
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of non-covalent self-dimerization on the spectroscopic and electrochemical properties of mixed Cu(I) complexes†
Joaquín Cáceres-Vásquez a,
Danilo H. Jarab,
Juan Costamagnaaf,
Fabián Martínez-Gómezaf,
Carlos P. Silvaf,
Luis Lemusf,
Eleonora Freirecde,
Ricardo Baggioc,
Cristian Vera*a and
Juan Guerrero*a
a,
Danilo H. Jarab,
Juan Costamagnaaf,
Fabián Martínez-Gómezaf,
Carlos P. Silvaf,
Luis Lemusf,
Eleonora Freirecde,
Ricardo Baggioc,
Cristian Vera*a and
Juan Guerrero*a
aLaboratorio de Compuestos de Coordinación y Química Supramolecular, Facultad de Química y Biología, Universidad de Santiago de Chile, Av. Libertador Bernardo O'Higgins 3363, Estación Central, Casilla 40, Correo 33, Santiago, Chile. E-mail: juan.guerrero@usach.cl; cristian.veraoy@usach.cl
bFacultad de Ingenieria y Ciencias, Universidad Adolfo Ibáñez, Av. Padre Hurtado 750, Viña del Mar, Chile
cGerencia de Investigación y Aplicaciones, Centro Atómico Constituyentes, Comisión Nacional de Energía Atómica, Avenida Gral. Paz 1499, 1650, San Martín, Buenos Aires, Argentina. E-mail: baggio@tandar.cnea.gov.ar
dEscuela de Ciencia y Tecnología, Universidad Nacional de San Martín, Argentina and Gerencia de Investigación y Aplicaciones, Centro Atómico Constituyentes, Comisión Nacional de Energía Atómica, Buenos Aires, Argentina
eMember of CONICET, Argentina
fFacultad de Química y Biología, Universidad de Santiago de Chile, Av. Libertador Bernardo O'Higgins 3363, Estación Central, Casilla 40, Correo 33, Santiago, Chile
First published on 3rd January 2023
Abstract
A series of six new Cu(I) complexes with ([Cu(N-{4-R}pyridine-2-yl-methanimine)(PPh3)Br]) formulation, where R corresponds to a donor or acceptor p-substituent, have been synthesized and were used to study self-association effects on their structural and electrochemical properties. X-ray diffraction results showed that in all complexes the packing is organized from a dimer generated by supramolecular π stacking and hydrogen bonding. 1H-NMR experiments at several concentrations showed that all complexes undergo a fast-self-association monomer–dimer equilibrium in solution, while changes in resonance frequency towards the high or low field in specific protons of the imine ligand allow establishing that dimers have similar structures to those found in the crystal. The thermodynamic parameters for this self-association process were calculated from dimerization constants determined by VT-1H-NMR experiments for several concentrations at different temperatures. The values for KD (4.0 to 70.0 M−1 range), ΔH (−1.4 to −2.6 kcal mol−1 range), ΔS (−0.2 to 2.1 cal mol−1 K−1 range), and ΔG298 (−0.8 to −2.0 kcal mol−1 range) are of the same order and indicate that the self-dimerization process is enthalpically driven for all complexes. The electrochemical profile of the complexes shows two redox Cu(II)/Cu(I) processes whose relative intensities are sensitive to concentration changes, indicating that both species are in chemical equilibrium, with the monomer and the dimer having different electrochemical characteristics. We associate this behaviour with the structural lability of the Cu(I) centre that allows the monomeric molecules to reorder conformationally to achieve a more adequate assembly in the non-covalent dimer. As expected, structural properties in the solid and in solution, as well as their electrochemical properties, are not correlated with the electronic parameters usually used to evaluate R substituent effects. This confirms that the properties of the Cu(I) complexes are usually more influenced by steric effects than by the inductive effects of substituents of the ligands. In fact, the results obtained showed the importance of non-covalent intermolecular interactions in the structuring of the coordination geometry around the Cu centre and in the coordinative stability to avoid dissociative equilibria.
Introduction
The high versatility of transition metal compounds is a well-known fact, and therefore several studies have been oriented towards an adequate control of their properties through suitable ligand selection. In particular, Cu(I) coordination complexes with bidentate nitrogen donor ligands have been given considerable attention due to their interesting photophysical and electrochemical behaviour with potential applications in solar energy conversion, luminescence-based sensing, display devices, molecular switches, probes of biological systems, and catalysis.1 Also, these complexes have played an important role in the construction of sophisticated molecular architectures and contributed to the development and great growth of supramolecular chemistry2 and material science.3Cu(I) is a structurally labile metal centre which can experience ligand exchange and also distortions of its ideal tetrahedral geometry, where both coordinative and conformational labilities of the Cu(I) complexes are ascribed to the lack of crystal field stabilization due to its d10 full shell configuration.4 Such dynamic behaviour has a direct impact on the redox stability of cuprous complexes or the establishment of equilibria in solution.4,5
The inclusion of bulky substituents adjacent to the coordinating nitrogen atoms in homoleptic [Cu(NN)2]+ and heteroleptic [Cu(NN)(PPh3)2]+ complexes, with NN = symmetrical chelating diimines ligands, for example neocuproine (Fig. 1a), has been the first reported strategy to improve its photophysical performance by way of restricting the tetrahedral to the square-planar flattening, in accordance with the structural requirements of the metal, formally Cu2+ in the excited state. The possibility of Cu(I) complexes to reach a more flattened conformation is also responsible for their oxidation to cupric species in solution.4d,6
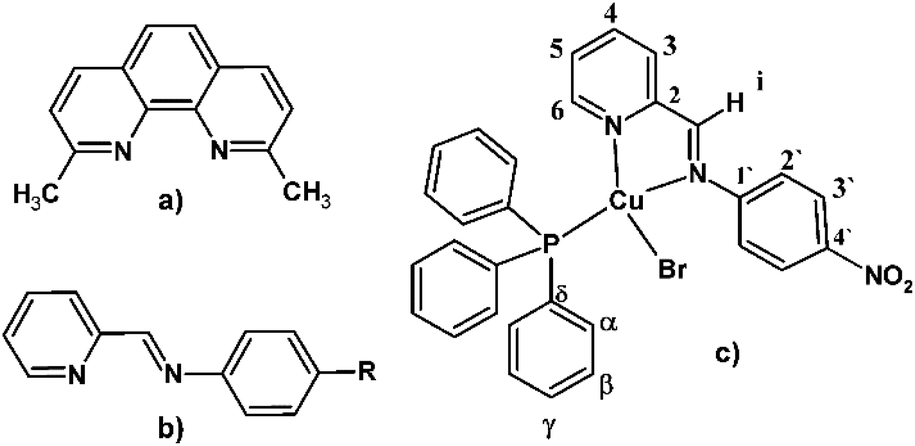 | ||
| Fig. 1 (a) Neocuproine. (b) Pyridine-imine type ligand. (c) General chemical structure of the [Cu(NN'-R)(PPh3)Br] complexes, including the labelling scheme used in the NMR assignments. | ||
Also, the influence of intramolecular non-covalent interactions on both chemical stabilization and electronic and emissive properties of Cu(I) complexes has been established, indicating that properties of Cu(I) complexes are not uniquely determined by structural effects of the ligand and that non-covalent interactions can play an important role on their behaviour.7
The presence of intermolecular π stacking aggregations in transition metal complexes with aromatic ligands has been well established in the solid state.8 However, the evidence of discrete dimers in solution is limited to a few examples.9 These structures have been identified by 1H-NMR in square-planar and octahedral metallic complexes where the supramolecular systems are supported by face–face (π–π) and face–edge (H⋯π) interactions between ligands possessing extensive aromatic systems.
On the other hand, bi-coordinating pyridine-imine type Schiff bases (Fig. 1b) are more flexible than polypyridines and rigid polyaromatic ligands and can be useful tools for the design of conformationally labile Cu(I) complexes that allow the establishment of intermolecular interactions. However, studies of pyridine-imine Cu(I) complexes as supramolecular materials as very limited, and most of the structural information on these systems has been obtained from studies regarding their catalytic,1p,q,10 and emissive properties.1a,10a,11
One of the few examples of supramolecular dimers of Cu(I) complexes with experimental evidence of self-association both in solution and in the solid phase by NMR spectroscopy and X-ray diffraction, respectively, has been reported for the mixed complex [Cu(N-{4-nitrophenyl}pyridine-2-yl-methanimine)(PPh3)Br],12 in which the imine ligand possess a small aromatic system (Fig. 2).
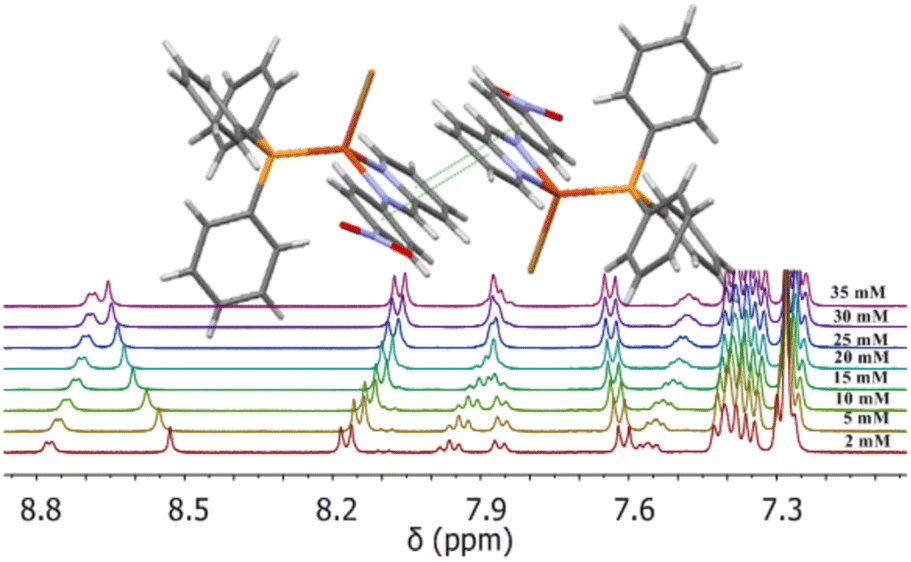 | ||
| Fig. 2 1H-NMR spectra of [Cu(N-{4-nitrophenyl}pyridine-2-yl-methan-imine)(PPh3)Br]2 complex as a function of concentration. (Insert) Scheme showing the dimer formed in the crystalline packing by both π–π and C–H⋯Br interactions around an inversion centre. | ||
For this complex, the monometallic structure corresponds to a slightly distorted tetrahedral complex. However, two non covalent forces, π-stacking and C–H⋯Br interactions are jointly acting to produce a discrete dimer. The NMR results showed the presence of an equilibrium between both monometallic and supramolecular bimetallic complexes. This dynamic behaviour in solution was interpreted as a self-dimerization process enthalpically driven through the formation of both π–π and C–H⋯Br interactions previously indicated where such a supramolecule retains a structure similar to that observed in the packing of its crystal structure.12
As a continuation of this work and with the aim of understanding more about this self-dimerization in neutral mixed Cu(I) complexes, a series of similar Cu(I) complexes, [Cu(NN'-R)(PPh3)Br], (PPh3 = triphenylphosphine, NN'-R = N-p-R-phenyl-pyridine-2-yl-me-thanimine, R = NO2, COCH3, Cl, H, CH3, OCH3 and N(CH3)2) were synthesized and characterized (Fig. 1c). These ligands include a series of substituents with different electronic characteristics in the para position so as not to cause a great conformational impact. The molecular structures and the presence of supramolecular dimers in the solid are discussed by interpretation of X-ray crystallographic data, while the evaluation of the structure and the presence of dimers in solution are studied by 1H-NMR. The dynamic behaviour of these systems was studied by 1H-NMR-VT for several concentrations and temperatures. The impact of this supramolecular behaviour on the electronic properties of this set of complexes is evaluated through electrochemical studies.
Results and discussion
Synthesis and characterization
The [Cu(NN'-R)(PPh3)Br] complexes were synthesized by the template condensation method from equimolar amounts of reagents as shown schematically in Scheme 1.2e,g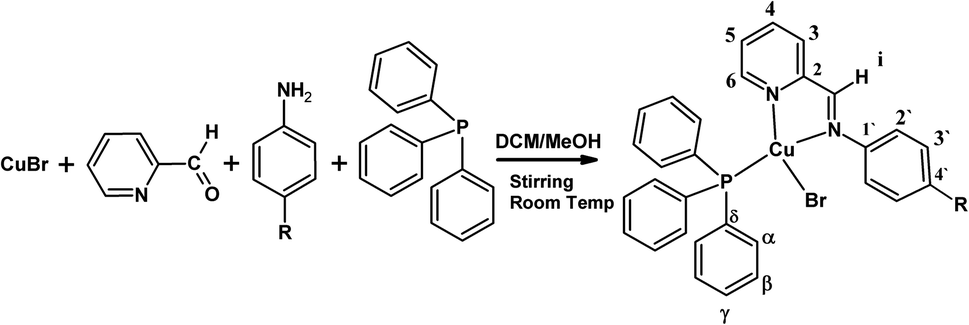 | ||
| Scheme 1 | ||
The structures of the complexes were unequivocally established by the concerted use of several NMR techniques from chloroform-d solutions, i.e. 1D NMR (1H-NMR, {1H}13C-NMR, {1H} 31P-NMR), and 2D NMR (COSY, HSQC, HMBC). The general and specific synthetic procedure and the NMR characterization data are reported in the Experimental section and ESI.†
Crystals of the complexes suitable for X-ray crystallographic analysis were obtained by slow ether diffusion into a dichloromethane solution of the complexes, and their crystal structures were solved for [Cu(NN'-R)(PPh3)Br] with R = COCH3, H, CH3, OCH3, and N(CH3)2. The crystal structure could not be obtained for the Cu(NN'-Cl)(PPh3)Br] complex, but its structure was determined by NMR techniques since all the complexes showed chemical stability in the time periods used for both NMR and electrochemical studies. All dynamic studies by 1H-NMR from chloroform solutions, and the electrochemical measurements were carried out from crystalline samples of the complexes. The accurate proton assignments for each temperature and concentration were confirmed by COSY experiments; in the same way, NOESY experiments for evaluating the structure of supramolecular adducts at some of the temperatures and concentrations used were carried out. The [Cu(N-p-NO2-phenyl-pyridine-2-yl-methanimine)(PPh3)Br] complex has been previously published, and the reported data were used for comparative analyses.12
Crystallographic structures
The structural drawings of the [Cu(N-p-dimethylamine-phenyl-pyridine-2-yl-methanimine)(PPh3)Br] complex are shown in Fig. 3 (a): displacement ellipsoid representation; (b): dimeric structure, and (c): packing organization. The same representations for the five other [Cu(NN'-R)(PPh3)Br] complexes are available as ESI (Fig. SI1a–c to SI5a–c†). Crystal and data collection parameters are available in Table SI1.† Selected coordination parameters are given in Table 1, while non-covalent interactions are given in Tables SI2 (π–π) and SI3† (C–H⋯X, X = Br, O, π). In these latter tables a #n code has been included to facilitate interaction identification and consequently, the packing description.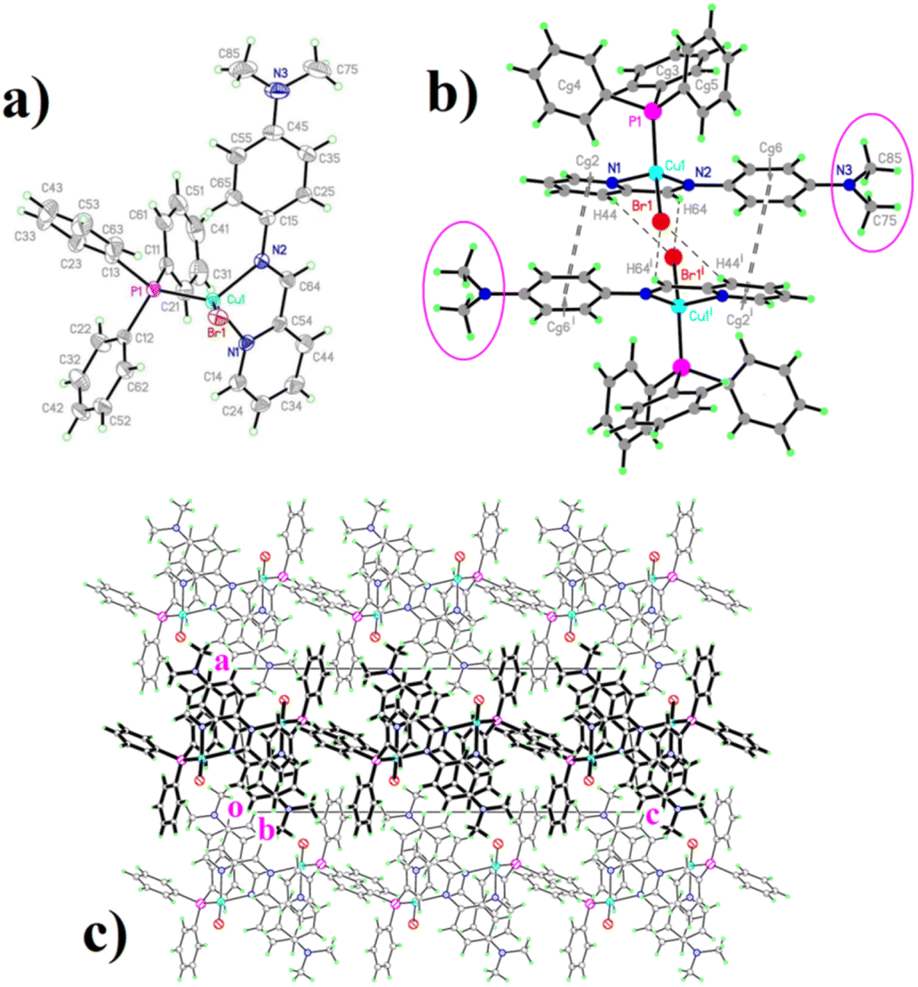 | ||
| Fig. 3 Molecular (a), dimers (b), and packing (c) diagrams for [Cu(NN′-N(CH3)2)(PPh3)Br] (a) displacement ellipsoids are drawn at a 30% level. (b) Bold dashed lines represent π–π bonds; simple dashed lines represent C–H⋯Br bonds. Circled is the N(CH3)2 substituent. (c) Viewed in projection, the (100) slabs generated by interaction #4 (One of these slabs has been highlighted for clarity). Non covalent interactions are omitted. | ||
| –R | (–NO2) | (–COCH3) | (–H) | (–CH3) | (–OCH3) | (–N(CH3)2) |
|---|---|---|---|---|---|---|
| Distances | ||||||
| Cu1–N1 | 2.096(3) | 2.115(5) | 2.1222(18) | 2.094(3) | 2.102(3) | 2.1015(17) |
| Cu1–N2 | 2.109(3) | 2.095(5) | 2.1169(18) | 2.101(3) | 2.097(2) | 2.1015(17) |
| Cu1–P1 | 2.1946(10) | 2.1833(16) | 2.1991(6) | 2.1848(9) | 2.1994(9) | 2.1906(6) |
| Cu1–Br1 | 2.421(6) | 2.4237(10) | 2.4376(4) | 2.4233(5) | 2.4499(5) | 2.3811(4) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Angles | ||||||
| N1–Cu1–N2 | 79.10(12) | 78.8(2) | 79.00(7) | 79.09(12) | 79.23(12) | 79.67(7) |
| N1–Cu1–P1 | 117.07(9) | 118.93(16) | 124.52(5) | 117.08(8) | 118.05(7) | 104.60(5) |
| N1–Cu1–Br1 | 106.34(8) | 105.63(16) | 102.94(5) | 106.16(7) | 112.15(7) | 113.56(5) |
| N2–Cu1–P1 | 124.56(9) | 127.36(16) | 119.59(5) | 122.42(7) | 125.12(7) | 112.02(5) |
| N2–Cu1–Br1 | 108.12(8) | 105.37(15) | 107.40(5) | 108.50(7) | 104.94(7) | 112.38(5) |
| P1–Cu1–Br1 | 115.45 (3) | 114.50(5) | 116.77(2) | 116.97(3) | 112.93(3) | 125.155(19) |
From a molecular point of view, the structures are quite similar (Fig. 3a and SI1a to SI5a†), with a tetracoordinate Cu(I) ion bonded to the corresponding pyridine-imine chelating ligand and the remaining coordination sites being completed by a PPh3 group and a bromine anion. Coordination bond distances and angles (Table 1) do not depart significantly from reported values in similar systems.10,11,14
In all complexes, the geometry around copper is that of a distorted tetrahedron, elongated along the common bisector of the small N–Cu–N angle and the larger P–Cu–Br one. The N–Cu–N angle is very similar to the one observed in complexes with polypyridine and pyridine-imine ligands (around 80°). As a reference, the bite angles of pyridine-imine ligands similar to those used in this work, in rhenium, rhodium, and iridium complexes are N–Re–N: 74.82,15 N–Rh–N: 79.38,16 N–Ir–N: 76.84.17
While the N–Cu–N angle remains relatively constant, the remaining coordination angles in the polyhedra show some differences through the series, but no correlation with the electronic effect of the substituent is observed.
The rather weak inter-molecular contacts are also similar in almost all complexes, and the most remarkable are the stacking contacts built around inversion centres of the phenyl/py fragments in the structures of all complexes with the exception of the py/py stacking exhibited by the [Cu(NN'-H)(PPh3)Br] complex (Table 2), which in all cases lead to the formation of some kind of weakly linked dimeric units (Fig. 3b and SI1b to SI4b, Table SI2†).
| R | Spatial groups | τ 4 | Selected torsion parameters in the monometallic units of complexes/° | Selected distances for intra dimer units of complexesa/Å | Selected interdimer distances/Å | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Orthogonal plane angles to metal centre | NCuN angle | NCCN torsion angle | Intraligand dihedral angle | Intra dimer⋯π–π⋯stacking | Br⋯H3a | Br⋯Hia | Br⋯H2a | Cu–Cu intra dimer | Br–Br intra dimer | Br⋯H4 | Br⋯H5 | Ph3⋯Hp-Ph3 π⋯T shape stacking | |||
| a Numbering of protons corresponds to that used for NMR assignment. b π–π stacking interaction. c π⋯T shape interaction, shown in the Fig. SI5b. | |||||||||||||||
| –NO2 | C2/c | 0.8388 | 88.88 (PCuBr,N2CuN1) 76.60 (BrCuN1,N2CuP) 73.49 (BrCuN2,N1CuP) | 79.07 | −0.1(6) | 4.63 | 3.738 | 3.127 | 3.020 | 3.550 | 6.69 | 7.438 | 2.896 | 3.431 | 3.380 |
| –COCH3 | C2/c | 0.8048 | 88.04 (PCuBr,N2CuN1) 74.34 (BrCuN1,N2CuP) 73.61 (BrCuN2,N1CuP) | 79.09 | 2(1) | 11.22 | 3.846 | 3.215 | 3.146 | 3.755 | 6.85 | 7.520 | 2.942 | 3.414 | 3.268 |
| –H | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
0.8219 | 86.15 (PCuBr,N2CuN1) 75.53(BrCuN2,N1CuP) 72.48 (BrCuN1,N2CuP) | 79.01 | −5.2(3) | 30.15 | 3.647b | 2.829c | 8.04 | ||||||
| –CH3 | C2/c | 0.8444 | 89.69 (PCuBr,N2CuN1) 73.49 (BrCuN1,N2CuP) 73.83 (BrCuN2,N1CuP) | 79.08 | 1.9(5) | 21.57 | 3.837 | 3.207 | 3.181 | 4.016 | 6.71 | 7.644 | 2.960 | 3.567 | 3.460 |
| –OCH3 | P |
0.8282 | 83.93 (PCuBr,N2CuN1) 75.15 (BrCuN1,N2CuP) 70.13 (BrCuN2,N1CuP) | 79.21 | −2.9(5) | 1.88 | 3.569 | 3.465 | 3.002 | 3.256 | 6.62 | 7.529 | 3.266 | 3.786 | 3.142 |
| –N(CH3)2 | P21/c | 0.8599 | 86.18 (PCuBr,N2CuN1) 76.43 (BrCuN1,N2CuP) 73.57(BrCuN2,N1CuP) | 79.65 | 3.5(3) | 18.57 | 3.814 | 3.165 | 3.102 | 3.589 | 6.38 | 7.636 | 3.097 | 3.158 | 2.796 |
Even if these π–π interactions between extended aromatic systems (shown in double dashed lines) are the main ones responsible for the dimeric organization, there are some further weak C–H⋯Br contacts (drawn in simple dashed lines) that reinforce this inter-molecular linkage. The way in which this is achieved is quite similar in five of these six structures, either through the C44–H44⋯Br1 or the C64–H64⋯Br1 contacts, or eventually through both.
Contrasting with these five structures, the dimer in the [Cu(NN'-H)(PPh3)Br] structure appears different, held together by a strong face–face py/py (π–π) and two extensive face-edge (CH⋯π) interactions18 (Fig. SI5b,†Table 2). Furthermore, this structure is the only one with a significant intra-molecular C–H⋯Br interaction.
The C–H⋯Xneighbour interactions left (X = Br, O, π), characterized in Table SI3† by the remaining sym-codes (i), are inter-dimeric in nature, and result in the formation of a diversity of packing motives shown in Fig. 4c and SI1c to SI5c of the ESI.†
Regarding the way in which this is achieved, there are some common features grouping structures together. Thus, structures for complexes with R = NO2, COCH3 and CH3, are nearly isostructural, and this shows up in the packing, where the C34–H34⋯Br1′ interactions (#4 in [Cu(NN'-NO2)(PPh3)Br] and [Cu(NN'-COCH3)(PPh3)Br], #3 in [Cu(NN'-CH3)(PPh3)Br]), give rise to columns directed along b, while the remaining ones link these 1D substructures transversally (along c) to form one-dimer-thick 2D arrays parallel to (100) (Fig. SI1c to SI3c†).
On the other hand, structures [Cu(NN'-OCH3)(PPh3)Br] and [Cu(NN'-H)(PPh3)Br] follow a similar pattern, in spite of the differences in the dimer's shape: there is a leading interaction (#3 and #5 in Table SI3†) defining columns along b, and the remaining ones linking them along c in [Cu(NN'-H)(PPh3)Br] and along a in [Cu(NN'-OCH3)(PPh3)Br], to define planar arrays parallel to (100) and (001), respectively (Fig. SI4c and SI5c†).
Finally, the structure of [Cu(NN'-N(CH3)2)(PPh3)Br] is slightly different, with interaction #4 (Table SI3†) defining “per se” a 2D array parallel to (100), the remaining ones interlinking these slabs into a complex 3D structure (Fig. 3c).
The eventual effect of the para-substituents on the phenyl ring over the dimeric structures was analyzed by comparison in the series of some relevant molecular parameters (e.g.), the interligand dihedral angles between phenyl and pyridine aromatic rings, the NCCN ligand torsion angles in the coordination environment, the N–Cu–N coordination angles, the τ4 parameters,19 and the angles between coordination planes (Table 2).
Even if these parameters differ somehow along the series, there is no clear trend ascribable to the electronic characteristics of the para-substituents groups, suggesting that this effect is not the only determinant of the final structure of the dimers, and these differences are probably due to one global influence of these substituents (steric, electronic, and their ability to generate non covalent interactions) i.e., they act in an unpredictable way over molecular structure as well as over dimer assembly and crystal packing.
The most important variation corresponds to the intraligand dihedral angle in the imine ligand. In this respect, inspection of Table 2 shows that those compounds with an extended stacking interaction present the smallest distortion, while compound [Cu(NN'-H)(PPh3)Br] (the one with an unsubstituted phenylimine fragment and one single π–π interaction), shows the largest distortion with a dihedral angle >30°.
In order to find out if this possible relationship (viz., the one between the extended stacking interaction and a reduction in the out-of-plane dihedral rotation in the imine ligand) was more general, we performed a systematic search in the CSD,20 looking for transition metal complexes similar to those reported here. For this purpose, the compounds considered were restricted to have a four-coordinate transition metal bound to an NN′-R imine ligand with an eventual para R substituent, not longer than four (non-H) atoms in length. A total of 54 structures of that sort were found in the CSD, and the resulting histograms of the number of appearances as a function of the distortion angle are presented in Fig. 4, where the blue entries correspond to complexes without extended stacking and the red ones to those with centrosymmetric dimeric structures.
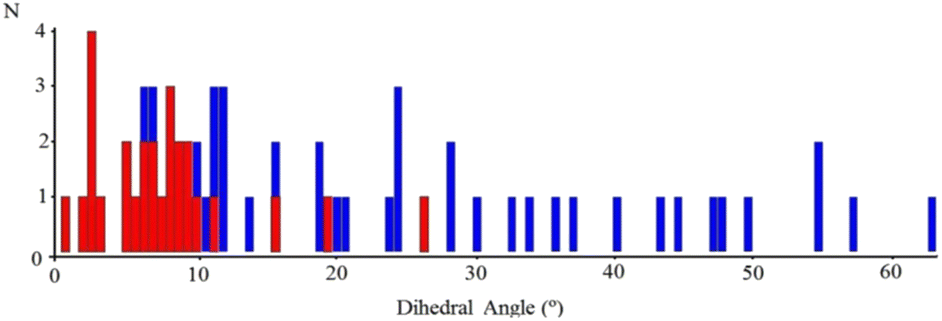 | ||
| Fig. 4 Histograms of the number of appearances as a function of the distortion angle of the NN′-R imine ligand in similar transition metal complexes. Red entries correspond to complexes with centrosymmetric dimeric structures. Blue entries to complexes where no extended stacking is present. Obtained from the CSD. | ||
These results strongly suggest a tendency to coplanarity of those NN′-R imine ligands that exhibit dimeric supramolecular interactions in the crystalline assembly, or, in other words, that due to the inter-molecular attractive force produced in the dimer, the imine ligand is forced to a more planar arrangement than expected for an ideal tetrahedral symmetry in single, not dimeric complexes.
Characterization of complexes and its self-assembly structures in solution by NMR spectroscopy
The 1H-NMR spectra of all complexes exhibit only one set of narrow signals which shifted as a function of concentration as shown in Fig. 5 and SI6a to SI12a.† These results indicate the presence of chemical exchange in solution, thus, the observed 1H NMR signals should be the consequence of the weighted average between more than one species in fast exchange on the NMR time scale at all the temperatures investigated (eqn (1)).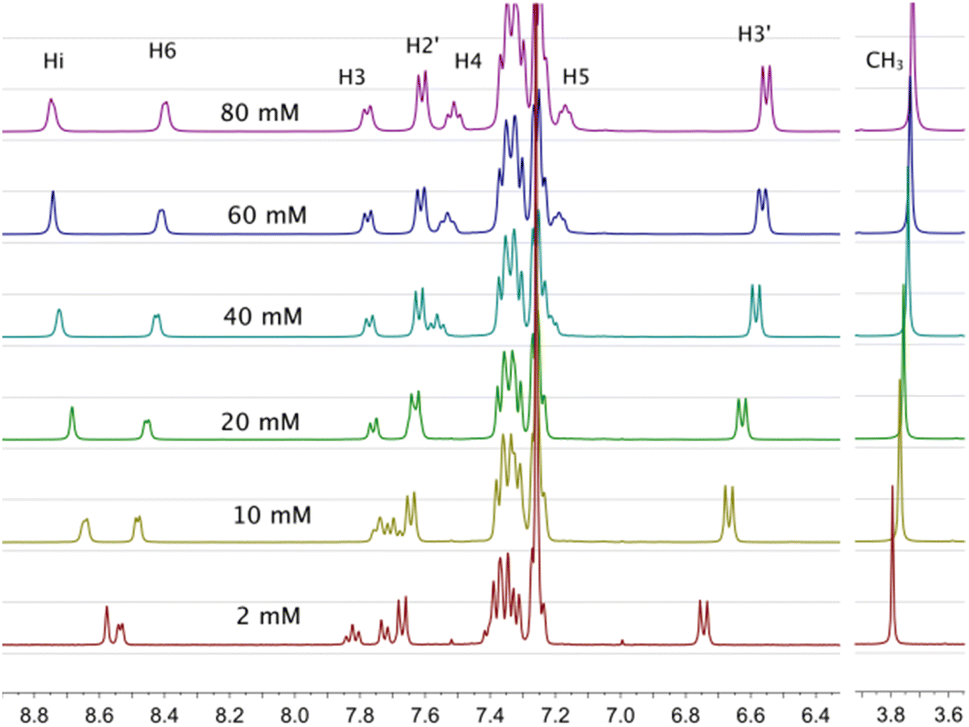 | ||
| Fig. 5 1H-NMR spectra of [Cu(NN′-OCH3)(PPh3)Br], T = 220 K, for several concentrations used to calculate self-association constants. Proton assignment is included. Similar behaviors were observed at different temperatures (see Tables SI3a to g†). | ||
In a previous paper we described the same behaviour for [Cu(NN′-NO2)(PPh3)Br] complex and established the presence of a self-dimerization phenomenon in solution which involves a fast equilibrium between a monomer complex and a supramolecular dimer supported by π–π stacking and C–H⋯Br interactions12 (eqn (2)). An analysis similar to those used in the above paper allowed us to establish that the same behaviour is followed by the series of complexes whose study we are reporting now.
| δ0 = λMδM + λDδD | (1) |
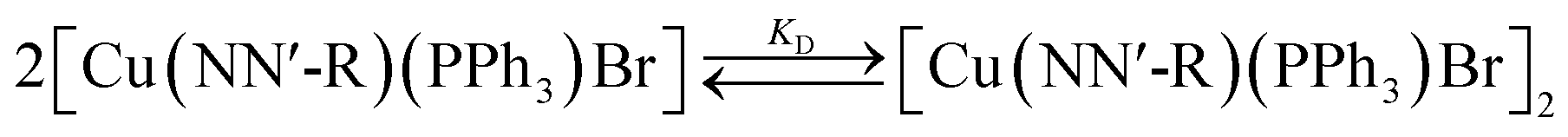 | (2) |
The exponential dependence between the change in the resonance frequency of each proton (Δδ) and the molar analytical concentration of the monomer is observed for most protons of this series of compounds (Fig. 6). Similar behaviour is seen for all the complexes at the different working temperatures studied, as shown in Fig. 6 for [Cu(NN′-COCH3)(PPh3)Br] and [Cu(NN′-Cl)(PPh3)Br] (As similar in ESI, Fig. SI6b, SI7b, c, SI8b, c, SI10b, c, SI11b, c and SI12c†).
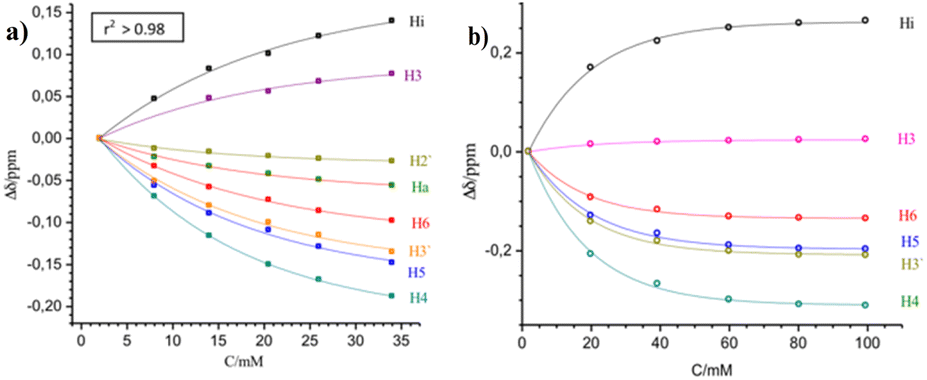 | ||
| Fig. 6 Concentration effect on the chemical shifts of the NN'-R ligand protons for the [Cu(NN′-COCH3)(PPh3)Br] complex at 235 K (a) and for [Cu(NN′-Cl)(PPh3)Br] at 220 K. (b) Δδ = δi − δ0, where δi correspond to δ observed at any concentration and δ0 corresponds to the lowest concentration measured. | ||
Dimerization was also confirmed by the presence of unexpected NOEs between protons which are distant from one another in the monomer structure. Indeed, NOESY crosspeaks can be observed between some protons of pyridine fragment (H3, H4, H5, H6) with the H2′ and H3′ protons of the 4-R-phenyl moiety of the NN′-R ligands (Fig. SI13†). These NOESY correlations are possible only when two ligands are assembled in an anti-conformation relative to an inversion centre placed between both complex units in a similar way to that in the crystal structures. This assembly of ligands resembles the one seen in the crystal packing and allows to establish that there is a great analogy between the assembly of the dimeric aggregates in the solid and those seen in the solution.
The exception is the [Cu(NN′-H)(PPh3)Br] complex, which presents the same behaviour as the rest of the complexes in solution, but showing a different assembly in the crystal (vide supra). We can explain this behaviour considering that the noncovalent interactions through which these dimers pack in the solid are substituted by complex-solvent interactions in solution. These forces lead to self-dimerization of Cu(NN′-H)(PPh3)Br] in the same structural shape as the other complexes.
This dimerization assembling symmetry does not change the proton signals pattern of the NN'-R ligand in the dimer relative to the monomer but modifies the proton chemical environments changing their resonance frequencies. This is well established by the resonance dependence on both concentration and temperature in the 1H-NMR spectra (Fig. 5, 6, and corresponding Fig. SI6 to SI12†).
As a result, increasing concentration produces progressive displacements of the resonance frequencies of the NN′-R ligand protons both, upfield as to downfield for the whole series of complexes at all the temperatures studied. In all the complexes, the H4, H5, H6, and H3′ protons of the NN′-R ligands show the more significant shift of proton frequencies to upfield (−Δδ) as the complex's concentration increases (Fig. 6 and corresponding SI figures).†
This behaviour is the expected one for a self-association process through “face to face” π type stacking interaction between two nearby NN-R ligands, due to the fact that these protons are placed close to the shielding currents of the aromatic rings of another ligand in the supramolecular [Cu(NN′-R)(PPh3)Br]2 dimers.21
On the contrary, a progressive downfield shift (+Δδ) occurs for Hi when the complex's concentration is increased. This deshielding effect is associated with the C–H⋯Br interaction,22 considering that this proton is closer to the bromine ligand in the dimeric complexes, in the same way as in the crystalline structure (Fig. 3b, vide supra).
Thus, the resonance frequency of each proton is determined mainly by two non-covalent forces which cause an opposite effect in the observed chemical shifts in the process of self-dimerization, i.e., AH⋯Br and π–π stacking. However, the relative contribution of these forces to the resonance frequency will be different for each proton depending on its position in the dimeric assembly.
However, H2′ and H3 protons move slightly towards lower or higher resonance frequencies, with temperature dependence (Tables SI3a to SI3g†) and not only toward the low fields as expected by their positions close to bromine in the crystalline packing. In those cases, we conclude that these protons undergo a compensatory influence of both π–π stacking and Br⋯H interactions and also suggest the presence of a large structural dynamism in the dimer.
The dimerization constants (KD) of the self-dimerization equilibrium between a monomer and a dimeric supramolecular complex (eqn (2)) in CDCl3 can be determined by the curve fitting method described by Horman and Dreux (eqn (3)),23a where the observed δ for a specific proton is a function of the analytical monomer concentration [M]0.
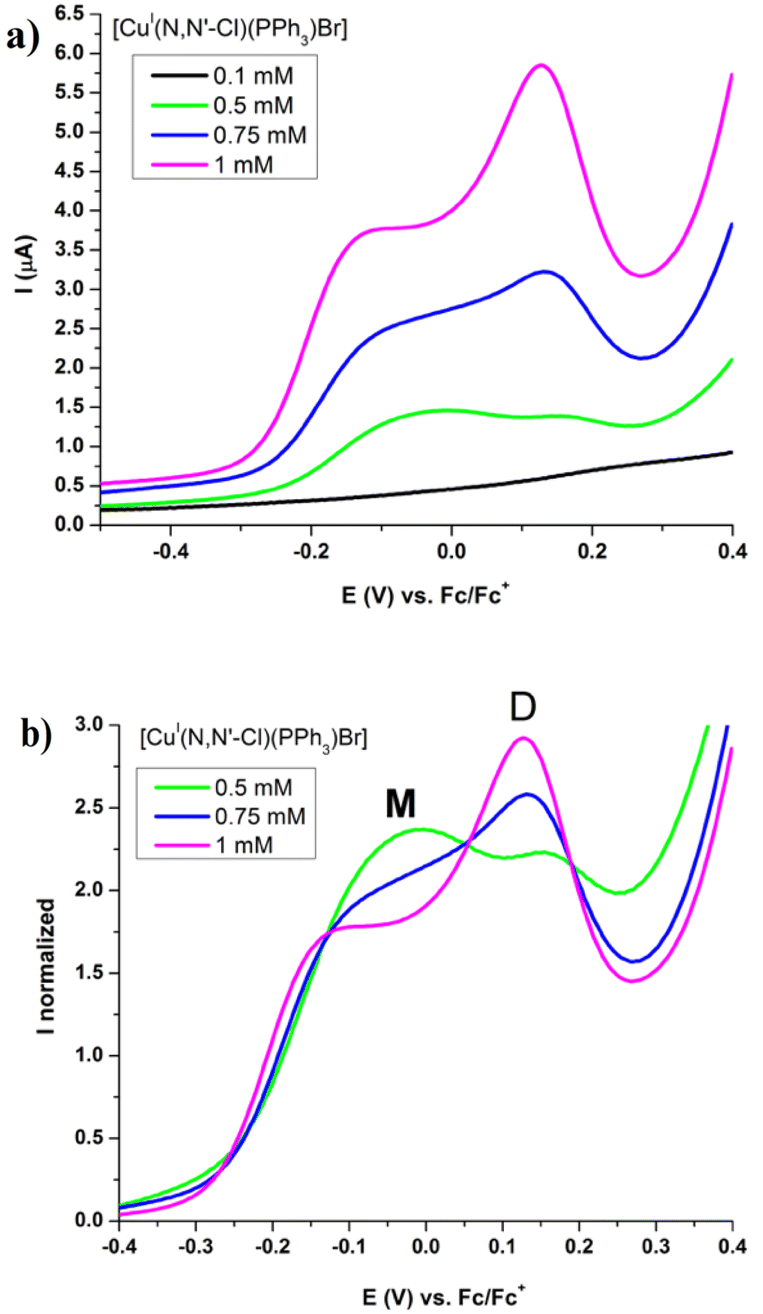
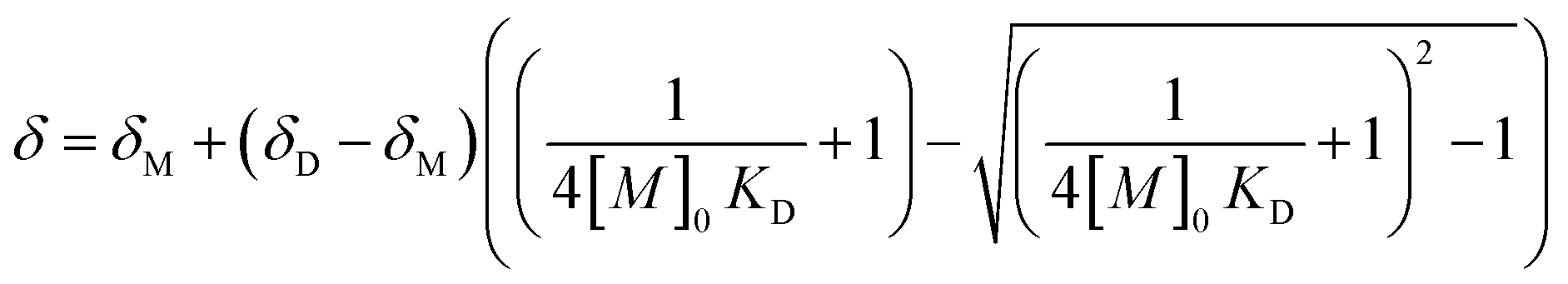 | (3) |
In this equation, δM and δD are the chemical shifts of the monomer and the dimer, respectively.
Using this self-dimerization model, we determine a set of KD values, one for each of protons of the NN'-R ligands in each complex with good fitting values.21,24
We consider getting a representative KD value for each complex averaging only the constants of the two protons that reach the highest |Δδ| in the measured concentration range (i.e. the greater −Δδ and +Δδ values at the highest concentrations) on the assumption that these protons should be influenced only by one of both assembly forces (AH⋯Br and stacking); but in fact, larger KD values were obtained for protons that exhibit lower |Δδ| as the concentration increases (see Tables SI3a to SI3g†).
However, this is a commonly observed phenomenon in several compounds that experience self-association in solution.9 As in those works, we decided to obtain a representative KD values by averaging all calculated KD's at each temperature, taking care that they were not very far from one another (Tables SI3a to g†). In addition, KD values for protons that remain almost unperturbed by the opposite effect of noncovalent forces were not considered, for example, the H2′ and H3 protons.
We associate this behavior with the self-association equilibrium involving simultaneous rearrangements of the NN'-R ligand coplanarity (see X-ray) and the metal coordination environment (see electrochemical). Thus, both internal molecular rearrangements would be the cause of the variability found in the ΔδH parameter and consequently, in the values of KD for different protons in each complex.
The KD average values for all complexes are given in Table 3, in the 210 to 298 K range and their values at all temperatures, are in the range 4 to 70 M−1. These values are in the same range or just slightly higher than those seen for non-covalent organic dimers formed by π stacking depending on how extended of their aromatic systems.21 Also, they have the same KD values range reported for a few examples of self-dimerization in octahedral metal complexes where the intermolecular π–π stacking between the ligands is also dependent of fused ring extension.9b,g However, they are smaller than self-association constants exhibit by square planar complexes, but in these, their planarity and free apical positions allow a better intra-ligand π stacking and favour other types of non-covalent interactions such cation-ligand or metal–metal to yielder larger aggregate species.9e,24,25
| –R | T/K | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 220 | 235 | 250 | 265 | 280 | 298 | ΔH kcal−1 mol−1 | ΔS cal mol−1 | ΔG298 kcal−1 mol−1 | |
| a Data from ref. 12. | |||||||||
| –NO2a | −2.00 | −0.67 | −1.79 | ||||||
| –COCH3 | 21.4 ± 3.0 | 20.2 ± 3.1 | 19.0 ± 3.1 | 12.8 ± 3.0 | 11.7 ± 2.0 | 9.7 ± 3.7 | −1.43 | −0.19 | −1.37 |
| –H | 9.0 ± 2.8 | 6.3 ± 1.4 | 6.4 ± 1.3 | 4.6 ± 0.5 | 4.0 ± 0.7 | 4.0 ± 0.8 | −1.40 | −2.09 | −0.77 |
| –CH3 | 67.4 ± 22.7 | 47.5 ± 1.9 | 43.5 ± 7.0 | 36.6 ± 7.7 | 31.7 ± 12.1 | 29.3 ± 15.3 | −1.35 | 2.08 | −1.97 |
| –OCH3 | 35.2 ± 9.0 | 21.3 ± 3.5 | 20.1 ± 12.5 | 14.4 ± 4.9 | 9.3 ± 2.2 | 8.2 ± 2.9 | −2.43 | −4.02 | −1.23 |
| –N(CH3)2 | 24.3 ± 4.0 | 16.4 ± 2.3 | 12.4 ± 2.8 | 11.2 ± 4.3 | 9.3 ± 2.0 | 9.3 ± 1.9 | −1.62 | −1.25 | −1.25 |
| –Cl | 69.4 ± 11.1 | 47.0 ± 3.5 | 31.3 ± 2.2 | 25.4 ± 4.7 | 18.6 ± 2.1 | 15.0 ± 2.4 | −2.58 | −3.35 | −1.58 |
As expected, these values decrease in each complex as temperature increases. Thus, from the dependence of the KD on temperature were determine the thermodynamic parameters ΔH and ΔS associated with the self-dimerization of [Cu(NN′-R)(PPh3)Br] complexes from van't Hoff plots (Fig. 7 and Table 3). The linear fitting of all [Cu(NN′-R)(PPh3)Br] complexes (R2 values over 0.89) are in good agreement with the presence of a single thermodynamic process for the self-dimerization assembly in the studied temperature range (Fig. 7).
 | ||
| Fig. 7 van't Hoff plot for complexes in range of work temperature, 220 to 298 K. | ||
Although the R2 = 0.89 in [Cu(NN′-COCH3)(PPh3)Br] can be considered a good linear fitting value for this system, two temperature regions with different slopes can also be assumed from the van't Hoff plot (Fig. 7). This behavior has been attributed to the thermodynamic phase change in the assembly geometry in supramolecular organic systems.21b,27 However, we find that the values of the thermodynamic parameters determined from each slope are not very far from each other. Therefore, we conclude that the association symmetry of the complex in the dimer remains unchanged in all temperature ranges, which is coherent with the frequency variation behaviour described previously (−Δδ and +Δδ). We assume that the slightly low linear correlation is caused by the versatility of the non-covalent interactions to adapt the assembly shape to the different structural requirements of both monomer and dimer in a fast exchange.
The values of ΔH (range −1.4 to −2.6 kcal mol−1), ΔS (range −0.2 to 2.1 cal mol−1 K−1), and ΔG298 (range −0.8 to −2.0 kcal mol−1) are of the same order for all complexes and are consistent with a spontaneous dimerization process enthalpically driven through the formation of both π–π and C–H⋯Br interactions (Table 3).
Electrochemical properties
Cyclic voltammetry (CV) and square wave voltammetry (SWV) experiments were performed pointing to the characterization of the effect of self-dimerization equilibrium observed by NMR (vide supra), on the electrochemical behavior of Cu(I) complexes in dichloromethane solutions. Unlike NMR, the electrochemical study was carried out in CH2Cl2 instead of CH3Cl because decomposition was observed in short periods of time under electrochemical conditions.To validate the use of this solvent in electrochemical studies, the 1H-NMR spectrum of the [Cu(NN′-OCH3)(PPh3)Br] complex in CD2Cl2 was performed to compare with those observed in CDCl3 finding a high similarity between their spectral profiles (Fig. SI13h to i†). Subsequently, we evaluated the effect of ionic strength on the stability of the supramolecular self-association equilibrium, for which we carried out 1H-NMR spectra measurements for both CDCl3 and CD2Cl2 solution of the Cu(I) complexes at concentrations similar to those used in electrochemical studies with increasing concentrations of supporting electrolyte (Fig. SI13j and k†). The results showed, (a) no spectral evidence of signals of species structurally different from those established in the NMR section (vide supra), (b) the presence of a chemical exchange process (c) that this chemical exchange follows a behavior similar to that observed without the electrolyte, (d) that the proton signals are shift as expected for an increasing of self-dimerization with an increase in ionic strength.
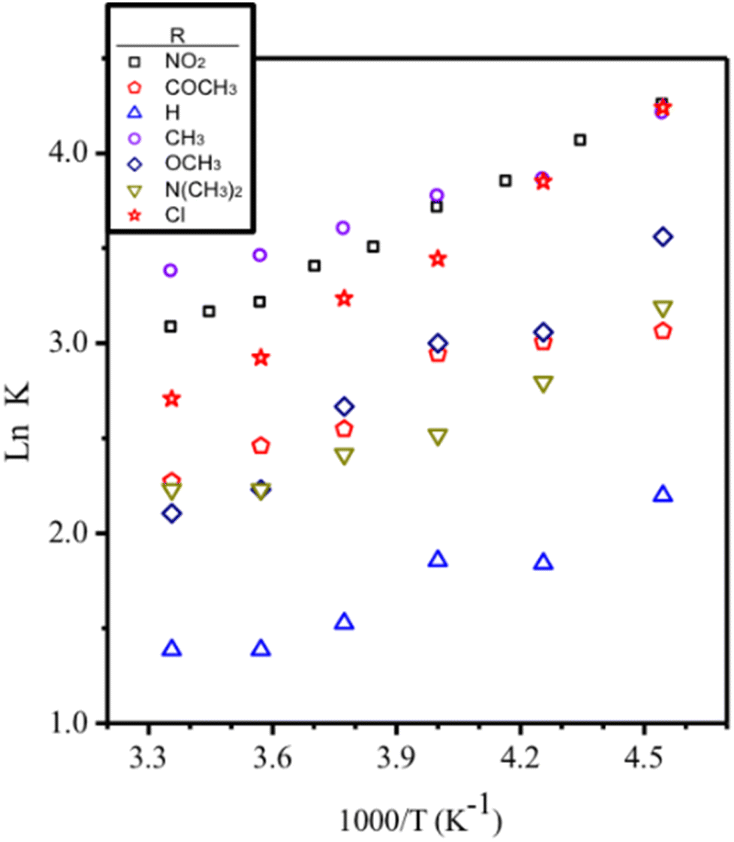
Both CV and SWV results show a similar electrochemical profile for all copper(I) complexes between −0.7 to +0.4 V potential range versus Fc/Fc+ process like internal reference. These correspond to two not well-defined redox processes at anodic sweep and one process at cathodic sweep (Fig. 8 and Table 4). The redox processes are electrochemically irreversible (ΔEp > 120 mV and Ipa/Ipc ≠ 1) and are diffusion controlled (see SI14 to SI17†).
 | ||
| Fig. 8 Cyclic voltammetry and square wave voltammetry of [Cu(NN′-N(CH3)2)(PPh3)Br] 1.0 mM, in CH2Cl2 and 0.1 M TBAP. SR = 50 mV s−1 at CV and 15 Hz to SWV. | ||
| –R | SWV. E/mV | CV. E/mV | ||||
|---|---|---|---|---|---|---|
| E pa M | E pa D | E pc | E pa M | E pa D | E pc | |
| a Sh: shoulder, EpaM: anodic peak potential of monomer, EpaD: anodic peak potential of dimer, Epc: cathodic peak potential. | ||||||
| –N(CH3)2 | −137 | 110 | −133 | Sh | Sh | −215 |
| –OCH3 | Sh | 138 | −181 | Sh | 182 | −232 |
| –CH3 | Sh | 142 | −101 | Sh | 196 | −210 |
| –H | Sh | 130 | −193 | Sh | 198 | −245 |
| –Cl | Sh | 126 | −113 | Sh | 172 | −165 |
| –COCH3 | Sh | 186 | −101 | Sh | 217 | −164 |
| –NO2 | Sh | 170 | −73 | Sh | 228 | −129 |
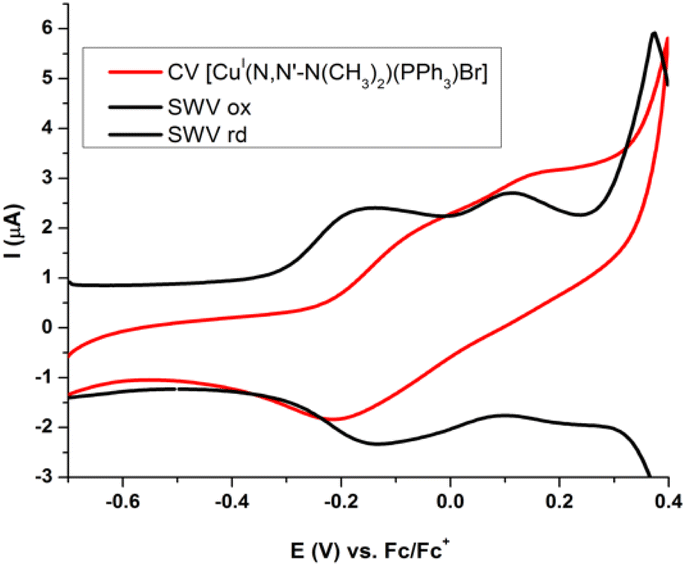
As expected, in SWV experiments an increase of the intensities for both anodic peaks as the initial concentration of complexes increases are seen. However, when these curves are normalized by the absolute areas (total charge for both processes), it can be seen that the second peak increases its intensity while the first one decreases as concentration increases (Fig. 9 and SI19†).
In this way, the dependence of the ratio of two anodic peak-areas with the initial concentration of complex can be ascribed to the monomer–dimer equilibrium established by NMR studies (vide supra) in solution.
The electrochemical results show similar peak intensities for monomer and dimer, suggesting that their concentrations are of the same order of magnitude (Fig. 8). On the contrary, the association constants obtained by NMR in CDCl3 without support electrolyte, indicate that the monomer concentration is more than one hundred times greater that of the dimer in solutions of same concentration as those used in electrochemical studies. This agrees with what was previously established through 1H-NMR spectra where medium's ionic strength displaced the equilibrium to the dimer (Fig. SI13j and k†).26
Since equilibrium tends toward dimer formation when the concentration of the complexes is increased, the first anodic peak at the electrochemical process can be assigned to monomer and the higher anodic peak to the oxidation of the supramolecular dimer (Fig. 9 and SI19†).
 | ||
| Fig. 9 (top a) SWV of [Cu(NN′-Cl)(PPh3)Br] in CH2Cl2, 0.1 M TBAP, 15 Hz, at different initial concentrations. (down b) SWV normalized at absolute area for [Cu(NN′-Cl)(PPh3)Br], at different initial concentrations. M = monomer; D = dimer. | ||
Unexpectedly, both monomer and dimer have different redox features, which make possible their detection by electrochemical methods. These electrochemical differences between monomer and dimer may be interpreted in terms of the structural lability of the Cu(I) center, allowing a structural rearrangement around this cation, due to the presence of previously discussed non-covalent interactions (π stacking and hydrogen bonding).
This structural lability has been associated with a d10 full electron configuration layer which does not provide crystal field stabilization of their complexes.4,30a Consequently, in the literature reports it is easily noted that the structures of Cu(I) complexes are far from corresponding to tetrahedral symmetry, and may take practically all four-coordinate structures, pseudo-tetrahedral, trigonal pyramidal, see-saw, and pseudo-square-planar.28
Since the oxidation of Cu(I) to Cu(II) is associated with a geometric flattening of the molecule from pseudotetrahedral to square or square pyramidal, this process would occur with more positive oxidation potentials in complexes with a more tetrahedral coordination geometry assuming no other significant electronic effects. Indeed, in a previous work we observed in [Cu(biq)(Ph2P-(CH2)n)-PPh2]ClO4 (biq = 2,2-biquinoline) complexes the dependence of both Cu(II)/Cu(I) potential and the MLCT band with the Cu(I) coordination geometry.28a We have also reported evidence of the ability of π-stacking intramolecular interactions to modulate a more tetrahedral geometry of a bimetallic helicate with respect to the corresponding bimetallic mesocate4c and the HOMO destabilization by the effect of intermolecular noncovalent interactions in a supramolecular adduct {[Cu(biq)2]-biq}.9a
So, considering that the anodic peaks of the dimer are located ca. 200 mV toward higher potential than those of the monomers in all complexes, it is possible to conclude that as self-assembly product, the ligands are forced towards a most orthogonal distribution among them in the dimer relative to the monomer.29 The τ4 crystal parameter for the complexes that are packed as supramolecular dimers, exhibit values over 0.8, indicating a metal centre structure closer to a tetrahedron.
The electrochemical irreversibility observed for the first oxidation process can be explained considering that this process is followed by a structural change, which is caused by the differences in the preferred coordination symmetry of copper ions, where Cu(I) complexes are closer to a tetrahedral symmetry, while Cu(II) prefers a more square-planar symmetry.30
Correspondingly, the dominant chemical irreversibility for the second Cu(II)/Cu(I) process can be explained considering that the anodic sweep could produce a Cu(II) dimer D2+ which undergoes flattening and loss of supramolecular interactions (H⋯Br and π-stacking) due to the very rapid generation of Cu(II) monomers M+, as shown in Fig. 10. In fact, a single dominant cathodic process instead, the cathodic peak associable to D2+ reduction is very faint in all complexes (Fig. 8 and SI14 to SI17†).
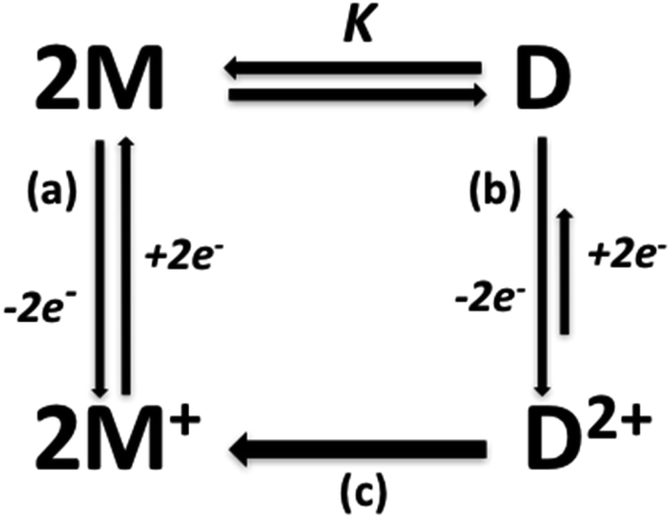 | ||
| Fig. 10 Proposed mechanism for electrochemical process. (a) Electron transfer followed by conformational changes, (b) electron transfer, (c) conformational change and loss of supramolecular interactions. | ||
Hammett parameter
Interested in rationalizing the electronic effects of peripheral p-substituents on the different properties of complexes, we plotted Hammett parameters for R (in NN'-R ligands)31versus several experimental parameters. For crystal data, Hammett parameters were plotted vs. τ4, intraligand dihedral angle, intradimer π–π stacking distance, and NCCN torsion angle. In the same way, we plotted Hammett parameters vs. KD constants at several temperatures and vs. ΔS and ΔH thermodynamic parameters obtained by NMR. Correlations were also investigated for the potential of Cu(II)/Cu(I) processes for dimer and monomer species. In all cases, poor correlation values (r2) were obtained. Thus, the strength and geometry of the assembly cannot be explained in terms of the electronic effect of the p-substituent. However, it can be rationalized by considering the cooperative contribution of both π–π stacking and Br⋯H interactions, while the electronic effect is usually determinant when only π–π interactions occur are predominant.Experimental
Materials
All reactions were carried out under purified nitrogen (99.9%, Linde-Chile S.A.). The diethyl ether was dried and distilled according to standard procedures prior to use, and the other solvents were synthesis grade and used as received. The pyridine-2-carbaldehyde, 4-chloroaniline, 4-aminophenol, 4-nitroaniline, 4-anilineacetophenone, N,N-dimethyl-1,4-phenylenediamine (Merck) and 4-methoxyaniline, 4-methylaniline (Aldrich) were used as purchased. CuBr was prepared according to the reported procedure.32 The [Cu{N-(4-nitrophenyl)pyridine-2-yl-methanimine} (PPh3)Br] complex was previously reported.12The specific synthetic procedures are reported in ESI.†
Instrumental
Preparation of complexes
The new series of [Cu(NN′-R)(PPh3)Br] complexes were synthesized by the template condensation method from equimolar amounts of reagents.2e,g,13To a dichloromethane/methanol (10/1) mixture solution of CuBr, dichloromethane/methanol solution of pyridine-2-carbaldehyde was added at room temperature, and after continuous stirring for 20 min, corresponding 4-substituted-aniline in the same solvents mixture was added and the solution mixture was stirred for 30 min. Subsequently, a solution of triphenylphosphine was added dropwise and stirred for 1 more hour at room temperature, forming a colored solution. The volume of solution was reduced in a rotary evaporator and the concentrate was precipitated with diethyl ether and washed with 2 × 5 mL of a diethyl ether/dichloromethane (9:1) mixture, and finally with 5 mL of diethyl ether.
The diffusion of diethyl ether vapour into a concentrated dichloromethane solution gave colored crystals ranging from orange to purple red which were adequate for crystallographic studies. Also, only crystallized materials were used for the spectroscopic and electrochemical studies.
General and specific synthetic procedure and NMR characterization data are reported in the experimental section and the ESI.†
Calculation of dimerization constants (KD)
The values of KD were determined by the method of Hormann and Dreux23a which relies on the gradual variation in the 1H-NMR chemical shifts as a function of concentration at constant temperature. The procedure involves an iterative KD, by fitting the observed chemical shift (δobs) of each proton using the mole fraction of dimer (eqn (3)) present at each concentration, starting from a reasonable guess of the association constant. The most accurate value of KD is defined as that which yields the best linear relationship between δobs and the molar fraction (xi)21,24 (Tables SI3a to g†).VT-NMR dimerization experiments
Solutions of complexes at different concentrations (in ranges of 2 to 100 mM according to each complex) were prepared in flasks of 1 and 2 mL with CDCl3 (Aldrich). Proton NMR spectra for all solutions were recorded between 220 and 298 K at 15 K intervals. Each measurement was recorded after thermal equilibrium was established (ca. 3 min). The KD values obtained at all temperatures were used to determine the thermodynamic parameter using the van't Hoff plot.Hammett constants (σ) for substituents in the p-position of the N-phenyl ring of the imine ligands used in this work correspond to those published elsewhere.31
Conclusions
The whole series of mixed neutral Cu(I) complexes studied crystallize in increasing organization, i.e. a tetrahedral distorted complex whose structure in a supramolecular dimer in turn packed in a crystal where the non-covalent interactions, π–π, and C–H⋯Br play the main role. The importance of these interactions is expressed in an enthalpy driven process of self-dimerization in solution between the monomer complex and discrete non-covalent dimers with a structure similar to that described in solid. The lack of correlation between Hammett constants of the p-substituents in N-phenyl fragments and several parameters obtained from NMR, X-ray, and electrochemical studies can be explained from the nature of the C–H⋯Br and π–π interactions. Since these interactions are more permissive of a spatial reordering of interacting fragments with respect to a covalent bond, they can remain linking the two complexes in a wide range of dimer conformations. Therefore, the noncovalent self-dimerization can involve the evolution of dimer from py/py assemblies toward one phenyl/py type in [Cu(NN′-H)(PPh3)Br].On the other hand, the significant Cu(II)/Cu(I) potential differences between both monomers and dimer complexes are consistent with the structurally labile character of Cu(I), demonstrating that this ion can adapt its coordination geometry to structural requirements of supramolecular interactions. Thus, both the C–H⋯Br and π–π, interactions that assemble the complexes in the dimers, modify the coordination symmetry of Cu(I) towards more tetrahedral geometry stabilizing the HOMO of the dimer with respect to the monomer.
Consequently, here we provide an experimental background of the remarkable influence of supramolecular interactions on the structure and properties of Cu(I) complexes through their ability to modulate the metal coordination symmetry and the coordinative stability to avoid dissociative equilibria. In addition, these results allow us to hypothesize that through supramolecular structuring it is possible to simultaneously obtain both effects, decrease the conformational mobility and increase the chemical stability of Cu(I) complexes as an alternative to intramolecular architectural molecular designs, which is the most researched strategy.
Author contributions
Conceptualization: JG, CV, RB. Investigation, experimental design, and formal analysis: JG, CV, RB, EF, JC-V, DHJ. Experimental data collection: DHJ, JC-V, FM, CV, CPS, EF, RB. Validation: JG, CV, RB, JC, LL. Funding acquisition: JG, CV. Writing (original draft): JG, CV, RB. Writing (review & editing): JG, CV, RB, JC, LL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of DICYT-USACH, FONDECYT 1101029, and FONDEQUIP-EQM 150106. C. V. acknowledges support from postdoctoral grant FONDECYT 3170661, J. C.-V. acknowledges a CONICYT Doctoral Fellowship 21141067.Notes and references
- (a) B. Bozic-Weber, E. C. Constable, C. E. Housecroft, M. Neuburger and J. R. Price, Dalton Trans., 2010, 39, 3585 RSC; (b) C. Cunningham, J. Moore, K. Cunningham, P. Fanwick and D. McMillin, Inorg. Chem., 2000, 39, 3638 CrossRef CAS PubMed; (c) M. Miller, P. Gantzel and T. Karpishin, Inorg. Chem., 1998, 37, 2285 CrossRef CAS PubMed; (d) M. Meyer, A. Albrecht-Gary, C. Dietrich-Buchecker and J. Sauvage, Inorg. Chem., 1999, 38, 2279 CrossRef CAS; (e) M. Eggleston, P. Fanwick, A. Pallenberg and D. McMillin, Inorg. Chem., 1997, 36, 4007 CrossRef CAS; (f) A. Pallenberg, K. Koenig and D. Barnhart, Inorg. Chem., 1995, 34, 2833 CrossRef CAS; (g) N. Navon, G. Golub, H. Cohen, P. Paoletti, B. Valtancoli, A. Bencini and D. Meyerstein, Inorg. Chem., 1999, 38, 3484 CrossRef CAS PubMed; (h) D. Scaltrito, D. Thompson, J. O'Callaghan and G. Meyer, Coord. Chem. Rev., 2000, 208, 243 CrossRef CAS; (i) D. Schultz, F. Biaso, A. Shahi, M. Geoffroy, K. Rissanen, L. Gagliardi, C. Cramer and J. R. Nitschke, Chem.–Eur. J., 2008, 14, 7180 CrossRef CAS PubMed; (j) M. Miller and T. Karpishin, Inorg. Chem., 1999, 38, 5246 CrossRef CAS; (k) N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, Top. Curr. Chem., 2007, 280, 69 CrossRef CAS; (l) Y. Liu, S. Yiu, C. Ho and W. Wong, Coord. Chem. Rev., 2018, 375, 514 CrossRef CAS; (m) S. Brauchli, F. Malzner, E. Constable and C. Housecroft, RSC Adv., 2014, 4, 62728 RSC; (n) M. Nishikawa, S. Kume and H. Nishihara, Phys. Chem. Chem. Phys., 2013, 15, 10549 RSC; (o) M. Nishikawa, Y. Takara, Y. Hattori, K. N omoto, T. Kusamoto, S. Kume and H. Nishihara, Inorg. Chem., 2013, 52, 8962 CrossRef CAS PubMed; (p) S. Sawant, G. Gaikwad, V. Sawant, B. Yamgar and S. Chavan, Inorg. Chem. Commun., 2009, 12, 632 CrossRef CAS; (q) G. Ardizzoia, S. Brenna, F. Castelli and S. Galli, Inorganica Chim. Acta, 2009, 362, 3507 CrossRef; (r) L. Ravaro, K. Zanoni and A. de Camargo, Energy Rep., 2020, 6, 37 CrossRef; (s) C. Smith, C. Branham, B. Marquardt and K. Mann, J. Am. Chem. Soc., 2010, 132(40), 14079 CrossRef CAS PubMed.
- (a) S. Saha, P. Biswas and M. Schmittel, Inorg. Chem., 2019, 58, 3466 CrossRef CAS PubMed; (b) J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci., 1987, 84, 2565 CrossRef CAS PubMed; (c) A. Goswami, S. Saha, P. Biswas and M. Schmittel, Chem. Rev., 2019, 120, 125 CrossRef PubMed; (d) S. Neogi, Y. Lorenz, M. Engeser, D. Samanta and M. Schmittel, Inorg. Chem., 2013, 52, 6975 CrossRef CAS PubMed; (e) J. Fan, J. Bats and M. Schmittel, Inorg. Chem., 2009, 48, 6338 CrossRef CAS PubMed; (f) S. Neogi, G. Schnakenburg, Y. Lorenz, M. Engeser and M. Schmittel, Inorg. Chem., 2012, 51, 10832 CrossRef CAS PubMed; (g) M. Schmittel and J. Fan, Org. Lett., 2011, 13, 3916 CrossRef CAS PubMed; (h) R. Forgan, J. Sauvage and J. Stoddart, Chem. Rev., 2011, 111, 5434 CrossRef CAS PubMed; (i) C. Piguet, M. Borkovec, J. Hamacek and K. Zeckert, Coord. Chem. Rev., 2005, 249, 705 CrossRef CAS; (j) M. Maekawa, T. Hayashi, K. Sugimoto, T. Okubo and T. Kuroda-Sowa, Inorganica Chim. Acta, 2019, 497, 119088 CrossRef CAS; (k) B. Schoentjes and J. M. Lehn, Helv. Chim. Acta, 1995, 78, 1 CrossRef CAS; (l) M. Schmittel, V. Kalsani and J. Bats, Inorg. Chem., 2005, 44, 4115 CrossRef CAS PubMed.
- (a) Y. Liu, Y. Ma, J. Yang, C. Diercks, N. Tamura, F. Jin and O. Yaghi, J. Am. Chem. Soc., 2018, 140, 16015 CrossRef CAS PubMed; (b) M. Saha, S. Pramanik and M. Schmittel, Chem. Commun., 2012, 48, 9459 RSC; (c) Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gándara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. Alshammari, X. Zhang, O. Terasaki and O. Yaghi, Science, 2016, 351, 365 CrossRef CAS PubMed; (d) C. Kepert, P. Southon, S. Brooker and R. Miller, Inorg. Chem., 2016, 55, 6195 CrossRef PubMed; (e) Q. L. Ni, X. F. Jiang, L. C. Gui, X. J. Wang, K. G. Yang and X. S. Bi, New J. Chem., 2011, 35, 2471 RSC; (f) Y. Liu, C. Diercks, Y. Ma, H. Lyu, C. Zhu, S. Alshmimri, S. Alshihri and O. Yaghi, J. Am. Chem. Soc., 2019, 141, 677 CrossRef CAS PubMed.
- (a) E. Leoni, J. Mohanraj, M. Holler, M. Mohankumar, I. Nierengarten, F. Monti, A. Sournia-Saquet, B. Delavaux-Nicot, J. Nierengarten and N. Armaroli, Inorg. Chem., 2018, 57, 15537 CrossRef CAS PubMed; (b) E. Riesgo, Y. Hu, F. Bouvier and R. Thummel, Inorg. Chem., 2001, 40, 2541 CrossRef CAS PubMed; (c) D. Díaz, L. Llanos, P. Arce, R. Lorca, J. Guerrero, J. Costamagna, D. Aravena, G. Ferraudi, A. Oliver, G. Lappin and L. Lemus, Chem.–Eur. J., 2018, 24, 13839 CrossRef PubMed; (d) A. Kaeser, M. Mohankumar, J. Mohanraj, F. Monti, M. Holler, J. Cid, O. Moudam, I. Nierengarten, L. Karmazin-Brelot, C. Duhayon, B. Delavaux-Nicot, N. Armaroli and J. Nierengarten, Inorg. Chem., 2013, 52, 12140 CrossRef CAS PubMed.
- (a) M. Miller, P. Gantzel and T. Karpishin, Angew. Chem., Int. Ed., 1998, 37, 1556 CrossRef CAS; (b) D. Datta and A. Chakravorty, Inorg. Chem., 1983, 22, 1085 CrossRef CAS; (c) L. Lemus, J. Guerrero, J. Costamagna, G. Estiu, G. Ferraudi, A. Lappin, A. Oliver and B. Noll, Inorg. Chem., 2010, 49, 4023 CrossRef CAS PubMed; (d) L. Lemus, J. Guerrero, J. Costamagna, R. Lorca, D. H. Jara, G. Ferraudi, A. Oliver and A. G. Lappin, Dalton Trans., 2013, 42, 11426 RSC.
- (a) M. Miller, P. Gantzel and T. Karpishin, J. Am. Chem. Soc., 1999, 121, 4292 CrossRef CAS; (b) C. Cunningham, K. Cunningham, J. Michalec and D. McMillin, Inorg. Chem., 1999, 38, 4388 CrossRef CAS PubMed; (c) M. Eggleston, D. McMillin, K. Koenig and A. Pallenberg, Inorg. Chem., 1997, 36, 172 CrossRef CAS; (d) M. Ruthkosky, F. Castellano and G. Meyer, Inorg. Chem., 1996, 35, 6406 CrossRef CAS PubMed; (e) D. McMillin, J. Kirchhoff and K. Goodwin, Coord. Chem. Rev., 1985, 64, 83 CrossRef CAS; (f) L. Llanos, C. Vera, A. Vega, D. Aravena and L. Lemus, Inorg. Chem., 2020, 59, 15061 CrossRef CAS PubMed.
- (a) P. Oguadinma, A. Rodrigue-Witchel, C. Reber and F. Schaper, Dalton Trans., 2010, 39, 8759 RSC; (b) T. Nicholls, C. Caporale, M. Massi, M. Gardiner and A. Bissember, Dalton Trans., 2019, 48, 7290 RSC; (c) J. Fan, Y. Zhang, K. Zhang, J. Liu, G. Jiang, L. Lin and C. Wang, Org. Electron., 2019, 71, 113 CrossRef CAS; (d) G. Filonenko, R. Fayzullin and J. Khusnutdinova, J. Mater. Chem. C, 2017, 5, 1638 RSC; (e) P. Papanikolaou, J. Mohanraj, A. Czapik, M. Gdaniec, G. Accorsi and P. Akrivos, Dalton Trans., 2013, 42, 3357 RSC; (f) M. Fraser, H. van der Salm, S. Cameron, J. Barnsley and K. Gordon, Polyhedron, 2013, 52, 623 CrossRef CAS; (g) L. Kohler, R. Hadt, D. Hayes, L. Chen and K. Mulfort, Dalton Trans., 2017, 46, 13088 RSC; (h) M. Fraser, H. van der Salm, S. Cameron, A. Blackman and K. Gordon, Inorg. Chem., 2013, 52, 2980 CrossRef CAS PubMed.
- (a) S. Kammer, A. Kelling, H. Baier, W. Mickler, C. Dosche, K. Rurack, A. Kapp, F. Lisdat and H. Holdt, Eur. J. Inorg. Chem., 2009, 31, 4648 CrossRef; (b) S. Kammer, I. Starke, A. Pietrucha, A. Kelling, W. Mickler, U. Schilde, C. Dosche, E. Kleinpeter and H. Holdt, Dalton Trans., 2012, 41, 10219 RSC; (c) M. Stollenz, H. Gehring, V. Konstanzer, S. Fischer, S. Dechert, C. Grosse and F. Meyer, Organometallics, 2011, 30, 3708 CrossRef CAS; (d) Y. Jahng, J. Hazelrigg, D. Kimball, E. Riesgo and F. Wu andR. P. Thummel, Inorg. Chem., 1997, 36, 5390 CrossRef CAS; (e) L. Zhang, B. Li and Z. Su, Langmuir, 2009, 25, 2068 CrossRef CAS PubMed; (f) S. Kuang, D. Cuttell, D. McMillin, P. Fanwick and R. Walton, Inorg. Chem., 2002, 41, 3313 CrossRef CAS PubMed.
- (a) N. Martínez, M. Isaacs, A. Oliver, G. Ferraudi, G. Lappin and J. Guerrero, Dalton Trans., 2018, 47, 13171 RSC; (b) D. Gut, A. Rudi, J. Kopilov, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2002, 124, 5449 CrossRef CAS PubMed; (c) N. De Tacconi, R. Chitakunye, F. MacDonnell and R. Lezna, J. Phys. Chem. A, 2008, 112, 497 CrossRef CAS PubMed; (d) R. Liu, M. Huang, X. Yao, H. Li, F. Yang and X. Li, Inorganica Chim. Acta, 2015, 434, 172 CrossRef CAS; (e) I. Kotzé, W. Gerber, J. Mckenzie and K. Koch, Eur. J. Inorg. Chem., 2009, 12, 1626 CrossRef; (f) S. Bergman, D. Reshef, S. Groysman, I. Goldberga and M. Kol, Chem. Commun., 2002, 2374 RSC; (g) S. Bergman, D. Reshef, L. Frish, Y. Cohen, I. Goldberg and M. Kol, Inorg. Chem., 2004, 43, 3792 CrossRef CAS PubMed.
- (a) S. Chavan, S. Sawant, S. Pawal and M. More, Polyhedron, 2016, 105, 192 CrossRef CAS; (b) S. Chavan, S. Sawant, V. Sawant and G. Lahiri, Inorg. Chem. Commun., 2011, 14, 1373 CrossRef CAS; (c) S. Chavan, S. Sawant, V. Sawant and G. Lahiri, Inorganica Chim. Acta, 2010, 363, 3359 CrossRef CAS; (d) D. Seth and S. Bhattacharya, Polyhedron, 2011, 30, 2438 CrossRef CAS; (e) D. Tzimopoulos, S. Brenna, A. Czapik, M. Gdaniec, A. Ardizzoia and P. Akrivos, Inorganica Chim. Acta, 2012, 383, 105 CrossRef CAS; (f) H. Zhao, B. Huang, Y. Wu and M. Cai, J. Organomet. Chem., 2015, 797, 21 CrossRef CAS.
- (a) S. Shakhatreh, J. Mohanraj, A. Czapik, D. Tzimopoulos, S. Kotoulas, M. Gdaniec and P. Akrivos, J. Mol. Struct., 2011, 1002, 51 CrossRef CAS; (b) T. Huang, J. Yan, H. Yang, H. Dum and M. Zhang, J. Coord. Chem., 2015, 68, 1514 CrossRef CAS; (c) T. Huang and M. Zhang, Aust. J. Chem., 2014, 67, 887 CrossRef CAS.
- D. Jara, L. Lemus, L. Farías, E. Freire, R. Baggio and J. Guerrero, Eur. J. Inorg. Chem., 2012, 10, 1579 CrossRef.
- (a) T. Ronson, S. Zarra, S. Black and J. Nitschke, ChemComm, 2013, 49, 2476 RSC; (b) M. Smulders, A. Jiménez and J. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681 CrossRef CAS PubMed; (c) K. Hänni and D. Leigh, Chem. Soc. Rev., 2010, 39, 1240 RSC.
- (a) S. Dehghanpour, A. H. Mahmoudkhani and M. Amirnasr, Struct. Chem., 2006, 17, 255 CrossRef CAS; (b) G. Saha, K. Sarkar, T. Mondal and C. Sinha, Inorganica Chim. Acta, 2012, 387, 240 CrossRef CAS; (c) A. Khalaji, S. Peyghoun, A. Akbari, N. Feizi, M. Dusek and V. Eigner, Polyhedron, 2016, 19, 429 CrossRef; (d) G. Saha, C. Patra, P. Datta, K. Sarkar, R. Saha, C. Chen and C. Sinha, Polyhedron, 2015, 87, 63 CrossRef CAS.
- (a) A. Hasheminasab, L. Wang, M. Dawadi, J. Bass, R. Herrick, J. Rack and C. Ziegler, Dalton Trans., 2015, 44, 15400 RSC; (b) A. Hasheminasab, J. Engle, J. Bass, R. Herrick and C. Ziegler, Eur. J. Inorg. Chem., 2014, 16, 2643 CrossRef; (c) R. Dominey, B. Hauser, J. Hubbard and J. Dunham, Inorg. Chem., 1991, 30, 4754 CrossRef CAS.
- (a) L. Gonsalvi, J. Gaunt, H. Adams, A. Castro, G. Sunley and A. Haynes, Organometallics, 2003, 22, 1047 CrossRef CAS; (b) D. Kanas, S. Geier, C. Vogels, A. Decken and S. Westcott, Inorg. Chem., 2008, 47, 8727 CrossRef CAS PubMed; (c) S. Mak, V. Yam, C. Che and T. Mak, J. Chem. Soc., Dalton Trans., 1990, 8, 2555 RSC.
- (a) R. Dhiman and C. Nagaraja, New J. Chem., 2019, 43, 13662 RSC; (b) J. Schöffel, N. Šušnjar, S. Nückel, D. Sieh and P. Burger, Eur. J. Inorg. Chem., 2010, 31, 4911 CrossRef; (c) G. Li, Y. Wu, G. Shan, W. Che, D. Zhu, B. Song, L. Yan, Z. Sua and M. Bryce, Chem. Commun., 2014, 50, 6977 RSC.
- (a) M. Nishio, CrystEngComm, 2004, 6, 130 RSC; (b) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 21, 3885 RSC; (c) C. Hunter, K. Lawson, J. Perkins and C. Urch, J. Chem. Soc., Perkin Trans. 1, 2001, 5, 651 RSC; (d) C. Hunter and J. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS; (e) M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757 RSC; (f) Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397 CrossRef CAS PubMed; (g) Y. Umezawa, S. Tsuboyama, K. Honda, J. Uzawa and M. Nishio, Bull. Chem. Soc. Jpn., 1998, 71, 1207 CrossRef CAS; (h) M. Nishio, Y. Umezawa, J. Fantini, M. Weiss and P. Chakrabarti, Phys. Chem. Chem. Phys., 2014, 16, 12648 RSC; (i) H. Brunner, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- L. Yang, D. Powell and R. Houser, Dalton Trans., 2007, 9, 955 RSC.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- (a) C. Xiao, M. Wu, Y. Zhang, X. Zhao, J. Yu and X. Zheng, Magn. Reson. Chem., 2014, 52, 460 CrossRef CAS PubMed; (b) H. Sun, K. Ye, C. Wang, H. Qi, F. Li and Y. Wang, J. Phys. Chem. A, 2006, 110, 10750 CrossRef CAS PubMed; (c) N. Marchettini, G. Valensin and E. Gaggelli, Biophys. Chem., 1990, 36, 65 CrossRef CAS PubMed; (d) A. Thakkar, L. Tensmeyer and W. Wilham, J. Pharm. Sci., 1971, 60, 1267 CrossRef CAS PubMed.
- (a) J. Del Bene, S. Perera and R. Bartlett, J. Phys. Chem. A, 1999, 103, 8121 CrossRef CAS; (b) K. Mizuno, T. Ochi and Y. Shindo, J. Chem. Phys., 1998, 109, 9502 CrossRef CAS; (c) R. Kaur, M. Ramesh, P. Bharatam and R. Kishore, J. Phys. Chem. B, 2014, 118, 9199 CrossRef CAS PubMed; (d) J. Purcell, H. Susi and J. Cavanaugh, Can. J. Chem., 1969, 47, 3655 CrossRef CAS; (e) M. Natalia, C. Zarycz and C. Fonseca, J. Phys. Chem. Lett., 2018, 9, 3720 CrossRef PubMed.
- (a) I. Horman and B. Dreux, Helv. Chim. Acta, 1984, 67, 754 CrossRef CAS; (b) R. Martin, Chem. Rev., 1996, 96, 3043 CrossRef CAS PubMed.
- K. Koch, C. Sacht and C. Lawrence, J. Chem. Soc., Dalton Trans., 1998, 4, 689 RSC.
- (a) I. Kotzé, W. Gerber, Y. Wu and K. Koch, Dalton Trans., 2013, 42, 3791 RSC; (b) V. Sivchik, E. Grachova, A. Melnikov, S. Smirnov, A. Ivanov, P. Hirva, S. Tunik and I. Koshevoy, Inorg. Chem., 2016, 55, 3351 CrossRef CAS PubMed.
- (a) V. Steullet and D. Dixon, J. Chem. Soc., Perkin Trans. 2, 1999, 1547 RSC; (b) H. Hwang, S. Lee, S. Lee and J. Park, J. Chem. Soc., Perkin Trans. 2, 1999, 1081 RSC.
- H. Sun, Y. Zhao, Z. Huang, Y. Wang and F. Li, J. Phys. Chem. A, 2008, 112, 11382 CrossRef CAS PubMed.
- (a) J. Guerrero, L. Cortez, L. Lemus, L. Farías, J. Costamagna, C. Pettinari, M. Rossi and F. Caruso, Inorganica Chim. Acta, 2010, 363, 380 Search PubMed; (b) M. Lazorski and F. Castellano, Polyhedron, 2014, 82, 57 CrossRef CAS; (c) M. Magni, A. Colombo, C. Dragonetti and P. Mussini, Electrochim. Acta, 2014, 141, 324 CrossRef CAS; (d) K. Kubicek, S. Veedu, D. Storozhuk, R. Kia and S. Techert, Polyhedron, 2017, 124, 166 CrossRef CAS.
- (a) T. Lan and W. Wang, Sens. Actuators, B, 2018, 254, 81 CrossRef CAS; (b) Y. Wang, S. Li, L. Wang, S. Qu and K. Liu, J. Lumin., 2017, 192, 269 CrossRef CAS; (c) Y. Zhao, Y. Van, D. Wang, L. Li and P. Qiu, J. Mol. Struct., 2019, 1181, 171 CrossRef CAS.
- (a) Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Coord. Chem. Rev., 2018, 356, 127 CrossRef CAS; (b) M. Mara, N. Jackson, J. Huang, A. Stickrath, X. Zhang, N. Gothard, M. Ratner and L. Chen, J. Phys. Chem. B, 2013, 117, 1921 CrossRef CAS PubMed; (c) S. Garakyaraghi, E. Danilov, C. McCusker and F. Castellano, J. Phys. Chem. A, 2015, 119, 3181 CrossRef CAS PubMed.
- (a) B. Jovanović, M. Mišić-Vuković, A. Marinković and V. Vajs, J. Mol. Struct., 2002, 642, 113 CrossRef; (b) D. McDaniel and H. Brown, J. Org. Chem., 1958, 23, 420 CrossRef CAS; (c) C. Hansch, A. Leo and R. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS; (d) R. Akaba, H. Sakuragi and K. Tokumaru, Bull. Chem. Soc. Jpn., 1985, 58, 1711 CrossRef CAS.
- (a) S. Bertz, E. Fairchild, I. Denissova and L. Barriault, Encyclopedia of Reagents for Organic Synthesis, 2001 Search PubMed; (b) R. Keller, H. Wrcoff and L. Marchi, Inorg. Synth., 1946, 2, 1 Search PubMed.
- G. M. Sheldrick, SHELXS97, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-2014, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. X-ray crystallographic files in cif format for the structures of the complexes of [Cu(NN′-R) (PPh3)Br] (R = NO2, COCH3, H, CH3, OCH3, N(CH3)2) have been deposited. CCDC 1559217–155922. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra05341a |
| This journal is © The Royal Society of Chemistry 2023 |