DOI:
10.1039/D2QO01664H
(Research Article)
Org. Chem. Front., 2023,
10, 193-202
Precise manipulation of electron transfers to enable the site-selective hydropyridylation of ynones†
Received
20th October 2022
, Accepted 13th November 2022
First published on 16th November 2022
Abstract
Precise manipulation of electron transfers for site-selective hydropyridylation of ynones for the synthesis of β-pyridyl ketones and pyridinated propargyl alcohols tends to be an attractive strategy but remains a challenging task. Herein, the site-tunable hydropyridylation of ynones was successfully delivered by precisely manipulating the electron transfer under electrochemical conditions using square wave voltammetry as a powerful tool. Mechanistic investigations revealed that solvation and suitable protonation reagents are crucial factors in controlling the electron transfer order of ynones to modulate active free radical sites. In addition, the kinetic studies demonstrate that the hydrogenation of ynones on the surface of the cathode should be the rate determining step.
Introduction
The ubiquity of pyridine-containing ketones and alkynols in natural products, pharmaceuticals, agrochemicals, and chiral ligands requires significant efforts to explore efficient methodologies for their preparation.1 Hydropyridylation, the addition of pyridine and hydrogen atoms across alkenes or carbonyl compounds, is a straightforward strategy for the construction of the pyridylated framework from abundant precursors, which can avoid the tedious steps of hydrogenation and functionalization with safe and effective features.2 As a result, new methods for the construction of pyridylated scaffolds from cyanopyridine derivatives in the presence of hydrogen donors mediated by transition metals, boron reagents, or photo/electrocatalysis have been developed to enable this type of process.3 These previous works have focused mainly on the hydropyridylation of alkenes and carbonyl groups, which are still limited to transition metals, because of their elusive conditions, poor selectivity, and electronic uncontrollability (Scheme 1a, left). However, the controllable electrochemical hydropyridylation of unsaturated ynone derivatives remain undeveloped (Scheme 1a, right). This might be because, ynones which contain two reactive centers, alkynyl, and carbonyl groups, easily undergo the multi-electron electrochemical reduction in a proton rich environment and the reaction site cannot be precisely controlled.4 Therefore, it is an imperative and challenging task to develop a simple, efficient, and site-tunable electrochemical platform to precisely regulate the hydropyridylation of ynones.
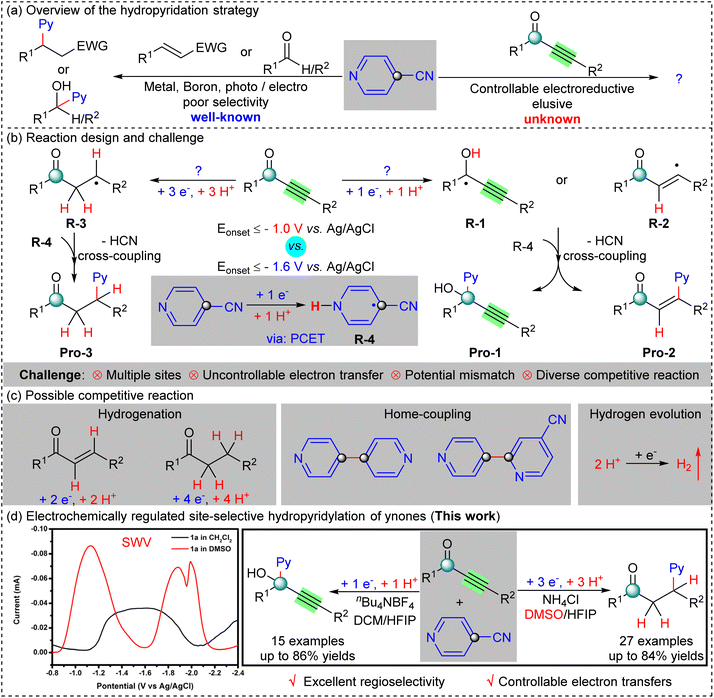 |
| | Scheme 1 (a) Overview of the hydropyridation strategy. (b) Reaction design and challenges. (c) Possible competitive reaction. (d) Research scheme of this work. | |
The reaction design and challenges of the hydropyridylation of ynones have been hypothesized, and we speculate that site-tunable hydropyridylation of ynones can be achieved by precise control of the substrate electron transfer (Scheme 1b and c). The radical intermediates R-1 and R-2 will be formed by the one-electron reduction of the ynones, and then cross-coupled with the radical R-4 to give the corresponding products, Pro-1 and Pro-2. On the other hand, the carbon radical intermediate R-3 will be generated when ynones undergo a three-electron reduction, followed by a radical cross-coupling with R-4 to generate the desired product, Pro-3. However, several challenges are unavoidable to achieve the site-selective hydropyridylation of ynones with cyanopyridines, including multiple sites, uncontrollable electron transfer, potential mismatch of substrates, and diverse competitive reactions.3f,h–j,5 Accordingly, we propose two strategies that should be able to effectively address these challenges. Firstly, suitable protonation reagents and solvation should be the most efficient strategy to precisely control the electron transfer of ynones to form the corresponding radical intermediates R-1, R-2, or R-3. Secondly, suitable use of the mechanism of proton-coupled electron transfer (PCET) in proton rich electrochemical systems can pull the potentials of the substrates closer to resolving the potential mismatch.3i,6
Electrochemical organic synthesis has received extensive attention as a powerful tool using traceless electrons as redox reagents.7 However, few methods can accurately and rapidly identify the electron transfer of organic compounds. Square wave voltammetry (SWV) is a versatile, fast, highly sensitive, efficient, and powerful electroanalytical method, which is widely used in quantitative analysis and kinetic studies of substances, and is rarely used in electro-organic synthesis.8 Herein, SWV was employed to identify the electron transfer of organic compounds under different circumstances to help elucidate the reaction mechanisms. To our delight, the site-tunable hydropyridylation of ynones using cyanopyridines as a precursor was successfully delivered by employing SWV (Scheme 1d, right). In particular, the SWV experiments confirmed that ynones are a three-electron transfer in dimethyl sulfoxide (DMSO), and a single-electron transfer in methylene chloride (DCM, CH2Cl2), which are the key determinants of site-selectivity (Scheme 1d, left). Kinetic studies have demonstrated that the hydrogenation of ynones at the cathode surface should be the rate-determining step. Guided by the established mechanism, the site-tunable hydropyridylation of ynones gave 42 instances with up to 86% yields with excellent regioselectivity, and enables the use of gram scale synthesis.
Results and discussion
Investigating the electron transfer properties of ynones
We speculated that the precise manipulation of the electron transfer of ynones is the key to achieving this site-selective transformation. Therefore, diphenylprop-2-yn-1-one (1a) was chosen as the benchmark to investigate the electron transfer in different solvents using SWV (Fig. S1, ESI†). First, the electron transfer of 1a in different solvents, such as MeCN, DMF, THF, acetone, toluene, and 1,4-dioxane, was investigated in the presence of NH4Cl and hexafluoroisopropanol (HFIP). However, smooth signal-free or puzzling multi-electron processes were observed when scanning 1a in these solvents (Fig. S1a, ESI†). To our delight, three peaks were presented when 1a was scanned in DMSO in the presence of NH4Cl and HFIP, suggesting that three-electron transfers should have occurred (Fig. S1d, red line, ESI†). Next, the electron transfer of 1a in different alcohols was investigated in the presence of NH4Cl (Fig. S1b, ESI†), and it was found that it should be a three-electron reduction in HFIP (Fig. S1d, black line, ESI†) and a four-electron process in methanol (CH3OH, which was consistent with our previous electrochemical hydrogenation of ynones). These data suggest that the presence of NH4Cl in the mixture of DMSO and HFIP is an opportunity for the hydropyridylation of the alkynyl site of 1a to give β-pyridyl ketones. Subsequently, the effect of solvent on the electron transfer of 1a in other systems was investigated, to allow it to capture one electron to enable hydropyridylation of the carbonyl site. After various investigations, we found that there was an opportunity for the desired hydropyridylation of the carbonyl position when the hydropyridylation of 1a was performed in the presence of tetrabutylammonium tetrafluoroborate (nBu4NBF4) as the electrolyte, and chloroalkane and HFIP as the mixed solvent, whereas using other solvents the results were incomprehensible (Fig. S1c, ESI†). As shown in Fig. S1d and f (ESI),† it was concluded that the radical intermediate R-3 will be obtained in DMSO via a three-electron transfer, and that the radical intermediates R-1 and R-2 can be generated in DCM via a single electron transfer in the presence of NH4Cl and/or HFIP. Importantly, the actual experimental data confirmed that the site-tunable hydropyridylation of ynones exhibited optimal regioselectivity and efficiency in DMSO and DCM, respectively (Fig. S1e and Table S1, ESI†).
Optimization conditions
Based on the previous analysis, and inspired by the mechanism of proton-coupled electron transfer, suitable proton sources and solvation effects were found to be the key factors in the precise manipulation of the electron transfer ynones to achieve site-selective hydropyridylation. Therefore, 1,3-diphenylprop-2-yn-1-one (1a) and 4-cyanopyridine (1b) were preferentially selected as the template substrates to investigate various key parameters such as protonation reagents, solvents, electrodes, and currents. After various optimizations, we found that the desired product 1c could be gained, with an 83% yield, in DMSO with NH4Cl and HFIP as protonation reagents and hydrogen donors, respectively, by employing carbon rods as the electrodes in an undivided cell under a constant current of 10 mA for 3 h (Table 1, entry 1). To our delight, the hydropyridylation of 1a could be delivered to 1d with a yield of 81% when the reaction was fitted with nickel plates as the cathode, carbon rods as the anode, DCM as the solvent, nBu4NBF4 as the electrolyte, and using HFIP as the protonation reagent and hydrogen donor with a galvanostatic reaction of 5 mA for 6 h at room temperature (Table 1, entry 14). Controlled experiments demonstrated that both the electricity, protonation reagents, and hydrogen donors were necessary for the site-selective hydropyridylation of 1a (Table 1, entries 2–4 and 16–25). Next, only 1c was able to give a 63% yield, and nothing was detected with 1d when using CH3OH instead of HFIP under the respective optimal conditions (Table 1, entries 5 and 17). Interestingly, the desired products 1c and 1d were not formed when DMSO and DCM were exchanged, suggesting that the solvent is a critical factor for the control of the site-selective hydropyridation of ynones (Table 1, entries 6 and 18). The yields of the corresponding products 1c and 1d were reduced when DMSO and DCM were replaced by other solvents (Table 1, entries 7 and 19, for detailed solvent data see Table S1, ESI†). The yields of both 1c and 1d were greatly reduced when the reactions were performed in other ammonium or quaternary ammonium salts, respectively, (Table 1, entries 8–10, 20 and 21). The presence of an appropriate amount of Na2CO3 could enhance the yield of 1d, whereas other types of bases such as NaHCO3 and triethylenediamine (DABCO) are ineffective (Table 1, entries 22 and 23). In addition, neither increasing nor decreasing the current contributed to the improvement of the corresponding product yield (Table 1, entries 11 and 24). Finally, the investigation of electrode materials shows that the combination of C(+)|C(−) and C(+)|Ni(−) is the best choice for the site-selective hydropyridylation of 1a to deliver 1c and 1d (Table 1, entries 12, 13, 25 and 26).
Table 1 Optimization of the reaction conditionsa
|

|
| Entry |
Standard condition Aa |
Yield of 1c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c [%] c [%] |
Entry |
Standard condition Bb |
Yield of 1dc [%] |
|
Reaction condition A: carbon rods as the anode, carbon rods as the cathode, constant current: 10 mA, 1a (0.25 mmol), 1b (0.5 mmol), NH4Cl (1.0 mmol), DMSO/HFIP (5.0 mL, v = 4:1), 55 °C, N2, 3 h.
Reaction condition B: carbon rods as the anode, Ni plates as the cathode, constant current: 5 mA, 1a (0.25 mmol), 1b (0.5 mmol), nBu4NBF4 (0.25 mmol), Na2CO3 (0.5 mmol), DCM/HFIP (10.0 mL, v = 9:1), N2, 6 h.
Isolated yields.
0.1 M nBu4NBF4.
|
| 1 |
None |
83 |
14 |
None |
81 |
| 2 |
Without current |
0 |
15 |
Without current |
0 |
| 3 |
Without HFIP |
37 |
16 |
Without HFIP |
Trace |
| 4 |
Without NH4Cl |
Traced |
17 |
CH3OH instead of HFIP |
n. d. |
| 5 |
CH3OH instead of HFIP |
63 |
18 |
DMSO instead of DCM |
Trace |
| 6 |
DCM instead of DMSO |
n. d. |
19 |
DCE, CHCl3 instead of DCM |
41, 68 |
| 7 |
DMF, CH3CN instead of DMSO |
21, 37 |
20 |
n
Bu4NOAc instead of nBu4NBF4 |
62 |
| 8 |
NH4I instead of NH4Cl |
8 |
21 |
n
Bu4NClO4, nBu4NNO3, nBu4NPF6 |
<50 |
| 9 |
NH4Br instead of NH4Cl |
39 |
22 |
Without Na2CO3 |
61 |
| 10 |
NH4OAc instead of NH4Cl |
67 |
23 |
NaHCO3, DABCO instead of Na2CO3 |
59, 54 |
| 11 |
5 mA, 12 mA instead of 10 mA |
34, 81 |
24 |
10 mA instead of 5 mA |
54 |
| 12 |
Pt(+) instead of C(+) |
56 |
25 |
Pt(+) instead of C(+) |
16 |
| 13 |
Pt(−), Ni(−) instead of C(−) |
40, 13 |
26 |
Pt(−), C(−) instead of Ni(−) |
53, 36 |
Mechanistic studies
Intrigued by the unusual reactivity and regioselectivity of this simple electrochemical hydropyridylation of ynones, we then focused on gaining insight into the mechanism of the reaction. It was speculated that suitable protonation reagents and solvation were the main factors governing the regioselectivity of the present transformation. Therefore, we decided to conduct a series of SWV and cyclic voltammetry (CV) experiments to thoroughly explore the effects of the protonation reagents and solvents on the reduction potential of the substrate (Fig. 1). Firstly, detailed SWV experiments were performed to further explore the reasons for the site-selectivity of this transformation from the perspective of proton and electron transfers. As shown in Fig. 1a, it can be seen that substrate 1a has obvious peak splitting with the decrease of frequency under condition A, which was consistent with the mechanism of proton-coupled electron transfer. In addition, the results of the SWV experiments of 1a and 1b in the presence of HFIP in DMSO suggested that the hydropyridylation of 1a with 1b to give 1c, undergoes four-electron transfer (Fig. 1b). To our delight, the hydropyridylation of 1a undergoes a three-electron transfer in DMSO and a single-electron transfer in DCM in the presence of NH4Cl and/or HFIP when performing SWV scans at low frequencies (Fig. 1c). Next, the reduction potentials of 1a and 1b were investigated to compare the effects of different conditions by employing CV to provide credible evidence for the selectivity of this transformation (Fig. 1d–i). It was found that the reduction potentials of 1a (from −1.16 to −0.9 V vs. Ag/AgCl) and 1b (from −1.6 to −1.02 V vs. Ag/AgCl) were positively shifted in the presence of NH4Cl and HFIP as protonation reagents, with DMSO as the solvent (Fig. 1d and e). Comparing the reduction potentials of 1a (−0.9 V vs. Ag/AgCl) and 1b (−1.02 V vs. Ag/AgCl) showed that the electrochemical hydrogenation of 1a should be preferentially initiated (Fig. 1f). Simultaneously, the reduction potentials of 1a (from −1.16 to −0.91 V vs. Ag/AgCl) and 1b (from −1.6 to −0.92 V vs. Ag/AgCl) were similarly shifted by employing DCM as the solvent and HFIP as the protonation reagent (Fig. 1g and h). It was found that the reduction potentials of 1a and 1b were pulled closer by employing DCM as solvent and HFIP as protonation reagent, which suggesting that their single-electron reduction procedures should proceed simultaneously (Fig. 1i).
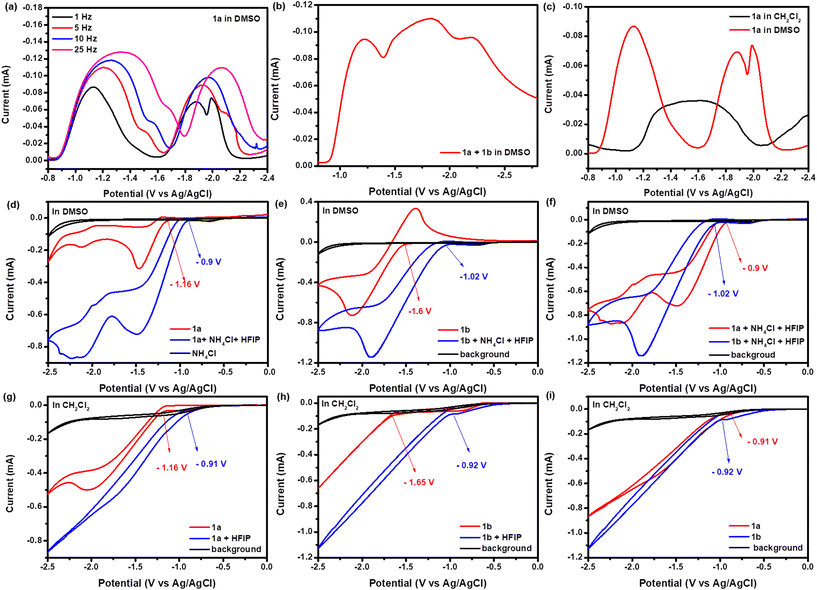 |
| | Fig. 1 Square wave voltammetry (SWV) was performed on solutions containing 0.1 M nBu4NBF4 in DMSO or DCM at room temperature in the presence of HFIP (1.0 mL) and/or NH4Cl (2.0 mM), the working electrode was glass carbon, the counter electrode was Pt (1.5 × 1.5 cm2), the reference electrode was Ag/AgCl, 1a (0.25 mM), 1b (0.5 mM), at a pulse height of 25 mV, and a step height of 4 mV. (a) Varying frequency, DMSO/HFIP (v = 10/1), NH4Cl (2.0 mM). (b) DMSO/HFIP (v = 10/1), NH4Cl (2.0 mM), 1 Hz. (c) DMSO/HFIP (v = 10/1), DCM/HFIP (v = 10/1), 1 Hz. Cyclic voltammetry with a working electrode of glass carbon, a counter electrode of Pt (1.5 × 1.5 cm2), a reference electrode of Ag/AgCl, scan rate of 100 mV s−1, nBu4NBF4 (0.1 M), 1a (0.25 mM), 1b (0.25 mM), HFIP (1.0 mL). (d)–(f), NH4Cl (2.0 mM), DMSO (10.0 mL). (g)–(i) DCM (10.0 mL). | |
Next, various control experiments were performed to determine more mechanistic details of this transformation (Scheme 2). Firstly, the desired products 1c and 1d were not observed in the presence of 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) or 2,6-di-tert-butyl-4-hydroxytoluene (BHT), suggesting that the site-selective hydropyridylation of ynones may undergo a radical process (Scheme 2a). In particular, this conclusion was further verified by the trapped products found: 1e (Fig. S2, ESI†) and 2e (Fig. S3, ESI†). Secondly, several experiments using deuterium demonstrated that both NH4Cl and alcohols act as hydrogen donors for the hydropyridylation of the alkynyl position of the ynones (Scheme 2b, and Fig. S4–S6, ESI†). We believe that HFIP should be a suitable hydrogen donor for the hydropyridylation of the carbonyl position of the ynones. Finally, intermediate validation experiments demonstrated that the preferential hydrogenation of ynones to enones is a necessary pathway for the site-selective delivery of β-pyridyl ketones (Scheme 2c). Moreover, various potentiostatic experiments were carried out under condition A to control one electron transfer, but none of them were able to monitor the formation of 1d, indicating the importance of solvent and protonation reagents for the site-selectivity control (Table S2, ESI†).
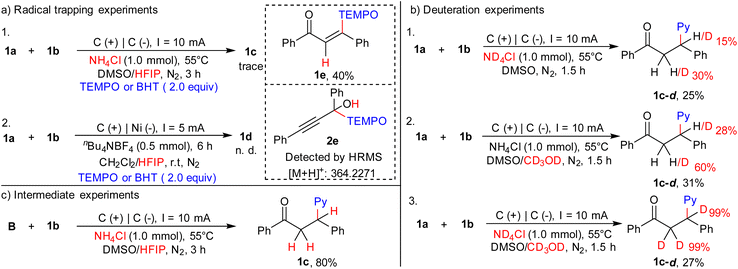 |
| | Scheme 2 Control experiments. | |
Subsequently, we focused on the kinetic exploration of site-selective cathodic reductive hydropyridylation and radical cross-coupling of 1a and 1b. Initially, the current was controlled at 5 mA, and the reaction was stopped at various time intervals, and then the yield of product 1c was monitored by GC with diphenylmethane as the internal standard. Similar experiments were performed under condition A when the operating current was changed to 10, 15, and 20 mA, and the initial rates were obtained by linearly fitting the different currents (Fig. S7, ESI†). As shown in Fig. 2a, a first-order kinetic behavior was presented between the initial rate and current. Meanwhile, the initial reaction rate was determined by employing a similar method when varying the concentrations of 1a, 1b, NH4Cl, and HFIP. The initial rate was negatively correlated with [1a] at relatively low concentrations (0.1, 0.2, and 0.3 M) and independent of [1a] at higher concentrations (0.4, 0.5, and 0.6 M) (Fig. 2b and Fig. S8, ESI†), that was most likely due to the consumption of reactive radical intermediates by side reactions (Fig. S9, ESI†). However, the initial rate vs.1b, NH4Cl, and HFIP showed zero-order kinetics (Fig. 2c–e and Fig. S10–S12, ESI†), that suggested cathodic reductive protonation of 1b and cross-coupling of radical intermediate C were excluded from the rate-determining step. Furthermore, a first-order kinetic behavior between the initial rate and current was also exhibited when the hydropyridylation of the 1a carbonyl site was performed under condition B (Fig. 2f and Fig. S13, ESI†). Thus, the previous kinetic experimental results demonstrated that the hydrogenation of 1a on the surface of the cathode should be the rate-determining step.
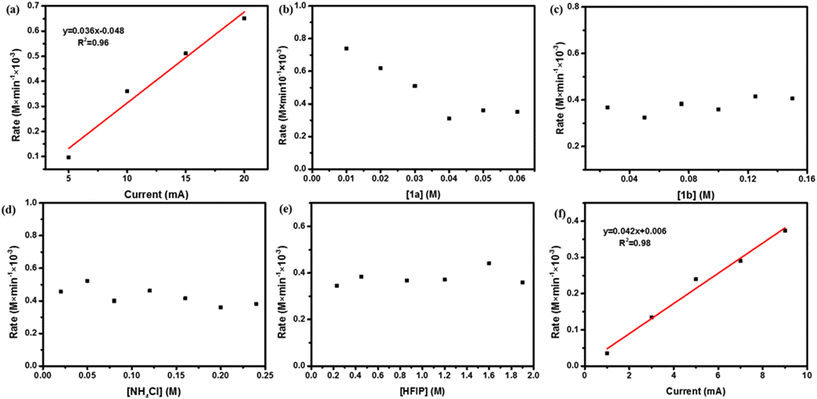 |
| | Fig. 2 Kinetics experiments. (a) Kinetic profiles obtained under different currents using condition A. (b)–(e) Kinetic profiles obtained under different concentrations of 1a, 1b, NH4Cl, and HFIP using condition A, respectively. (f) Kinetic profiles obtained under different currents using condition B. For more data on these experiments, please check Fig. S1–S6 (ESI†). | |
Based on the previous experimental observations and previous reports,3h,i,4f,5a,d,6 a plausible mechanistic pathway for site-selective hydropyridylation of ynones was proposed and is shown in Scheme 3 (with 1a and 1b as an example). We believe that solvent and protonation reagents are crucial factors for the control of the site-selective hydropyridylation of ynones. The following steps may be involved in the hydropyridylation of the alkynyl site of 1a with NH4Cl and HFIP as protonation reagents in DMSO. (1) 1a should preferentially deliver the radical intermediate Cvia a sequential hydrogenation reduction on the surface of the cathode, (2) intermediate D or E were derived from the protonation of 1b and reduced on the surface of the cathode to generate free radical intermediate F, (3) radical intermediates F and C deliver the desired product 1cvia radical cross-coupling and dehydrocyanation. In addition, the mechanism for the hydropyridylation of the carbonyl site of 1a to access the 1-pyridyl propargyl alcohols using HFIP as a protonation reagent in DCM involves the following procedure. (a) The protonated intermediates E and 1a undergo simultaneous single-electron reduction to generate radical intermediates F and G, (b) radical cross-coupling of intermediates F and G followed by dehydrocyanation give the desired product 1d. The anodic half-reactions in this site-selective transformation may involve the oxidation of chloride ions, DMSO, and HFIP.
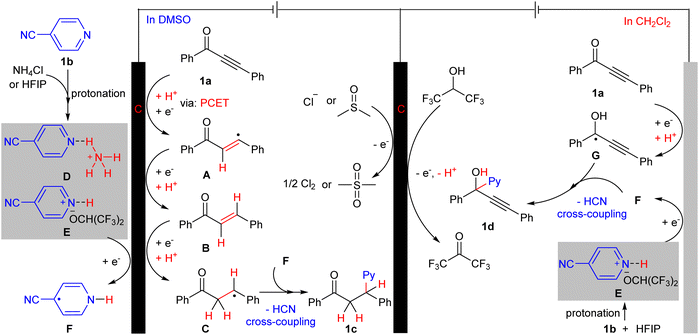 |
| | Scheme 3 Proposed mechanism. | |
Substrate scope
Guided by mechanistic studies, the substrate scope and limitations of the established protocol were examined by employing the optimal conditions shown in Table 1. Initially, 4-cyanopyridine (1b) was chosen as the benchmark to investigate the scope and limitations of the substrate. As shown in Scheme 4, various functional groups such as methyl, ethyl, tertiary butyl, methoxyl, fluoro, chloro, and bromo substituents at the p-positions of 1-aryl rings can be compatible in the present protocol (1c–8c), giving good to excellent yields (60%–83%). The corresponding products can be obtained with good yield and selectivity when fluoro or chloro group substituents were at the b m- and o-positions of the 1-aryl rings (9c–11c). Other types of ynones can also be applied as substrates in this site-selective hydropyridylation transformation. All of 12a to 14a were capable of delivering the desired product under the given conditions with high efficiency and selectivity (12c–14c). Next, the functional group tolerance at the 3-position of the aryl group was also selectively investigated. We found that p-substituted ynones on the 3-positions of aryl rings could give the corresponding products in moderate yields, whereas the yields for the o-substituents decreased slightly (15c–20c). Subsequently, the substrate applicability and limitations of the cyanopyridine derivatives were also investigated. Both pyridine-2,4-dicarbonitrile (2b) and phthalazine-1-carbonitrile (3b) gave the desired products 21c and 22c with a yield of 67% and 47%, respectively. Notably, the corresponding products (23c–26c) were delivered in moderate yields when the 3-position of 4-cyanopyridine contained Br, Cl, F, and Me substituents. To our delight, the desired product 27c could be produced with a 47% yield when 3,5-difluoroisonicotinonitrile was used with the established protocol.
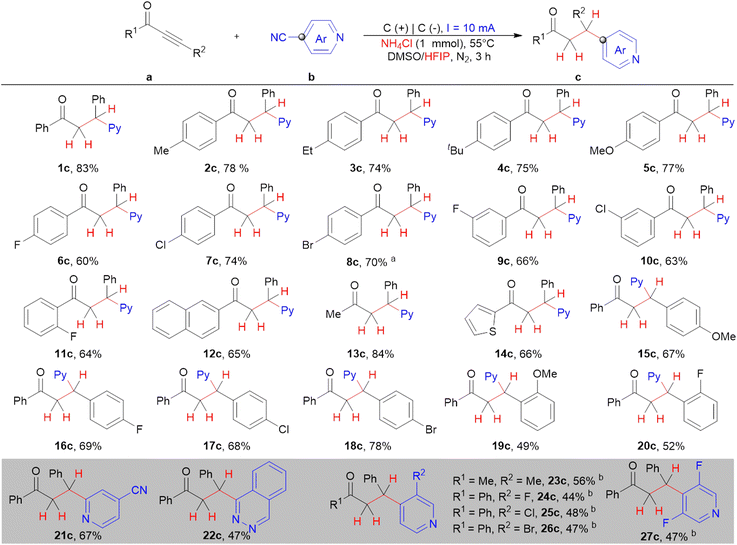 |
| | Scheme 4 The hydropyridylation of the alkynyl sites of ynones, performed with condition A (a) 15 mA. (b) DMSO/HFIP (v = 6:0.5 mL). All cited yields are isolated yields. | |
Guided by the putative reaction mechanism, the substrate scope and limitations of hydropyridylation of carbonyl sites of ynones were investigated by employing the reaction conditions shown in Table S2, entry 14 (ESI†), see Scheme 5. To our delight, the established protocol can be applied to diverse ynones with electron-donating and electron-withdrawing groups (EWG) at varying positions, and the corresponding products can be obtained with excellent regioselectivity and yield (1d–10d). In particular, the desired product 7d can be obtained in yields up to 86% when 3-phenyl-1-(thiophen-2-yl)prop-2-yn-1-one was used under the present conditions. Importantly, the substrate scope of the established controllable electrochemical strategy can be extended to the hydropyridylation of aromatic ketones, and a variety of tetrasubstituted pyridyl carbinols can be obtained with yields of 47%–76% (11d–15d).
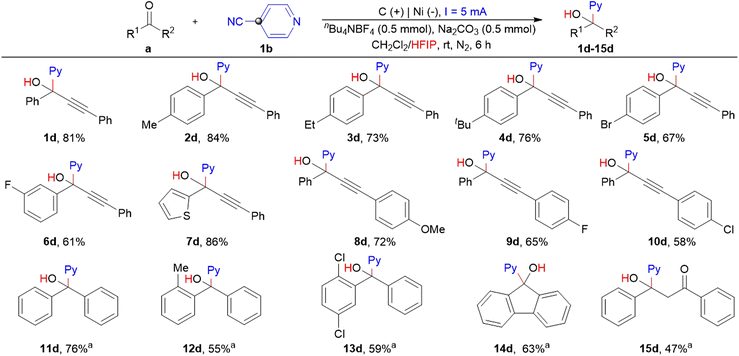 |
| | Scheme 5 The hydropyridylation of the carbonyl sites of ynones and aromatic ketones, performed with condition B. (a) 12 mA, nBu4OAc (0.5 mmol), without Na2CO3, 4 h. All cited yields are isolated yields. | |
Gram scale synthesis
The synthetic applicability of the current protocol was investigated using a gram scale reaction between 1a and 2a, that provided the desired products 1c and 1d under the established conditions. As shown in Scheme 6, the reaction gave 1c and 1d with 68% and 65% yields, respectively. The results demonstrate that the present protocol can serve as a straightforward and practical strategy to access the desired products via site-selective hydropyridylation of ynones.
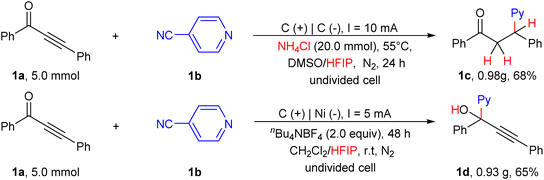 |
| | Scheme 6 Gram scale synthesis. | |
Conclusions
In summary, the site-tunable hydropyridylation of ynones using cyanopyridines as precursors has been successfully revealed by precisely manipulating the electron transfer under electrochemical conditions. Detailed CV and SWV experimental results have confirmed that solvation and suitable protonation reagents are the most critical factors for regulating the site-selective hydropyridylation of ynones, and the mechanism of PCET is the most effective method to resolve the potential mismatch. In addition, the results of the kinetic experiments demonstrate that the hydropyridylation of ynone is a first-order kinetic relationship with current. This simple method features controllable electron transfers, excellent regioselectivity, simple gram scale synthesis, and is environmentally friendly. It was confirmed that NH4Cl and HFIP can play multiple roles such as protonation reagent, hydrogen donor, electrolyte, and solvent in this site-selective transformation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21902083), the Natural Science Foundation of Shandong Province (No. ZR2020QB130 and ZR2021QB159). This work was also supported by the Talent Program Foundation of Qufu Normal University (No. 6132 and 6125), and the Talent Program Foundation of Dezhou University (No. 2021xjrc102).
References
-
(a) F. A. Cotton and R. A. Schunn, Metal Salts and Complexes of Dialkoxyphosphonylacetylmethanide Ions, J. Am. Chem. Soc., 1963, 85, 2394 CrossRef CAS;
(b) D. J. McCabe, E. N. Duesler and R. T. Paine, Monodentate Coordination by a Tripodal Ligand System: Synthesis and Crystal and Molecular Structure of Bis[diisopropyl [1,2-bis(diethylcarbamoyl)ethyl]phosphonate]erbium(III) Nitrate Monohydrate, Inorg. Chem., 1985, 24, 4626 CrossRef CAS;
(c) M. Kitamura, M. Tokunaga and R. Noyori, Asymmetric Hydrogenation of β-Keto Phosphonates: A Practical Way to Fosfomycin, J. Am. Chem. Soc., 1995, 117, 2931 CrossRef CAS;
(d) S. K. Perumal, S. A. Adediran and R. F. Pratt, β-Ketophosphonates as β-lactamase I#nhibitors: Intramolecular Cooperativity Between the Hydrophobic Subsites of a class D β-lactamase, Bioorg. Med. Chem., 2008, 16, 6987 CrossRef CAS;
(e) N. Auberger, C. Gravier-Pelletier and Y. Le Merrer, Synthesis of a β-Ketophosphonate Bioisostere of UDP-N-acetylglucosamine, Eur. J. Org. Chem., 2009, 3323 CrossRef CAS;
(f) W. Sittiwong, E. L. Cordonier, J. Zempleni and P. H. Dussault, β-Keto and β-hydroxyphosphonate Analogs of Biotin-5′-AMP are Inhibitors of Holocarboxylase Synthetase, Bioorg. Med. Chem. Lett., 2014, 24, 5568 CrossRef CAS.
-
(a) Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, Selective C-4 Alkylation of Pyridine by Nickel/Lewis Acid Catalysis, J. Am. Chem. Soc., 2010, 132, 13666 CrossRef CAS;
(b) Y. Yamamoto, Synthesis of Heterocycles via Transition-metal-catalyzed Hydroarylation of Alkynes, Chem. Soc. Rev., 2014, 43, 1575 RSC;
(c) R. Manikandan and M. Jeganmohan, Recent Advances in the Ruthenium-catalyzed Hydroarylation of Alkynes with Aromatics: Synthesis of Trisubstituted Alkenes, Org. Biomol. Chem., 2015, 13, 10420 RSC;
(d) S. W. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins, Chem. Rev., 2016, 116, 8912 CrossRef CAS;
(e) X. Ma and S. B. Herzon, Intermolecular Hydropyridylation of Unactivated Alkenes, J. Am. Chem. Soc., 2016, 138, 8718 CrossRef CAS;
(f) R. A. Aycock, H. Wang and N. T. Jui, A Mild Catalytic System for Radical Conjugate Addition of Nitrogen Heterocycles, Chem. Sci., 2017, 8, 3121 RSC;
(g) A. J. Boyington, M. Y. Riu and N. T. Jui, Anti-Markovnikov Hydroarylation of Unactivated Olefins via Pyridyl Radical Intermediates, J. Am. Chem. Soc., 2017, 139, 6582 CrossRef CAS;
(h) X. Ma, H. Dang, J. A. Rose, P. Rablen and S. B. Herzon, Hydroheteroarylation of Unactivated Alkenes Using N-Methoxyheteroarenium Salts, J. Am. Chem. Soc., 2017, 139, 5998 CrossRef CAS.
-
(a) S. Bordi and J. T. Starr, Hydropyridylation of Olefins by Intramolecular Minisci Reaction, Org. Lett., 2017, 19, 2290 CrossRef CAS;
(b) G. Wang, J. Cao, L. Gao, W. Chen, W. Huang, X. Cheng and S. Li, Metal-Free Synthesis of C-4 Substituted Pyridine Derivatives Using Pyridine-boryl Radicals via a Radical Addition/Coupling Mechanism: A Combined Computational and Experimental Study, J. Am. Chem. Soc., 2017, 139, 3904 CrossRef CAS;
(c) D. Chen, L. Xu, T. Long, S. Zhu, J. Yang and L. Chu, Metal-free, Intermolecular Carbopyridylation of Alkenes via Visible-light-induced Reductive Radical Coupling, Chem. Sci., 2018, 9, 9012 RSC;
(d) R. C. Betori and K. A. Scheidt, Reductive Arylation of Arylidene Malonates Using Photoredox Catalysis, ACS Catal., 2019, 9, 10350 CrossRef CAS;
(e) S. Zhang, L. Li, X. Li, J. Zhang, K. Xu, G. Li and M. Findlater, Electroreductive 4-Pyridylation of Electron-deficient Alkenes with Assistance of Ni(acac)2, Org. Lett., 2020, 22, 3570 CrossRef CAS PubMed;
(f) H. Xu, J. Liu, F. Nie, X. Zhao and Z. Jiang, Metal-free Hydropyridylation of Thioester-activated Alkenes via Electroreductive Radical Coupling, J. Org. Chem., 2021, 86, 16204 CrossRef CAS PubMed;
(g) S. Zhang, L. Li, J. Li, J. Shi, K. Xu, W. Gao, L. Zong, G. Li and M. Findlater, Electrochemical Arylation of Aldehydes, Ketones, and Alcohols: from Cathodic Reduction to Convergent Paired Electrolysis, Angew. Chem., Int. Ed., 2021, 60, 7275 CrossRef CAS;
(h) X. Zhang, C. Yang, H. Gao, L. Wang, L. Guo and W. Xia, Reductive Arylation of Aliphatic and Aromatic Aldehydes with Cyanoarenes by Electrolysis for the Synthesis of Alcohols, Org. Lett., 2021, 23, 3472 CrossRef CAS PubMed;
(i) J. Yang, J. Ma, K. Yan, L. Tian, B. Li and J. Wen, Electrochemical Ammonium Cation-Assisted Hydropyridylation of Ketone-Activated Alkenes: Experimental and Computational Mechanistic Studies, Adv. Synth. Catal., 2022, 364, 845 CrossRef CAS;
(j) H.-J. Zhou and J.-M. Huang, Hydropyridylation of α,β-Unsaturated Esters through Electroreduction of 4-Cyanopyridine, J. Org. Chem., 2022, 87, 5328 CrossRef CAS;
(k) Y. Li, C. Han, Y. Wang, X. Huang, X. Zhao, B. Qiao and Z. Jiang, Catalytic Asymmetric Reductive Azaarylation of Olefins via Enantioselective Radical Coupling, J. Am. Chem. Soc., 2022, 144, 7805 CrossRef CAS;
(l) W. Ding, M. Li, J. Fan and X. Cheng, Palladium-catalyzed Asymmetric Allylic 4-pyridinylation via Electroreductive Substitution Reaction, Nat. Commun., 2022, 13, 5642 CrossRef CAS PubMed.
-
(a) J. Wen, W. Shi, F. Zhang, D. Liu, S. Tang, H. Wang, X.-M. Lin and A. Lei, Electrooxidative Tandem Cyclization of Activated Alkynes with Sulfinic Acids To Access Sulfonated Indenones, Org. Lett., 2017, 19, 3131 CrossRef CAS;
(b) W. Zhang, G. M. Johnson, Z. Guan and Y.-H. He, Regio- and Stereoselective Hydrosulfonylation of Electron-Deficient Alkynes: Access to Both E- and Z-β-Sulfonyl-α,β-Unsaturated Carbonyl Compounds, Adv. Synth. Catal., 2018, 360, 4562 CrossRef CAS;
(c) C. Chen, Q. Xiong and J. Wei, Synthesis of 2-sulfenylindenones by Visible-light-Mediated Addition of Sulfur-centered Radicals to 1,3-diarylpropynones, Synth. Commun., 2019, 49, 869 CrossRef CAS;
(d) X.-X. Wang, X. Lu, Y. Li, J.-W. Wang and Y. Fu, Recent Advances in Nickel-Catalyzed Reductive Hydroalkylation and Hydroarylation of Electronically Unbiased Alkenes, Sci. China: Chem., 2020, 63, 1586 CrossRef CAS;
(e) L. Zeng, H. Li, J. Hu, D. Zhang, J. Hu, P. Peng, S. Wang, R. Shi, J. Peng, C.-W. Pao, J.-L. Chen, J.-F. Lee, H. Zhang, Y.-H. Chen and A. Lei, Electrochemical Oxidative Aminocarbonylation of Terminal Alkynes, Nat. Catal., 2020, 3, 438 CrossRef CAS;
(f) H. Qin, J. Yang, K. Yan, Y. Xue, M. Zhang, X. Sun, J. Wen and H. Wang, Electrochemical-Induced Hydrogenation of Electron-Deficient Internal Olefins and Alkynes with CH3OH as Hydrogen Donor, Adv. Synth. Catal., 2021, 363, 2104 CrossRef CAS;
(g) Y. Wu, L. Zeng, H. Li, Y. Cao, J. Hu, M. Xu, R. Shi, H. Yi and A. Lei, Electrochemical Palladium-Catalyzed Oxidative Sonogashira Carbonylation of Arylhydrazines and Alkynes to Ynones, J. Am. Chem. Soc., 2021, 143, 12460 CrossRef CAS.
-
(a) D. Lehnherr, Y.-H. Lam, M. C. Nicastri, J. Liu, J. A. Newman, E. L. Regalado, D. A. DiRocco and T. Rovis, Electrochemical Synthesis of Hindered Primary and Secondary Amines via Proton-Coupled Electron Transfer, J. Am. Chem. Soc., 2020, 142, 468 CrossRef CAS PubMed;
(b) S. Zhang, L. Li, X. Li, J. Zhang, K. Xu, G. Li and M. Findlater, Electroreductive 4-Pyridylation of Electron-deficient Alkenes with Assistance of Ni (acac)2, Org. Lett., 2020, 22, 3570 CrossRef CAS;
(c) S. Tong, K. Li, X. Ouyang, R. Song and J. Li, Recent Advances in the Radical-mediated Decyanative Alkylation of Cyano (hetero) arene, Green Synth. Catal., 2021, 2, 145 CrossRef;
(d) J. Wen, X. Yang, K. Yan, H. Qin, J. Ma, X. Sun, J. Yang and H. Wang, Electroreductive C3 Pyridylation of Quinoxalin-2(1H)-ones: An Effective Way to Access Bidentate Nitrogen Ligands, Org. Lett., 2021, 23, 1081 CrossRef CAS.
- C. Niu, J. Yang, K. Yan, J. Xie, W. Jiang, B. Li and J. Wen, Electrochemical Ammonium-cation-assisted Pyridylation of Inert N-heterocycles via Dual-proton-coupled Electron Transfer, iScience, 2022, 25, 104253 CrossRef CAS PubMed.
-
(a) M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230 CrossRef CAS;
(b) Y. Jiang, K. Xu and C. Zeng, Use of Electrochemistry in the Synthesis of Heterocyclic Structures, Chem. Rev., 2018, 118, 4485 CrossRef CAS;
(c) Y. Liu, H. Yi and A. Lei, Oxidation-Induced C–H Functionalization: A Formal Way for C—H Activation, Chin. J. Chem., 2018, 36, 692 CrossRef CAS;
(d) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Recent Advances in Oxidative R1-H/R2-H Cross-Coupling with Hydrogen Evolution via Photo-/Electrochemistry, Chem. Rev., 2019, 119, 6769 CrossRef CAS PubMed;
(e) P. Xiong and H. C. Xu, Chemistry with Electrochemically Generated N-Centered Radicals, Acc. Chem. Res., 2019, 52, 3339 CrossRef CAS;
(f) K. J. Jiao, Y. K. Xing, Q. L. Yang, H. Qiu and T. S. Mei, Site-Selective C−H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis, Acc. Chem. Res., 2020, 53, 300 CrossRef CAS;
(g) K. Yamamoto, M. Kuriyama and O. Onomura, Anodic Oxidation for the Stereoselective Synthesis of Heterocycles, Acc. Chem. Res., 2020, 53, 105 CrossRef CAS;
(h) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Electrocatalysis as an Enabling Technology for Organic Synthesis, Chem. Soc. Rev., 2021, 50, 7941 RSC;
(i) J. Yang, H. Qin, K. Yan, X. Cheng and J. Wen, Advances in Electrochemical Hydrogenation Since 2010, Adv. Synth. Catal., 2021, 363, 5407 CrossRef CAS.
-
(a) J. Hoyos-Arbeláez, M. Vázquez and J. Contreras-Calderón, Electrochemical Methods as a Tool for Determining the Antioxidant Capacity of Food and Beverages: A review, Food Chem., 2017, 221, 1371 CrossRef PubMed;
(b) B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-ion Battery Chemistry, Science, 2019, 363, 838 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data, and copies of NMR spectra. See DOI: https://doi.org/10.1039/d2qo01664h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Bingwen
Li
b,
Boju
Luo
a,
Meirou
Jing
a,
Yumeng
Zhou
a,
Ziling
Wu
a,
Laijin
Tian
a and
Jiangwei
Wen
a,
Bingwen
Li
b,
Boju
Luo
a,
Meirou
Jing
a,
Yumeng
Zhou
a,
Ziling
Wu
a,
Laijin
Tian
a and
Jiangwei
Wen








