
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of the radical polymerization mechanism and its application in the preparation of high-performance PMMA by reactive extrusion†
Han Shi ,
Qixin Zhuang,
Anna Zheng*,
Yong Guan*,
Dafu Wei and
Xiang Xu
,
Qixin Zhuang,
Anna Zheng*,
Yong Guan*,
Dafu Wei and
Xiang Xu
Key Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China
First published on 6th March 2023
Abstract
In this study, the mechanism of radical polymerization was further explored by pre-dissolving different polymers and studying the kinetics of the bulk polymerization of methyl methacrylate (MMA) under shear-free conditions. Based on the analysis of the conversion and absolute molecular weight, it was found that, contrary to the shearing effect, the inert polymer with viscosity was the key factor to preventing the mutual termination of radical active species and reducing the termination rate constant kt. Therefore, pre-dissolving the polymer could increase the polymerization rate and molecular weight of the system simultaneously, making the polymerization system enter the automatic acceleration zone faster and greatly reducing the generation of small molecular weight polymers, leading to a narrower molecular weight distribution. When the system entered the auto-acceleration zone, kt decreased rapidly and greatly and entered the second steady-state polymerization stage. Then, with the increase in the polymerization conversion, the molecular weight gradually increased, while the polymerization rate gradually decreased. In shear-free bulk polymerization systems, kt can be minimized and radical lifetimes maximized, but the polymerization system is at best a long-lived polymerization rather than a living polymerization. On this basis, by using MMA to pre-dissolve ultrahigh molecular weight PMMA and core–shell particles (CSR), the mechanical properties and heat resistance of the PMMA with pre-dissolved polymer obtained by reactive extrusion polymerization were better than for pure PMMA obtained under the same conditions. Compared with pure PMMA, the flexural strength and impact strength of PMMA with pre-dissolved CSR were up to 166.2% and 230.5%. With the same quality of CSR, the same two mechanical properties of the samples obtained by the blending method were just improved by 29.0% and 20.4%. This was closely related to the distribution of CSR in the pre-dissolved PMMA-CSR matrix with a distribution of spherical single particles 200–300 nm in diameter, which enabled PMMA-CSR to exhibit a high degree of transparency. This one-step process for realizing PMMA polymerization and high performance shows extremely high industrial application prospects.
1. Introduction
There exists a particular auto-acceleration phenomenon in radical polymerization, with a significant increase in the polymerization rate and molecular weight at the same time, called the “Norrish–Smith1”, “Auto-acceleration”, or “Trommsdorff2” (gel) effect. The first widely accepted qualitative explanation was documented by Flory,3 indicating the termination rate constant (kt) dropped significantly in a medium with increasing viscosity.Viscosity plays a crucial role in radical polymerization. Polymethyl methacrylate (PMMA), polystyrene (PS), and polyvinyl alcohol (PVVA) were pre-dissolved in methyl methacrylate (MMA) or St solvent to prepare a mixture of monomers with high zero-shear viscosity by Brooks.4 Then, the kinetics were tested and the kinetics results revealed that the polymerization could be divided into two regions according to the viscosity, and until reaching the high-viscosity region, kt started to reduce. Therefore, during the initial stage of polymerization, the pre-dissolved polymers slightly influenced the termination rate constant kt, and the micro-viscosity directly, controlling the segmental diffusion and termination reaction at a low level. O'Shaughnessy5 pointed out that the termination rates were dominated by different short-long chain events. Long-chain mobility was reduced because of the high bulk viscosity and chain entanglements, while short chains provided a faster termination mechanism despite their small numbers, and the rate constant between a pair was determined by the short chains. More recently, fluorescent probes were introduced in polymer science and characteristic fluorescent macromolecular compounds or molecules were formed in order to investigate the segment and chain diffusion during polymerization at different viscosities.6,7 Also, the fluorescent spectra showed that the structural heterogeneity of the macro- and micro-radical chains had a significant impact on kt.
Considering the viscosity gradient effect, numerous experiments have been performed to determine the critical viscosity of the Trommsdorff effect. Dvornić8 controlled the molecular weight of products via utilizing the chain transfer agent dodecyl mercaptan during the suspension polymerization of MMA. The results revealed that the Trommsdorff effect was reduced with the increase in the chain transfer agent content. In addition, there existed a critical viscosity when the Trommsdorff effect happened and the value was independent of the growth chain length. In our previous study,9,10 polymerizations were performed under different shear rates, initiator concentrations, and temperature in a concentric-cylinder kinetic apparatus. It was concluded that 1000 ± 100 Pa s viscosity could be a criterion for the occurrence of the Trommsdorff effect.
Although the Trommsdorff effect is inhibited in conventional reactors as far as possible according to the characteristics of auto-catalyzation and uncontrollability, it can be fully utilized in reactive extrusion. Zhan and co-workers10 performed the free radical polymerization of MMA in a short residence time and utilized the Trommsdorff effect by adding a new monomer in the intermediate stage (the Trommsdorff effect region, as shown in Fig. 1b). The crucial viscosity in the occurrence of the Trommsdorff effect can be used to determine the content of extra added monomers. On the other hand, the conversion curve in Fig. 1a implied another method to utilize the Trommsdorff effect, by which polymerization was conducted with the initial viscosity, approaching crucial viscosity, via pre-dissolving polymers. As a result, the system would reach the Trommsdorff effect region beforehand and the polymerization rate would be significantly increased.
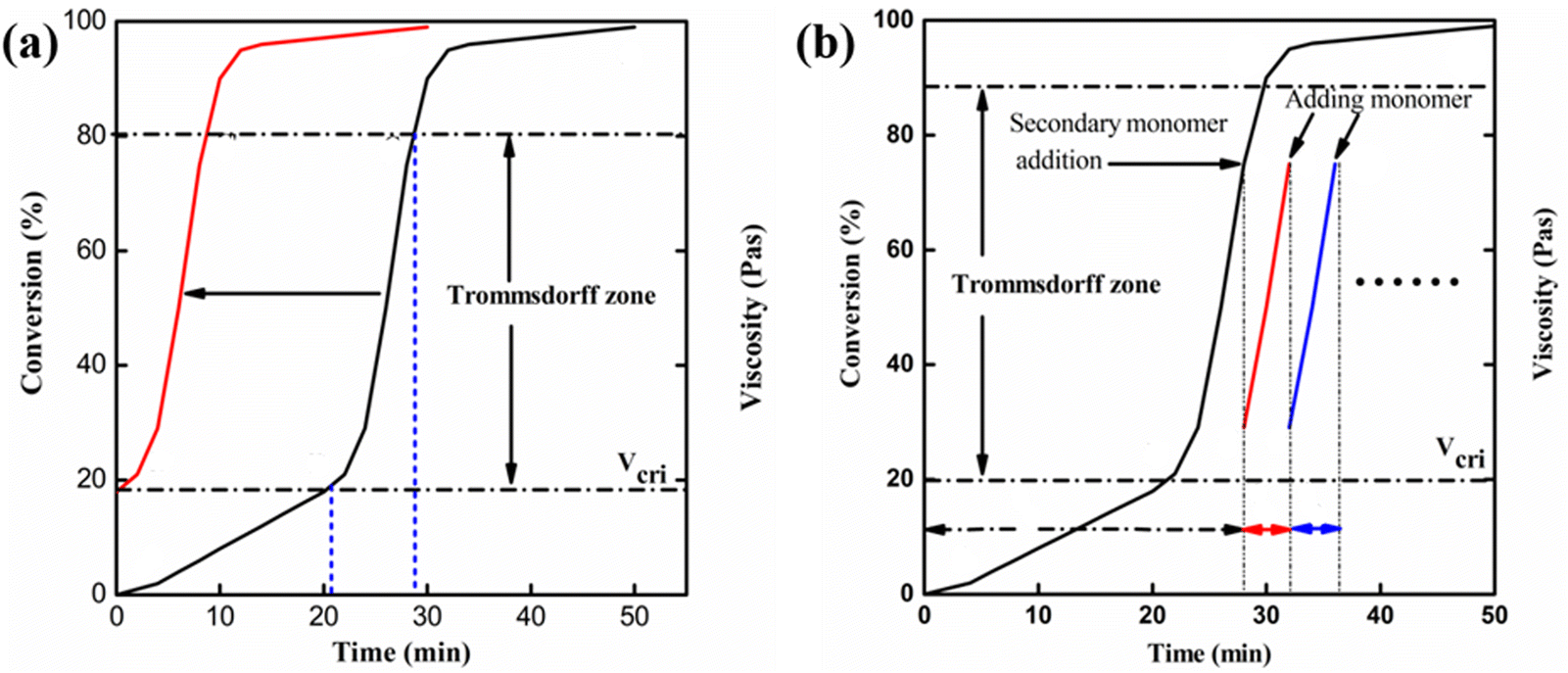 | ||
| Fig. 1 Schematic diagram for utilizing the Trommsdorff effect. | ||
PMMA is an important engineering plastic, with high transparency, excellent weather resistance, and a high dielectric constant and hardness.11–13 However, the application of PMMA is limited because of some poor mechanical performances, especially its impact resistance. Hence, the requirement for strengthened PMMA is significantly increasing in different industrial fields, such as construction, automotive, medical devices, lighting tools, and aerospace.14–17 Physical blending is one of the main methods for strengthening PMMA. Adding specific additives, like rubbers,18 nanoparticles,19,20 core–shell particles,21,22 carbon nanotubes (CNT),23 and graphene,24,25 is common in the physical blending method. It should be noted that the compatibility of additives with the matrix is especially important, and poor compatibility can not only decrease the effectiveness of modification, but can even weaken the strength of polymers.
Based on the above discussion, high molecular weight PMMA (Mn = 1.2 × 106, molecular weight distribution (MWD) ≈ 1.2) was chosen to build pre-dissolved polymer systems and to further strengthen PMMA. For toughening PMMA, core–shell particles (CSR) were introduced to a pre-dissolved polymers system. Despite CSR being widely used for toughening engineering plastics like PMMA, polycarbonate (PC) and ABS, the toughening mechanism could be attributed to shear yielding,26 crazing,27,28 rubber particles' cavities,29 and so on. However, in reactive extrusion, a one-step method could help reduce the costs and improve environmental protection. Also, the use of pre-dissolved polymers systems to improve the initial viscosity would help utilization of the available Trommsdorff effect and improve production efficiency.
In this work, the preparation of toughened PMMA with high performance was performed via the reactive extrusion of MMA containing pre-dissolved polymers. First, the kinetics of the free radical polymerization with pre-dissolved polymer was investigated. Then the “critical viscosity” at which point the Trommsdorff effect occurs during polymerization with pre-dissolved polymer is discussed. Also, polymerization with pre-dissolved polymer was carried out via reactive extrusion. Finally, the reactive extrusion products with pre-dissolved HWM-PMMA or CSR were characterized. This one-step method for preparing strengthened PMMA could shorten production cycles, and significantly reduce the costs and resource consumption, and consequently has important industrial application prospects.
2. Experimental section
2.1 Materials
Methyl methacrylate (MMA), analytical reagent, was provided by China's Oil Sunup Group Co., Ltd (China). Tetrahydrofuran, methanol, ethanol, and hydroquinone were analytical grade and supplied by Sinopharm Chemical Reagent Co., Ltd. AIBN analytical reagent was provided by Shanghai Ling Feng Chemical Reagent Co. Ltd. (Shanghai, China). Homo-polymethyl methacrylate (PMMA-C, VH001, Mn = 4.26 × 104, MWD = 1.61) was purchased from Mitsubishi Rayon Polymer Co., Ltd. (Nantong, China). High molecular weight PMMA (HWM-PMMA, Mn = 1.23 × 106, MWD = 1.2) was purchased from Taixing Donchamp Acrylic Co., Ltd. (Taixing, China). Core–shell particles (CSR) were purchased from Kaneka Chemical Industry Co., Japan, which comprised a shell of PMMA and elastic core of MBS. All the reagents were used as received.2.2 Kinetic experiments of the free radical bulk polymerization with pre-dissolved polymers
First, HWM-PMMA and PMMA-C were fully dissolved in MMA according to set mass ratios, respectively. The mixed monomers were poured into test tubes (about 5 ml per tube), and an initiator (AIBN, 0.7% wt) was added to fully dissolve the monomers, and the solution was then placed in a 40 °C water bath for polymerization. After each predetermined time, a small aliquot was pipetted from the polymer solution at the bottom of the tube. When the viscosity of the solution was too high to be pipetted out, the test tube was broken, and a small amount of the viscous solution at the bottom of the test tube was taken out as a sample. For the sample taken out, it was first accurately weighed, and quickly dissolved in tetrahydrofuran (THF) containing the terminator, then precipitated with methanol, filtered, and fully dried to obtain a refined polymer. After accurate weighing, the polymerization conversion was calculated. Finally, the absolute molecular weight of the sample was obtained by analysis in a waters 515 multi-detection gel permeation chromatography (MGPC) system equipped with a Wyatt Technology DAW NEOS small-angle light scatterer. Spectroscopically pure THF was used as the solvent in the analysis, and the sample concentration was 5 mg ml−1, and the test was carried out at 25 °C.2.3 Preparation of high-performance PMMA by reactive extrusion
HWM-PMMA and CSR were completely dissolved in MMA and placed in tank A, and then 0.4 wt% AIBN initiator was added. The mixed monomers were then fed into two-stage twin-screw extruders in series through a precisely metered gear pump for reactive extrusion polymerization (as shown in Fig. 2). The first-stage extruder was a TDE-40 co-rotating close-meshing twin-screw extruder (D = 41.3 mm, L/D = 68), and the second-stage extruder was a TDY-40 counter-rotating close-meshing twin-screw extruder (D = 41 mm, L/D = 60), produced by Nanjing Yuesheng Extrusion Machinery Co., Ltd. A vacuum devolatilization device was installed at the 10th screw barrel of the second-stage extruder near the die to remove residual monomers in the polymerization system.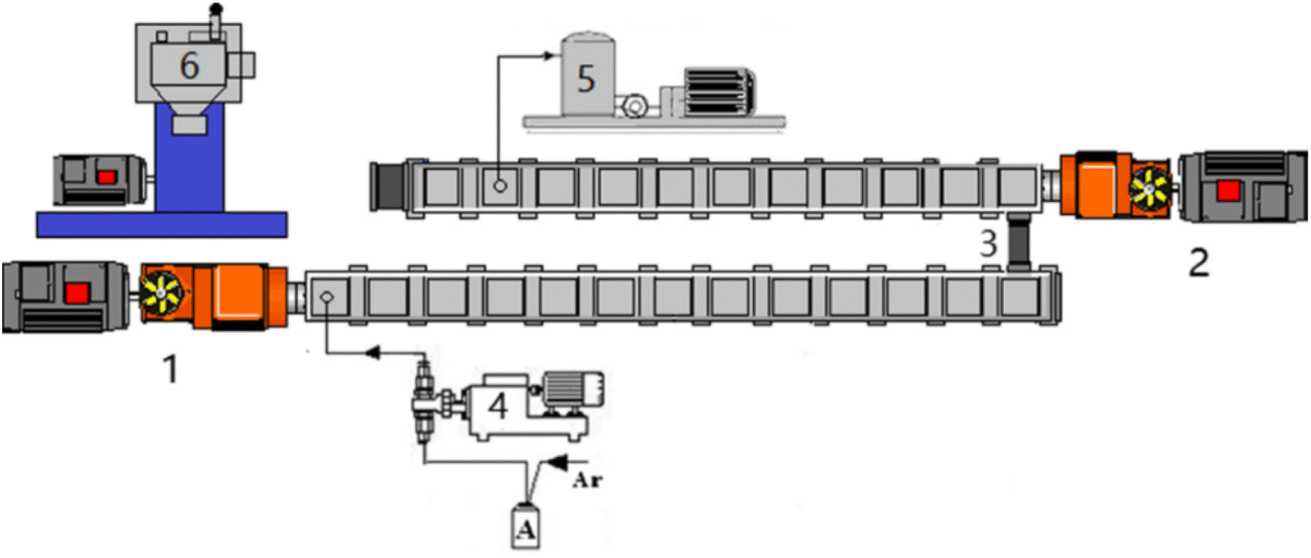 | ||
| Fig. 2 Experimental setup for reactive extrusion of the copolymers. 1. TDE-40 extruder, 2. TDY-40 extruder, 3. connector, 4. metering pumps, 5. vacuum pump, 6. granulator. | ||
The extruder was heated to 220 °C before polymerization, and the impurities in the extruder were removed with dry argon. The mixed monomer was then pumped into the extruder and the screw speed was slowly increased. It was initially set at 78–82 rpm and then gradually decreased to a “steady state”. At this time, the working current was basically stable, the product distribution was uniform, and no bubbles were generated. Residual monomers were removed using a vacuum devolatilizer. After the extrusion was stable, granulation was performed to obtain the product, and the corresponding parameters, such as screw barrel temperature, screw speed, current, and product quality were recorded at the same time. The residence time was about 35–40 min from injection of the monomers to the completion of the reactive extrusion polymerization.
2.4 Characterization of the reactive extrusion-polymerized PMMA
The absolute molecular weight of the reactive extrusion-polymerized PMMA was analyzed by MGPC, and the equipment and test conditions were the same as those outlined in Section 2.2.The glass transition temperature, Tg, of the samples was determined using a Diamond DSC system (Perkins-Elmer, USA). The samples were scanned from 25 °C to 180 °C in a nitrogen atmosphere at a heating rate of 5 K min−1.
The melt flow rate (MFR) of the samples was detected using a ZRZ1452 melt flow rate tester in accordance with the GB/T 3682-2000 standard (China). The tensile strength and flexural strength of the polymers were in accordance with the GB/T 11997-2008 and GB/T 15597.2-2010 standards (China), and were injection molded into the standard specimen, and then tested at the rate of 5 mm min−1 and 2 mm min−1, respectively, using the CMT4204 microcomputer control electronic universal material testing machine (China).
For the impact strength testing of the polymers, first, they were injection molded into a sample of 80 × 10 × 4 mm3. Before testing, a V-shaped A-notch with an angle of 45° at 21 mm from the middle to the edge of the specimen was made. Then, using the CEAST9050 Izod impact tester (China), the Charpy method was used to test the impact strength. Also, the Izod impact strength acN (kJ m−2) of the A-notch specimen was calculated using eqn (1):
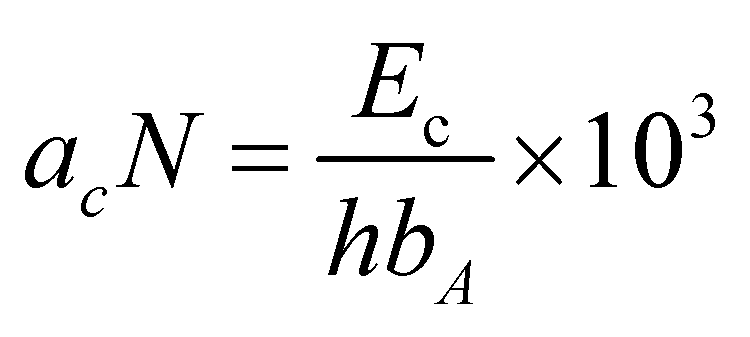 | (1) |
For the observation of the impact fracture surface, the fracture surface of the sample was sprayed with gold for 30 s, and then the fracture toughness of the sample was observed by field emission scanning electron microscopy (FESEM, Hitachi, Japan) at an accelerating voltage of 15 kV.
Transmission electron microscopy (TEM) was used to observe the distribution of CSR in the PMMA matrix. The samples were cut to a thickness of 60–100 nm with a microtome at −100 °C, and exposed to 1% OsO4 solution for 8 h for staining. Observations were performed with a Talos F200X TEM instrument at 100 kV.
3. Results and discussion
3.1 Kinetics of radical polymerization (RP)
First, different polymers were pre-dissolved in MMA monomer, and then bulk, non-stirring room temperature polymerization was carried out. The pre-dissolved PMMA/MMA and HWM-PMMA/MMA systems were first subjected to shear rheology at a frequency of 0.1 Hz, respectively, and the apparent viscosity (approximately zero-shear viscosity) of each system was tested, as shown in Tables 1 and 2. After deducting the mass of the pre-dissolved polymer, the polymerization conversion rate–time curve of each system was obtained, as shown in Fig. 3. The curves were typical “S”-shaped curves of free radical polymerization, and the final conversion rates were all over 85%, showing the polymerization conversion versus time for the pure monomers was completely independent of the pre-dissolved polymer. However, it could be seen that the introduction of the pre-dissolved polymer shortened the time for the system to achieve the auto-acceleration effect and accelerate the conversion rate. Since the initiator and reaction temperature of all the systems were the same, there was no shearing effect. It can be found from the classical polymerization rate [eqn (2)] that the only reason for the change in the polymerization rate can be a result of the change in kt. This shows that an inert polymer with viscosity in the system can effectively hinder the collision termination of active species, and play a role in reducing kt. However, it could be seen that simply increasing the apparent viscosity did not directly correspond to the increasing rate of conversion, especially for the pre-dissolved ultrahigh molecular weight polymer system. For example, although the apparent viscosity of 4% HWM-PMMA/MMA system was similar to that of the 20% PMMA-C/MMA system, the latter significantly increased the polymerization rate and entered the auto-acceleration effect zone at about 180 min, while the former entered the auto-acceleration effect zone after 248 min. In contrast, increasing the mass of the pre-dissolved polymer showed a corresponding relationship with increasing the polymerization rate. For example, the 5%, 10%, and 20% PMMA-C/MMA series, and 2%, 4%, and 6% HWM-PMMA/MMA series all showed increased polymerization rated according to the increase in the quality of the pre-dissolved polymer.
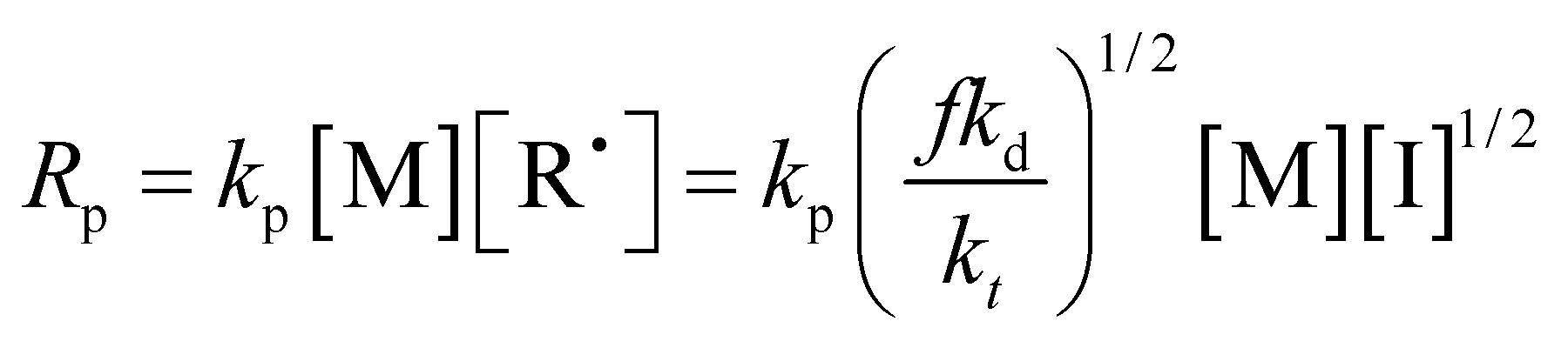 | (2) |
| Mass ratio | 0 | 5% | 10% | 20% | 30% |
| Viscosity (Pa S) | 0 | 79 | 425 | 894 | 1783 |
| Mass ratio | 0 | 2% | 4% | 6% |
| Viscosity (Pa S) | 0 | 568 | 975 | 1765 |
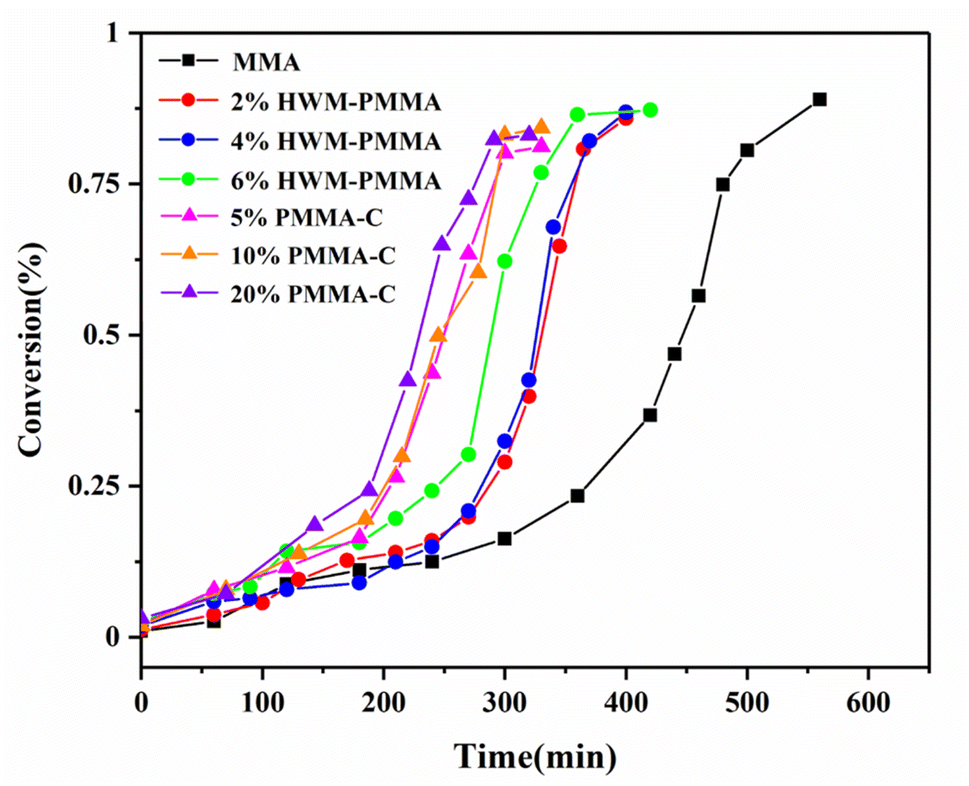 | ||
| Fig. 3 Curves of conversion vs. time during the bulk polymerization of MMA with different pre-dissolved polymers. | ||
In addition, it could be seen that although the initial apparent viscosity of the pre-dissolved ultrahigh molecular weight polymer system was high, it did not contribute much to the increase in the polymerization rate. In fact, the auto-acceleration effect could only appear in the polymerizing system, and had nothing to do with the pre-dissolved polymer. The function of the pre-dissolved polymer was only to inhibit the mutual termination of active species through its viscosity and steric hindrance, thereby reducing the chain termination rate constant kt. It could be seen that the better the compatibility between the pre-dissolved polymer and the monomer, the more this effect could be exerted. It is well known that the higher the molecular weight of the polymer, the greater the viscosity of the system, but the worse the compatibility with monomers, then even a phase separation state may occur. This means that the higher the molecular weight of the pre-dissolved polymer, the less influence it has on the monomer polymerization than lower molecular weight polymers of the same mass fraction. Thus, although their high viscosity could better hinder the termination of the two active species, their compatibility with the system was poor, and hence they canceled each other out. It could be seen that the pre-dissolved polymer inhibited the mutual termination of active species and reduced the chain termination rate constant kt, which should be due to the combined effect of the amount and the viscosity of the pre-dissolved polymer.
Based on the above-mentioned basic knowledge, further results can be obtained by further analyzing Fig. 3. It could be clearly seen that the conversions of almost all the polymerization systems were about 25% for them to enter the auto-acceleration effect area, regardless of whether the system included pre-dissolved polymers, or the type of pre-dissolved polymers, the amount of pre-dissolved polymers, or the speed of the initial polymerization rate. It seems that the polymerization behavior was completely independent of the pre-dissolved polymer. In fact, under the condition of no stirring or shearing, the initially pre-dissolved polymer was gradually excluded from the system and became a heterogeneous phase with the decrease in monomers during the polymerization process, which meant it would no longer affect the pure monomer polymerization system. Therefore, Fig. 3 only reflects the polymerization behavior of the polymerization system of pure monomers. Naturally, under the conditions of the same initiator concentration, the same temperature, and no shear, the results of the polymerization were the same. So in the pre-dissolved polymer system, it is also easy to understand why the polymer had a great influence on the initial stage of polymerization of the system. However, in the polymerization system with shearing and stirring, the situation was different. Due to the existence of shearing and stirring, the pre-dissolved polymer could not be phase-separated from the system, and always became entangled with the pure monomer polymerization system. This would affect the polymerization of the system from the beginning to the end. Therefore, regarding the influence of kt, this should be the result of the combined effect of the amount and the viscosity of the pre-dissolved polymer.
The above results also confirmed our previous conclusions about the polymerization kinetics at different shear rates, different initiator concentrations, and different temperatures; that is, in bulk polymerization systems with shear, the criterion is that the zero-shear viscosity of the polymerization system must reach 1000 ± 100 Pa s for the system to enter the auto-acceleration effect zone.14 Based on the above research, the criterion is also easy to understand. In a bulk polymerization system with shear, the main factors that inhibit the mutual termination of active species and reduce the chain termination rate constant kt are the quality of the inert polymer and its viscosity; that is, the polymerization conversion and the molecular weight of the polymer. Since in this case there was no pre-dissolved polymer, the source of the inert polymer was the polymerization itself. Therefore, with the increase in the polymerization conversion, that is, the continuous accumulation of inert polymers, the viscosity of the system also continuously increased. When the viscosity of the system reached this characteristic value, it could finally break through the classical steady state, and the chain termination rate constant kt suddenly and significantly dropped, resulting in the generation of the auto-acceleration effect. If the molecular weight of the system polymerization is relatively small, then a higher conversion is required to make up the mass due to the lack of molecular weight or viscosity. After reaching the automatic acceleration zone, with the large increase in the number of free radical active species, the chain termination rate Rt in turn increases with the square rate of the number of free radical active species, just like the formation of the steady-state polymerization stage in the classical free radical polymerization theory, such that Rt can rapidly reach a new equilibrium value.
The molecular weights of the pre-dissolved 20% PMMA-C samples at different conversion rates were tested by MGPC, as shown in Fig. 4. The absolute molecular weight of the polymer was obtained by calculation of the small-angle laser light scattering (LS) detection curve and the refractive index (dRI) detection curve. It can be seen from Fig. 4 that the MGPC curves all showed a bimodal distribution, in which the low molecular weight peak was contributed by the pre-dissolved polymer, and the high molecular weight peak was the peak formed by the polymerization system. It can be seen that with the increase in the conversion, the position of the low molecular weight peak, that is, the molecular weight, did not change, and only due to the continuous increase in high molecular weight polymer did its proportion in the total polymer gradually decrease. Therefore, after normalization, the peak intensity gradually decreased. This indicated that the molecular weight of the polymer formed by polymerization throughout the process was concentrated in the high molecular weight region. It was precisely because of the introduction of a pre-dissolved polymer with a low molecular weight as a reference that the process of an increasing polymerization conversion could be observed more clearly. Similarly, the macromolecular unimodal and adjacent bimodal morphologies of the pure MMA sample and the pre-dissolved HWMA-PMMA sample at different conversions are shown in Fig. S1 and S2,† respectively.
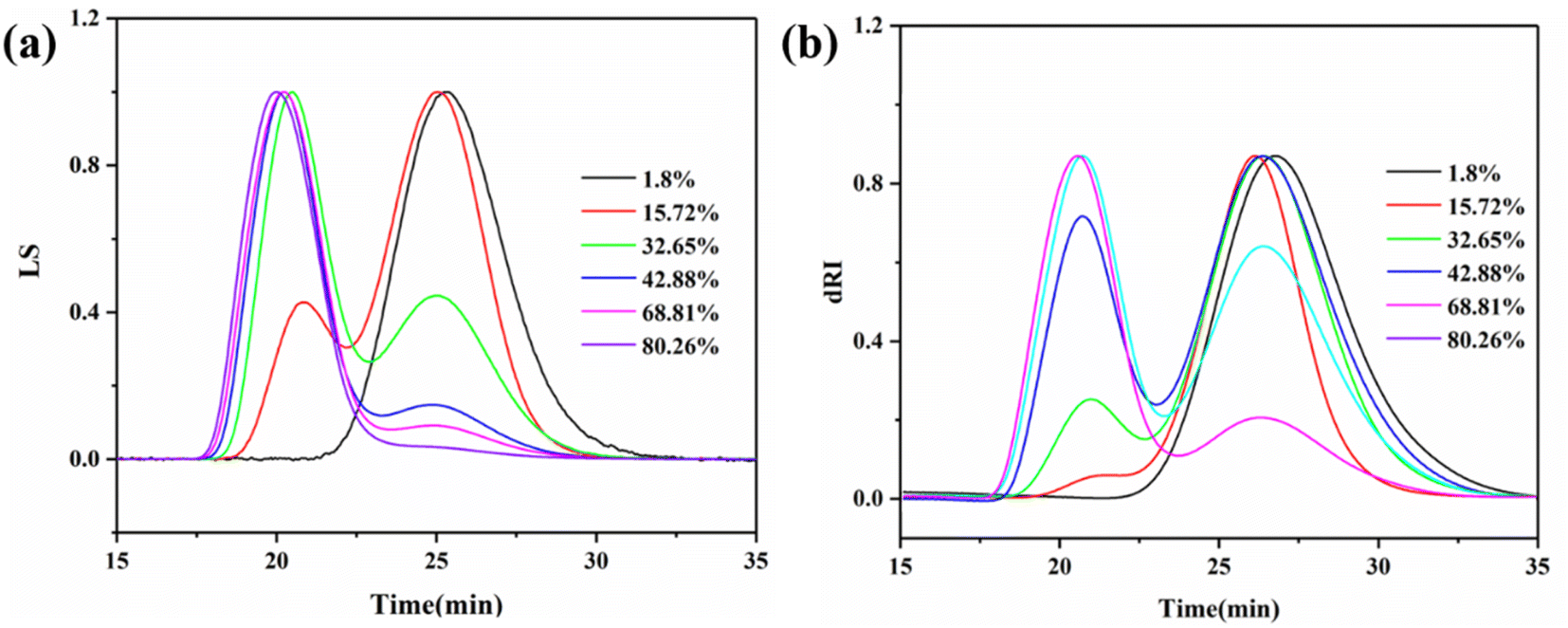 | ||
| Fig. 4 MGPC curves of bulk polymers of MMA with 20% PMMA-C at different conversions: LS curve (a) and dRI curve (b). | ||
The absolute molecular weight of the polymer formed by removing the pre-dissolved polymer in Fig. 4 was obtained by calculating the data for the LS and dRI curves, and its variation curve relative to the polymerization conversion is shown in Fig. 5. At the same time, the change curve of the polymerization rate of the compound against the polymerization conversion rate is also shown in Fig. 5. It can be seen that the polymerization conversion from 25% to 40% showed a high-speed increase in the polymerization rate and the polymerization molecular weight at the same time. Then the molecular weight of the system entered a relatively stable stage. It can be seen from Fig. 3 that this polymerization stage was just the polymerization stage after the polymerization system entered the auto-acceleration effect zone. The simultaneous increase in the polymerization rate and molecular weight in free radical polymerization is only possible when kt decreases, see eqn (2) and the kinetic chain length in classical free radical polymerization theory, as shown in eqn (3). This further confirmed the above inference; that is, the increased polymerization conversion and viscosity of the system would make the classical steady state become broken, and result in the generation of the auto-acceleration effect; further, the increase in free radical active species would prompt Rt to rapidly reach a new equilibrium value in the so-called second steady-state stage after entering the auto-acceleration effect region. In practice, the reason for the continuous increase in molecular weight is the result of the continuous decrease in kt due to the continuous increase in the inert polymer. But what is very interesting is that although the monomer and the polymerization rate were greatly reduced, the molecular weight still continued to rise. The only explanation is that the lifetime of the radical reactive species was greatly extended, and it could even be stagnant there. In full accord with what Liu et al.30 pointed out, it was believed that the chain polymerization was different from the classical mechanism, and it did not involve the instantaneous formation of the polymer chain, but rather involved a step-by-step or jumping process.
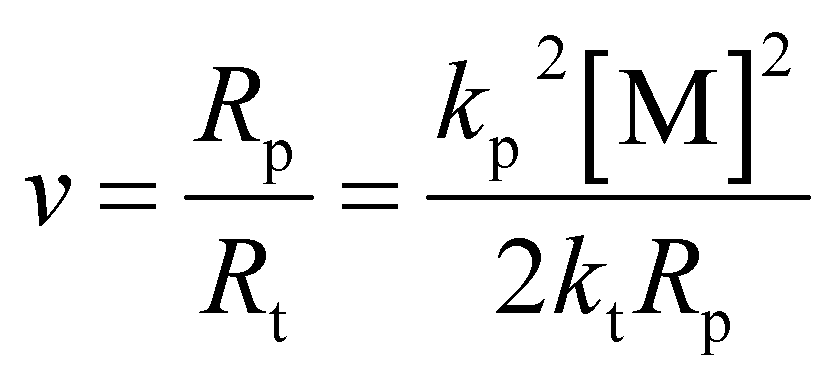 | (3) |
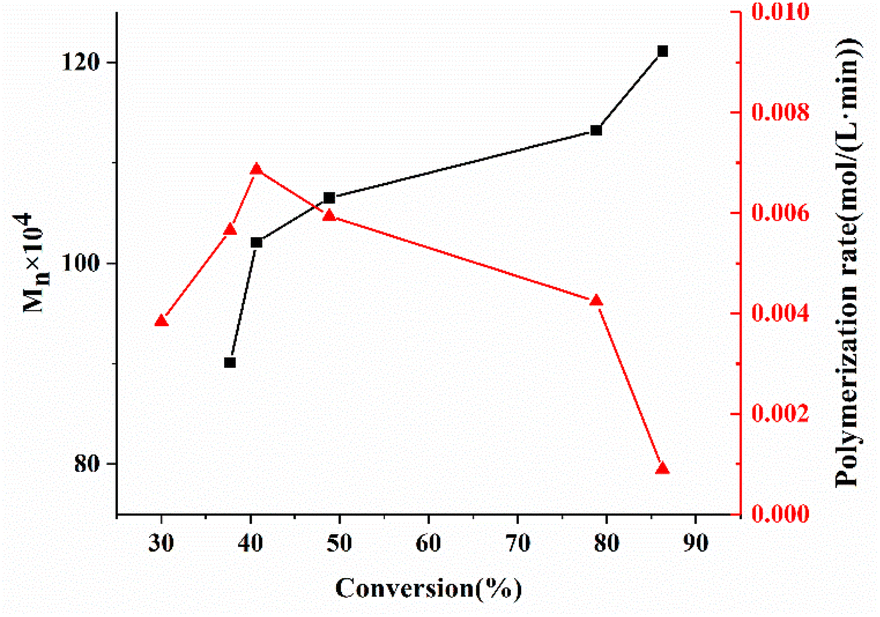 | ||
| Fig. 5 Absolute molecular weight and polymerization rate vs. conversion of the MMA bulk polymerization pre-dissolved 20% PMMA-C. | ||
The above results show that under the condition of no shearing at all and the initiator and polymerization temperature being the same, although the preparation of each polymerization sample was completely different, the molecular weights obtained by the polymerization of each system were all nearly the same. Also, even if the conversion rate was close to 90%, the molecular weight of the polymerization would not decrease even in the glass state. This point cannot be explained by classical radical polymerization theory. It can only confirm the conclusion put forward by Chunming et al.,30 who suggested that radical polymerization does not necessarily form a polymer chain instantaneously, but can be carried out a step-by-step or jumping process. In addition, the molecular weights of the polymers all reached the ultrahigh molecular weight level higher than 100 × 104, while the MWD remained around 1.2, which was almost comparable to that of anionic living polymerization.31,32 The most critical factor for this is that there was no shearing effect at all, so that the termination rate constant kt of the active species was reduced to the lowest level. As pointed out in previous studies, kt increases with increasing the shear rate, so the molecular weight would also decrease.9 Furthermore, under the premise of a completely shear-free condition, this radical polymerization was almost comparable to anionic living polymerization. However, it can be seen from Fig. 5 that the corresponding relationship between the molecular weight and polymerization time was completely contradictory with that of the living polymerization, in which the molecular weight of the later increased linearly with the polymerization time. So this shear-free radical polymerization was at best a long-lived polymerization, not a living polymerization.
3.2 Preparation of reinforced PMMA via reactive extrusion
Previous studies have shown that PMMA and its copolymers with higher molecular weight can be obtained by reactive extrusion polymerization in a short residence time by making full use of the auto-acceleration effect.10,33 It is known from the above studies that by pre-dissolving the polymer, the initial viscosity of the polymerization can be increased, and thus the polymerization rate can be increased, and further, that it increases with the increase in the quality of the pre-dissolved polymer. Furthermore, in addition to the initiator and temperature, the shear rate can directly control the molecular weight of the polymer. These results have extremely important guiding significance for the following preparation of polymers by reactive extrusion polymerization. Although reactive extrusion technology has been widely used for polymerization modification, there are few industrial examples of its direct use in the polymerization of olefin monomers, so there are certain differences in equipment and control.First, since the raw material was a liquid monomer, the propulsion mainly depended on the pressure of the delivery pump and the delivery of the smaller closely meshed elements, so the pressure in the front section of the barrel was extremely low, even close to a non-full state. Therefore, a rapid increase in viscosity could change the filling degree of the screw, which would be manifested in the running current. In addition, since the boiling point of MMA was about 100 °C, in order to avoid monomer vaporization, the front part of the first-stage screw was only set at 90 °C. The definition of the sample and the polymerization parameters after the reaction reached a steady state are shown in Table 3.
| Samples | First-stage extruder | Second-stage extruder | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fast-growth zone temperaturea (°C) | Current (A) | Screw speed (rpm) | Fast-growth zone temperatureb (°C) | Current (A) | Screw speed (rpm) | |||||||
| a The 10th–13th screw sections in the first-stage extruder.b The 1st–4th screw sections in the second-stage extruder. | ||||||||||||
| PMMA (the control) | 90 | 90 | 90 | 90 | 18.0 | 66.0 | 90 | 90 | 120 | 140 | 17.3 | 76.2 |
| PMMA-2HPM | 90 | 90 | 90 | 90 | 18.2 | 67.5 | 90 | 120 | 120 | 140 | 18.2 | 75.5 |
| PMMA-4HPM | 90 | 90 | 90 | 100 | 19.1 | 68.5 | 100 | 120 | 140 | 140 | 18.9 | 73.2 |
| PMMA-6HPM | 100 | 100 | 100 | 120 | 20.1 | 69.3 | 120 | 120 | 140 | 140 | 19.9 | 72.6 |
| PMMA-5CSR | 90 | 90 | 90 | 90 | 18.1 | 65.0 | 90 | 90 | 120 | 140 | 18.7 | 67.9 |
| PMMA-10CSR | 90 | 90 | 90 | 90 | 18.3 | 66.8 | 90 | 100 | 120 | 140 | 18.5 | 68.8 |
| PMMA-15CSR | 90 | 90 | 90 | 120 | 18.7 | 68.1 | 120 | 120 | 140 | 140 | 17.9 | 69.0 |
It can be seen from Table 3 that the driving current of the first-stage extruder of the MMA system containing the pre-dissolved polymer and no initiator was similar to that of the no-load case, i.e., only 17 A. Thus it was shown that the pre-dissolved polymer was not sufficient to cause an increase in the driving current of the first extruder. Second, compared with the control sample of the pure MMA monomer polymerization system, the driving current of the first extruder obviously increased whether it was the system of pre-dissolved ultrahigh molecular weight HWM-PMMA or the CSR system with the pre-dissolved conventional molecular weight, and increased with increasing the pre-dissolved polymer content. As shown in Fig. 3, due to the pre-dissolving of the polymer, the arrival time of the auto-acceleration effect could be greatly shortened. The viscosity of the system rapidly increased, and then the filling degree of the screw rapidly increased, and the driving current then naturally increased. Of course, the driving current of the pre-dissolved HWM-PMMA system was larger than that of the pre-dissolved CSR system, which was related to the high viscosity of the ultrahigh molecular weight pre-dissolved polymer.
The molecular weight and distribution of the products were analyzed by MGPC, and the results are shown in Table 4, while the LS curve is shown in Fig. 6. The Mn of pure PMMA was only 6.04 × 104, and the MWD was 1.95. With the introduction of the pre-dissolved polymer, the molecular weight of the product increased significantly and its distribution was narrower. However, as shown by the LS curve, which is more sensitive to high molecular weight, with the introduction of the pre-dissolved polymer, the molecular weight distribution of the product tended to be more normal, which confirmed that the dissolution of the high-viscosity inert polymer caused the kt to drop rapidly, shortening the arrival time of the auto-acceleration effect, and thereby greatly reducing the generation of small molecular weight polymers in the initial stage of polymerization.
| Samples | Conversion (%) | Mn × 10−4 | MWD | Capacity (kg h−1) |
|---|---|---|---|---|
| PMMA | 98.9 | 6.04 | 1.95 | 3.1 |
| PMMA-2HPM | 98.3 | 6.44 | 1.88 | 4.0 |
| PMMA-4HPM | 98.7 | 6.74 | 1.80 | 4.5 |
| PMMA-6HPM | 98.9 | 7.03 | 1.73 | 4.9 |
| PMMA-5CSR | 97.9 | 7.83 | 1.75 | 5.2 |
| PMMA-10CSR | 97.8 | 7.27 | 1.77 | 5.4 |
| PMMA-15CSR | 98.6 | 7.85 | 1.76 | 5.1 |
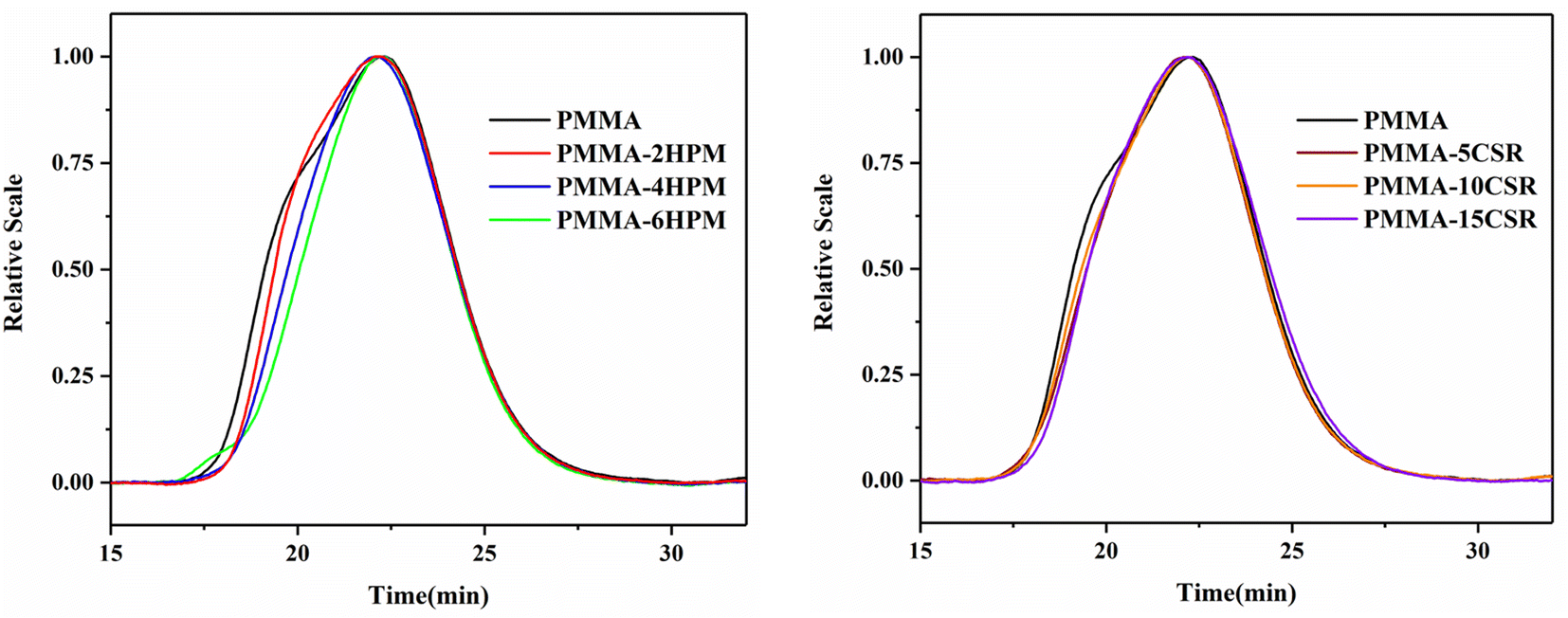 | ||
| Fig. 6 GPC-LS curves for the reactive extrusion polymerization product PMMA. | ||
3.3 Characterization of the pre-dissolved polymer PMMA made by reactive extrusion
The thermal properties of all reactive extrusion polymer products were analyzed by DSC and the results are shown in Fig. 7. It can be seen that all samples showed a step-like change in heat flow at around 100 °C, and the intersection of the tangents of the two curves in the descending step was taken as the Tg. The Tg of pure PMMA was 95.81 °C, while the Tg values of the pre-dissolved polymer samples were all higher than that of pure PMMA, apparently due to the differences in molecular weight. Among them, the Tg of the pre-dissolved HWM-PMMA samples increased by 2.5 °C, 3.3 °C, and 5.2 °C with the increase in the pre-dissolved mass, respectively (Fig. 7a). However, the Tg of the pre-dissolved CSR samples was not proportional to the pre-dissolved amount (Fig. 7b); whereby, when the pre-dissolved amount reached 10%, the Tg was the highest, reaching 102 °C. After the pre-dissolved amount increased again, the Tg then decreased. However, the molecular weight of the polymer was the highest at this time, see Table 4. This was because the shells of the CSR was made of PMMA, but the flexible MBS rubber in the core had a very low Tg. Under the premise that the rubber does not form an obvious separated phase region (see Fig. 7), the movement of the rubber molecular chain will affect the movement of PMMA molecular chains, so Tg would decrease with the increase in rubber units.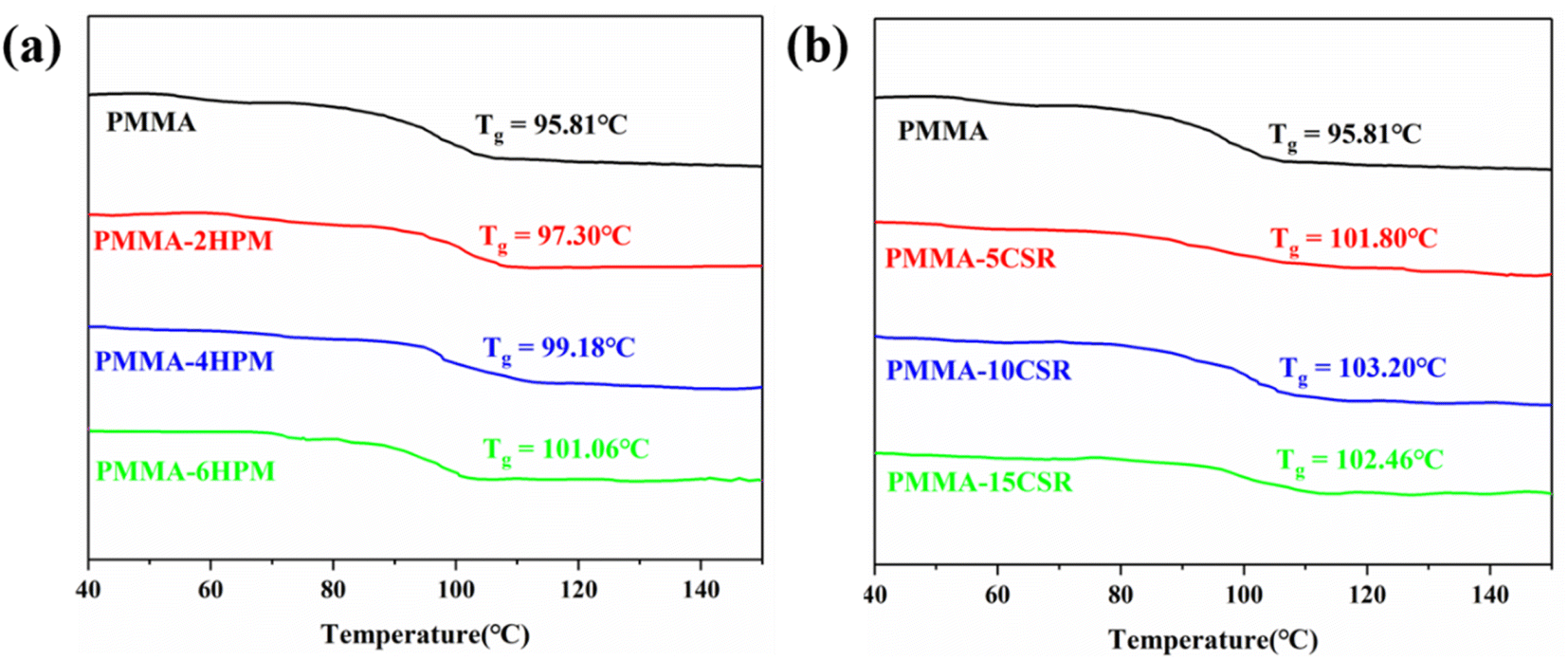 | ||
| Fig. 7 DSC curves for the reactive extrusion polymerization product PMMA. | ||
The mechanical properties and melt flow rate (MFR) of all the reactive extrusion polymers are shown in Table 5. It can be seen that pure PMMA had the highest MFR (2.1 g/10 min). However, the MFR of all the samples with the pre-dissolved polymer decreased, which was obviously related to the increase in the molecular weight of the polymerized product. Therefore, the products of the pre-dissolved HWM-PMMA series were further affected by the pre-dissolved HWM-PMMA. Although the amount of pre-dissolved HWM-PMMA was small, it still made a great contribution to the mechanical properties of the products. Their tensile and flexural strengths were higher than those of pure PMMA, and both increased with the pre-dissolved mass. The notched impact strength was also slightly improved compared to pure PMMA. It can be seen that the PMMA prepared by the pre-dissolved HWM-PMMA reactive extrusion method had better comprehensive mechanical properties.
| Samples | MFR (g/10 min) | Tensile strength (MPa) | Flexural strength (MPa) | Notched impact strength (kJ m−2) |
|---|---|---|---|---|
| PMMA | 2.1 ± 0.2 | 75.0 ± 1.8 | 118.70 ± 3.01 | 10.5 ± 0.6 |
| PMMA-2HPM | 1.9 ± 0.3 | 83.2 ± 2.2 | 145.76 ± 4.32 | 11.5 ± 1.3 |
| PMMA-4HPM | 2.0 ± 0.4 | 85.5 ± 3.5 | 156.42 ± 3.91 | 12.1 ± 2.6 |
| PMMA-6HPM | 1.7 ± 0.3 | 88.9 ± 2.5 | 162.67 ± 5.52 | 12.6 ± 2.5 |
| PMMA-5CSR | 1.9 ± 0.3 | 87.8 ± 2.1 | 178.44 ± 4.51 | 15.3 ± 2.7 |
| PMMA-10CSR | 1.9 ± 0.4 | 90.2 ± 2.5 | 183.67 ± 5.95 | 19.5 ± 2.1 |
| PMMA-15CSR | 1.7 ± 0.2 | 88.9 ± 3.4 | 197.32 ± 5.82 | 24.2 ± 1.6 |
| Blend PMMA/5%CSR | — | 74.8 ± 4.4 | 123.43 ± 4.32 | 13.2 ± 2.0 |
| Blend PMMA/10%CSR | — | 70.6 ± 5.6 | 105.67 ± 4.65 | 17.4 ± 1.6 |
| Blend PMMA/15%CSR | — | 68.9 ± 3.6 | 99.85 ± 6.57 | 20.1 ± 2.8 |
However, the modified PMMA with pre-dissolved CSR showed better mechanical properties, especially the flexural strength and impact strength. As the mass of pre-dissolved CSR increased from 5% and 10% to 15%, the flexural strength and impact strength increased from 178.44, 183.67, to 197.32 MPa, respectively and 15.31, 19.5, to 24.2 kJ m−2, respectively. Compared with pure PMMA, the highest increase rates of the two kinds of mechanical properties reached 166.2% and 230.5%, respectively. This was most likely due to the role of the flexible rubber links in CSR, which was also closely related to the extremely uniform distribution of CSR in the PMMA matrix.
In order to confirm the important influence of the uniform distribution of CSR in PMMA, pure PMMA was selected to be blended with different mass fractions of SCR to obtain PMMA/5%CSR, PMMA/10%CSR, and PMMA/15%CSR blend samples, respectively. Their mechanical properties are also shown in Table 5. It could be clearly seen that the mechanical properties of the blended samples were greatly reduced compared with the samples pre-dissolved with the same mass fraction of CSR. Among them, in terms of the flexural strength and impact strength, the maximum decrease rate of the blended samples compared with the samples with the same mass fraction of pre-dissolved CSR reached 29.0% and 20.4%, respectively. Such a huge gap was because the modified PMMA obtained by the reaction extrusion of the pre-dissolved polymer could make the CSR distribution more uniform, and there was also a better interfacial bond strength between PMMA and CSR. Also, it could be obtained by only a one-step reaction extrusion polymerization, which probably offers stronger industrial competitiveness.
To evaluate the substantial difference between pre-dissolved polymer modification and blending modification, SEM observation was performed. Consequently, the cross-section of the impact sample was selected, treated with gold spray, and then observed by SEM. The SEM photos are shown in Fig. 8. Fig. 8a shows the morphology of a smooth section of the pure PMMA sample, clearly indicating the nature of its brittle fracture. Second, the CSR contents of 5% and 15% were selected for typical comparison, and the SEM images of the cross-section of the blended sample and the pre-dissolved polymer sample were compared, as shown in Fig. 8b, d and 8c, e. It could be seen that when the content of CSR was the same, the cross-section of the blended sample was layered and fractured, indicating that the uniformity of the system was poor and there was a relatively easy-to-destruct cross-section. Also with the increase in CSR content, the thickness of this sheet-like fault was larger; that is, the uniformity of the system was worse. In contrast, the cross-sections of the pre-dissolved CSR samples did not change the cross-section morphology due to the amount of CSR content, and they all uniformly presented the shape of rough and ductile cross-sections. This further confirmed the inherent essence that the pre-dissolved polymer modification greatly improved the mechanical properties of the product more than the secondary blending modification.
 | ||
| Fig. 8 SEM photographs of the fracture surface of the reactive extrusion PMMA impact specimens: (a) PMMA, (b) blend PMMA/5%CSR, (c) PMMA-5CSR, (d) blend PMMA/15%CSR, (e) PMMA-15CSR. | ||
Based on the pre-judgment that the use of pre-dissolved CSR to carry out reactive extrusion polymerization can make CSR extremely evenly distributed, test samples were prepared in ultrathin sections after freezing, and then the double bonds in the rubber phase were stained with OsO4 vapor, and then observed by TEM. The results are shown in the Fig. 9. It could be clearly seen that the CSR distribution was extremely uniform, which was also true even for the 15% pre-dissolved CSR. In addition, the CSR balls ranged in size from 200–300 nm and were smooth round or oval in shape.
 | ||
| Fig. 9 TEM micrographs of CSR dispersed in PMMA matrix: (a) PMMA-5CSR, ×14 K, (b) PMMA-5CSR, ×22.5 K, (c) PMMA-15CSR, ×14 K, (d) PMMA-15CSR, ×22.5 K. | ||
For PMMA, the most important aspect is the transparency performance. We used UV–vis spectrophotometry to analyze the light transmittance of each sample, and the results are shown in Fig. 10. Pure PMMA had the best transmittance, with 91% visible-light transmittance. With the increase in pre-dissolved HWM-PMMA or CSR, the transparency decreased. The visible-light transmittances of PMMA-2, 4 and 6-HPM were 89%, 88% and 86%, respectively. This could be caused by the difference in the refractive index between high molecular weight and low molecular weight PMMA. It could be seen that even for the same polymer, the size difference in molecular weight will also obviously affect their optical performance, and even lead to a certain degree of phase separation. On the other hand, compared with the visible-light transmittance of pure PMMA, that of PMMA-5, 10, and 15-CSR decreased to 87%, 86%, and 84%, respectively. This result was because, as shown in Fig. 9, the pre-dissolved CSR in the matrix of PMMA appeared as uniformly dispersed balls at the scale of 200–300 nm, which meant it had not yet reached the nanometer scale. Therefore, although the modified PMMA was still transparent, the transmittance of visible light was decreased.
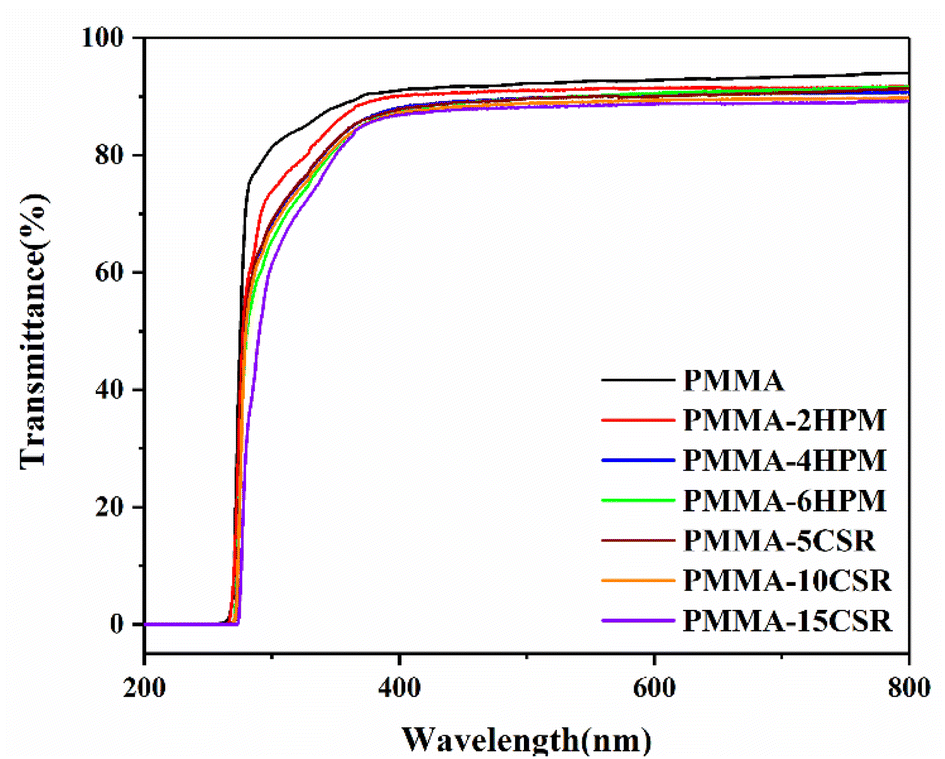 | ||
| Fig. 10 Transmittance curves of the reactive extrusion samples. | ||
4. Conclusion
The mechanism of radical polymerization was further explored through studying the kinetics of the bulk polymerization of MMA under shear-free conditions with different pre-dissolved polymers. The results indicated that, contrary to the shearing effect, viscosity was the key factor to reduce the termination rate constant kt. In detail, the degree of reducing the kt effect was influenced by the combined effect of the amount and the viscosity of the pre-dissolved polymer, and the compatibility between the pre-dissolved polymer and the monomer. Therefore, pre-dissolving the polymer could make the polymerization system enter the automatic acceleration zone faster, and greatly make the molecular weight distribution narrower. When the system entered the auto-acceleration zone, kt decreased rapidly and greatly and entered the second steady-state polymerization stage. Under shear-free conditions, kt could be minimized and the radical lifetimes maximized. However, the polymerization system was at best a long-lived polymerization rather than a living polymerization. On this basis, by pre-dissolving ultrahigh molecular weight PMMA and the core–shell particles CSR, the mechanical properties and heat resistance of the modified PMMA obtained by reactive extrusion were better than those of pure PMMA obtained under the same conditions. Compared with pure PMMA, the flexural strength and impact strength of PMMA with pre-dissolved CRS were up to 166.2% and 230.5%. With the same quality of CSR, the two mechanical properties of the samples obtained by blending were 29.0% and 20.4% worse than those of the pre-dissolved samples, respectively. It was the distribution of CSR in the pre-dissolved PMMA-CSR matrix with spherical single particle distribution (Diameter = 200–300 nm), which made PMMA-CSR exhibit a high degree of transparency. This one-step process of realizing high-performance PMMA polymerization shows extremely high industrial application prospects.Author contributions
H. S. carried out the kinetic tests and reactive extrusion, performed the characterization, and prepared the manuscript; Q. X. Z., A. N. Z., Y. G., D. F. and X. X. W helped design and supervise the experiments and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (no. 50573020).References
- R. Norrish and R. R. Smith, Nature, 1942, 150, 336–337 CrossRef CAS.
- V. E. Trommsdorff, H. Köhle and P. Lagally, Macromol. Chem. Phys., 1948, 1, 169–198 CrossRef.
- P. J. Flory, Principles of polymer chemistry, Cornell university press, 1953 Search PubMed.
- B. W. Brooks, Proc. R. Soc. London, Ser. A, 1977, 357, 183–192 CAS.
- B. O'Shaughnessy and J. Yu, Macromolecules, 1994, 27, 5067–5078 CrossRef.
- J. G. Victor and J. M. Torkelson, Macromolecules, 1987, 20, 2241–2250 CrossRef CAS.
- D. Wöll, E. Braeken, A. Deres, F. C. De Schryver, H. Uji-i and J. Hofkens, Chem. Soc. Rev., 2009, 38, 313–328 RSC.
- P. R. Dvornić and M. S. Jaćović, Polym. Eng. Sci., 1981, 21, 792–796 CrossRef.
- P. Zhan, J. Chen, A. Zheng, H. Shi, F. Chen, D. Wei, X. Xu and Y. Guan, Eur. Polym. J., 2020, 122, 109272 CrossRef CAS.
- P. Zhan, J. Chen, A. Zheng, T. Huang, H. Shi, D. Wei, X. Xu and Y. Guan, Mater. Res. Express, 2019, 6, 025315 CrossRef.
- E. Jacobs, K. Saralidze, A. K. Roth, J. J. A. de Jong, J. P. W. van den Bergh, A. Lataster, B. T. Brans, M. L. W. Knetsch, I. Djordjevic, P. C. Willems and L. H. Koole, Biomaterials, 2016, 82, 60–70 CrossRef CAS PubMed.
- R. Y. Hong, L. L. Chen, J. H. Li, H. Z. Li, Y. Zheng and J. Ding, Polym. Adv. Technol., 2007, 18, 901–909 CrossRef CAS.
- S. Saha and S. Pal, J. Biomed. Mater. Res., 1984, 18, 435–462 CrossRef CAS PubMed.
- H. Itokawa, T. Hiraide, M. Moriya, M. Fujimoto, G. Nagashima, R. Suzuki and T. Fujimoto, Biomaterials, 2007, 28, 4922–4927 CrossRef CAS PubMed.
- F. Pahlevanzadeh, H. R. Bakhsheshi-Rad, A. F. Ismail, M. Aziz and X. B. Chen, Mater. Lett., 2019, 240, 9–12 CrossRef CAS.
- X. Weng, Y. Bian, J. Zhang, Z. Zheng and T. Li, Chin. Pat., CN109316627-A, 2019 Search PubMed.
- E. T. Kopesky, G. H. McKinley and R. E. Cohen, Polymer, 2006, 47, 299–309 CrossRef CAS.
- C. Zhou, Q.-B. Si, Y.-H. Ao, Z.-Y. Tan, S.-L. Sun, M.-Y. Zhang and H.-X. Zhang, Polym. Bull., 2006, 58, 979–988 CrossRef.
- A. Alhotan, J. Yates, S. Zidan, J. Haider and N. Silikas, Materials, 2021, 14, 4127 CrossRef CAS PubMed.
- S. Zidan, N. Silikas, A. Alhotan, J. Haider and J. Yates, Materials, 2019, 12, 1344 CrossRef CAS PubMed.
- S. Nooma and R. Magaraphan, Polym. Bull., 2018, 76, 3329–3354 CrossRef.
- Y. Gui, S. L. Sun, Y. Han, H. X. Zhang and B. Y. Zhang, J. Appl. Polym. Sci., 2010, 115, 2386–2393 CrossRef CAS.
- H. Runqin, N. Fenglian and C. Qiuxiang, J. Exp. Nanosci., 2017, 12, 308–318 CrossRef.
- S. Q. Song, C. Y. Wan and Y. Zhang, RSC Adv., 2015, 5, 79947–79955 RSC.
- S. G. Peng, M. He, Z. Yang, K. Zhang, B. Xue, S. H. Qin, J. Yu and G. M. Xu, RSC Adv., 2020, 10, 28527–28535 RSC.
- M. Todo, K. Takahashi, B. Y. Jar and P. Beguelin, JSME Int. J., Ser. A, 1999, 65, 432–438 CAS.
- C. B. Bucknall and R. R. Smith, Polymer, 1965, 6, 437–446 CrossRef CAS.
- A. M. Donald and E. J. Kramer, J. Mater. Sci., 1982, 17, 1765–1772 CrossRef CAS.
- A. F. Yee, J. Mater. Sci., 1977, 12, 757–765 CrossRef CAS.
- C. Liu, K. Kubo, E. Wang, K. Han, G. Chen, F. A. Escobedo, G. W. Coates and P. Chen, Science, 2017, 358, 352–355 CrossRef CAS PubMed.
- J. Smid, M. V. Beylen and T. E. Hogen-Esch, Prog. Polym. Sci., 2006, 31, 1041–1067 CrossRef CAS.
- D. Baskaran and A. Muller, Prog. Polym. Sci., 2007, 2, 32 Search PubMed.
- P. Zhan, J. Chen, A. Zheng, H. Shi, T. Wu, D. Wei, X. Xu and Y. Guan, Mater. Res. Express, 2020, 7, 095305 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06441c |
| This journal is © The Royal Society of Chemistry 2023 |