds through a metal-nitrene-containing reversible [2 + 2] cycloaddition followed by a cycloreversion reaction (Scheme 1a).4 This strategy has been leveraged for the development of transition metal-catalyzed imine metathesis, which was reported by Bergman,4a,b Meyer,4c–e Mountford,4f Espenson4g and Bruno.4h In strategy B, two new amide C–N bonds can also be constructed via reversible carbonyl insertion/β-N elimination (Scheme 1b).5 Under the guidance of this principle, Gellman and Stahl documented the successful and elegant Zr-catalyzed C–N/C–N metathesis of tertiary amides.5 Moreover, a hydrogen-borrowing strategy, proceeding via the removal of hydrogen from an amine, transimination and return of the hydrogen from the catalyst, can also be applied for N–H/C–N metathesis reactions between two amines (Scheme 1c).6 Notably, nucleophilic addition of an amine to an imine, 1,3-proton transfer and elimination are involved in this transimination step. Recently, this hydrogen-borrowing strategy has attracted close attention for C–N metathesis. For example, Concilio et al. demonstrated the formation of a secondary amine from the parent primary amine using an Ru catalyst.6a,b Beller's group also developed successful N–H/C–N metathesis reactions between arylamines and primary amines.6c In addition, Williams and co-workers found that primary amines could be selectively alkylated with diisopropylamine in the presence of an Ir catalyst.6d Among these strategies, we found that the processes involved in C–N metathesis are usually reversible and the oxidation state of the transition metal remains unchanged during this step.
N metathesis transfers an amine moiety in a manner analogous to alkene metathesis, which proceeds through a metal-nitrene-containing reversible [2 + 2] cycloaddition followed by a cycloreversion reaction (Scheme 1a).4 This strategy has been leveraged for the development of transition metal-catalyzed imine metathesis, which was reported by Bergman,4a,b Meyer,4c–e Mountford,4f Espenson4g and Bruno.4h In strategy B, two new amide C–N bonds can also be constructed via reversible carbonyl insertion/β-N elimination (Scheme 1b).5 Under the guidance of this principle, Gellman and Stahl documented the successful and elegant Zr-catalyzed C–N/C–N metathesis of tertiary amides.5 Moreover, a hydrogen-borrowing strategy, proceeding via the removal of hydrogen from an amine, transimination and return of the hydrogen from the catalyst, can also be applied for N–H/C–N metathesis reactions between two amines (Scheme 1c).6 Notably, nucleophilic addition of an amine to an imine, 1,3-proton transfer and elimination are involved in this transimination step. Recently, this hydrogen-borrowing strategy has attracted close attention for C–N metathesis. For example, Concilio et al. demonstrated the formation of a secondary amine from the parent primary amine using an Ru catalyst.6a,b Beller's group also developed successful N–H/C–N metathesis reactions between arylamines and primary amines.6c In addition, Williams and co-workers found that primary amines could be selectively alkylated with diisopropylamine in the presence of an Ir catalyst.6d Among these strategies, we found that the processes involved in C–N metathesis are usually reversible and the oxidation state of the transition metal remains unchanged during this step.
Although the above three C–N metathesis modes are commonly used as effective strategies in transition metal-catalyzed C–N bond formation reactions, the lack of a new type of reaction mode for C–N metathesis has hampered the development of constructing new C–N bonds in organic synthesis.2 In 2020, Huang and co-workers developed a new mode, proceeding through a reversible reductive elimination, 1,3-proton transfer, and oxidative addition sequence, to realize C–N bond metathesis.7 But the detailed mechanism and computational evidence for this new type of C–N bond metathesis is still not clear, which limits the development of such C–N bond metathesis reactions. In this reaction, the C–N bond metathesis was proposed to occur from an active Huang-complex. Notably, the iminium-coordinated Pd(0) cationic complex was one limiting resonance form of the active Huang-complex, which was confirmed by our previous DFT calculations.8 Thus, we propose that this reaction may also proceed via a transimination mechanism to realize C–N and N–H exchange (Scheme 2).9 With this in mind, we performed density functional theory (DFT) calculations to investigate the mechanism of this new Pd-catalyzed C–N bond metathesis and ring-closing reaction. The competition between the proposed reversible reductive elimination/oxidative elimination-induced C–N bond metathesis and transimination was investigated. A detailed reaction mechanism and reactivity profile for this reaction were revealed. Furthermore, the origin of enantioselectivity established by a chiral P–N ligand was also elucidated. The calculated results presented herein would provide a theoretical guide for further experimental investigations of C–N bond metathesis reactions.
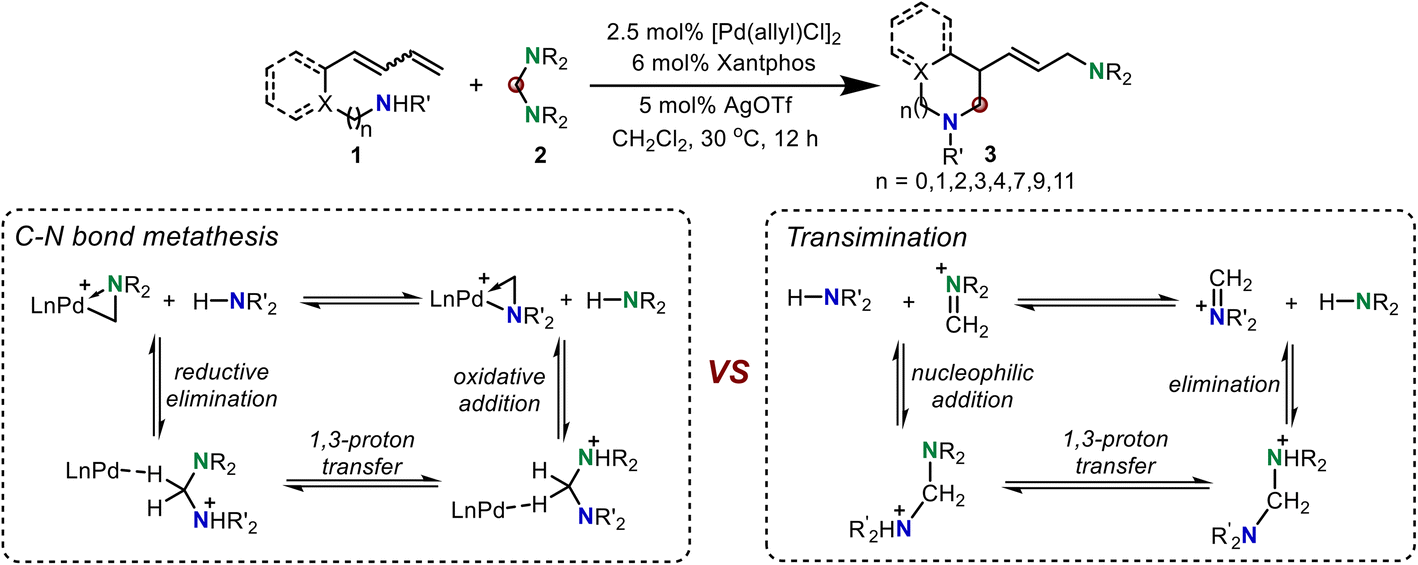 | ||
| Scheme 2 Palladium-catalyzed C–N bond metathesis and ring-closing reaction. | ||
Computational methods
All DFT calculations were carried out using the GAUSSIAN 09 series of programs.10 The density functional B3-LYP11 with a standard 6-31G(d) basis set (SDD basis set for Pd) was used for geometry optimizations. Harmonic frequency calculations were performed for all stationary points to confirm them as local minima or transition structures and to derive the thermochemical corrections for the enthalpies and free energies. All minima have zero imaginary frequency and all transition states have only one imaginary frequency. Based on the gas-phase optimized structures, the single-point energies and solvent effects of dichloromethane (the solvent used in the experiment) were calculated with the M06![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 functional, the SDD basis set for palladium, and the larger 6-311+G(d) basis set for other atoms, using the SMD13 implicit solvent model. The energies given in this report are the M06 calculated Gibbs free energies in the dichloromethane solvent. The values of ΔGM06/solvent in free energy profiles were obtained by eqn (1), in which ΔGcorrection/gas is the thermochemical correction for the Gibbs free energy calculated B3-LYP/6-31G(d)/SDD level in the gas phase, and ΔEM06/solvent is the single point energy calculated at the M06/6-311+G(d)/SDD level in the solvent phase based on the gas-phase stationary point.
12 functional, the SDD basis set for palladium, and the larger 6-311+G(d) basis set for other atoms, using the SMD13 implicit solvent model. The energies given in this report are the M06 calculated Gibbs free energies in the dichloromethane solvent. The values of ΔGM06/solvent in free energy profiles were obtained by eqn (1), in which ΔGcorrection/gas is the thermochemical correction for the Gibbs free energy calculated B3-LYP/6-31G(d)/SDD level in the gas phase, and ΔEM06/solvent is the single point energy calculated at the M06/6-311+G(d)/SDD level in the solvent phase based on the gas-phase stationary point.| ΔGM06/solvent = ΔEM06/solvent + ΔGcorrection/gas | (1) |
We also performed benchmarks for some frequently used calculation methods (see the ESI for details†). The calculated results indicated that this method used herein is reliable. The optimized structures were shown using CYLview.14
Results and discussion
The Huang-complex (Xantphos)Pd-(CH2NBn2)+CP1, which is the active catalyst in this C–N bond metathesis reaction,7 was quickly generated from an allyl palladium complex and an aminal (Scheme 3). The overall activation free energy for this transformation is 28.4 kcal mol−1, while the Huang-complex CP1 was generated with an exothermic energy of 0.7 kcal mol−1. The detailed calculated free energy profiles for this step are provided in the ESI.† The calculated structural information of this active Huang-complex CP1 is shown in Scheme 3. In CP1, the lengths of the Pd–C and Pd–N bonds were 2.05 and 2.24 Å, respectively. According to our previous DFT calculations, the Pd(0)-iminium cation complex was one limiting resonance form of this active Huang complex. Therefore, we first considered the possibility of a transimination mechanism.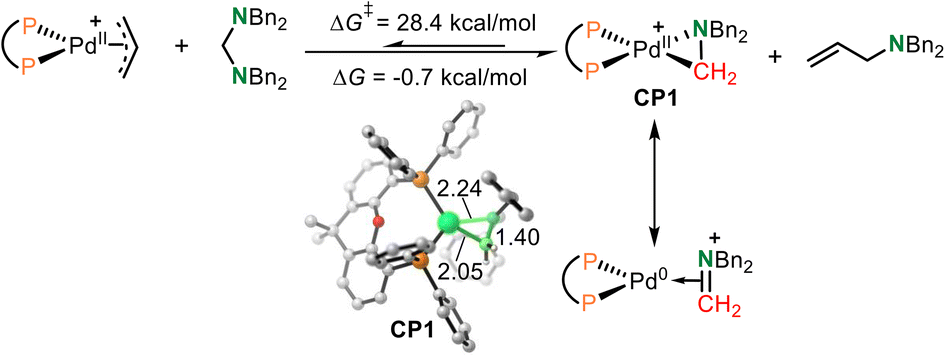 | ||
| Scheme 3 The formation of active Huang-complex CP1 from an allyl palladium complex. | ||
The calculated free energy profiles for the transimination mechanism are shown in Fig. 1. The dissociation of palladacycle-complex CP1 into an imine cation and Pd(0) occurs through two steps via transition states TS1 and TS2. The energy barrier for the dissociation of the amino from the Pd center was 11.3 kcal mol−1. In TS1, the length of the C–N bond being broken was 2.80 Å. From TS1, alkyl-Pd(II) intermediate CP2 was generated. The subsequent dissociation of the C–Pd bond occurs via transition state TS2 with an energy barrier of 12.2 kcal mol−1 to give CP3. In CP3, the Pd(0) was bonded to imine cations by two hydrogen bonds. After the dissociation of Pd(0), the imine cation was generated. The free energy of imine cation CP4 and Pd(0) was 24.3 kcal mol−1. The subsequent nucleophilic addition of the diene-tethered amine substrate to the imine cation occurred through transition state TS3 with an activation free energy of 31.6 kcal mol−1 to give amino cation intermediate CP5. Another pathway, the nucleophilic addition of the diene-tethered amine substrate to the imine in CP3, was also analyzed. The nucleophilic addition transition state TS4 has an energy barrier of 8.1 kcal mol−1 relative to CP3, and gave the amino cation Pd(0) intermediate CP6. Due to the high activation free energy of TS3 and TS4, the transimination process does not readily occur under the current reaction conditions at a temperature of 30 °C. The calculated results also indicate that no imine cation exists in the reaction system, and palladacycle-complex CP1 is the active catalyst in the catalytic cycle.
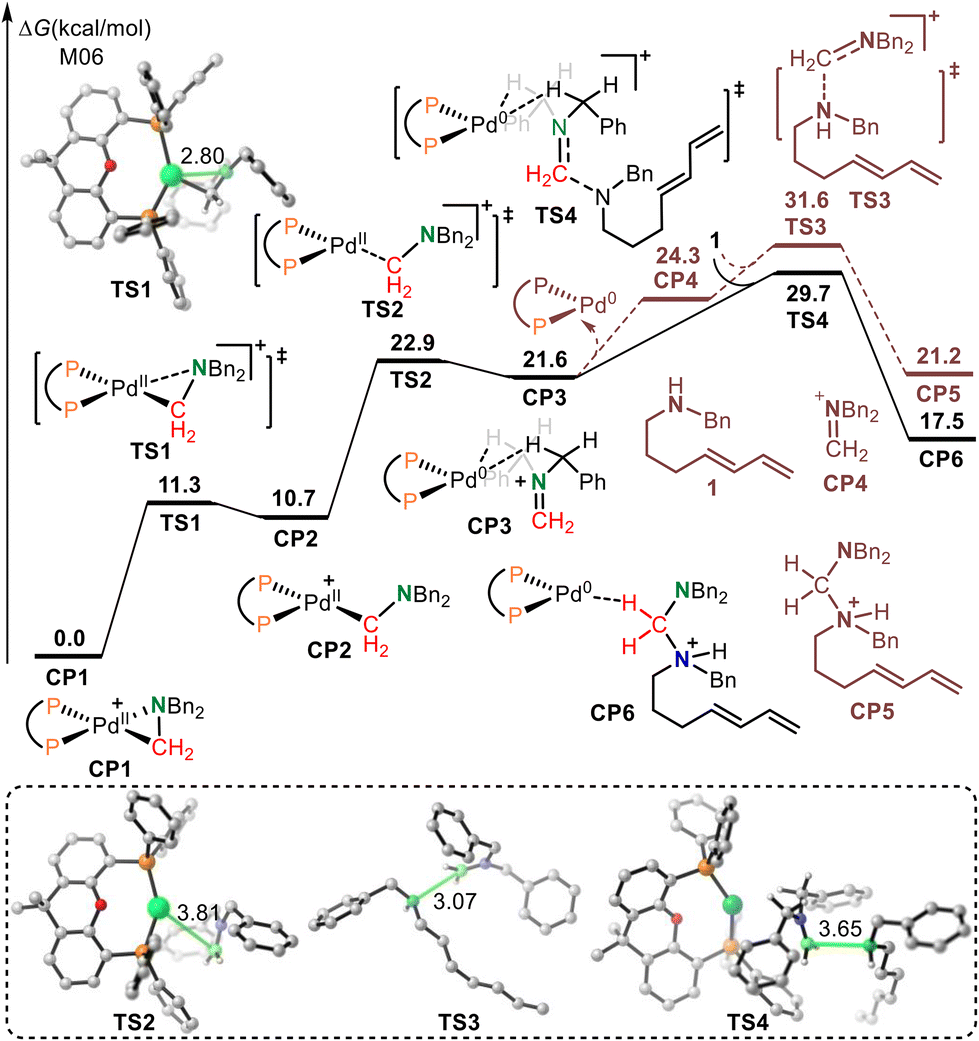 | ||
| Fig. 1 The calculated free energy profiles for transimination. | ||
The calculated Gibbs energy profiles for the Pd-catalyzed reductive elimination/oxidative addition-induced C–N bond metathesis are presented in Fig. 2, where the (Xantphos)Pd-(CH2NBn2)+ Huang-complex CP1 was also set as the relative zero point. Because CP1 can be described as a resonance form of the square-planar nitrogen-coordinated aminomethyl-Pd(II) and triangular iminium-coordinated Pd(0) complex, the iminium cation group in CP1 is electrophilic enough to be attacked by exogenous secondary amine 1via transition state TS5 to form amino cation Pd(0) intermediate CP6. In CP6, there is a hydrogen bond between the hydrogen atom in the methylene group and the Pd(0) center. The calculated free energy barrier of this step was 23.9 kcal mol−1. The structural information of TS5 shows that the lengths of the C–N bond being formed and the C–Pd bond being broken were 2.27 and 3.15 Å, respectively. Notably, TS5 can also be considered as a reductive elimination transition state because Pd(0) was generated. The rapid proton transfer in CP6 gave another amino cation Pd(0) intermediate CP7. Subsequent oxidative addition of CP7 to the Pd(0) center occurred via transition state TS6 with an energy barrier of only 6.9 kcal mol−1 to generate CP8 with a concomitant release of an amine. In TS6, the lengths of the C–N bond being broken and the C–Pd bond being formed were 2.14 and 3.13 Å, respectively. The calculated results show that the desired C–N bond metathesis is furnished through a reversible process involving reductive elimination, hydrogen transfer, and oxidative addition. The overall activation free energy for this type of C–N bond metathesis was only 23.9 kcal mol−1, which is 5.8 kcal mol−1 lower than that of the transimination pathway via transition state TS4. This indicates that the reductive elimination/oxidative addition-induced C–N bond metathesis pathway is more favorable than transimination. Our theoretical studies also indicated that due to the participation of palladium, the imine cation generated online becomes stable and Huang-complex CP1 was generated. Because CP1 can be described as a triangular iminium-coordinated Pd(0) complex, the iminium cation group in CP1 can be attacked by a secondary amine directly, without dissociation of Pd(0), which results in this reaction via the C–N bond metathesis mechanism rather than transamination.
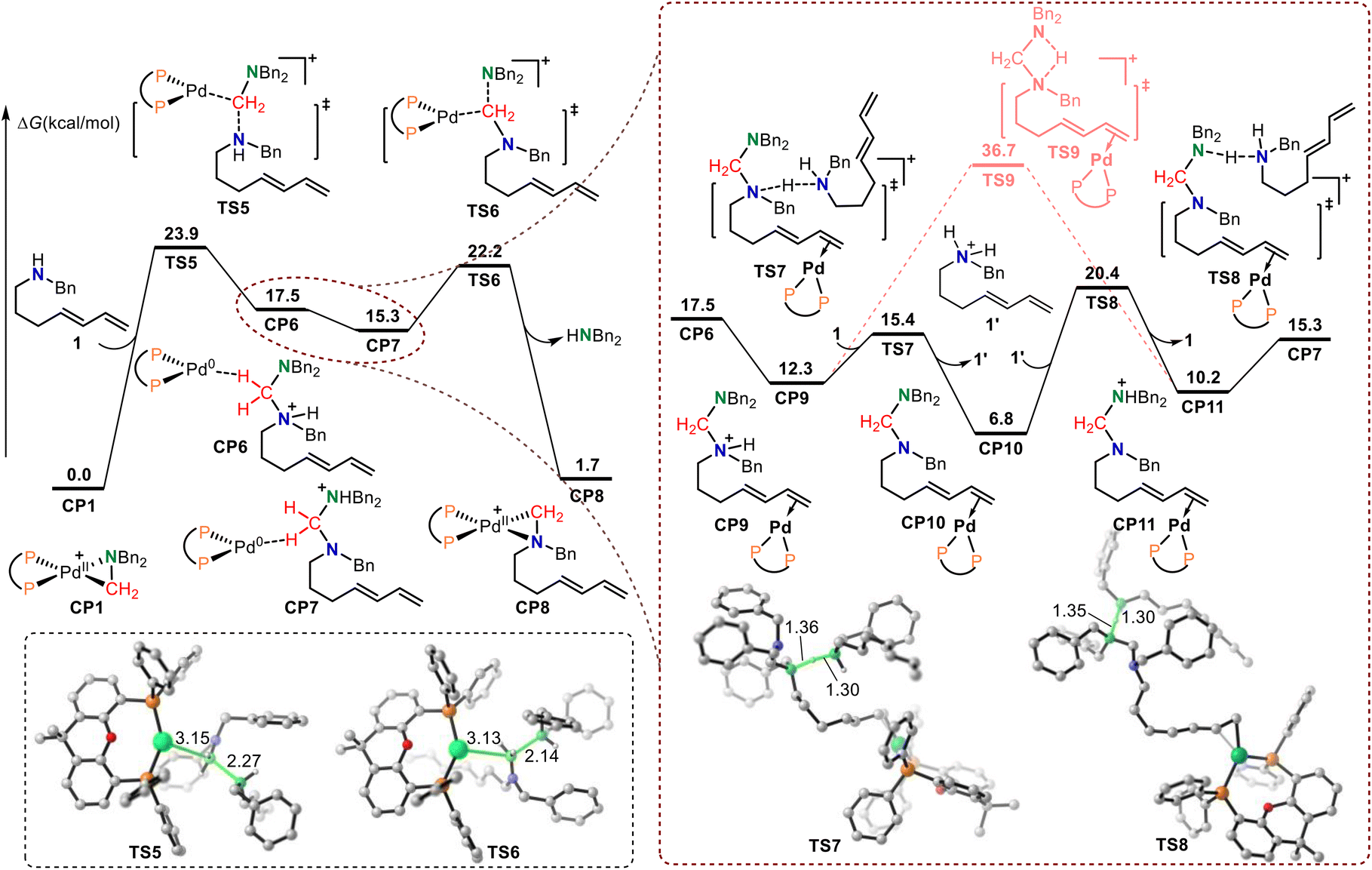 | ||
| Fig. 2 The calculated free energy profiles for C–N bond metathesis. | ||
Owing to the stronger basicity of the secondary amine compared with that of the aminal, the amine substrate 1 would promote a rapid hydrogen transfer process (Fig. 2). The isomerization of amino cation Pd(0) intermediate CP6 gives intermediate CP9, in which the terminal alkene coordinates with the Pd(0) center. Then, intermolecular hydrogen transfer from CP9 to secondary amine substrate 1 occurs via transition state TS7 with an energy barrier of only 3.1 kcal mol−1. In this process, complex CP10 and secondary amine cation 1′ are generated. The subsequent intermolecular hydrogen transfer from 1′ to the terminal amino in CP10 gives the amino cation intermediate CP11via transition state TS8 with a free energy barrier of 13.6 kcal mol−1. CP11 then isomerizes to amino cation Pd(0) intermediate CP7. The direct intramolecular hydrogen transfer of CP8 has also been calculated. The results also show that the free energy of the direct intramolecular hydrogen transfer transition state TS9 is 16.3 kcal mol−1 higher than that of transition state TS8, which indicates that secondary amine substrate 1 promoted rapid hydrogen transfer process is more favorable. The calculated results also show that this secondary amine-promoted rapid hydrogen transfer process is reversible.
Based on the above-calculated results, we proposed a catalytic cycle for the palladium-catalyzed C–N bond metathesis for the ring-closing aminomethylamination reaction, where the Huang-complex A is set as the active catalytic complex (Scheme 4). The reversible reductive elimination, hydrogen transfer, and oxidative addition first induce the C–N bond metathesis. The generated palladacycle-complex intermediate D isomerizes to E, in which the internal alkene coordinates with the Pd(II) center. The subsequent intramolecular alkene migratory insertion generates the π-allylpalladium species F, which is intercepted by an aminal to form intermediate Gvia an SN2-type reductive elimination process. Finally, the SN2-type oxidation addition of the amino cation to the Pd(0) center delivers the saturated N-heterocycle together with regenerating the active palladium-complex A to complete the catalytic cycle.
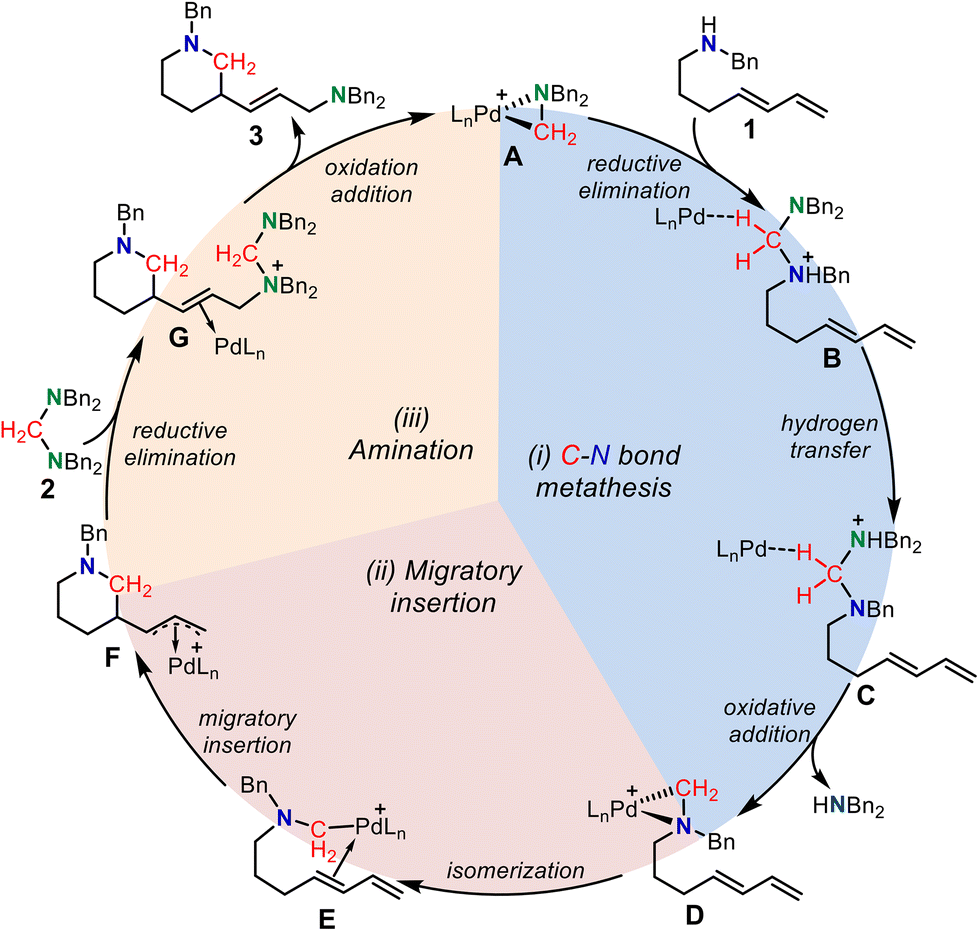 | ||
| Scheme 4 The proposed mechanism for the palladium-catalyzed C–N bond metathesis for ring-closing aminomethylamination reaction. | ||
Fig. 3 shows the free energy profiles for the migratory insertion of an internal alkene. The dissociation of the amino group and coordination of the internal alkene in palladacycle-complex intermediate CP8 forms cationic Pd(II) intermediate CP12. Migratory insertion of the internal alkene into the C(alkyl)–Pd bond was calculated to proceed via transition state TS10 with an energy barrier of 21.9 kcal mol−1. Alternatively, dissociation of the amino group and coordination of the terminal alkene in CP8 generate cationic Pd(II) intermediate CP13. Subsequent 1,4-insertion of the conjugated diene into the C(alkyl)–Pd bond gives the π-allylpalladium species CP14via transition state TS11. In transition state TS11, the lengths of the C–Pd bond being broken and the C–C bond being formed were 3.45 and 3.28 Å, respectively. The calculated free energy for TS11 was 16.7 kcal mol−1 lower than that of TS10, which indicates that the 1,4-insertion pathway is favorable. In addition, 1,2-insertion of the terminal alkene was also calculated. The calculated free energy for the 1,2-insertion of terminal alkene transition state TS12 was 48.4 kcal mol−1, which is 33.2 kcal mol−1 higher than that of TS11. Therefore, the 1,2-insertion of the terminal alkene pathway is also unfavorable.
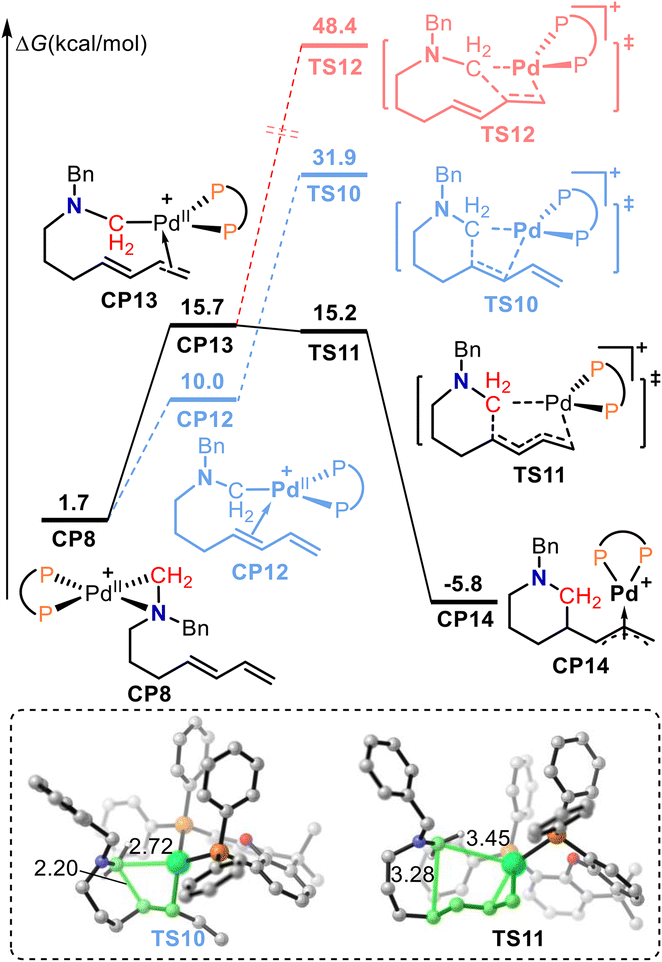 | ||
| Fig. 3 The calculated free energy profiles for migratory insertion of the internal alkene. | ||
The aminals used in this reaction system contain two tertiary amine moieties; each end of the nucleophile can potentially act as a nucleophile. Therefore, although the basicity of the aminal is lower than that of Bn2NH and aminodiene, the nucleophilicity of the aminal should be stronger than that of Bn2NH and aminodiene. It is reasonable that the more nucleophilic aminal should be the preferred agent to trap the π-allylpalladium intermediate to give the desired product. As shown in Fig. 4, π-allylpalladium species CP14 would be intercepted by an aminal via SN2-type reductive amination transition state TS13 with an energy barrier of 26.5 kcal mol−1 to form the alkene-coordinated Pd(0) complex CP15. In TS13, the length of the C–N bond being formed was 1.91 Å. The subsequent oxidative addition of the C–N bond to the Pd(0) center via transition state TS14 delivers saturated N-heterocycle product 3 together with regenerating the active palladium-complex CP1 to complete the catalytic cycle. The lengths of the C–N bond being broken and the C–Pd bond being formed in TS14 were 2.15 and 3.42 Å, respectively. The calculated results show that the reductive amination is the rate-limiting step, which is consistent with the inverse secondary kH/kD = 0.86 ± 0.2 observed in the competition experiments between aminodiene and deuterated aminodiene. The overall activation free energy for this reaction was 26.5 kcal mol−1, corresponding to the free energy difference between transition state TS13 and intermediate CP14. The π-allylpalladium species CP14 is the resting state in the catalytic cycle, which is also consistent with the dynamic experimental results.
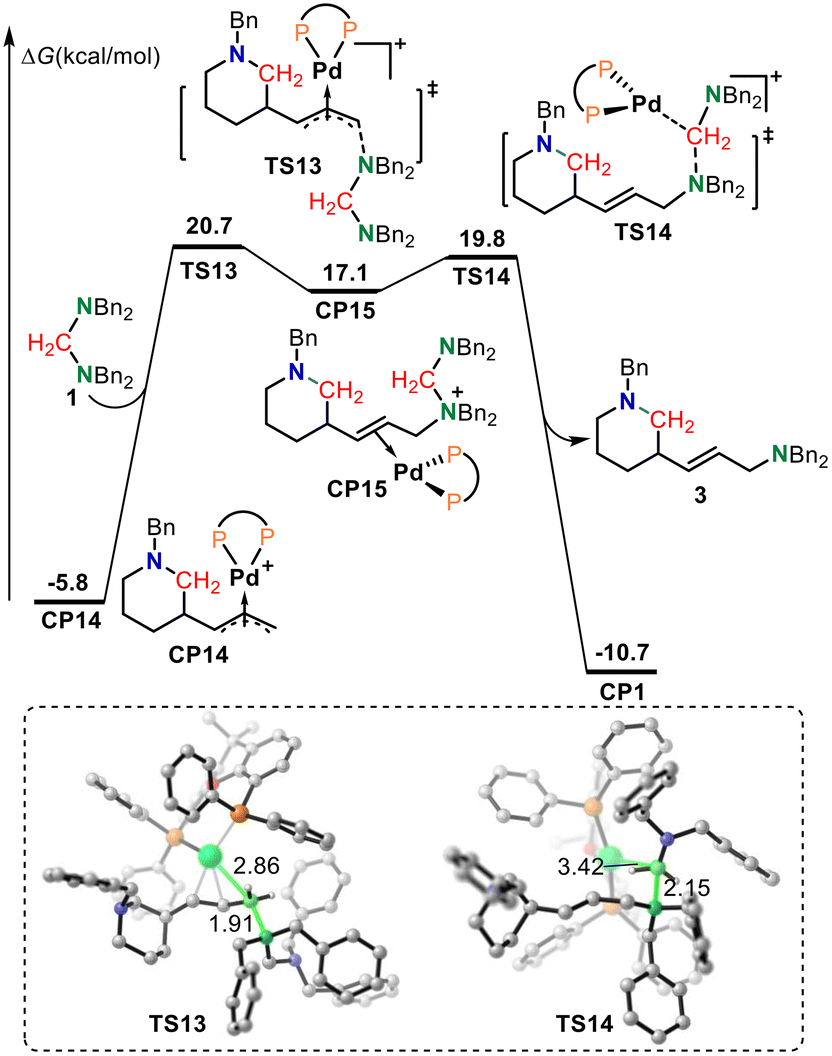 | ||
| Fig. 4 The calculated free energy profiles for amination. Values for bond lengths are given in angstroms. | ||
A primary enantioselective reaction was also established using chiral Pd/ligand complexes. As shown in Scheme 5, when a chiral phosphinamide ligand was used, chiral N-heterocyclic products were generated in 78% yield and 81% ee. DFT calculations were further performed to illustrate the enantiocontrol in this reaction.
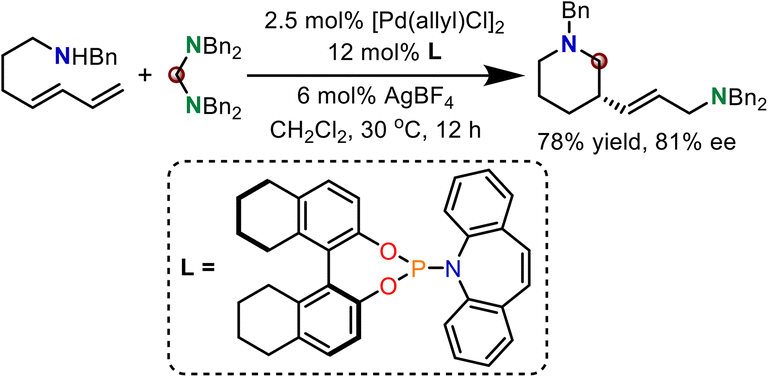 | ||
| Scheme 5 Primary enantioselective reaction. | ||
The above calculations indicate that the stereoselectivity for this Pd-catalyzed C–N bond metathesis and ring-closing reaction is determined by the 1,4-insertion step. The calculated enantiotopic 1,4-insertion patterns of the two transition states (TS11-Si and TS11-Re) were examined. As shown in Fig. 5, the calculated relative energy of TS11-Si was 2.3 kcal mol−1 lower than that of TS11-Re, which is in accordance with the (R) selectivity observed experimentally. The structural information for TS11-Si and TS11-Re indicate that TS11-Re is generated posterior to TS11-Si as the C–C bond being formed in TS11-Si was 2.57 Å, while the C–C bond being formed in TS11-Re was 2.36 Å. The late transition state for TS11-Re results in its high relative energy.
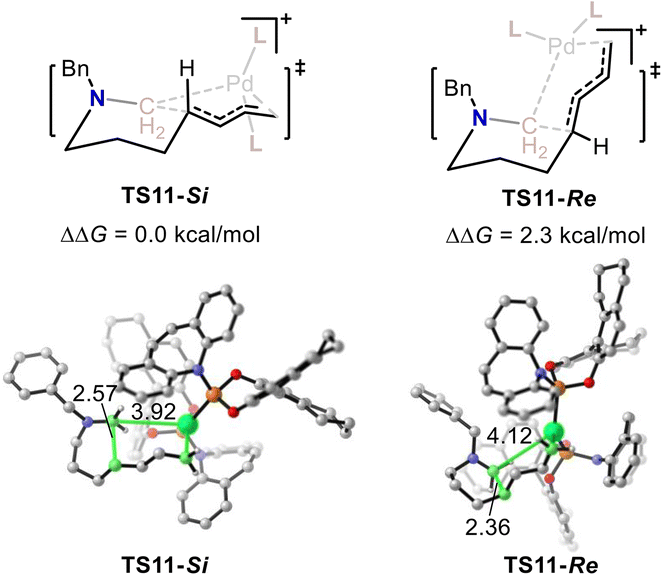 | ||
| Fig. 5 The calculated relative free energy for TS11-Si and TS11-Re. Values for bond lengths are given in angstroms. | ||
To better understand the origin of stereoselectivity in this type of reaction, NCI analysis was performed for the 1,4-insertion patterns of diene transition states TS11-Re and TS11-Si. As shown in Fig. 6, NCI calculations indicate that the steric repulsion of the terminal methylene in the chiral phosphinamide ligand in TS11-Re is stronger than that in TS11-Si, which contributes to the free energy difference between TS11-Re and TS11-Si. The structure information of TS11-Re and TS11-Si also indicated that the butadiene group is located at an axial bond in TS11-Re, and an equatorial bond in TS11-Si. The conformation of TS11-Re limits the position of the butadiene, which leads to the steric repulsion of the terminal methylene in the chiral phosphinamide ligand in TS11-Re stronger than that in TS11-Si.
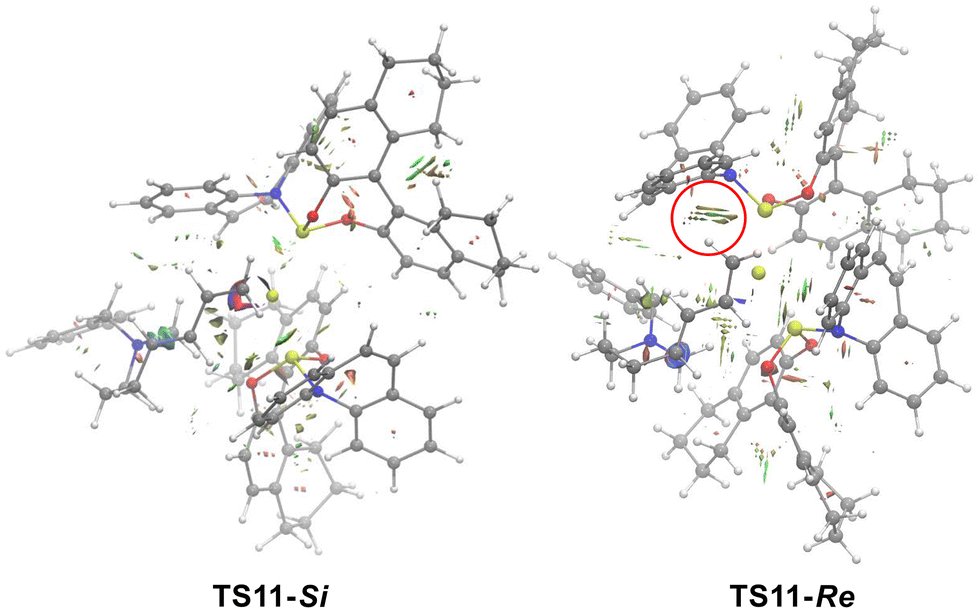 | ||
| Fig. 6 NCI calculations of TS11-Si and TS11-Re (blue is the attraction; green is the weak interaction; red is the steric effect). | ||
Conclusions
In this work, a reversible reductive elimination/oxidative addition-induced C–N metathesis is proposed for a Pd-catalyzed C–N bond metathesis and ring-closing reaction. Different from typical C–N bond metathesis, the active Huang-complex precedes the reversible reductive elimination, amine-assisted hydrogen transfer and oxidative addition to accomplish C–N bond metathesis and form a new C–N bond. Compared with the reductive elimination/oxidative addition-induced C–N metathesis process, the transimination pathway does not readily occur due to the high activation free energy of this step. The reductive amination was shown to be the rate-determining step in this reaction; the calculated overall activation free energy for this step was 26.5 kcal mol−1. Furthermore, the stereoselectivity is controlled by the diene 1,4-migratory insertion step, from which the energy difference is determined by the steric repulsion of the terminal methylene with the chiral phosphinamide ligand. The calculated results also indicate that this C–N bond metathesis requires the participation of palladium, which facilitates the subsequent ring-closing process and can control the regioselectivity and stereoselectivity. The calculated results presented herein contribute to a better understanding of C–N bond metathesis in transition metal-mediated C–N bond formation and provide a theoretical guide for further experimental investigations.Author contributions
S. Liu and R. Bai conceived and designed the project. S. Liu designed the computational studies. D. Zhang, M. Xiao, C. Pu, X. Zhang and X. Yang performed the DFT calculations. S. Liu and T. Zhang prepared the manuscript, and D. Zhang and M. Xiao prepared the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Basic and Frontier Research Project of Chongqing Science and Technology Commission (no. cstc2021jcyj-msxmX0593) and the Scientific Research Fund of Chongqing University of Arts and Sciences (no. R2020SHH01 and P2021HH03) for financial support. This project was also funded by the Project of Scientific and Technological Research Program of Chongqing Municipal Education Commission (no. KJQN202101301).References
- (a) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge Univ. Press, 2004 Search PubMed; (b) A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley, 2008 Search PubMed; (c) V. G. Chandrashekhar, W. Baumann, M. Beller and R. V. Jagadeesh, Nickel-catalyzed hydrogenative coupling of nitriles and amines for general amine synthesis, Science, 2022, 376, 1433–1441 CrossRef CAS; (d) J. A. Garduño and J. J. García, Toward Amines, Imines, and Imidazoles: A Viewpoint on the 3d Transition-Metal-Catalyzed Homogeneous Hydrogenation of Nitriles, ACS Catal., 2020, 10, 8012–8022 CrossRef; (e) T. Irrgang and R. Kempe, Transition-Metal-Catalyzed Reductive Amination Employing Hydrogen, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS; (f) O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Reductive Amination in the Synthesis of Pharmaceuticals, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS; (g) T. Irrgang and R. Kempe, 3d-Metal Catalyzed N- and C-Alkylation Reactions via Borrowing Hydrogen or Hydrogen Autotransfer, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS; (h) P. Ruiz-Castillo and S. L. Buchwald, Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS.
- (a) B. N. Bhawal and B. Morandi, Catalytic Isofunctional Reactions—Expanding the Repertoire of Shuttle and Metathesis Reactions, Angew. Chem., Int. Ed., 2019, 58, 10074–10103 CrossRef CAS; (b) B. Yu, S. Zou and H. Huang, Palladium-Catalyzed Ring-Closing Reaction for the Synthesis of Saturated N-Heterocycles with Aminodienes and N,O-Acetals, J. Org. Chem., 2021, 86, 7849–7863 CrossRef CAS; (c) H. Yu, B. Yu, H. Zhang and H. Huang, Palladium-Catalyzed Aminomethylation and Cyclization of Enynol to O-Heterocycle Confined 1,3-Dienes, Org. Lett., 2021, 23, 3891–3896 CrossRef CAS; (d) S. Zou, B. Yu and H. Huang, Enantioselective ring-closing aminomethylamination of aminodienes enabled by modified Trost ligands, Chem. Catal., 2022, 8, 2034–2048 CrossRef.
- (a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (b) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Dynamic Combinatorial Chemistry, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (c) M. H. Chisholm, E. E. Delbridge, A. R. Kidwell and K. B. Quinlan, Nitrogen atom exchange between molybdenum, tungsten and carbon. A convenient method for N-15 labeling, Chem. Commun., 2003, 126–127 RSC; (d) E. Bon, D. C. H. Bigg and G. Bertrand, Aluminum Chloride-Promoted Transamidation Reactions, J. Org. Chem., 1994, 59, 4035–4036 CrossRef CAS; (e) C. Bui-The-Khai, Concilio and G. Porzi, Cyclization of α,ω Aliphatic Diamines and Conversion of Primary Amines to Symmetrical Tertiary Amines by a Homogeneous Ruthenium Catalyst, J. Org. Chem., 1981, 46, 1759–1760 CrossRef CAS; (f) S.-I. Murahashi, N. Yoshimura, T. Tsumiyama and T. Kojima, Catalytic Alkyl Group Exchange Reaction of Primary and Secondary Amines, J. Am. Chem. Soc., 1983, 105, 5002–5011 CrossRef CAS.
- (a) K. E. Meyer, P. J. Walsh and R. G. Bergman, Zirconium-Mediated Imine Metathesis. Synthesis of 2,4-Diaza-1-zirconiacyclobutanes and the Mechanism of Their Reactions with Imines and Alkynes, J. Am. Chem. Soc., 1994, 116, 2669–2670 CrossRef CAS; (b) R. L. Zuckerman, S. W. Krska and R. G. Bergman, Zirconium-Mediated Metathesis of Imines: A Study of the Scope, Longevity, and Mechanism of a Complicated Catalytic System, J. Am. Chem. Soc., 2000, 122, 751–761 CrossRef CAS; (c) G. K. Cantrell and T. Y. Meyer, Catalytic C=N Bond Formation by Metal-Imide-Mediated Imine Metathesis, J. Am. Chem. Soc., 1998, 120, 8035–8042 CrossRef CAS; (d) M. C. Burland, T. W. Pontz and T. Y. Meyer, Role of Trace Amine in the Metathesis of Imines by CpTa(=NR)Cl2, Organometallics, 2002, 21, 1933–1941 CrossRef CAS; (e) M. C. Burland and T. Y. Meyer, Iminophosphorane Mediated Imine Metathesis, Inorg. Chem., 2003, 42, 3438–3444 CrossRef CAS PubMed; (f) J. M. McInnes and P. Mountford, Transition metal imide/organic imine metathesis reactions: unexpected observations, Chem. Commun., 1998, 1669–1670 RSC; (g) W.-D. Wang and J. H. Espenson, Metathesis Reactions of Tris(adamantylimido)methylrhenium and Aldehydes and Imines, Organometallics, 1999, 18, 5170–5175 CrossRef CAS; (h) J. W. Bruno and X. J. Li, Use of Niobium(III) and Niobium(V) Compounds in Catalytic Imine Metathesis under Mild Conditions, Organometallics, 2000, 19, 4672–4674 CrossRef CAS.
- (a) S. E. Eldred, D. A. Stone, S. H. Gellman and S. S. Stahl, Catalytic Transamidation under Moderate Conditions, J. Am. Chem. Soc., 2003, 125, 3422–3423 CrossRef CAS; (b) C. M. Bell, D. A. Kissounko, S. H. Gellman and S. S. Stahl, Catalytic Metathesis of Simple Secondary Amides, Angew. Chem., Int. Ed., 2007, 46, 761–763 CrossRef CAS; (c) N. A. Stephenson, J. Zhu, S. H. Gellman and S. S. Stahl, Catalytic Transamidation Reactions Compatible with Tertiary Amide Metathesis under Ambient Conditions, J. Am. Chem. Soc., 2009, 131, 10003–10008 CrossRef CAS PubMed.
- (a) Bui-The-Khai, C. Concilio and G. Porzi, A facile synthesis of symmetrical secondary amines from primary amines promoted by the homogeneous catalyst RuCl2(Ph3P)3, J. Organomet. Chem., 1981, 208, 249–251 CrossRef CAS; (b) Bui-The-Khai, C. Concilio and G. Porzi, Cyclization of α,ω Aliphatic Diamines and Conversion of Primary Amines to Symmetrical Tertiary Amines by a Homogeneous Ruthenium Catalyst, J. Org. Chem., 1981, 46, 1759–1760 CrossRef CAS; (c) D. Hollmann, S. Bähn, A. Tillack and M. Beller, N-Dealkylation of aliphatic amines and selective synthesis of monoalkylated aryl amines, Chem. Commun., 2008, 3199–3201 RSC; (d) O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Selective Amine Cross-Coupling Using Iridium-Catalyzed “Borrowing Hydrogen” Methodology, Angew. Chem., Int. Ed., 2009, 48, 7375–7378 CrossRef CAS; (e) Y. Shvo and R. M. Laine, Homogeneous Catalytic Activation of C-N Bonds. Alkyl Exchange between Tertiary amines, J. Chem. Soc., Chem. Commun., 1980, 753–754 RSC; (f) R. B. Wilson Jr. and R. M. Laine, Transalkylation Reaction. Homogeneous Catalytic Formation of C-N Bonds, J. Am. Chem. Soc., 1985, 107, 361–369 CrossRef.
- B. Yu, S. Zou, H. Liu and H. Huang, Palladium-Catalyzed Ring-Closing Reaction via C−N Bond Metathesis for Rapid Construction of Saturated N-Heterocycles, J. Am. Chem. Soc., 2020, 142, 18341–18345 CrossRef CAS PubMed.
- X. Qi, S. Liu and Y. Lan, Computational Studies on an Aminomethylation Precursor: (Xantphos)Pd(CH2NBn2)+, Organometallics, 2016, 35, 1582–1585 CrossRef CAS.
- (a) J. E. Dander and N. K. Garg, Breaking Amides using Nickel Catalysis, ACS Catal., 2017, 7, 1413–1423 CrossRef CAS; (b) J. E. Dander, E. L. Baker and N. K. Garg, Nickel-catalyzed transamidation of aliphatic amide derivatives, Chem. Sci., 2017, 8, 6433–6438 RSC; (c) E. L. Baker, M. M. Yamano, Y. Zhou, S. M. Anthony and N. K. Garg, A two-step approach to achieve secondary amide transamidation enabled by nickel catalysis, Nat. Commun., 2016, 7, 11554 CrossRef.
- M. J. Frisch, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed. The full author list is shown in the ESI.†.
- (a) A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetticorrelation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (c) H. L. Schmider and A. D. Becke, Optimized density functionals from the extended G2 test set, J. Chem. Phys., 1998, 108, 9624–9631 CrossRef CAS.
- (a) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Ab initio study of solvated molecules: a new implementation of the polarizable continuum model, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS; (b) E. Cances, B. Mennucci and J. Tomasi, A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, Geometry optimization of molecular structures in solution by the polarizable continuum model, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS; (d) A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Ab initio study of solvated molecules: a new implementation of the polarizable continuum model, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- C. Y. Legault, CYLView, 1.0b, Université de Sherbrooke, Quebec, Canada, 2009. https://www.cylview.org (accessed 1/10/2017) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qo01638a |
| This journal is © the Partner Organisations 2023 |