
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization, and in vitro anti-cholinesterase screening of novel indole amines†
Humaira Yasmeen Gondala,
Sobia Tariqa,
Shahzaib Akhter a,
Abdul Rauf Raza*a,
Muhammad Fayyaz ur Rehman*a and
Syeda Laila Rubabb
a,
Abdul Rauf Raza*a,
Muhammad Fayyaz ur Rehman*a and
Syeda Laila Rubabb
aInstitute of Chemistry, Ibn e Sina Block, University of Sargodha, Sargodha-40100, Pakistan. E-mail: rauf.raza@uos.edu.pk; humaira.yasmeen@uos.edu.pk; sobiatariq66@gmail.com; shahzaibakhter740@gmail.com; muhammad.fayyaz@uos.edu.pk; Tel: +92-48-9230-546
bDepartment of Chemistry, Division of Science and Technology, University of Education, Lahore-54770, Pakistan. E-mail: laila.rubab@ue.edu.pk
First published on 4th January 2023
Abstract
The present study involved the targeted synthesis and characterization of novel indole amines with anti-acetylcholinesterase profiling. A series of proposed indole amines was virtually screened against human acetylcholinesterase. A few indole amines (23, 24, and 25) showing strong enzyme binding in the in silico studies were synthesized in the laboratory and characterized using spectroscopic (IR, UV, NMR, single crystal XRD) and spectrometric (EIMS, HR-EIMS) methods. The indole amine 23 was crystallized from EtOH and analyzed with single crystal XRD. These ligands interacted with the PAS site in the enzyme, and their binding may disrupt the activity. The in vitro acetylcholinesterase inhibition studies revealed that the IC50 values for indole amines 25 and 24 (4.28 and 4.66 μM, respectively) were comparable to that of galantamine (4.15 μM) and may be studied further as cost-effective acetylcholinesterase inhibitors.
1. Introduction
The indole [2,3-benzo-(1H)-azole or 2,3-benzo-(1H)-pyrrole] is a remarkable class of heteroaromatic compounds that has been found in many natural products and has been a focus of numerous synthetic studies. The indole moiety found in natural products and synthetic compounds has gained popularity due to its various pharmacological activities, such as anti-cancer,1 anti-tumor,2 anti-proliferative,3 anti-HIV,4 anti-ulcer,5 anti-parkinsonian,6 anti-malarial,7 anti-oxidant,8 anti-inflammatory,9 anti-viral,10 anti-bacterial,11 anti-fungal,12 anti-MRSA,13 anti-emetic,14 anti-asthmatic, opioid agonist, sexual dysfunction,15 anti-convulsant,16 anti-migraine,17 anti-depressant,18 anti-obesity,19 anti-hypertensive,20 acetylcholinesterase inhibitory,21 butyryl-cholinesterase inhibitory,22 α-glucosidase inhibitory,23 β-glucuronidase inhibitory,24 α-amylase inhibitory,25 and urease inhibitory.26 It has recently been discovered that the indole acts as a signaling molecule in various roles, including plasmid stability, drug resistance, extracellular signaling, biofilm formation, and virulence control.27Alzheimer's Disease (AD) is the condition of progressive dementia diagnosed by the loss of cognitive function and memory. It has been found that there is an accumulation of plaque within the brain of AD patients due to the accumulated beta-amyloids and neurofibrillary tangles (NFTs).28 Some enzymes, such as acetylcholinesterase (AChE), glycogen synthase kinase 3 (GSK3), cyclin-dependent kinase 5 (CDK5), and secretase, as well as the N-methyl-D-aspartate (NMDA) receptor, play a role in the progression of this disease.29 AD symptoms are alleviated if the levels of the neurotransmitter acetylcholine (ACh) are increased by inhibiting AChE. Higher AChE activity promotes the aggregation of Aβ plaques and neurofibrillary tangles within the brain.30 FDA-approved drugs such as memantine (N-methyl-D-aspartate (NMDA) inhibitor) and tacrine, donepezil, galantamine, and rivastigmine (acetylcholinesterase inhibitors) are used to alleviate AD symptoms.
The indole nucleus is not only limited to pharmaceutical research and biological systems, but it is also a common moiety in material science (organic photovoltaic devices)31 and agricultural applications, including fungicidal efficiency,32 plant growth regulators,33 and spore formation,34 and has been used in industrial applications in dyes,35 flavour enhancers,36 food supplements,37 fragrances, and photographic materials.38
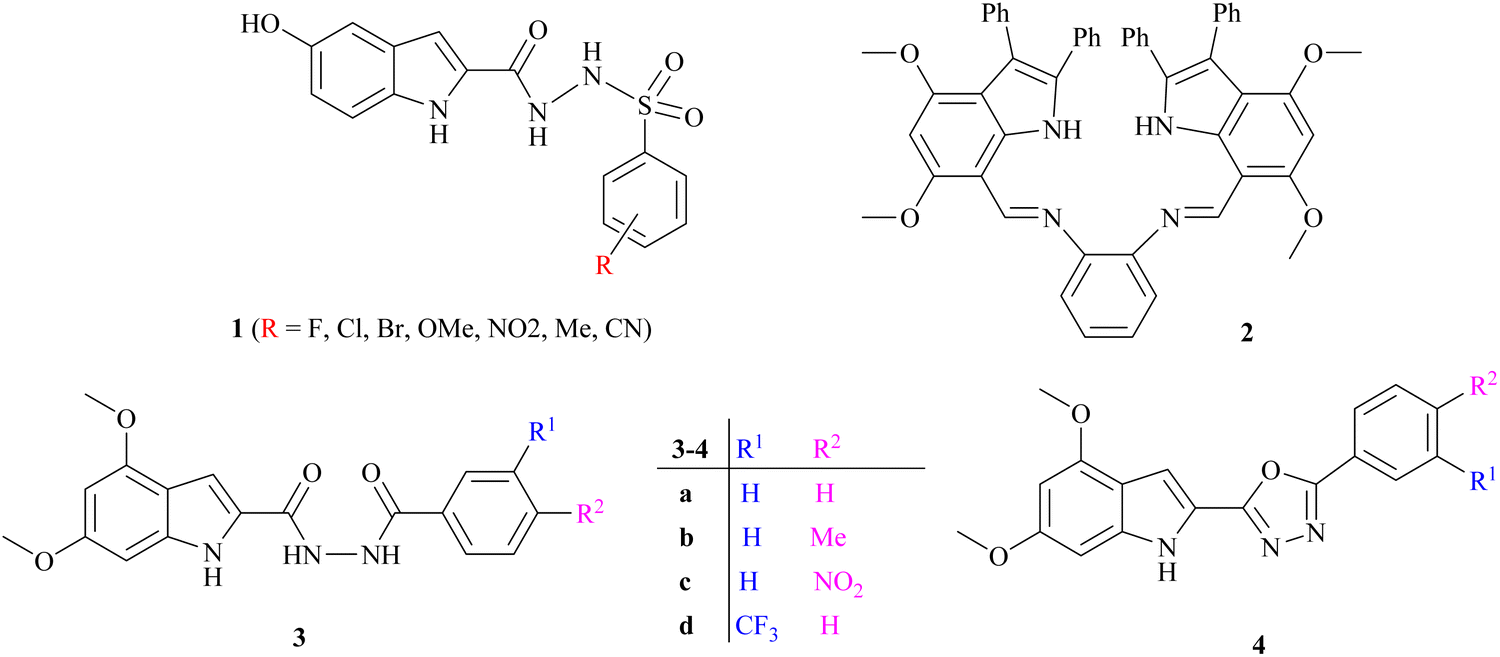
Several indole alkaloids are effective anti-cholinesterase and memory enhancers, and therefore, they are intriguing candidates for anti-AD drug development.39 The reported indole-based sulfonamide derivatives 1 have exhibited potent inhibition (IC50 = 0.17 ± 0.02 to 8.53 ± 0.32 μM) against acetylcholinesterase enzyme under the positive control of donepezil (IC50 = 0.014 ± 0.01 μM) as a standard drug.21 It has been reported that the anti-cholinesterase potency of bis-indolyl imine 2 is the highest, with a percentage inhibition value of 89.21 ± 1.39 and 96.06 ± 1.41 at 200 μM, which is higher than that of standard galantamine for AChE (78.76 ± 0.94) and BChE (79.72 ± 0.33), respectively.40 Bingul et al. reported the synthesis and anticholinesterase activity of novel indole-2-carbohydrazides 3a–d and 2-(indol-2-yl)-1,3,4-oxadiazoles 4a–d. The % inhibition shown by these compounds is in the range of 36.85 ± 0.20 to 90.60 ± 1.56 at 200 μg cm−3 concentration compared with standard galantamine.41
2. Results and discussion
Our research goal is to synthesize enantiopure heterocycles following the Chiron approach of asymmetric synthesis. We have successfully synthesized and reported a number of enantiomerically pure fused heterocycles.42,43 In considering the biological and other importance of natural and synthetic indoles, our goal is to produce indolic diazepine derivatives 36a–p and determine their pharmacological potential (Scheme 1).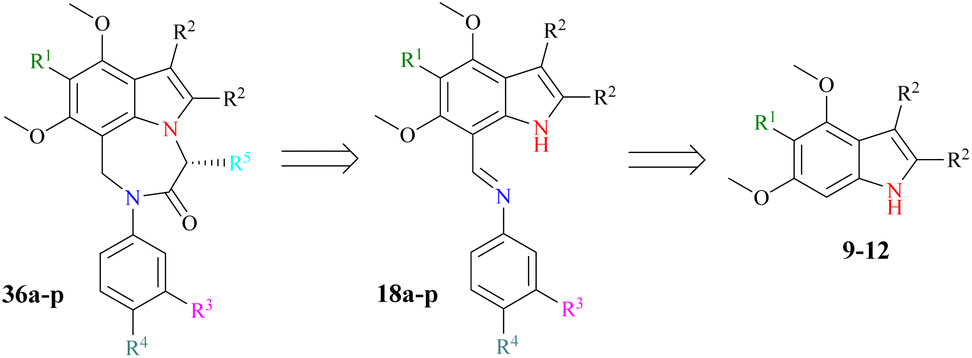 | ||
| Scheme 1 Retrosynthesis of designed indole-based diazepines 36a–p. | ||
The synthetic strategy (Scheme 2) starts with the reaction of commercially available 3,5-dimethoxyaniline 5 or 3,4,5-trimethoxyaniline 6 with benzoin 7 (±-2-hydroxy-1,2-diphenyl ethanone) or acetoin 8 (±-3-hydroxy butan-2-one) to afford indoles 9–12 following a modified Bischler indole protocol.44–46 The substituted indoles 9–12 would be formylated, following the Vilsmeier–Haack sequence (the addition of POCl3 to DMF under anhydrous conditions to produce chloroiminium ion as an intermediate) to furnish indole aldehydes 13–16. The resulting indole aldehydes 13–16 would be coupled with substituted anilines 17a–d to produce indole imines (Schiff bases) 18a–p. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of these imines 18a–p may be reduced with NaBH4, preferably to result in the formation of indole amines 19–34. The resulting amines 19–34 are a satisfactory synthon in our hand, and may be used in the formation of a variety of indolo-fused-diazepines 36a–p or 37a–p and indolo-fused-diazines 38a–p (Scheme 2). The work on all three indole-based targets (36–38) is in progress in our laboratory.
N of these imines 18a–p may be reduced with NaBH4, preferably to result in the formation of indole amines 19–34. The resulting amines 19–34 are a satisfactory synthon in our hand, and may be used in the formation of a variety of indolo-fused-diazepines 36a–p or 37a–p and indolo-fused-diazines 38a–p (Scheme 2). The work on all three indole-based targets (36–38) is in progress in our laboratory.
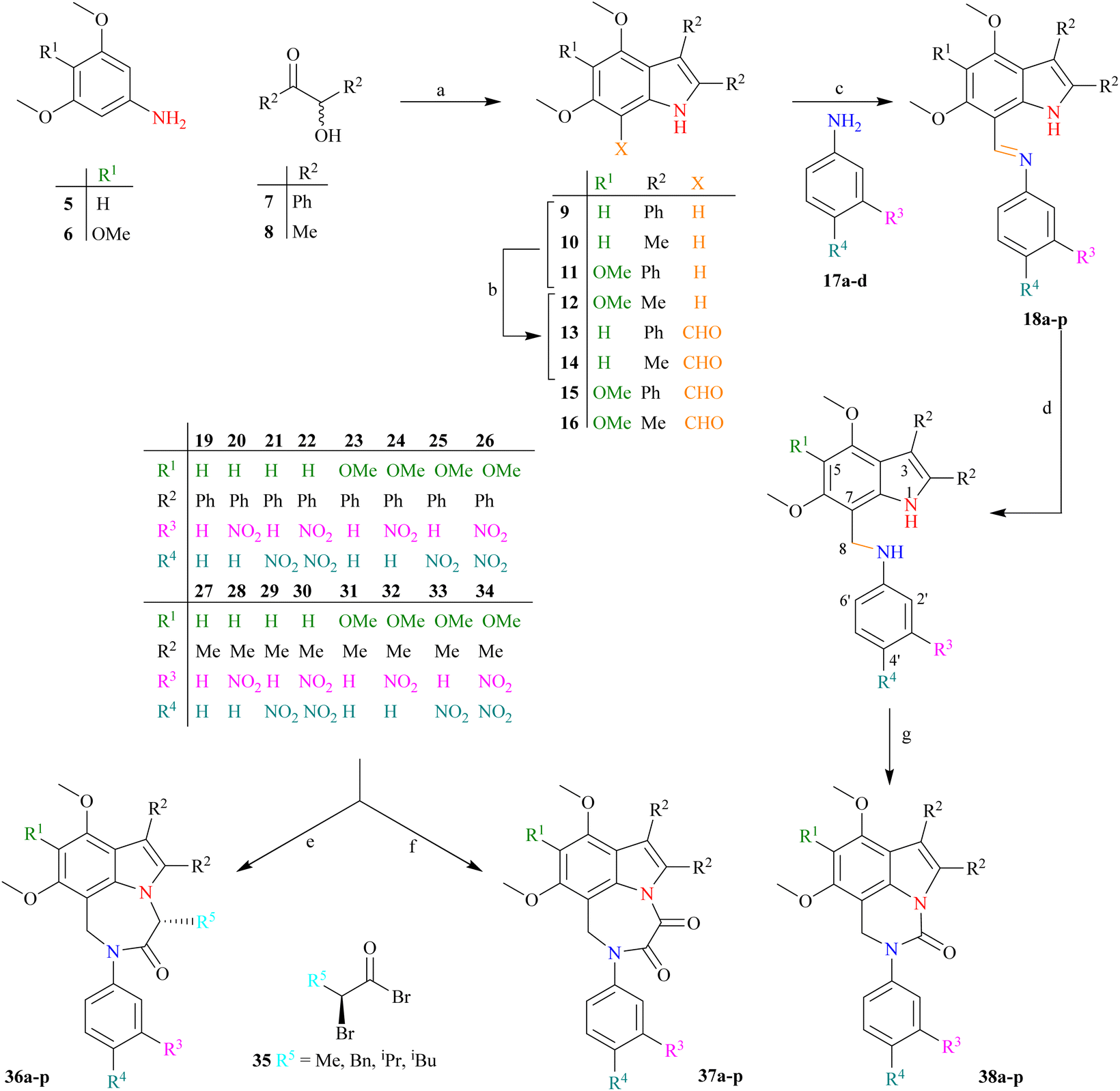 | ||
Scheme 2 (a) AcOH, PhNH2 (cat.), 130 °C; (b) POCl3, Me2NCHO, ambient; (c) EtOH, reflux; (d) NaBH4, dry THF/EtOH (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), reflux; (e) (i). LDA (−78 °C), (ii). 35; (f) (i). LDA (−78 °C), (ii). (COCl)2; (g) (i). LDA (−78 °C), (ii). (MeO)2CO. :1), reflux; (e) (i). LDA (−78 °C), (ii). 35; (f) (i). LDA (−78 °C), (ii). (COCl)2; (g) (i). LDA (−78 °C), (ii). (MeO)2CO. | ||
The bathochromic shift in λmax of the synthesized indoles (9–12), the appearance of an N–H as a single spike at 3353 ± 10 cm−1, and the disappearance of any CO stretching (corresponding to benzoin 7 or acetoin 8) indicate the formation of the indolic ring. Both indoles (9 and 11) are obtained as colourless crystalline solids. The low λmax values of indoles (9 and 11) indicate that both phenyl rings at position 2 and 3 are not in conjugation with the remainder of the indolic chromophore. In fact, both phenyl groups are oriented perpendicular to the chromophore of the indole ring to minimize steric repulsion, which is supported by single crystal XRD studies (Fig. 1 and Table 1).
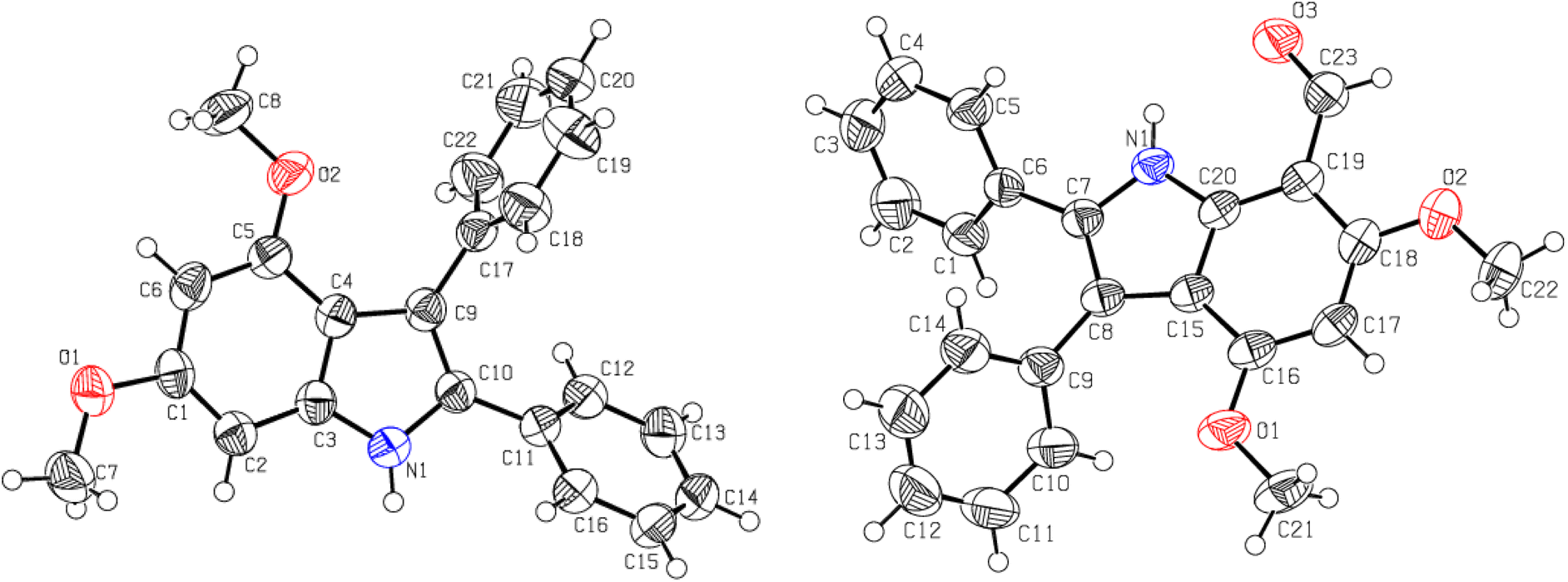 | ||
| Fig. 1 The ORTEP presentations of 4,6-dimethoxy-2,3-diphenylindole 9 (left) and 7-formyl-4,6-dimethoxy-2,3-diphenylindole 13 (right). | ||
| Parameters | 9 | 13 |
|---|---|---|
| Molecular formula | C22H19NO2 | C23H21NO3 |
| Molecular mass (amu) | 329.38 | 357.39 |
| Crystal system, space group | Monoclinic, P21/n | Monoclinic, P21/c |
| Temperature (K) | 296 | 296 |
| a, b, c (Å) | 11.7435 (16), 9.4480 (12), 15.940 (2) | 32.036 (2), 7.2010 (4), 17.3062 (8) |
| α, β, γ (°) | 90, 106.682 (7), 90 | 90, 112.060 (2), 90 |
| Volume of crystal (Å3) | 1694.2 (4) | 3700.1 (4) |
| Z | 4 | 8 |
| μ (MoKamm−1) | 0.08 | 0.085 |
| Crystal size (mm) | 0.30 × 0.26 × 0.24 | 0.35 × 0.18 × 0.16 |
The molecular ion of indoles (9 and 11) does not undergo considerable fragmentation, indicating high stability due to high resonance energy. The [M]+˙ appears as a base peak in the EIMS, which verifies the construction of the indolic ring. The ESI MS of indole 11 showed [M + Na]+˙ at 382.1677 amu, which is clear evidence of the successful formation of the indole nucleus. The 1H-NMR of indole 9 showed a pair of singlets (3.73 and 3.92 ppm) corresponding to the surrounding OMe groups, and two doublets of H5 and H7 appeared at 6.26 (J = 1.8 Hz) and 6.57 ppm (J = 1.8 Hz), respectively. The +R effect of OMe functionalities results in an upfield shift of H5 and H7. The aromatic protons of two phenyl rings emerged as multiplets of 10H integration. This concrete evidence was provided by single crystal XRD studies (Fig. 1 and Table 1).47,48
The indoles (9 and 11) were formylated by the reported Vilsmeier–Haack protocol, in which the formylating agent is the chloroiminium ion intermediate, generated by the interaction of DMF with POCl3. No prominent change in the N–H stretching was observed between indoles 9 and 11 (3343 and 3363 cm−1, respectively) and 7-formylindoles 13 and 15 (3298 and 3347 cm−1, respectively) in the infra-red (IR) spectra, which suggests that no H-hydrogen bonding exists between formylic O and indolic H. The CO bending in the 7-formylindoles (13 and 15) appears at a very low value (1608 and 1647 cm−1, respectively), which reveals that the aldehydic functional group is attached to a highly conjugated system. The bathochromic shift in the λmax of 7-formylindoles 13 and 15 (372, 358 nm, respectively) was observed in contrast to indoles 9 and 11 (322, 318 nm, respectively), which indicates an increase in conjugation due to the induction of a formyl moiety to the indolic chromophore.
The disappearance of doublets of H7 in the aromatic region (6.57/6.78 ppm) along with the rise of other singlets at 10.41/10.43 ppm with no broadening upon D2O exchange also indicated the formylation of indoles (9 and 11) to 7-formylindoles (13 and 15), respectively. The 13C-NMR showed the C5 and C7 of indoles (9 and 11) as doublet and quaternary carbons, respectively, proving the occurrence of formylation at C7. Furthermore, the doublet carbon of aldehydic functionality (H–CO) arose at 188.2 and 190.0 ppm, respectively, in both indoles (13 and 15); the δH values indicate the connectivity of formyl functionality with a highly conjugated system. The −I effect of formyl functionality shifted C7 to downfield. The single crystal XRD studies of 7-formylindoles (13 and 15) depict a perpendicular orientation of both phenyl groups, resulting in a minimum steric repulsion (Fig. 1 and Table 1).47,48 The [M]+˙ in the EIMS of both 7-formylindoles (13 and 15) is the base peak due to higher stability with no further considerable fragmentation.
The 7-formylindoles (13 and 15) would be condensed with a variety of substituted anilines 17a–d to furnish a variety of indole imines 18a–p, followed by the reduction of CN to afford a variety of indole amines 19–34. However, we limited ourselves to targeting the synthesis of the most potent (revealed after the in silico study) anti-cholinesterase indole amines (20–22, 24–26). The in silico study revealed that the selected six indole amines (20–22, 24–26) possessed great anti-cholinesterase potential. Interestingly, all six compounds contain NO2 group(s) at either the meta-/para- or both positions in the aminic phenyl ring and two phenyl substituents at the 2- and 3-position of the indole ring.
The other similar indole amine derivatives (28–30, 32–34) with two methyl groups at the 2- and 3-positions of the indole ring and a NO2 group at either the meta- or para-position in the aminic phenyl ring exhibited a relatively low anti-cholinesterase potential with a relatively higher concentration of targeted indole amines (20, 21, 24, 25) (Table 2 and S1†). Hence, only 2,3-diphenyl indole amines (20–22, 24–26) were synthesized and characterized.
| Indole amines | R1 | R2 | R3 | R4 | Binding energy (kcal mol−1) | Dissociation constant (nM) |
|---|---|---|---|---|---|---|
| 25 | OMe | Ph | H | NO2 | −13.37 | 0.16 |
| 26 | OMe | Ph | NO2 | NO2 | −12.81 | 0.4 |
| 24 | OMe | Ph | NO2 | H | −12.43 | 0.77 |
| 22 | H | Ph | NO2 | NO2 | −12.39 | 0.83 |
| 21 | H | Ph | H | NO2 | −12.34 | 0.9 |
| 20 | H | Ph | NO2 | H | −12.19 | 1.17 |
| 19 | H | Ph | H | H | −11.67 | 2.81 |
| 23 | OMe | Ph | H | H | −11.67 | 2.81 |
| 33 | OMe | Me | H | NO2 | −10.3 | 28.02 |
| 30 | H | Me | NO2 | NO2 | −10.21 | 32.58 |
| 34 | OMe | Me | NO2 | NO2 | −9.93 | 52.58 |
| 29 | H | Me | H | NO2 | −9.85 | 60.13 |
| 32 | OMe | Me | NO2 | H | −9.57 | 97.32 |
| 28 | H | Me | NO2 | H | −9.14 | 198.53 |
| 27 | H | Me | H | H | −8.72 | 405.14 |
We attempted to synthesize eight derivatives of indole imines (18a–h) by coupling aniline 17a/nitroanilines 17b–d with 7-formylindoles (13/15). We already reported the synthesis of imines (Schiff bases) involving a variety of reagents; however, the reflux of anilines with 7-formylindoles in dry EtOH afforded the best yields.48 Therefore, the coupling of nitroanilines 17b–d with 7-formylindoles (13/15) was carried out by refluxing in dry EtOH.
It was observed that a longer reaction was required for the coupling of 7-formylindoles (13/15) with 4-nitroaniline 17c, as compared to 17b, which would be due to the −M effect of a para-NO2 group that creates a positive charge on the ipso-C to NH2 group. In fact, the lone pair on the NH2 group exhibits poor basicity because it is delocalized from the NO2 group, while in aniline 17b, the lone pair on the NH2 group is not delocalized from the NO2 group. Surprisingly, the coupling of 3,4-dinitroaniline 17d with 7-formylindoles (13/15) showed very poor transformation to the desired imines (18d, 18h) while refluxing in dry EtOH for a week. This may be due to the strong −M effect of the para-NO2 group and the reinforcing −I effect of the meta-NO2 group.
The two nitro groups on a phenyl ring caused additional electron withdrawal from the aminic (NH2) group, decreasing its nucleophilicity (Scheme 3). We failed to synthesize two targeted indole imines (18d, 18h), whereas the other four targeted indole imines (18b, 18c, 18f, 18g) were successfully synthesized in excellent yield (76–88%) upon solidification from refluxing EtOH (Table 3). To crystallise these indole imines, a variety of solvents was used, but without success.
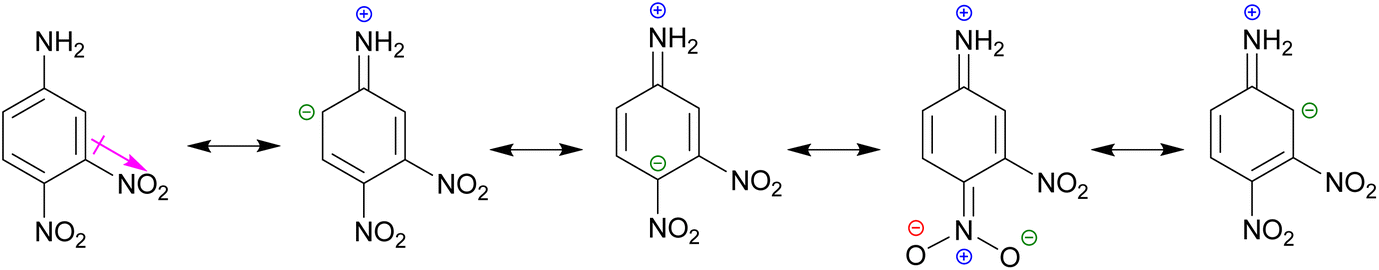 | ||
| Scheme 3 The −M and −I effect of 4-nitro and 3-nitro groups, respectively, in aniline 17d for decreasing its nucleophilicity. | ||
| Imine | % Yield | M.P. (°C) | λmax (nm) | logεa (L.M−1 cm−1) |
νb (cm−1) | δc (ppm) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| CN |
N–H | ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) CNAr CNAr |
N– |
H5 | H![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) NAr NAr |
|||||
| a Recorded in MeOH.b Recorded as anhydrous KBr disc.c Recorded at 400 MHz NMR machine in CDCl3. | ||||||||||
| 18b | 76 | 246 | 347 | 4.46683 | 1578 | 3310 | 9.13 | 11.26 | 6.22 | 157.2 |
| 18c | 76 | 268 | 351 | 4.15902 | 1583 | 3345 | 9.14 | 11.25 | 6.23 | 158.9 |
| 18f | 88 | 152 | 375 | 4.78249 | 1584 | 3356 | 9.10 | 11.14 | — | 156.9 |
| 18g | 85 | 179 | 388 | 5.16388 | 1576 | 3350 | 8.96 | 10.98 | — | 159.1 |
Four synthesized imines (18b, 18c and 18f, 18g) showed no absorbance in the carbonyl (1600–1850 cm−1) range, whilst a new signal at 1580 ± 4 cm−1 appeared in IR that indicated the transformation of CO to CNAr. Indolic N–H stretching was observed at 3333 ± 23 cm−1. A slight bathochromic shift in the λmax was exhibited by imines 18f, 18g, while a hypsochromic shift in the λmax resulted for imines 18b, 18c, which indicated nothing. However, the presence of a singlet of most shielded aromatic protons (H5) in only imines 18b, 18c and its absence in imines 18f, 18g, the appearance of an additional sharp singlet (did not show broadening upon D2O exchange) at 9.06 ± 0.08 ppm, the appearance of a mostly deshielded new broad singlet (exhibited broadening upon D2O exchange) at 11.12 ± 0.14 ppm in 1H NMR, and upfield shifting of an aldehydic methine C (188.2 and 190.0 ppm) to an iminic methine C (158.0 ± 1.1 ppm) in 13C NMR confirmed the formation of indole imines (18b, 18c and 18f, 18g) (Table 3).
Because the synthesized indole imines (18b, 18c and 18f, 18g) exist in an amorphous state, their single crystal XRD was not possible, although there were utmost efforts to crystallize these compounds with variable gradients. The single crystal XRD studies of N-phenyl(4,5,6-trimethoxy-2,3-diphenyl-1H-indol-7-yl)methanimine highlighted the perpendicular orientation of all three phenyl substituents and indicated the successful synthesis of the targeted compounds (Fig. 2 and Table S2†).
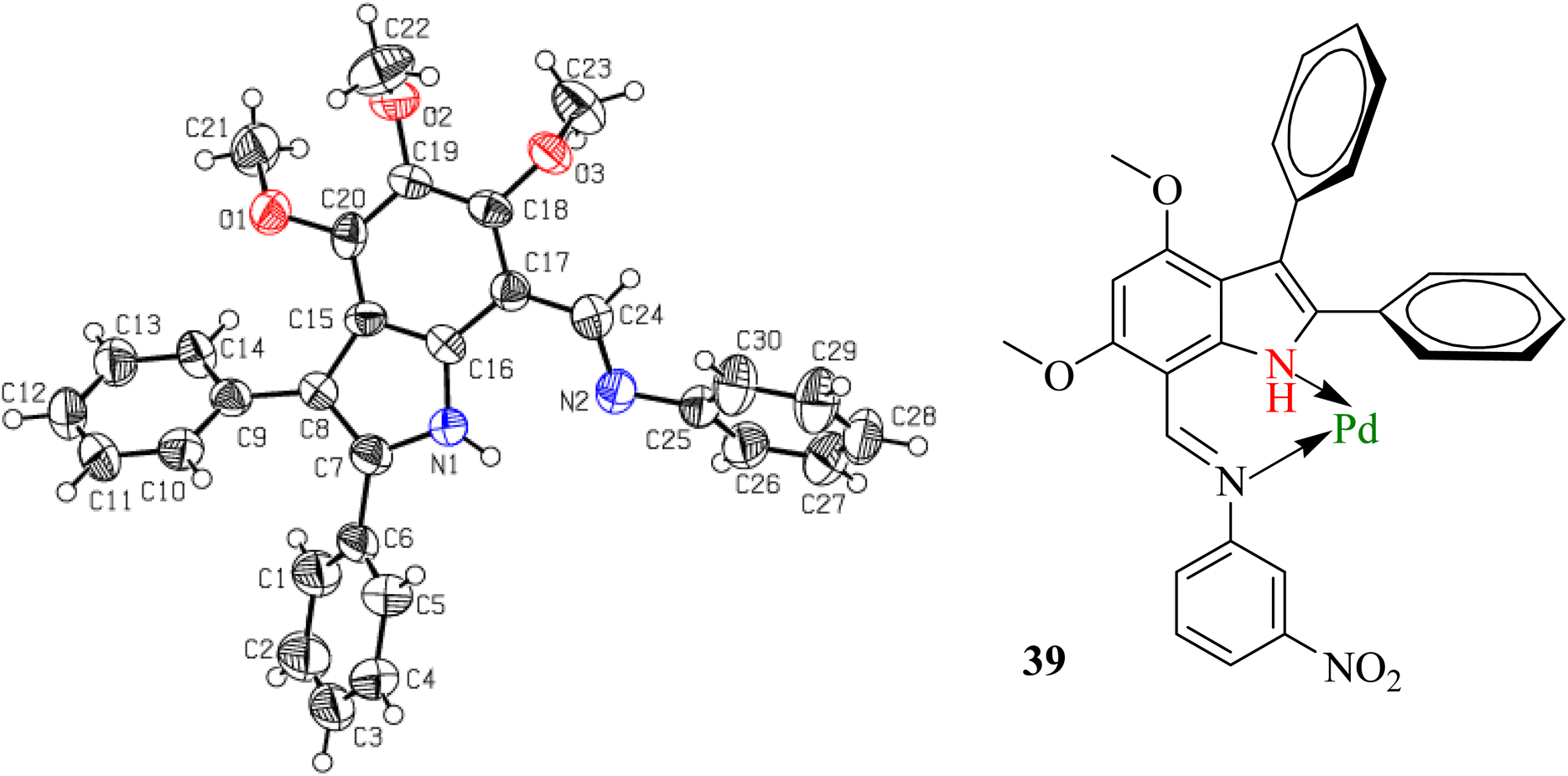 | ||
| Fig. 2 The ORTEP presentation of N-phenyl(4,5,6-trimethoxy-2,3-diphenyl-1H-indol-7-yl)methanimine (left) and coordination of imine 18b with Pd(C) to poison the catalyst. | ||
The reduction of imine 18b to corresponding amine 20 was initially carried out under an H2 atmosphere in the presence of a catalytic amount (5 mol%) of Pd(C), which resulted in the formation of a small amount of product. Prolonging the reaction duration improved nothing, which indicated that our imine 18b acted as a bidentate ligand and coordinated with Pd[0] adsorbed on activated charcoal to result in transition metal complex 39 (Fig. 2). Alternatively, other common reducing agents such as NaBH4 and LiAlH4 were tried. A suspension of imine and NaBH4 in MeOH was stirred at ambient temperature for an hour, but did not result in a reduction.
The excess NaBH4 in dry EtOH:THF (1:1) solvent at ambient temperature resulted in incomplete conversion to products. Refluxing an imine and NaBH4 in dry THF/EtOH (3:1) yielded amines 20, 21 and 24, 25. The reduction with LiAlH4 was useless, as it resulted in many inseparable products. The strong electron-releasing (+M) effect of OMe at indolic C4 and C6 rendered a prominent decrease in the electrophilicity of N, thus resulting in quite problematic reduction (Table 4).
| Reagentsa | Temperature (Time in h) | Dry solvent | Product(s) | Yield | |
|---|---|---|---|---|---|
| a Equivalents to respective imines.b mixture of reactant and product. | |||||
| 1 | NaBH4 (1.0 eq.) | rt (1) | MeOH | — | — |
| 2 | NaBH4 (3.0 eq.) | rt (36) | MeOH | b | — |
| 3 | NaBH4 (excess) | Reflux (3) | MeOH | b | — |
| 4 | NaBH4 (excess) | Reflux (5–48) | THF:EtOH (3:1) |
20 | 86 |
| 21 | 82 | ||||
| 24 | 89 | ||||
| 25 | 92 | ||||
| 5 | LiAlH4 (1.0 eq.) | rt (48) | THF:EtOH (3:1) |
b | — |
The appearance of a signal of N–H at approximately 3368 ± 12 cm−1 in reduced candidates (20, 21, 24 and 25) as compared to their corresponding substrates (which displayed only one N–H absorption at 3333 ± 23 cm−1) and the disappearance of a characteristic CN absorption at approximately 1580 ± 4 cm−1 clearly indicated the successful reduction. The hypsochromic shift of λmax from 368 ± 20 nm (imines) to 317 ± 3 nm was another clue as to the reduction of CN functionality to C–N (Fig. 3).
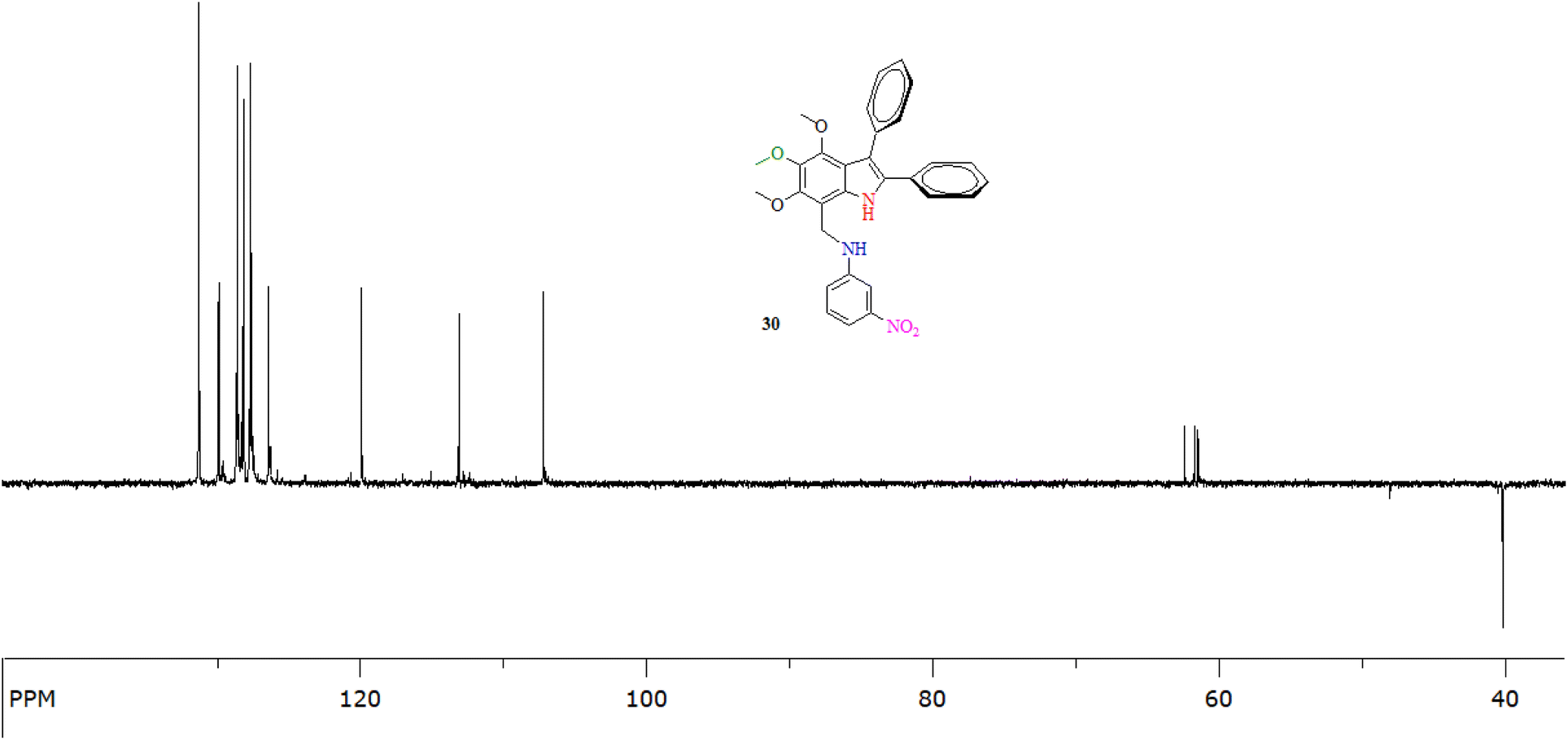 | ||
| Fig. 3 The 13C-NMR (DEPT-135°) of an indole amine 24 (at 100 MHz in CDCl3) showing the presence of a methylene carbon (C8) at 40.0 ppm. | ||
The disappearance of a singlet of 1H corresponding to the iminic functionality (CN) at 9.06 ± 0.08 ppm and the appearance of a singlet of 2H integration corresponding to the amine (2C–NH) at 4.67 ± 0.02 ppm in 1H-NMR confirmed the successful reduction of HCN to H2C–NH. The broad singlet of NH showed an upfield shift from 11.12 ± 0.14 ppm (imines) to 8.66 ± 0.03 ppm (reduced compounds), which is mainly due to the absorption in the anisotropic effect in later cases and a minor decrease in the electronegativity on N due to the change in hybridization from sp2 to sp3. The upfield shift of protons of aniline fragments also verified the conversion of HCN to H2C–NH due to a decrease in conjugation and reversal of the −M to +M effect of N.
The 13C-NMR of indole amines verified the successful reduction of imines to amines, depicting the abolition of a doublet carbon of HN (C8) at 158.0 ± 1.1 ppm and the emergence of a triplet C of H2–NH (C8) at 40.7 ± 0.7 ppm. The upfield shift of aromatic protons in the indole amines compared to the indole imines also authenticated an increase in the +M effect of aminic N due to the transformation of HC![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) into H2–NH (Fig. 4).
into H2–NH (Fig. 4).
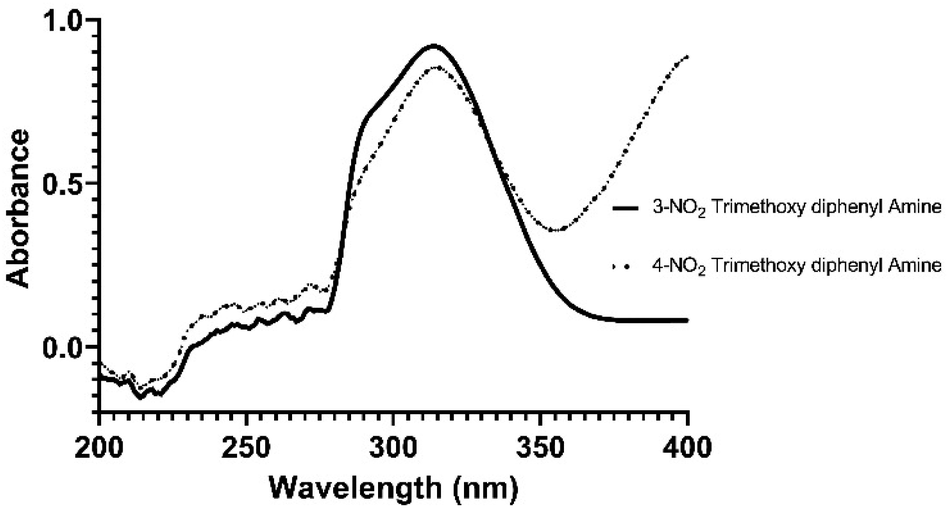 | ||
| Fig. 4 The overlay UV spectra of indole amines 24 and 25 (recorded in MeOH at 26 °C). | ||
Utmost efforts were made to crystallize the targeted indole amines (20, 21, 24 and 25) with variable gradients, but were without success. Another derivative (4,5,6-trimethoxy-2,3-diphenyl-7-phenyl aminomethyl-1H-indole) was successfully crystallized from MeOH/CHCl3. The single crystal XRD analysis of this indole amine indicated the successful reduction of the imine functional group to an amine (Fig. 5 and Table S3†).
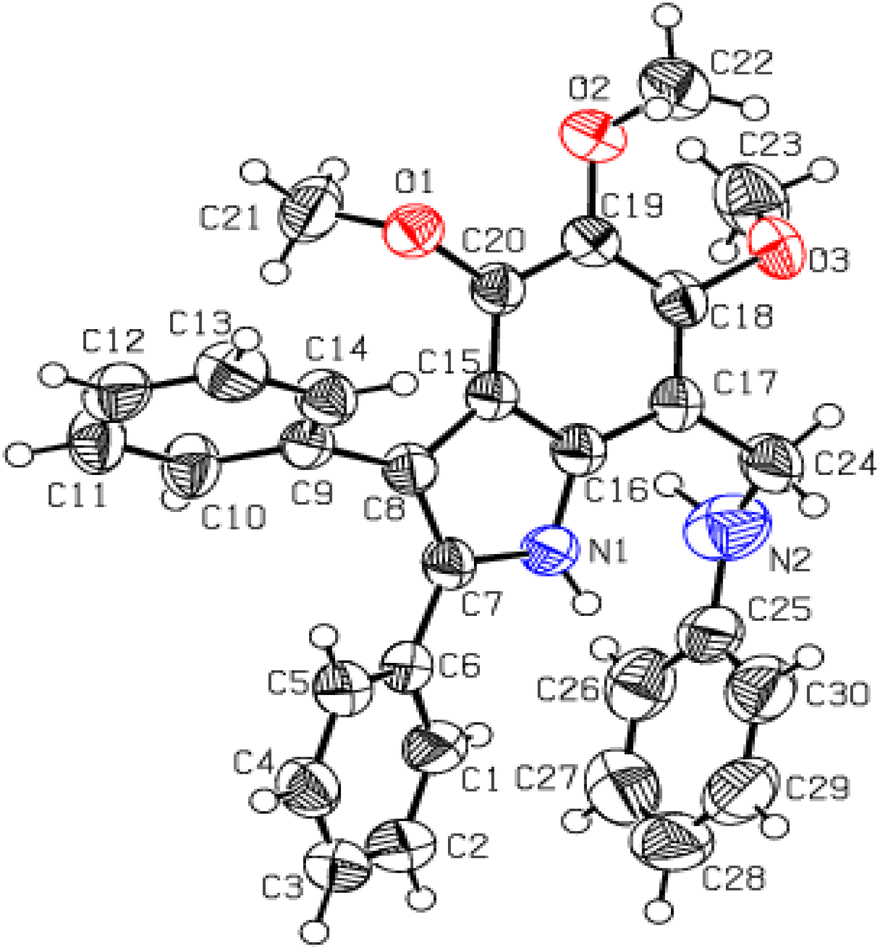 | ||
| Fig. 5 The ORTEP presentation of 4,5,6-trimethoxy-2,3-diphenyl-7-phenylaminomethyl-1H-indole 23. | ||
2.1 In silico anti-cholinesterase activity
Initially, all the proposed compounds were virtually screened against acetylcholinesterase. In previous studies, it was observed that target ligands specifically interact either at the active catalytic site (CAS) or the peripheral anionic site (PAS) of acetylcholinesterase. The catalytic triad in CAS consists of Ser203, Glu334, and His447 surrounded by an anionic site with aromatic residues Trp86, Tyr130, Tyr337, and Phe338 involved in binding to the quaternary trimethylammonium choline moiety of ACh through cation–π interaction. The entrance of the catalytic gorge contains the peripheral anionic site (PAS) with Tyr72, Asp74, Tyr124, Trp286, and Tyr341 residues that allosterically perform catalysis. The CAS and PAS are connected by a narrow groove.49The binding mode of the proposed indole amine 25 is characterized by a binding energy of −13.37 kcal mol−1 and a dissociation constant of 0.16 nM (Table 2). The hydrogen bonds of 3.68 and 3.01 Å with Asp74 of the PAS active site and 3.15 and 2.34 Å with Tyr337 of an anionic domain were observed in the stable binding. The –OH of Tyr337 and –OH from COOH of Asp74 formed a hydrogen bond with –NO2 of compound 25. The phenyl and pyrroline rings of indole amine 25 showed π–π stacking with Trp286 in the PAS region. The interaction of an indole moiety with Trp286 may lead to degenerative AChE activity.50,51
The following hydrophobic interactions occurred: phenyl ring with Gly292, Leu289, and Tyr72; the –CH3 of methoxy groups with Val294, Phe338 (anionic domain), and Tyr341 (PAS); and phenyl ring of indole moieties with Gly121 (oxyanion hole) and Phe297 (acyl pocket) (Fig. 6). The acyl pocket (Trp236, Phe295, and Phe297) is involved in selective binding with ACh by stabilizing the acetyl group. The oxyanion hole consists of one water molecule and Gly121, Gly122, and Ala204 residues, which stabilize the negative transition state during acylation and deacylation.
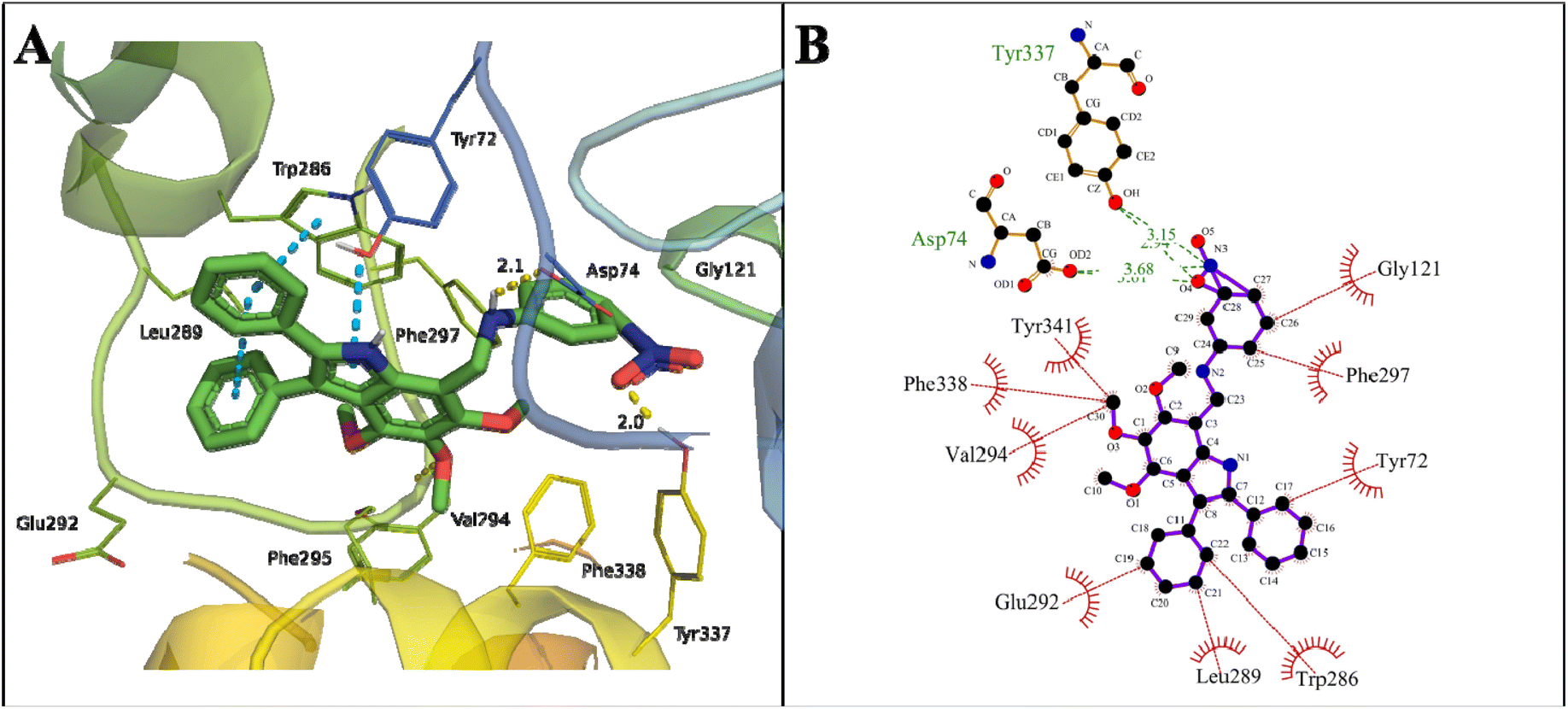 | ||
| Fig. 6 Binding mode of indole amine 25 with AChE. (A) 3D interaction of key residues of 4M0E, where pi–pi stacking (blue dotted line), hydrogen bond (yellow dotted line), and π–cation (green dotted line) are represented. (B) 2D interaction of key residues of 4M0E, where hydrophobic interactions (red lines) and hydrogen bond (green lines) are represented. | ||
Another proposed candidate, 24, showed a binding energy of −12.43 kcal mol−1 with a dissociation constant of 0.77 nM against AChE (Table 2). Here, the oxygen atom of the methoxy group attached to the C2 atom of the phenyl ring of indole formed a hydrogen bond of 2.98 Å with the NH2 of Phe295 in the acyl pocket of AChE. The oxygen atom of NO2 at C28 of the phenyl ring of compound 24 formed a hydrogen bond of 3.69 Å with the NH2 of Tyr124 located in the PAS, the allosteric binding site of AChE. The phenyl ring attached to the pyrroline and pyrroline ring formed π–π stacking with Trp286 of the PAS site (Fig. 7).
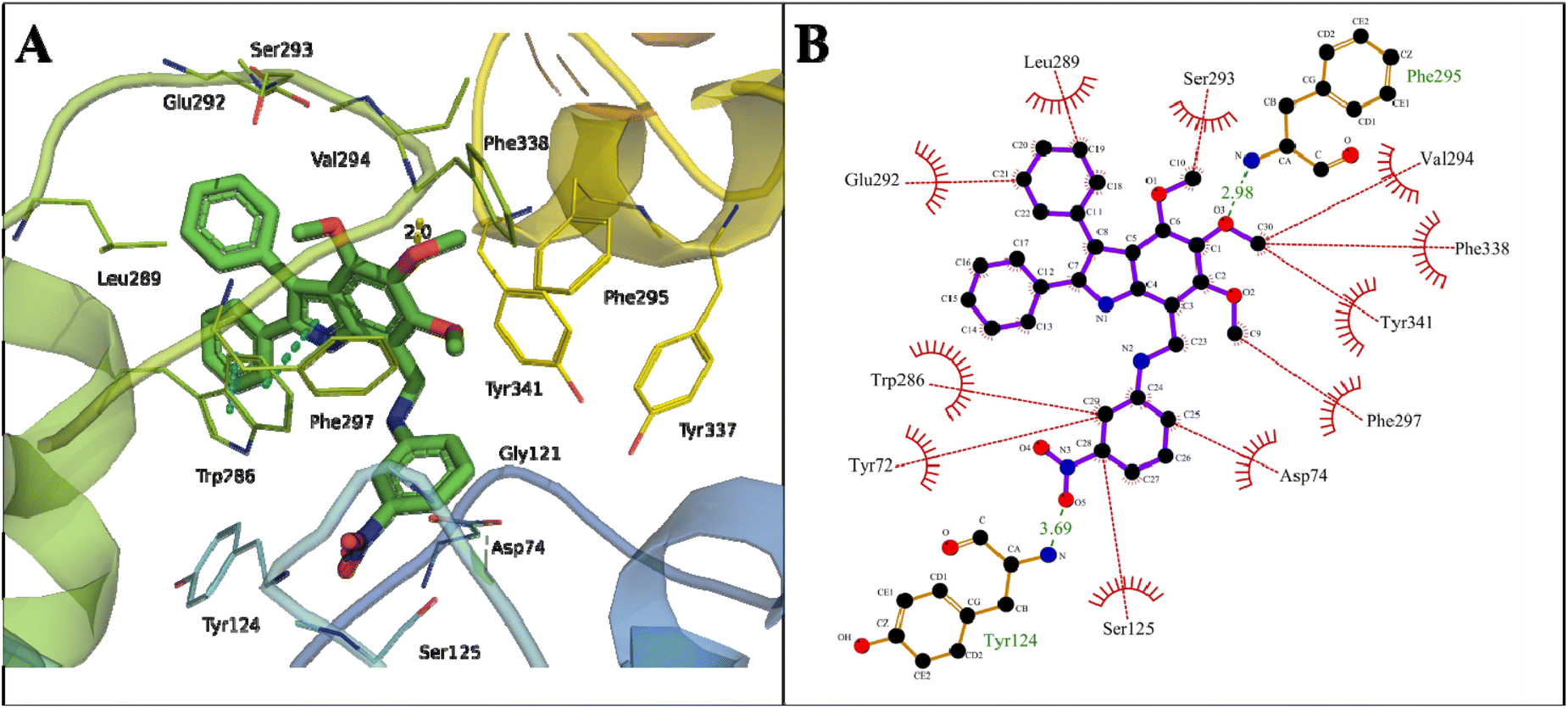 | ||
| Fig. 7 Binding mode of indole amine 24 with AChE. (A) 3D interaction of key residues of AChE, where pi–pi stacking (blue dotted line), hydrogen bond (yellow dotted line), and π–cation (green dotted line) are represented. (B) 2D interaction of key residues of AChE, where hydrophobic interactions (red lines) and hydrogen bond (green lines) are represented. | ||
The indole amine 21 showed a binding energy of −12.34 kcal mol−1 with a dissociation constant of 0.9 nM against AChE (Table 2). The oxygen atom of NO2 at C27 stabilized the docked complex by extensive hydrogen bonds of 3.03 and 3.73 Å with the OH of COOH in Asp74 of the PAS site and the H-bond of 3.19 Å with OH in the Tyr337 of anionic domain. Here, Trp286 also stabilized the docked complex by π–π stacking (Fig. 8).
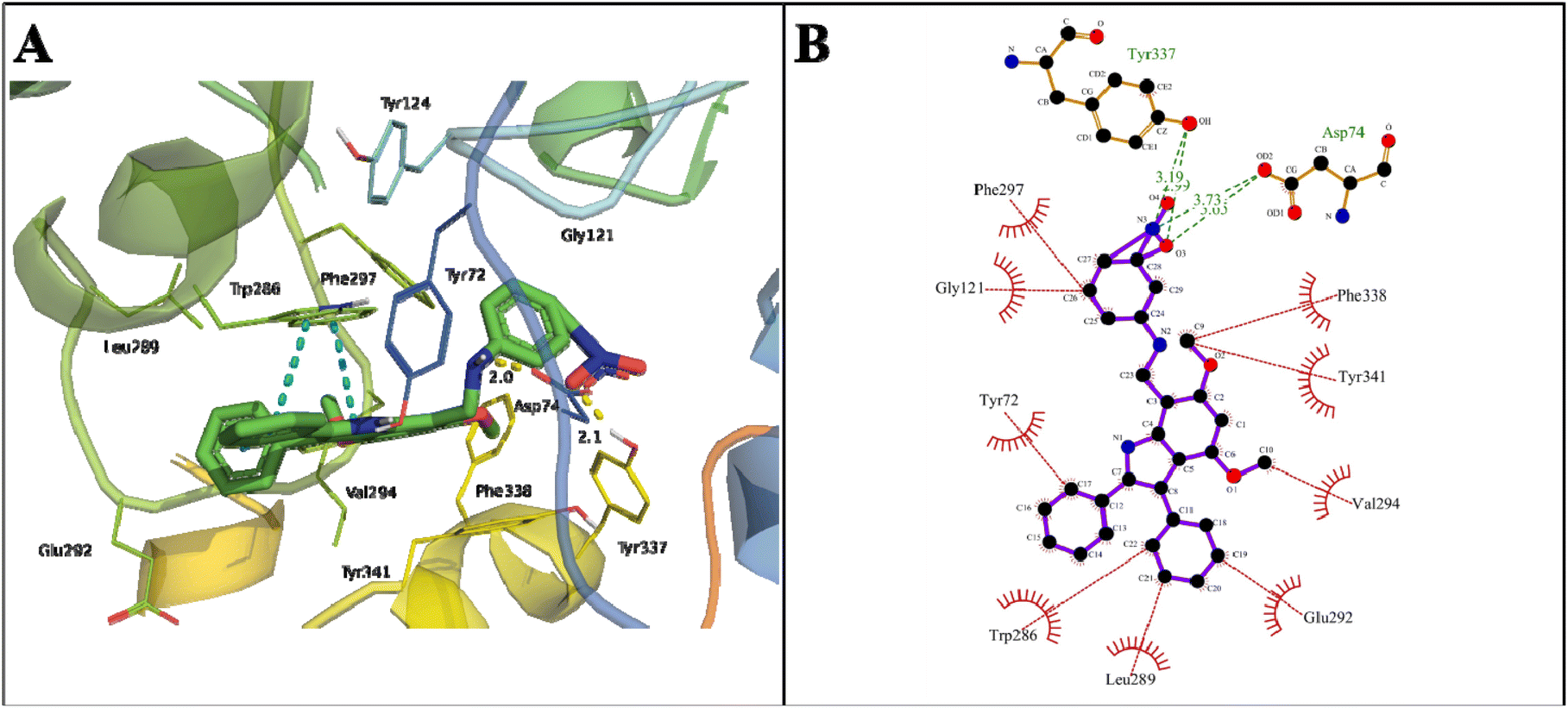 | ||
| Fig. 8 Binding mode of indole amine 21 with AchE. (A) 3D interaction of key residues, where π–π stacking (blue dotted line), hydrogen bond (yellow dotted line), and π–cation (green dotted line) are represented. (B) 2D interaction of key residues of AChE with 21, where hydrophobic interactions (red lines) and hydrogen bond (green lines) are represented. | ||
A binding energy of −12.19 kcal mol−1 with a dissociation constant of 1.17 nM for indole amine 20 against AChE was found (Table 2). The carbonyl oxygen of COOH of Tyr341 in the PAS site formed a hydrogen bond of 3.75 Å with N1 of the pyrroline ring of 20. The OH of COOH of Ser293 formed a hydrogen bond of 2.80 Å with the O4 of NO2 in compound 20 in the acyl pocket of AChE. Here, Trp286 and Tyr341 formed π–π stacking with a phenyl ring attached to the indole moiety and the 4-NO2 derivative phenyl ring of the compound (Fig. 9).
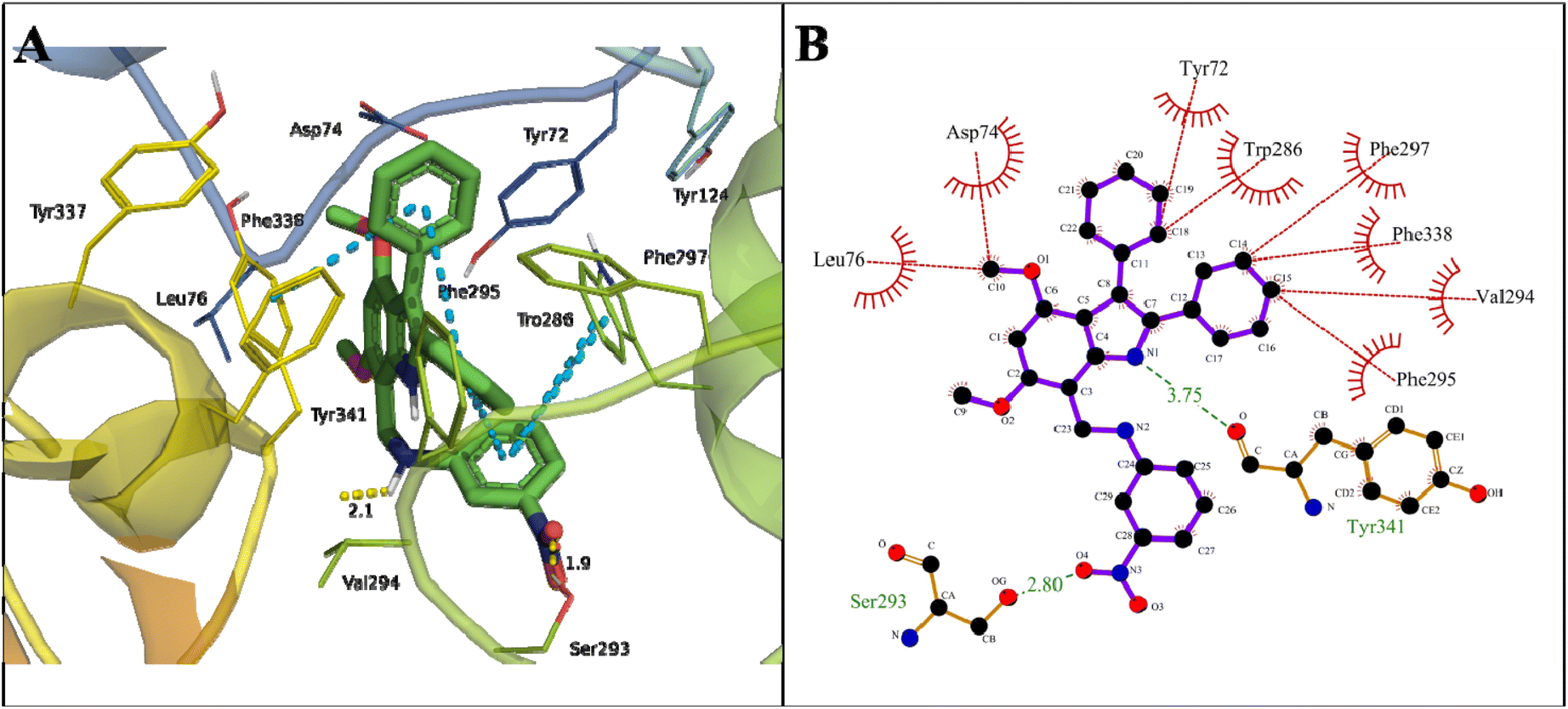 | ||
| Fig. 9 Binding mode of indole amine 20 with AChE (4M0E). (A) 3D interaction of key residues of 4M0E, where pi–pi stacking (blue dotted line), hydrogen bond (yellow dotted line), and π–cation (green dotted line) are represented. (B) 2D interaction of key residues of 4M0E, where hydrophobic interactions (red lines) and hydrogen bond (green lines) are represented. | ||
2.2 In vitro anti-cholinesterase activity
The synthesized indole amines (23, 24 and 25) were subjected to anti-acetylcholinesterase activity. Two amines (24 and 25) exhibited acetylcholinesterase inhibition comparable to that of galantamine, with IC50 values of 4.28 and 4.66 μM, respectively (Table 5). These inhibition potentials agree with the in silico results. The IC50 values of galantamine and donepezil were 4.15 and 0.028 μM, respectively, and are established cholinesterase inhibitors52,53 that are being used as anti-AD therapeutics.54,55 The natural indole amines (e.g., serotonin) inhibit the acetylcholinesterase activity in plaques and tangles involved in the progression of AD.562.3 ADMET parameters
Online web servers, including pkCSM (https://structure.bioc.cam.ac.uk/pkcsm) and SwissADME (https://www.swissadme.ch/index.php), were used to predict the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the novel synthesized indole derivatives. All synthesized indole-based derivatives have the highest intestinal absorption with high skin permeability as logKp > −2.5 cm h−1. However, it was predicted that the Caco2 permeabilities of indole amines (20, 21, 24 and 25) were satisfactory, with Papp values greater than 0.90 cm s−1.
All compounds were substrates of P-glycoprotein and were inhibitors of P-glycoprotein I and P-glycoprotein II. The indole amines (24 and 25) exhibited slightly low values of volume of distribution (Vd), while others showed the same values. All indole amines showed a fraction unbound value within the range of 0.239–0.312. The fraction unbound value represents compounds not bound to serum proteins, and thus, affects renal glomerular filtration and hepatic metabolism. If compounds show a high affinity for such proteins in the blood, they cannot cross the cell membrane.57
The indole amines (24 and 25) showed low values of blood–brain barrier (BBB) permeability. The BBB is the structure that controls the transfer of materials from blood to the brain and vice versa.58 There were low values of central nervous system (CNS) permeability (logPS) for indole amines (20 and 21). All compounds were not substrates of CYP2D6 or CYP3A4. Each compound was an inhibitor of CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 enzymes, which are the main cytochromes (CYP) in the P450 superfamily. The inhibition of CYP leads to the enhanced toxicity of drugs and decreases their efficacy.59 The bioavailability of the drugs is related to hepatic and renal clearance in excretion. It is crucial for predicting the dosing rate of the drugs to study the steady-state concentration. All compounds showed total clearance within the range of 0.609–0.688 mL min−1 kg−1 (Table S4†).
3. Experimental
3.1 General information
All reagents (analytical grade) and chemicals were obtained from Sigma-Aldrich, Fluka, or Merck. Glass columns packed with silica gel (0.6–0.2 mm, 60 Å mesh size, Merck) were used for purification. Pre-coated silica gel (0.25 mm thick layer over Al sheet, Merck, Darmstadt, Germany) thin-layer chromatography (TLC) plates were used for reaction monitoring.The Thermo Spectronic (UV-1700) spectrophotometer and Shimadzu (Prestige 21) FT-IR spectrometer were utilized for recording UV-Vis and IR spectra (as anhydrous KBr discs), respectively, at the High-Tech Laboratory, University of Sargodha, Sargodha (Pakistan). 1H-NMR and 13C-NMR were performed using a Bruker AVANCE DPX (300, 400, or 500 MHz) spectrometer in CDCl3 at the ICCBS, H. E. J. Research Institute of Chemistry, University of Karachi, Karachi (Pakistan). For MS Q-TOF Ultima API (Micromass), the Biomedical Mass Spectrometry Facility (BMSF) at UNSW, Sydney (Australia) was utilized.
The single crystal XRD was performed using a Bruker Kappa APEX 11 CCD diffractometer at the University of Sargodha, Sargodha (Pakistan). Virtual screening for molecular docking and ADME prediction was performed using YASARA (Yet Another Scientific Artificial Reality Application) software version 20.7.4,60 with a modified AutoDock LGA algorithm and AMBER03 force field and parameters that have been previously described. Docking poses were visualized and analysed by LigPlus61 and PyMol software.
The pharmacokinetic properties and drug-likeness predictions for the top 10 compounds were performed by the SwissADME server (https://www.swissadme.ch/). The acetylcholinesterase inhibition was determined using a modification of the Ellman assay,62 as described by Gholivand et al.63
A 96-well plate was used for the assay with 0.1 mM DTNB, 0.135 mM, acetylthiocholine iodide (AChI), and 70 mM phosphate-buffered saline (PBS) pH 7.4, for a total volume of 200 μL. In the reaction mixture, ATChI was added at the end to start the reaction. Positive and negative controls were also used. The absorbance was noted at 405 nm at 1 min intervals for at least ten minutes using a microwell plate reader (BioTek) at 25 °C. The reaction rates were calculated to obtain enzymatic activity and IC50. All reactions were performed in triplicate. The ELISA reader ELx800 (BioTek, USA) and Gen5 3.02 software were used to calculate the absorbance of the solutions at variable wavelengths (in nm).
3.2 Synthesis of 2,3-diphenyl-(1H)-indoles (9, 11)
A mixture of aniline 5 or 6 (13.1 mmol, 3 eq.) with benzoin 7 (13.1 mmol, 3 eq.) was stirred at 120 °C for 2 h. The mixture was cooled to ambient temperature and stirred upon the addition of PhNH2 (4.4 mmol, 1 eq.) and AcOH (8.1 mL, 8.5 g, 0.141 mol, 32 eq.). The resulting mixture was converted to a solution upon stirring (5 h) at 130 °C. Precipitation resulted after gradual cooling to ambient temperature. The precipitate was filtered, and the crude product was washed with chilled MeOH to afford a white solid (65–67%). A small portion of this amorphous solid was crystallized from EtOAc for spectroscopic and spectrometric characterization.3.3 Synthesis of 2,3-diphenyl-(1H)-indole-7-carbaldehydes (13, 15)
The indole 9 or 11 (3 mmol, 1 eq.) was added to a stirred solution of POCl3 (0.85 mL, 1.4 g, 9 mmol, 3 eq.) in DMF (20 mL) at ambient temperature. The resulting solution was stirred at room temperature for 2½ h before being quenched with chilled H2O (50 mL) and was basified with aq. NaOH solution (50 mL of 1 M). The resulting yellow precipitate was filtered, washed with chilled H2O, and dried over anhydrous silica in a desiccator under reduced pressure to afford targeted aldehyde (80–92%) as a yellow amorphous solid. A small portion of this amorphous solid was crystallized from EtOAc/MeOH for spectroscopic and spectrometric characterization.3.4 Synthesis of indole imines (18b, 18c, 18f, 18g)
The 4,6-dimethoxy-2,3-diphenyl-(1H)-indole-7-carbaldehyde 13 or 4,5,6-trimethoxy-2,3-diphenyl-(1H)-indole-7-carbaldehyde 15 (1.00/1.08 g, 2.8 mmol, 1 eq.) and aniline 17b or 17c (502 mg, 3.64 mmol, 1.3 eq.) were collectively dissolved in dry EtOH (25 mL). The resulting solution was refluxed for a variable duration until the disappearance of the aldehyde spot on the neutralized (after elution with n-hexane/Et3N 1:1) TLC plate, which was chemically spotted with 2,4-dinitrophenylhydrazine (2,4-DNP) dip. The solvent was evaporated under reduced pressure, and the resulting solid product was flushed with chilled MeOH (3 × 20 mL) to afford an orange amorphous solid (18b, 18c, 18f, 18g). It was attempted to further purify this orange solid by crystallization from different solvents without success.
:1); M.P. 268 °C; ν (cm−1): 1578 (CN), 3310 (N–H); logε (λmax, nm): 4.15902 (347); δH in ppm (500 MHz): 3.80, 3.99 (3H each, s, OC3), 6.22 (1H, s, H5), 7.22–7.58 (12H, m, 3 Ph), 8.03 (1H, d, J = 7.8 Hz, H4′), 8.10 (1H, t, J = 2.0 Hz, H2′), 9.13 (1H, s, D2O non-exchangeable, CN), 11.26 (1H, bs, D2O exchangeable, N); δC in ppm (125 MHz): 55.4, 56.5 (q, OH3), 87.5 (d, C5), 101.7 (s, C7), 113.0, 114.8 (s, C3 & C3a), 115.6 (d, C5′), 119.5 (d, C6′), 126.2, 127.1 (d, C4′′ & C4′′′), 127.5, 127.8, 128.6, 131.4 (all 2×, d, C2′′, C3′′, C2′′′ & C3′′′), 128.1 (d, C4′), 129.7 (d, C2′), 132.8, 133.0 (s, C1′′ & C1′′′), 135.7, 136.6 (s, C2 & C7a), 149.1 (s, C3′), 154.2 (s, C1′), 157.2 (d, C8), 159.8, 160.2 (s, C4 & C6); CHNS analysis: found for C29H23N3O4: C (69.59%), H (4.57%), N (7.84%), requires: C (72.94%), H (4.85%), N (8.80%), O (13.40%).:1); M.P. 266 °C; ν (cm−1): 1583 (CN), 3345 N–H; logε (λmax, nm): 3.98706 (351); δH in ppm (500 MHz): 3.82, 3.98 (3H each, s, OC3), 7.08–7.29 (10H, m, 2 Ph), 7.36 (2H, d, J = 5.0 Hz, H2′), 8.25 (2H, d, J = 5.0 Hz, H3′), 9.14 (1H, s, D2O non-exchangeable, CN), 11.25 (1H, bs, D2O exchangeable, N).:1); M.P. 168 °C; ν (cm−1): 1584 (CN), 3356 (N–H); logε (λmax, nm): 4.04059 (375); δH in ppm (500 MHz): 3.59, 3.88, 4.07 (3H each, s, OC3), 7.22–7.38 (12H, m, 2 Ph, H5′ & H6′), 7.40 (1H, d, J = 7.8 Hz, H4′), 8.34 (1H, t, J = 2.0 Hz, H2′), 9.10 (1H, s, D2O non-exchangeable, CN), 11.14 (1H, bs, D2O exchangeable, N).:1); M.P. 179 °C; ν (cm−1): 1576 (CN), 3350 (N–H); logε (λmax, nm): 5.16388 (388); δH in ppm (500 MHz): 3.53, 3.84, 4.00 (3H each, s, OC3), 7.19–7.29 (10H, m, 2 Ph), 7.36 (2H, d, J = 5.0 Hz, H2′), 8.25 (2H, d, J = 5.0 Hz, H3′), 8.96 (1H, s, D2O non-exchangeable, CN), 10.98 (1H, bs, D2O non-exchangeable, N); δC in ppm (125 MHz): 61.3, 61.5, 62.9 (q, OH3), 107.8 (s, C7), 114.6, 118.7 (s, C3 & C3a), 121.8 (2×, d, C2′), 125.2 (2×, d, C3′), 127.6, 127.7 (d, C4′′ & C4′′′), 127.8, 127.9, 128.7, 131.2 (2×, d, C2′′, C3′′, C2′′′ & C3′′′), 131.2, 131.8 (s, C1′′ & C1′′′), 134.7, 135.4 (s, C2 & C7a), 145.4 (s, C4/C5/C6), 153.2 (2×, s, any two of C4, C5 & C6), 154.1 (s, C4′), 159.1 (d of C8 and s of C1′ merged).3.5 Reduction of imines to indole amines (20, 21, 24 and 25)
To a solution of indole imines (18b, 18c, 18f, 18g) (1 mmol, 1 eq.) in THF/EtOH (3:1, 40 mL), NaBH4 (151 mg, 4 mmol, 4 eq.) was added, and the suspension was refluxed for a variable duration. The solvent was partially removed under reduced pressure upon completion of the reaction (highlighted via TLC by the disappearance of imine's spot), and the reaction mixture was partitioned between chilled H2O and CHCl3 (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4, rotary concentrated, and left standing overnight to afford indole amines (20, 21, 24 and 25) as a colourless solid in appreciable yield. The crystallization from different solvents failed.
:2); M.P. 179 °C; ν (cm−1): 3379 (N–H, broad s); logε (λmax, nm): 4.3613 (314); δH in ppm (400 MHz): 3.47, 3.96, 3.99 (3H each, s, OCH3), 4.69 (2H, s, H8), 7.05 (1H, dd, J = 8.2, 1.8 Hz, H6′), 7.20–7.47 (14H, m, remainder of the signals of 3 Ph rings and aminic N–H), 8.69 (1H, bs, D2O exchangeable, indolic N–H); δC in ppm (100 MHz): 40.0 (t, C8), 61.3, 61.6, 62.2 (all q, OCH3), 107.1 (d, C6′), 108.4 (s, C7), 113.0 (d, C4′), 114.9 (s, C3a), 118.9 (s, C3), 119.8 (d, C2′), 126.3 (d, C5′), 127.5 (d, C4′′′), 127.6, 128.1, 128.5 (all 2×, d, C2′′′, C3′′′ & C2′′), 129.8 (d, C4′′), 131.2 (2×, d, C3′′), 131.9, 132.3, 134.5, 135.3 (all s, C1′′′, C1′′, C7a, C2), 140.8 (s, C1′), 147.1, 148.8, 149.2 (all s, C4, C5 & C6), 149.3 (s, C3′); HR ESIMS (m/z, amu): 510.2036 [M + H]+.:1); M.P. 191 °C; ν (cm−1): 3356 (N–H, broad s); logε (λmax, nm): 4.2490 (320); δH in ppm (400 MHz): 3.53, 3.84, 4.00 (3H each, s, OC3), 4.65 (2H, s, H8), 7.19–7.29 (11H, m, 2 Ph rings and aminic N–H), 7.41 (2H, d, J = 6.4 Hz, H2′), 8.35 (2H, d, J = 6.4 Hz, H3′), 8.63 (1H, bs, D2O exchangeable, indolic N–H); δC in ppm (100 MHz): 41.3 (t, C8), 61.0, 61.5, 62.9 (q, OH3), 107.8 (s, C7), 110.4 (2×, s, C2′), 114.9, 118.5 (s, C3 & C3a), 125.2 (2×, d, C3′), 127.1, 128.2 (d, C4′′ & C4′′′), 127.6, 128.7, 129.7, 130.6 (2×, d, C2′′, C3′′, C2′′′ & C3′′′), 131.7, 132.9 (s, C1′′ & C1′′′), 135.5, 136.4 (s, C2 & C7a), 149.4 (2×, s, C4 & C6), 153.2 (s, C5), 154.8 (s, C1′), 158.8 (s, C4′).4. Conclusion
Our in vitro and in silico studies revealed that two indole amines (24 and 25) showed a strong acetylcholinesterase inhibition potential. These indole amines bind to the PAS site and may lead to enzyme inactivity. The IC50 values of reported compounds for acetylcholinesterase are comparable to the standard compound, galantamine. These can be further studied for the development of cost-effective and one-step synthetic alternatives to current AD therapeutics.Author contributions
Conceptualization, ARR and MFR; methodology, ST, SA, and SLR; software, MFR and SA; validation, ARR, SLR and HYG; formal analysis, SA and ST; investigation, SA, ST. and SLR; resources, MFR, ARR and SLR; writing—original draft preparation, HYG and MFR, and ARR; writing—review and editing, HYG, SA, MFR, and ARR; visualization, SA, MFR, and ARR; supervision, MFR and ARR; funding acquisition, ARR and MFR.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge the Higher Education Commission (HEC) of Pakistan for generous financial support for research funding (HEC-20-3873). We are grateful to the University of Sargodha for the provision of basic instruments and the XRD facility.References
- A. S. Kuzmich, S. N. Fedorov, V. V. Shastina, L. K. Shubina, O. S. Radchenko, N. N. Balaneva, M. E. Zhidkov, J.-I. Park, J. Y. Kwak and V. A. Stonik, Bioorg. Med. Chem., 2010, 18, 3834–3840 CrossRef CAS PubMed.
- A. Andreani, S. Burnelli, M. Granaiola, A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, L. Varoli, L. Landi, C. Prata, M. V. Berridge, C. Grasso, H. H. Fiebig, G. Kelter, A. M. Burger and M. W. Kunkel, J. Med. Chem., 2008, 51, 4563–4570 CrossRef CAS PubMed.
- K. Lalit, B. Shashi and J. Kamal, Int. J. Res. Pharm. Sci., 2012, 2, 23–33 Search PubMed.
- L. Ding, J. Munch, H. Goerls, A. Maier, H. H. Fiebig, W. H. Lin and C. Hertweck, Bioorg. Med. Chem. Lett., 2010, 20, 6685–6687 CrossRef CAS PubMed.
- H. de Sousa Falcao, J. A. Leite, J. M. Barbosa-Filho, P. F. de Athayde-Filho, M. C. de Oliveira Chaves, M. D. Moura, A. L. Ferreira, A. B. de Almeida, A. R. Souza-Brito, M. de Fatima Formiga Melo Diniz and L. M. Batista, Molecules, 2008, 13, 3198–3223 CrossRef PubMed.
- F. Zhang, Y. Zhao, L. Sun, L. Ding, Y. Gu and P. Gong, Eur. J. Med. Chem., 2011, 46, 3149–3157 CrossRef CAS PubMed.
- S. T. Chan, A. N. Pearce, M. J. Page, M. Kaiser and B. R. Copp, J. Nat. Prod., 2011, 74, 1972–1979 CrossRef CAS PubMed.
- A. Longeon, B. R. Copp, E. Quevrain, M. Roue, B. Kientz, T. Cresteil, S. Petek, C. Debitus and M. L. Bourguet-Kondracki, Mar. Drugs, 2011, 9, 879–888 CrossRef CAS PubMed.
- M. Verma, M. Tripathi, A. K. Saxena and K. Shanker, Eur. J. Med. Chem., 1994, 29, 941–946 CrossRef CAS.
- S. Cai, X. Kong, W. Wang, H. Zhou, T. Zhu, D. Li and Q. Gu, Tetrahedron Lett., 2012, 53, 2615–2617 CrossRef CAS.
- P. Devi, C. Rodrigues, C. G. Naik and L. D'Souza, Indian J. Microbiol., 2012, 52, 617–623 CrossRef CAS PubMed.
- Y. Takahashi, T. Kubota, A. Shibazaki, T. Gonoi, J. Fromont and J. Kobayashi, Org. Lett., 2011, 13, 3016–3019 CrossRef CAS PubMed.
- Y. Yamamoto and M. Kurazono, Bioorg. Med. Chem. Lett., 2007, 17, 1626–1628 CrossRef CAS PubMed.
- J. E. Macor, D. Gurley, T. Lanthorn, J. Loch, R. A. Mack, G. Mullen, O. Tran, N. Wright and J. C. Gordon, Bioorg. Med. Chem. Lett., 2001, 11, 319–321 CrossRef CAS PubMed.
- S. Biswal, U. Sahoo, S. Sethy, H. K. S. Kumar and M. Banerjee, Asian J. Pharm. Clin. Res., 2012, 5, 1–6 CAS.
- N. Kumar, P. K. Sharma, V. I. K. Garg and P. Singh, Curr. Res. Chem., 2011, 3, 114–120 CrossRef CAS.
- M. S. Chambers, L. J. Street, S. Goodacre, S. C. Hobbs, P. Hunt, R. A. Jelley, V. G. Matassa, A. J. Reeve, F. Sternfeld and M. S. Beer, J. Med. Chem., 1999, 42, 691–705 CrossRef CAS PubMed.
- A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed.
- H. P. Chang, M. L. Wang, M. H. Chan, Y. S. Chiu and Y. H. Chen, Nutrition, 2011, 27, 463–470 CrossRef CAS PubMed.
- A. A. El-Gendy and H. A. El-Banna, Arch. Pharmacal Res., 2001, 24, 21–26 CrossRef CAS PubMed.
- M. Taha, F. J. Alshamrani, F. Rahim, N. Uddin, S. Chigurupati, N. B. Almandil, R. K. Farooq, N. Iqbal, M. Aldubayan and V. Venugopal, J. King Saud Univ., Sci., 2021, 33, 101401 CrossRef.
- M. Bingul, S. Ercan and M. Boga, J. Mol. Struct., 2020, 1213, 128202 CrossRef CAS.
- M. Alomari, M. Taha, F. Rahim, M. Selvaraj, N. Iqbal, S. Chigurupati, S. Hussain, N. Uddin, N. B. Almandil and M. Nawaz, Bioorg. Chem., 2021, 108, 104638 CrossRef CAS PubMed.
- M. Taha, H. Ullah, L. M. R. Al Muqarrabun, M. N. Khan, F. Rahim, N. Ahmat, M. Ali and S. Perveen, Eur. J. Med. Chem., 2018, 143, 1757–1767 CrossRef CAS PubMed.
- M. Taha, M. S. Baharudin, N. H. Ismail, S. Imran, M. N. Khan, F. Rahim, M. Selvaraj, S. Chigurupati, M. Nawaz, F. Qureshi and S. Vijayabalan, Bioorg. Chem., 2018, 80, 36–42 CrossRef CAS PubMed.
- M. Taha, H. Ullah, L. M. R. Al Muqarrabun, M. N. Khan, F. Rahim, N. Ahmat, M. T. Javid, M. Ali and K. M. Khan, Bioorg. Med. Chem., 2018, 26, 152–160 CrossRef CAS PubMed.
- R. Pandey, K. V. Swamy and M. B. Khetmalas, Ind. J. Biotech., 2013, 12, 297–310 CAS.
- L. Minati, T. Edginton, M. G. Bruzzone and G. Giaccone, Am. J. Alzheimer's Dis. Other Dement., 2009, 24, 95–121 CrossRef PubMed.
- A. Alzheimer's, Alzheimers. Dement., 2015, 11, 332–384 CrossRef PubMed.
- M. P. Mattson, Nature, 2004, 430, 631–639 CrossRef CAS PubMed.
- Z. R. Owczarczyk, W. A. Braunecker, A. Garcia, R. Larsen, A. M. Nardes, N. Kopidakis, D. S. Ginley and D. C. Olson, Macromolecules, 2013, 46, 1350–1360 CrossRef CAS.
- A. European Food Safety, EFSA J., 2012, 10, 2686 CrossRef.
- I. M. Padilla, I. Vidoy and C. L. Encina, Plant Cell Rep., 2009, 28, 1411–1420 CrossRef CAS PubMed.
- K. Gerth, R. Metzger and H. Reichenbach, Microbiology, 1993, 139, 865–871 CrossRef CAS.
- R. B. Mujumdar, L. A. Ernst, S. R. Mujumdar, C. J. Lewis and A. S. Waggoner, Bioconjug. Chem., 1993, 4, 105–111 CrossRef CAS PubMed.
- B. Stavric, B.-Y. Lau, T. I. Matula, R. Klassen, D. Lewis and R. H. Downie, Food Chem. Toxicol., 1997, 35, 185–197 CrossRef CAS PubMed.
- M. Suzui, M. Inamine, T. Kaneshiro, T. Morioka, N. Yoshimi, R. Suzuki, H. Kohno and T. Tanaka, Int. J. Oncol., 2005, 27, 1391–1399 CAS.
- J. Buback, M. Kullmann, F. Langhojer, P. Nuernberger, R. Schmidt, F. Wurthner and T. Brixner, J. Am. Chem. Soc., 2010, 132, 16510–16519 CrossRef CAS PubMed.
- P. Williams, A. Sorribas and M.-J. R. Howes, Nat. Prod. Rep., 2011, 28, 48–77 RSC.
- M. F. Saglam, M. Bingul, E. Şenkuytu, M. Boga, Y. Zorlu, H. Kandemir and I. F. Sengul, J. Mol. Struct., 2020, 1215, 128308 CrossRef CAS.
- M. Bingul, M. F. Saglam, H. Kandemir, M. Boga and I. F. Sengul, Monatsh. Chem., 2019, 150, 1553–1560 CrossRef CAS.
- B. Nisar, A. R. Raza, D. S. Black, N. Kumar and M. N. Tahir, Chirality, 2013, 25, 865–870 CrossRef CAS PubMed.
- S. L. Rubab, B. Nisar, A. R. Raza, N. Ullah and M. N. Tahir, Molecules, 2013, 19, 139–148 CrossRef.
- D. S. Black, M. C. Bowyer, P. K. Bowyer, A. J. Ivory, M. Kim, N. Kumar, D. B. McConnell and M. Popiolek, Aust. J. Chem., 1994, 47, 1741–1750 CrossRef CAS.
- D. S. C. Black, N. Kumar and L. C. H. Wong, Aust. J. Chem., 1986, 39, 15–20 CrossRef CAS.
- D. S. C. Black, B. Gatehouse, F. Theobald and L. C. H. Wong, Aust. J. Chem., 1980, 33, 343–350 CrossRef CAS.
- S. Tariq, A. R. Raza, M. Khalid, S. L. Rubab, M. U. Khan, A. Ali, M. N. Tahir and A. A. C. Braga, J. Mol. Struct., 2020, 1203, 127438 CrossRef CAS.
- B. Nisar, S. L. Rubab, A. R. Raza, S. Tariq, A. Sultan and M. N. Tahir, Mol. Diversity, 2018, 22, 709–722 CrossRef CAS PubMed.
- R. J. Kitz, L. M. Braswell and S. Ginsburg, Mol. Pharmacol., 1970, 6, 108–121 CAS.
- N. C. Inestrosa, A. Alvarez, C. A. Perez, R. D. Moreno, M. Vicente, C. Linker, O. I. Casanueva, C. Soto and J. Garrido, Neuron, 1996, 16, 881–891 CrossRef CAS PubMed.
- D. Barak, C. Kronman, A. Ordentlich, N. Ariel, A. Bromberg, D. Marcus, A. Lazar, B. Velan and A. Shafferman, J. Biol. Chem., 1994, 269, 6296–6305 CrossRef CAS.
- Z. Rakonczay, Acta Biol. Hung., 2003, 54, 183–189 CrossRef CAS.
- C. A. Barnes, J. Meltzer, F. Houston, G. Orr, K. McGann and G. L. Wenk, Neuroscience, 2000, 99, 17–23 CrossRef CAS PubMed.
- L. Na-Young and K. Young-Sook, Biomol. Ther., 2010, 18, 65–70 CrossRef.
- A. S. M. Ali Reza, M. S. Hossain, S. Akhter, M. R. Rahman, M. S. Nasrin, M. J. Uddin, G. Sadik and A. H. M. Khurshid Alam, BMC Complementary Altern. Med., 2018, 18, 123 CrossRef CAS PubMed.
- C. I. Wright, C. Guela and M. M. Mesulam, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 683–686 CrossRef CAS PubMed.
- R. Watanabe, T. Esaki, H. Kawashima, Y. Natsume-Kitatani, C. Nagao, R. Ohashi and K. Mizuguchi, Mol. Pharmaceutics, 2018, 15, 5302–5311 CrossRef CAS PubMed.
- M. A. Malkiewicz, A. Szarmach, A. Sabisz, W. J. Cubala, E. Szurowska and P. J. Winklewski, J. Neuroinflammation, 2019, 16, 15 CrossRef PubMed.
- P. Manikandan and S. Nagini, Curr. Drug Targets, 2018, 19, 38–54 CAS.
- E. Krieger and G. Vriend, Bioinformatics, 2014, 30, 2981–2982 CrossRef CAS PubMed.
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS PubMed.
- M. Hoenicka, Anal. Biochem., 1968, 25, 192–205 CrossRef.
- K. Gholivand, M. Abdollahi, F. Mojahed, A. M. Alizadehgan and G. Dehghan, J. Enzyme Inhib. Med. Chem., 2009, 24, 566–576 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05105b |
| This journal is © The Royal Society of Chemistry 2023 |