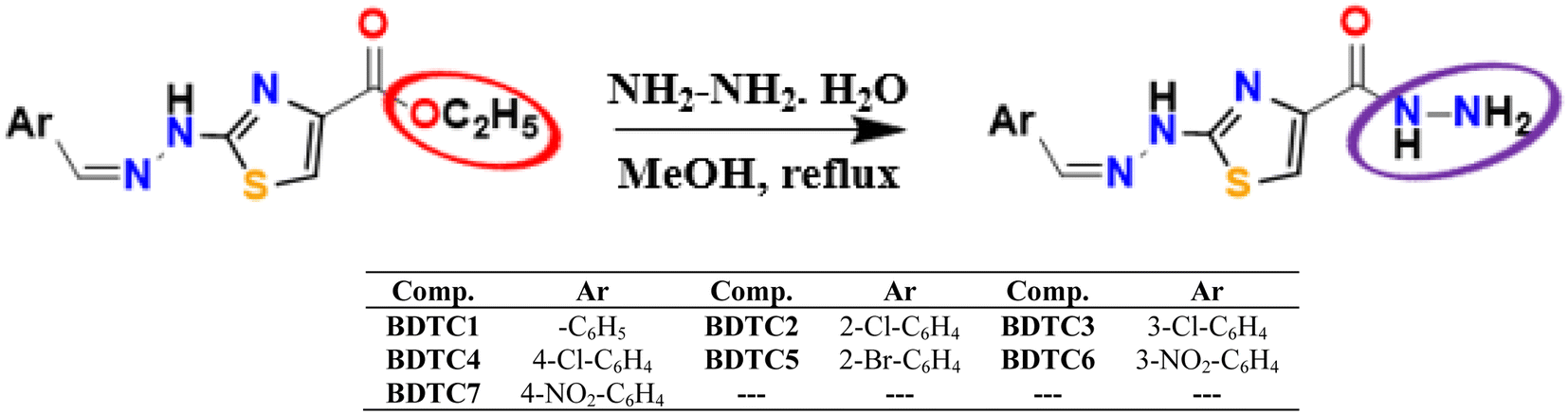DOI:
10.1039/D3RA00431G
(Paper)
RSC Adv., 2023,
13, 7237-7249
Synthesis, characterization and exploration of photovoltaic behavior of hydrazide based scaffolds: a concise experimental and DFT study†
Received
20th January 2023
, Accepted 16th February 2023
First published on 6th March 2023
Abstract
Solar energy being a non-depleting energy resource, has attracted scientists' attention to develop efficient solar cells to meet energy demands. Herein, a series of hydrazinylthiazole-4-carbohydrazide organic photovoltaic compounds (BDTC1–BDTC7) with an A1–D1–A2–D2 framework was synthesized with 48–62% yields, and their spectroscopic characterization was accomplished using FT-IR, HRMS, 1H and 13C-NMR techniques. Density functional theory (DFT) and time dependent DFT analyses were performed utilizing the M06/6-31G(d,p) functional to calculate the photovoltaic and optoelectronic properties of BDTC1–BDTC7 via numerous simulations of the frontier molecular orbitals (FMOs), transition density matrix (TDM), open circuit voltage (Voc) and density of states (DOS). Moreover, the conducted analysis on the FMOs revealed efficient transference of charge from the highest occupied to the lowest unoccupied molecular orbitals (HOMO → LUMO), further supported by TDM and DOS analyses. Furthermore, the values of binding energy (Eb = 0.295 to 1.150 eV), as well as reorganization energy of the holes (−0.038–0.025 eV) and electrons (−0.023–0.00 eV), were found to be smaller for all the studied compounds, which suggests a higher exciton dissociation rate with greater hole mobility in BDTC1–BDTC7. Voc analysis was accomplished with respect to HOMOPBDB-T–LUMOACCEPTOR. Among all the synthesized molecules, BDTC7 was found to have a reduced band gap (3.583 eV), with a bathochromic shift and absorption maximum at 448.990 nm, and a promising Voc (1.97 V), thus it is regarded as a potential candidate for high performance photovoltaic applications.
Introduction
Recently, organic solar cells (OSCs) have emerged at the forefront of technology to convert solar energy to electricity.1,2 This remarkable strategy utilizes solar cells to produce electricity by solar energy consumption through the photoelectric effect.3,4 Because of this, photovoltaic (PV) technology has rapidly developed and is providing a sustainable power supply.5 Some major factors responsible for its wide usage include its lightweight nature, low cost, simple device framework, good flexibility, and easy fabrication.6,7 The foremost photovoltaic devices are inorganic silicon-based solar cells, which developed rapidly due to the excellent proficiency of silicon, best power conversion efficiencies (PCEs), stability at high temperature and eco-friendly nature.8,9 Nevertheless, conventional silicon-based solar cells (Si-SCs) have disadvantages such as non-tunable energy levels, high costs, and a brittle nature. Consequently, dye-sensitized solar cells (DSSCs) have gained attention over Si-SCs due to their strong stability, simple modulation, tunable optical characteristics, environment-friendly nature, and visual properties such as transparency and a colored nature.10–13 However, DSSCs also have some major failings, i.e. difficulty in minimizing energy loss while enhancing electron injection at the dye interface.14 Thus, organic photovoltaics (OPVs), also known as bulk heterojunction (BHJ) solar cells, have been widely accepted and highly investigated owing to their mechanical stiffness, light transparency, economical nature, color alteration, and lightweight property.2,15–19 Over the past 20 years, fullerene acceptors have gained the attention of researchers and improved the PCE to 8.3% in junction with polymeric donors.20,21 They have remained as presiding acceptors in BHJ solar cells owing to their 3-D structure, high electron affinities, and electronic delocalization in the lowest unoccupied molecular orbitals (LUMOs), hence effectively generating charge separation states with donor moieties. In a bulk heterojunction (BHJ), 11–12% power conversion efficiency was gained using fullerene-based organic solar cells (FOSCs such as PC71BM, PC61BM, and ICBA). Fullerene-based organic solar cells can withstand a high Voc, and this model competes with donor-accepting heteroatoms, while the trajectories of electrons and holes are in their respective phases of transport. The two elements that control the transfer of current, the HOMO difference between acceptor and donor, and recombining strength, agreed with the experimental results and kinetic Monte Carlo simulation. Evidence proved that the difference in HOMO levels between the acceptor and the donor was the same as the space between the donor and acceptor system.22 However, these fullerene photovoltaic devices possess some limitations: least absorption in the visible area, large band gaps, a small Voc, etc. Recently, an emerging class of non-fullerene acceptors (NFAs) has become a foremost part of the research on BHJ-SCs and has enabled a major breakthrough in the power conversion efficiencies of OSCs. A few drawbacks, such as non-tunable LUMO energies and unreliable morphologies, have rendered the fullerene acceptors less efficient materials as compared to the NFAs.23 With the emergence of NF chromophores, the power conversion efficiencies (PCEs) of OSCs have been massively growing and have achieved a value of 18%.24–27 Among the NFAs, fused-ring electron acceptors with acceptor1–donor1–acceptor2–donor2 (A1–D1–A2–D2) molecular configurations are tremendously exciting.28–33 In the literature, we found many reports in which NFAs with various configurations such as D–π–D–π–A, A–π–D–π–A, A–π–A–π–A, D–π–A, A–π–A, etc. were widely utilized to tune the photovoltaic properties of organic solar cells. It is well-known that utilization of a strong electron withdrawing group on acceptor moieties at the terminal of a molecule and core with some donating units like benzothiophene, diphenylamine, naphthalene diamine etc. significantly reduces band gaps along with binding energies, widens the absorption spectra and improves the exciton dissociation rate and open-circuit voltage of OSCs. They have also shown exceptional attributes contributing to promising PCEs in OSCs.34–36 Khan and co-authors reported a Z series of novel Z1–Z6 acceptor materials with fused chrysene designed using FClC. A reduction in HOMO–LUMO energy gap was observed with a lower binding energy (0.272–0.353 eV) than that of the parent chromophore (0.394 eV). Low excitation energy values mean that maximum charge transfer occurs during excitation. DOS, TDM, and FMO analyses showed that end capped modification in acceptors was significant in tuning the photovoltaic properties of photovoltaic materials.37 Fullerene-free acceptor chromophores (MOL-1–MOL-4) with an A2–π–A1–π–A2 architecture containing benzothiazole core units and bridged with four different end-capped acceptors were reported to have excellent photovoltaic properties.38 All the chromophores exhibited reduced band gaps with broader absorption responses than their parent (RCAM) owing to an electron withdrawing effect of the terminal acceptor units. Furthermore, MOL-1 exhibits a strong absorption λmax = 545.02 nm in solvent with a narrow Eg and notable photovoltaic response. Aqil and co-workers reported (BTM1–BTM5) benzothiazole based fullerene-free acceptor chromophores to enhance the efficiency of OSCs. They designed molecules using an efficient acceptor named BTP-Cl and recently reported a 16.5% PCE. The designed molecules were found to be much better in structural configurations and their bandgap decreased by 10 times as compared to R. Optoelectronic investigations revealed that BTM-3 and BTM-4 were superior with respect to their absorption responses.39 Herein, a series of hydrazide containing NF-based small molecular acceptors (BDTC1–BDTC7) was synthesized, and their photovoltaic responses were studied using DFT in order to use them as OSCs.
Materials and methods
The synthesis of hydrazides (BDTC1–BDTC7) was accomplished from hydrazinylthiazole esters via commercially available solvents and reagents. The solvents and reagents were purchased from internationally well-reputed chemical suppliers; glacial acetic acid, thiosemicarbazide, 3-nitrobenzaldehyde, 2-chlorobenzaldehyde, 4-chlorobenzaldehyde (Sigma Aldrich, USA), methanol, absolute ethanol, ethyl bromopyruvate, 3-chlorobenzaldehyde, 2-bromobenzaldehyde, 4-nitrobenzaldehyde (Merck, Germany), n-hexane, acetone, ethyl acetate (Riedel-de-Haen) and hydrazine monohydrate (Dae Jung, Korea).
The synthesis was initially determined by monitoring different physical parameters, like color change, melting point (MP), and retardation factor (Rf). Different spectro-analytical methods like Fourier-transform infrared (FT-IR) spectroscopy, high-resolution mass spectrometry (HRMS), and 1H- and 13C nuclear magnetic resonance (NMR) spectroscopies were used for structural confirmation. The purity and progress of the reaction product were monitored using TLC with silica gel 60 HF-254 pre-coated aluminum sheets (Merck, Germany). The approximate MP was determined with DMP-300 (A&E Lab, UK) apparatus. The absorptions in the IR spectra were used to determine functional groups and were recorded on a FT-IR spectrophotometer using attenuated total reflectance (ATR). 1H and 13C NMR data were recorded using Bruker Advance 300 MHz and Varian VNMRS 400 MHz spectrometers. A Bruker Micro TOF-ESI system was used to obtain the mass spectrometry data.
Results and discussion
The targeted hydrazinylthiazole-4-carbohydrazides (BDTC1–BDTC7) were synthesized as shown in Scheme 1. The condensation reaction of hydrazinylthiazole-4-carboxylates with excess hydrazine monohydrate in methanol solvent under reflux (overnight) led to the formation of hydrazinylthiazole-4-carbohydrazides (BDTC1–BDTC7). The condensed hydrazides (BDTC1–BDTC7) were obtained as solid products in moderate yields (48–62%).
 |
| | Scheme 1 Systematic representation of the synthesis process of BDTC1–BDTC7. | |
The formation of the purified hydrazides (BDTC1–BDTC7) was initially indicated by simple changes in physical parameters. The color change from yellow to light green, increase in melting point, and reduction in Rf values initially confirmed the formation of the respective hydrazides. Different spectro-analytical data confirmed the synthesized structures of the desired hydrazides (BDTC1–BDTC7).
In the FT-IR spectra of BDTC1–BDTC7, the carbonyl group absorptions were recognized in the 1608–1735 cm−1 range. The characteristic absorptions in the ranges of 3272–3310 cm−1 and 3375–3433 cm−1 were attributed to –NH and –NH2 functional groups, respectively, indicating the conversion of esters to their respective hydrazides.
In the 1H NMR spectra of BDTC1–BDTC7, the appearance of two additional signals for –NH2–CO and –NH confirmed the formation of hydrazides, compared with the parent esters. The two-proton signal as singlet observed at 4.50–4.54 ppm was assigned to the –NH2 moiety. The signal appearing as one proton singlet observed in the range of 9.03–9.09 ppm was ascribed to –CO–NH– protons. These two signals evidenced the successful conversion of hydrazinylthiazole esters to hydrazinylthiazole hydrazides. Another proton signal as a singlet at 8.04–8.43 ppm arose due to the azomethine (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) functionality. Additionally, the proton signals within the aromatic region did not show any significant shift and authenticate the formation of hydrazides.
N–) functionality. Additionally, the proton signals within the aromatic region did not show any significant shift and authenticate the formation of hydrazides.
In the 13C NMR spectra of BDTC1–BDTC7, the thiazole ring carbons at positions 5 and 4 appeared at 113.5–113.7 and 145.8–145.9 ppm, respectively. The most downfield carbon signal at 168.3–168.6 ppm was assigned to the ipso carbon 2 of the thiazole ring. The appearance of a new carbon signal at 160.9 ppm was seen as a confirmation of the carbonyl carbon of the hydrazide functionality. There was no significant change in the aromatic carbon signals when the esters converted into their respective hydrazides.
HRMS data finally confirmed the structures of the hydrazinylthiazole-4-carbohydrazides (BDTC1–BDTC7). The calculated masses of the hydrazides were found to be in harmony with the observed masses (see ESI† and experimental data). The synthesized compounds based on their structural features were anticipated as an important motif for solar cell applications to enhance the PCE of photovoltaic devices. Therefore, a DFT approach was taken to verify the solar cell efficiencies by analyzing the band gap, inertness, chemical reactivity, charge density transfer, and spectral absorption. The optimized structures for the studied compounds are also displayed in Fig. 1.
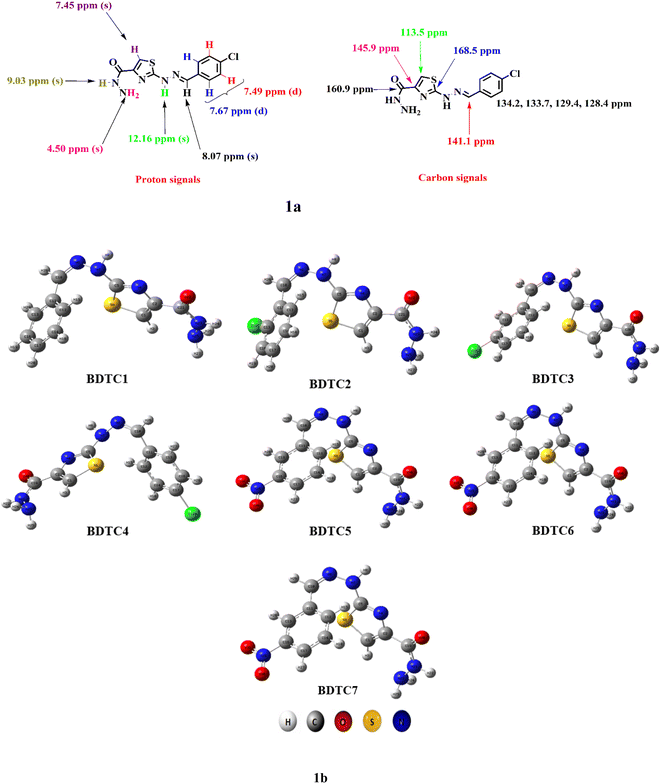 |
| | Fig. 1 (a) Proton and carbon signal interpreted structure of BDTC4; (b) the optimized structures of BDTC1–BDTC7. | |
Experimental section
Synthesis of 2-(2-(arylidene) hydrazinyl) thiazole-4-carbohydrazides (BDTC1–BDTC7)
The targeted hydrazides (BDTC1–BDTC7) were obtained from a mixture of hydrazinylthiazole esters (0.2 g, 1 mmol), hydrazine monohydrate in excess (3.0 equivalent), and methanol (20 mL) by heating under reflux, overnight. TLC was used to monitor the conversion of esters into their respective hydrazides. TLC was repeated every 30 min in the solvent system (acetone/n-hexane: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). Completion of the reaction was observed as a single spot on the TLC plate. The mixture was left to reach room temperature for almost an hour. Then, the reaction mixture was transferred into ice. The chilled reaction mixture was put in a refrigerator to obtain precipitates of the hydrazinylthiazole-4-carbohydrazides. The precipitates formed were filtered. The mother liquor was removed by washing the precipitated solids with plenty of water. The pure hydrazides were dried at room temperature. The 1H- and 13C-NMR spectra of all the hydrazides were recorded in DMSO-d6 solvent. Spectroscopic data of all the synthesized compounds (BDTC1–BDTC7) are presented in the ESI† (Tables S1–S18).
:2). Completion of the reaction was observed as a single spot on the TLC plate. The mixture was left to reach room temperature for almost an hour. Then, the reaction mixture was transferred into ice. The chilled reaction mixture was put in a refrigerator to obtain precipitates of the hydrazinylthiazole-4-carbohydrazides. The precipitates formed were filtered. The mother liquor was removed by washing the precipitated solids with plenty of water. The pure hydrazides were dried at room temperature. The 1H- and 13C-NMR spectra of all the hydrazides were recorded in DMSO-d6 solvent. Spectroscopic data of all the synthesized compounds (BDTC1–BDTC7) are presented in the ESI† (Tables S1–S18).
Computational procedure
In this study, all the DFT calculations of BDTC1–BDTC7 were accomplished using the Gaussian 09 program40 and for, input files, GaussView 5.0 (ref. 41) was utilized. The results obtained from the output files were interpreted using Avogadro,42 Chemcraft,43 PyMOlyze,44 Multiwfn 3.7,45 and GaussSum46 software. The true minima of BDTC1–BDTC7 were obtained using the M06 (ref. 47 and 48) functional along with the 6-31G(d,p) basis set.49 Other quantum chemical computations, including density of states (DOS), open circuit voltage (Voc) and reorganization energy (RE) analyses, were also performed using the aforesaid functional of DFT. Some analyses: transition density matrix (TDM), frontier molecular orbitals (FMOs), and UV-vis were performed using TD-DFT at the above-mentioned level of theory. RE is divided into two main types: internal reorganization energy (λint) and external reorganization energy (λext). Internal reorganization energy can be easily identified due to sudden modifications in the internal environment. In contrast, external reorganization energy reflects the effect of polarizability on the surroundings as well as relaxation of the external environment. In the current study, λext effects were ignored, and only λint was considered. eqn (1) and (2) were used to calculate the energies of electrons (λe) and holes (λh).50–52| | |
λe = [E0 − E−] + [E0− − E0]
| (1) |
| | |
λh=[E0+ − E+] + [E0+ − E0]
| (2) |
In eqn (1) and (2), E0− and E0+ represent the energies of neutral molecules in their anionic and cationic states, respectively. E+ and E− specify the energies of the cation and anion obtained via the optimized geometry of the cation and anion molecules, and E0 is the energy of the neutral molecules in the ground state.53
Frontier molecular orbital (FMO) analysis
FMO investigation is helpful in understanding the intramolecular charge transfer (ICT) and optoelectronic characteristics of compounds via assessing the electronic charge transition between the HOMO and LUMO.54–63 Band theory describes the HOMO and LUMO as conduction and valence bands, accordingly, and the energy gap between orbitals is described as the band gap (ΔE).64,65 The efficiency of OSCs can be elucidated with the help of Egap as a photovoltaic material having a smaller energy gap will have a high power conversion efficiency (PCE).30,66,67 Herein, we have examined the conducting nature of electronic charge density followed by photon properties of BDTC1–BDTC7. The results in Table S8† show the energies of orbitals and energy gaps while the pictographic representation of the molecular orbitals of the aforesaid chromophores along with their energies is shown in Fig. 2.
 |
| | Fig. 2 Graphical representation of ICT and energies of molecular orbitals of BDTC1–BDTC7. | |
The values of EHOMO of BDTC1–BDTC7 were found to be −5.843, −5.954, −6.010, −5.979, −5.935, −6.385, and −6.249 eV, while the values of ELUMO are noted as −1.365, −1.450, −1.636, −1.619, −1.389, −2.327, and −2.666 eV, respectively. The band gap is noted to be 4.478, 4.504, 4.374, 4.360, 4.546, 4.058, and 3.583 eV for BDTC1–BDTC7, respectively. The data from Table S8† show that all the synthesized compounds (BDTC1–BDTC7) exhibit comparable bandgaps. Nevertheless, among all these studied compounds (BDTC1–BDTC7), the highest band gap is seen for BDTC5 (4.546 eV), which has the least electron-withdrawing groups among bromo (–Br), chloro (–Cl), and nitro (–NO2) groups. However, the smallest band gap value (3.583 eV) was found for BDTC7 among all the synthesized compounds because of the –NO2 group at the para position. This narrow band gap might be due to the resonance effect because, in terms of resonance, the nitro group facilitates the sharing of its non-bonded electron pair charge with acceptors. Overall, the descending trend of Egap of the investigated compounds was found to be: BDTC5 > BDTC2 > BDTC1 > BDTC3 > BDTC4 > BDTC6 > BDTC7. Besides the determination of energies of the orbitals, charge transference can also be examined from FMO study. Therefore, charge transference between orbitals of BDTC1–BDTC7 was also studied, and their pictographs are shown in Fig. 2. From the FMO diagrams, it is noted that the electronic cloud of the HOMO is significantly concentrated over the entire molecule for BDTC1–BDTC7. However, for the LUMO, the major portion of charge density is located over the end-capped acceptor (A2), and a minute portion of it is present over the donor 1 and acceptor 1 regions in all the synthesized compounds (BDTC1–BDTC7). In terms of charge transfer, compounds with a lower Egap exhibit a high charge transference rate and show greater photovoltaic properties.
Density of states (DOS)
Density of states (DOS) study is considered to be an effective tool to investigate the electronic cloud transference in compounds and also support the results of FMO analysis.68,69 Herein, the DOS maps of BDTC1–BDTC7 at the M06/6-31G(d p) level were plotted, and their pictographs are shown in Fig. 3. In order to perform DOS analysis of BDTC1–BDTC7, each compound is split into four respective segments: i.e., acceptor 1 (A1), donor 1 (D1), acceptor 2 (A2), and donor 2 (D2), which are represented by red, green, blue, and pink lines, accordingly. In the DOS plots, the peaks (negative values) on the right side along the x-axis show the electronic cloud on the HOMOs while the peaks on the left side represent the charge on the LUMOs of the investigated compounds. The distance between these right and left peaks indicates the energy difference. From the DOS results, it was found that the charge contribution of donor 1 is 91.8, 88.8, 93.4, 92.5, 88.5, 37.6, and 47.0% at the HOMO and 51.1, 49.7, 49.7, 50.5, 48.1, 57.2, and 48.3% at the LUMO in BDTC1–BDTC7, respectively. For acceptor 1, the electronic cloud contribution was 1.2, 0.3, 2.1, 1.6, 60.6, and 51.0% at the HOMO, and 0.2, 0.1, 0.6, 0.2, 0.1, and 0.4% at the LUMO for BDTC2–BDTC7, respectively. Donor 2 contributes 0.4, 0.0, 0.0, 0.0, 0.1, 0.0, and 0.0% to the HOMO and 2.1, 1.4, 1.4, 1.3, 1.5, 1.4 and 1.5% to the LUMO in BDTC1–BDTC7, respectively. Acceptor 2 contributes 7.7, 10.0, 6.3, 5.4, 9.8, 1.7, and 2.0% to the HOMO, and 46.7, 48.7, 48.9, 47.6, 50.3, 41.3, and 49.9% to the LUMO in BDTC1–BDTC7, respectively. The plots in Fig. 3 show that for the HOMO the highest peak (blue) is exhibited by A2 at nearly −4 eV while for the LUMO the highest peak is observed for D1 (green) at −1.5 eV for BDTC1–BDTC5. However, in BDTC6 and BDTC7, the most prominent LUMO peak (red) is observed for A1 at −2.5 eV, demonstrating efficient ICT for BDTC6 and BDTC7. From the above discussion, it was concluded that the maximum charge density in the HOMOs is provided by the donor while in the LUMOs the charge is mainly located over the acceptor region as already elucidated in the FMO analysis. Overall, a massive ICT from donor to acceptor is clear from the DOS spectra, which indicates that the studied compounds are good photonic materials.
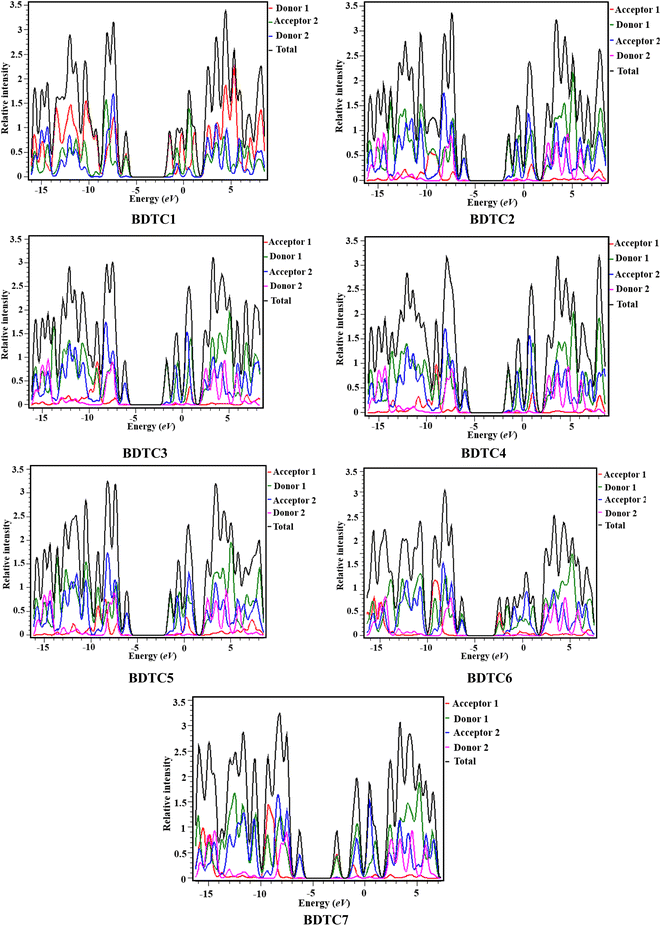 |
| | Fig. 3 DOS plots of the investigated molecules (BDTC1–BDTC7). | |
UV-vis analysis
To understand the effects of the donor, bridging core, and acceptor moieties on the optical properties of the studied compounds, excited state absorption spectra were investigated in dichloromethane (DCM). It has been anticipated that the polar medium is included in stability of π–π* level corresponds to the n–π* level via a suitable electronic state.34,70 Generally, the interaction energy of a compound in DCM solvent is governed by the polarity and non-covalent interactions (NCIs).71 This means that the dipolar interactions and hydrogen bonding play an essential role in stabilizing the first singlet electronic state of the compounds.72 It is considered that the excited level is more polar than the ground level. Hence, the ground state is less stabilizing than the excited state in DCM.71 The significant results, including the transition energy (E), maximum absorption wavelength (λmax), oscillator strength (fos), and major molecular orbital contributions for the investigated compounds, are presented in Table 1 and their detailed analysis is shown in Tables S1–S7† while a graph of the absorption properties is shown in Fig. 4.
Table 1 Computed absorption properties of BDTC1–BDTC7a
| Comp. |
λmax(nm) |
E (eV) |
fos |
MO contributions |
| MO = molecular orbital, H = HOMO, L = LUMO, fos = oscillator strength. |
| BDTC1 |
372.591 |
3.328 |
0.064 |
H → L (96%) |
| BDTC2 |
362.888 |
3.417 |
0.087 |
H → L (97%) |
| BDTC3 |
380.180 |
3.261 |
0.075 |
H → L (96%) |
| BDTC4 |
381.197 |
3.253 |
0.076 |
H → L (96%) |
| BDTC5 |
360.902 |
3.435 |
0.067 |
H → L (97%) |
| BDTC6 |
382.809 |
3.239 |
0.038 |
H → L (93%), H → L + 1 (5%) |
| BDTC7 |
448.990 |
2.761 |
0.070 |
H → L (97%), H → L + 1 (2%) |
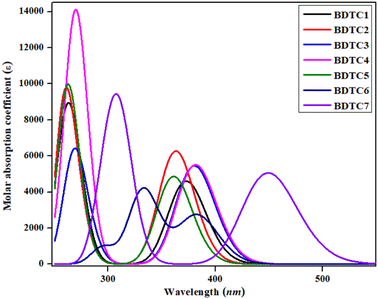 |
| | Fig. 4 Simulated absorption spectra of BDTC1–BDTC7. | |
Table 1 shows that all the synthesized molecules exhibit maximum absorbance in the range of 360.902–448.990 nm with an excitation energy of 2.761–3.435 eV. All the molecules exhibit comparable results of λmax when the position of the electron-withdrawing units on benzene is changed (see Table 1). However, a hypochromic shift (λmax = 360.902 and 362.888 nm) with the highest transition energy (ΔE = 3.435 and 3.417 eV) was noted for BDTC5 and BDTC2, respectively. The highest bathochromic shift (448.990 nm) was seen in BDTC7 with a smaller transition energy (ΔE = 2.761 eV) because of the presence of an electron-withdrawing nitro (–NO2) unit at the end capped acceptor moiety. Overall, the descending trend of λmax of the investigated compounds is in the following order: BDTC7 > BDTC6 > BDTC4 > BDTC3 > BDTC1 > BDTC2 > BDTC5, which is inversely related to the FMO results. Literature surveys revealed that molecules with smaller energy gaps show better absorption properties leading to larger HOMO to LUMO charge transfer that results in efficient photovoltaic behavior with a high-power conversion efficiency (PCE).73 The prior discussion reports that a small band gap with significant charge transference was found in the red-shifted compound BDTC7 leading to a high PCE and making it an excellent material for organic solar cells (OSCs).
Reorganization energy (RE)
Reorganization energy is the ability of holes and electrons to recombine in a molecule. Through this analysis, the rate of mobility of electrons and holes in an organic system can be examined.53,74 The smaller the reorganization energy, the greater the charge carrier ability and vice versa.61,75,76 There are a number of factors such as external environmental factors (temperature, solvent media etc.), and cationic and anionic geometries that influence reorganization energy.51,67,77 Here, the main focus was to determine the mobility of charges (hole and electron) within a molecule.
The data from Table 2 show that almost all the compounds exhibit comparable values for λe (−0.024 to −0.014 eV) except BDTC3 and BDTC7. These λe results indicate that except for BDTC3 and BDTC7, all the other synthesized compounds have good electron transportation rates. Similarly, all the compounds exhibit comparable values of λh (0.025–0.038 eV), when the electron withdrawing groups change position on the benzene ring, except BDTC2 and BDTC5. The highest value of λh is seen in BDTC5 (−0.273 eV), which might be due to the bromo unit located at the benzene ring that deactivates the ring and lowers the charge transference rate. The lowest value of λh is noted in BDTC2 (0.0025 eV), indicating a high hole transference rate in this molecule. The aforementioned analysis shows that λh values are lower than λe values for the compounds. This decrease in the values of λh suggests that all the molecules (BDTC1–BDTC7) are promising hole transfer candidates and could behave as effective photovoltaic materials.
Table 2 Computed RE parameters of BDTC1–BDTC7 in eVa
| Compounds |
λe |
λh |
| λe = RE of electrons; λh = RE of holes. |
| BDTC1 |
−0.023 |
−0.038 |
| BDTC2 |
−0.024 |
0.0025 |
| BDTC3 |
0.00 |
−0.025 |
| BDTC4 |
−0.021 |
−0.027 |
| BDTC5 |
−0.024 |
−0.273 |
| BDTC6 |
−0.014 |
−0.033 |
| BDTC7 |
0.00 |
−0.025 |
Hole–electron and transition density matrix (TDM) analysis
The hole–electron investigation found almost the same index values for the investigated hydrazide-based scaffolds, as shown in Table 3. As can be seen from Table 3, among all the chromophores, BDTC6 possesses the highest D index at 4.101 Å, which diminished to 2.863 and 2.802 Å in BDTC4 and BDTC3, respectively. These values further reduce to 2.686 and 2.611 Å in BDTC1 and BDTC5, respectively.
Table 3 Hole–electron analysis indices for the S0 → S1 excitation (with the highest fosc) of the studied compoundsa
| Compound |
Excitation |
λmax(nm) |
fos |
E (eV) |
D (Å) |
ECoul (eV) |
Sr (a.u) |
H (Å) |
t (Å) |
HDI |
EDI |
| Electron delocalization index (EDI) and hole delocalization index (HDI). |
| BDTC1 |
S0 → S1 |
372.591 |
0.064 |
3.328 |
2.686 |
4.3125 |
0.48759 |
2.422 |
1.099 |
9.96 |
8.69 |
| BDTC2 |
S0 → S1 |
362.888 |
0.087 |
3.417 |
2.547 |
4.3651 |
0.52521 |
2.449 |
0.993 |
9.66 |
8.30 |
| BDTC3 |
S0 → S1 |
380.180 |
0.075 |
3.261 |
2.802 |
4.2040 |
0.48147 |
2.425 |
1.228 |
9.85 |
8.64 |
| BDTC4 |
S0 → S1 |
381.197 |
0.076 |
3.252 |
2.863 |
4.2142 |
0.47441 |
2.470 |
1.228 |
9.86 |
8.68 |
| BDTC5 |
S0 → S1 |
360.902 |
0.067 |
3.435 |
2.611 |
4.2812 |
0.50120 |
2.432 |
1.106 |
9.70 |
8.31 |
| BDTC6 |
S0 → S1 |
382.809 |
0.038 |
3.239 |
4.101 |
4.2510 |
0.38413 |
2.568 |
2.419 |
9.97 |
9.81 |
| BDTC7 |
S0 → S1 |
448.990 |
0.070 |
2.761 |
3.851 |
4.2713 |
0.42878 |
2.692 |
2.151 |
9.73 |
8.59 |
However, BDTC2 possesses a medium D index value of 2.547 Å, which implies that the S0 → S1 excitations involve a small distance between electrons and holes and consequently a medium ECoul value of 4.3651 eV, which suggests a weak hole–electron interaction. Similarly, among all compounds, BDTC2 demonstrates the highest Sr index value of 0.52521 a.u. along with a H index of 2.449 Å. These hydrazide scaffolds (BDTC1–BDTC7) undergo π–π* excitations owing to their high Sr index values (>0.6), and consequently they have high H index values of 2.422, 2.449, 2.425, 2.470, 2.432, 2.568, and 2.692 3.32 Å, evidencing the presence of extended hole and electron distributions in these compounds. All the aforesaid compounds exhibit positive t values, indicating that the main excitation is local excitation (LE) in the ascending order of BDTC2< BDTC1 < BDTC5 < BDTC6< BDTC3 = BDTC4. The excitation also showed relatively small and similar HDI and EDI values, indicating a localized excitation, as illustrated in Table 3. TDM analysis is an essential method for investigating the extent of transitions and their nature in a molecule. It is utilized to evaluate interactions between acceptor and donor units, electronic excitation, and electron–hole pair localization.78,79 TDM analysis of the studied compounds (BDTC1–BDTC7) was performed in the first excitation state (S1) by employing the above-mentioned DFT functional. The influence of hydrogen atoms was neglected due to their negligible contribution to transitions. The TDM heat maps of all the studied compounds show the nature of transitions and are displayed in Fig. 5.
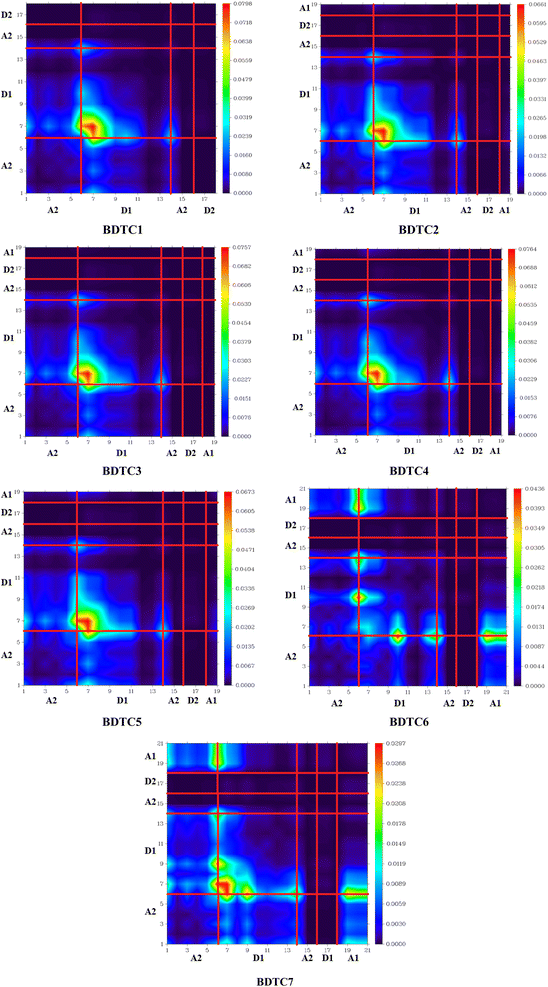 |
| | Fig. 5 Simulated TDM graphs of BDTC1–BDTC7. | |
For the simulation of heat maps, the examined molecules were divided into four parts: A1, D1, A2, and D2. Efficient ICT is driven from D2 towards A1 because of the excellent electron withdrawing nature of the terminal acceptor moieties. The most prominent ICT is observed in BDTC6 and BDTC7, as both contain a nitro group as the terminal acceptor part. In the maps of BDTC1–BDTC5, higher electronic cloud density can be seen over D1, as indicated by the green and red regions. This electronic charge density is diagonally shifted from donor to acceptor through the central structure in BDTC6 and BDTC7. In the heat map of BDTC7, obvious electronic charge is shifted diagonally from the donor towards the acceptor, as indicated by the green and red spots (Fig. 5). This means that the strong terminal electron-withdrawing segment attracts the electronic density towards itself, hence efficient movement of electronic cloud occurs from donor to acceptor in BDTC7, acting as a charge facilitator without charge entrapping.
Similarly, BDTC6 also shows effective charge coherence and proliferation of electron density in the diagonal path. In fact, all the compounds show charge coherence in the diagonal and off-diagonal regions. From the above discussion, it is clear that TDM analysis provided excellent results for all the studied compounds and proved them to be highly efficient solar cell materials.
Exciton-binding energy (Eb)
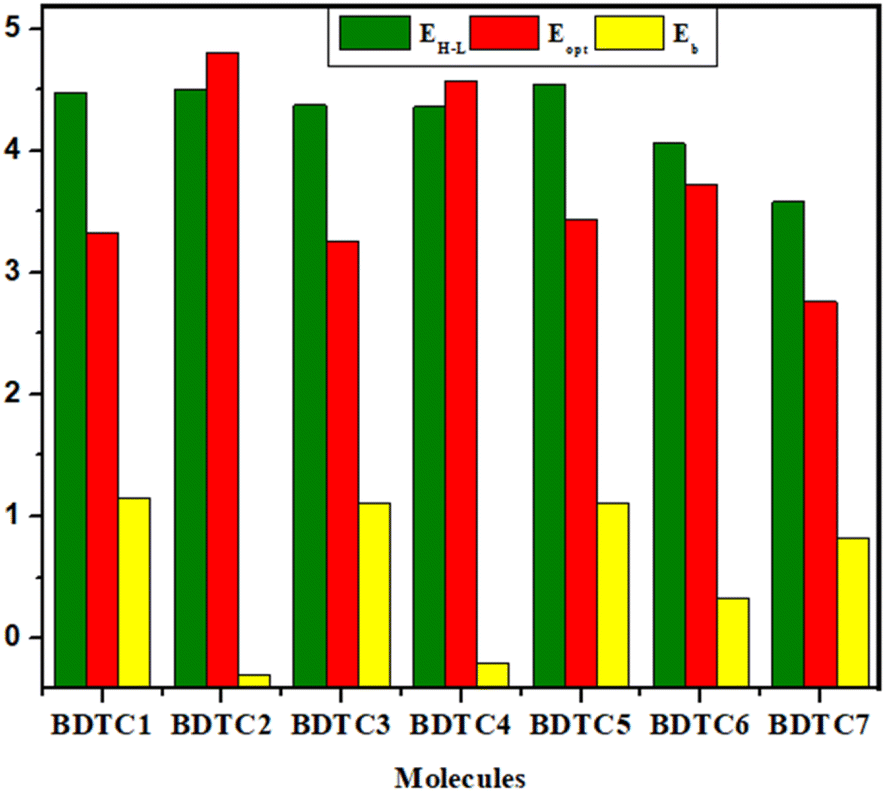
Binding energy or exciton binding energy (Eb) is the difference between the electrical (EH–L) and optical (Eopt) energy band gap and is a key factor for the evaluation of optoelectronic properties of a compound.80,81 A higher rate of electronic charge shift is obtained due to lower Eb values.82 eqn 3 was used for the calculation of the binding energies of BDTC1–BDTC7.Here, Eb is the binding energy, EH–L is the bandgap, and Eopt is the first excitation energy.83,84 All the calculated values of binding energy are presented in Table 4, and their pictographic illustration is shown in Fig. 6.
Table 4 Computed exciton binding energy (Eb) of BDTC1–BDTC7
| Comp. |
EH–L (eV) |
Eopt (eV) |
Eb (eV) |
| BDTC1 |
4.478 |
3.328 |
1.150 |
| BDTC2 |
4.504 |
3.417 |
−0.295 |
| BDTC3 |
4.374 |
3.261 |
1.113 |
| BDTC4 |
4.360 |
3.253 |
−0.211 |
| BDTC5 |
4.546 |
3.435 |
1.111 |
| BDTC6 |
4.058 |
3.239 |
0.334 |
| BDTC7 |
3.583 |
2.761 |
0.822 |
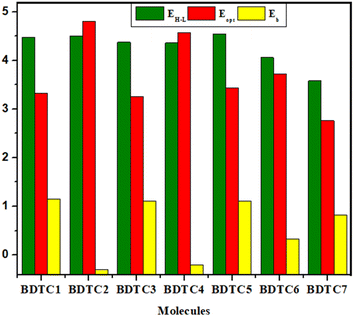 |
| | Fig. 6 Energy gap = EH–L, Eopt and (Eb) of the investigated compounds. | |
BDTC1 has the highest value of binding energy (1.150 eV) with a band gap of 4.478 eV. All the molecules exhibited Eb values in the range of (−0.295 to 1.150 eV). A smaller Eb with higher excitation enables greater exciton dissociation and considerably greater mobility of charges with significant optoelectronic properties.78 In the investigated compounds, the smaller Eb values correlate with the TDM heat maps where effective charge transfer is seen. The overall descending order of Eb is as follows: BDTC1 > BDTC3 > BDTC5 > BDTC7 > BDTC6 > BDTC4 > BDTC2 with values of 1.150, 1.113, 1.111, 0.822, 0.334, −0.211, and −0.295 eV, respectively (Table 4). The smallest value of Eb was found for BDTC2, which corresponds with the TDM results and hence it might possess excellent optoelectronic properties for use in OSCs.
Open circuit voltage (Voc)
The operational mechanism and performance of OSCs can be predicted using open-circuit voltage analysis. In essence, it measures the total current drawn from an optical instrument. Voc is measured at zero voltage in solar devices. In order to evaluate Voc, some scaling factors have been developed that can be applied to accurately estimate the Voc of a solar cell. With the LUMO of an acceptor material, the HOMO of the donor material can be scaled. For higher open circuit voltage levels, the LUMO of the donor materials ought to have a higher energy than the LUMO of the acceptor material. The donor polymer molecule PBDB-T was selected for open circuit voltage measurements since all of the reference and synthesized molecules used in this investigation are acceptors by nature. The difference in HOMO and LUMO energies between D and A molecules is directly related to the Voc.85 In the current study, we calculated the Voc values using the well-known donor polymer PBDB-T, whose simulated EHOMO value is −4.936 eV. Theoretically, eqn (4) was used to calculate the Voc of the OSCs,86 and the results are tabulated in Fig. 7.| | |
Voc = (|EDHOMO| − |EALUMO|) − 0.3
| (4) |
where E denotes energy, and the constant, i.e. 0.3, was derived from the resolution factors of voltage drop.66 The Voc lines up the HOMO of a well-known donor polymer, PBDB-T, with the LUMO of the acceptor of the investigated compounds (BDTC1–BDTC7).
 |
| | Fig. 7 The Voc values of BDTC1–BDTC7 with respect to the donor material PBDB-T. | |
The above data reveal that all the derivatives (BDTC1–BDTC7) exhibit Voc values in the range of 1.97–3.27 V. ΔE between the HOMO of the donor/LUMO of the acceptor complexes was found to be 4.478, 4.504, 4.374, 4.360, 4.546, 4.058, and 3.583 eV for BDTC1–BDTC7, respectively (Table 5). The Voc values of BDTC1–BDTC7 with respect to the HOMOPBDB-T–LUMOacceptor difference of energy were found to be 3.27, 3.19, 3.00, 3.02, 3.25, 2.31, and 1.97 V, respectively. BDTC1 displays the maximum Voc (3.27 V) among all the synthesized molecules. The diminishing order of Voc of the designed chromophores with respect to HOMOPBDB-T–LUMOacceptor is as follows: BDTC1 (3.27 V)> BDTC5 (3.25 V) > BDTC2 (3.19 V) > BDTC4 (3.02 V) > BDTC3 (3.00 V) > BDTC6 (2.31 V) > BDTC7 (1.97 V). Voc depends on the energy gap, and HOMO of the donor and LUMO of the acceptor. A low-lying LUMO results in high Voc values with improved optoelectronic properties. The orbital energy diagram for every component in relation to PBDB-T is shown in Fig. 7. It can be seen that the LUMO levels of the acceptor molecules (BDTC1–BDTC7) are lower than those of the donor PBDB-T polymer. Such a molecular orbital alignment makes the electron density move more quickly from a donor polymer to an acceptor, thus improving optoelectronic performance.
Table 5 Computed Voc values and energy driving force of BDTC1–BDTC7
| Compounds |
VOC (V) |
ΔE (eV) |
| BDTC1 |
3.27 |
4.478 |
| BDTC2 |
3.19 |
4.504 |
| BDTC3 |
3.00 |
4.374 |
| BDTC4 |
3.02 |
4.360 |
| BDTC5 |
3.25 |
4.546 |
| BDTC6 |
2.31 |
4.058 |
| BDTC7 |
1.97 |
3.583 |
Conclusion
Hydrazinylthiazole-4-carbohydrazide based photovoltaic materials (BDTC1–BDTC7), with an A1–D1–A2–D2 framework, were synthesized, and structural elucidation was done using various spectroscopic (HRMS, FT-IR, 1H, and 13C-NMR) techniques. The photovoltaic properties of BDTC1–BDTC7 were calculated via DFT. In all the aforesaid compounds, lower reorganization energy values were found for holes (λh = −0.273 to −0.038 eV) than for electrons (λe = 0.00 to −0.014 eV) except for BDTC2, which indicated higher hole mobility rates in BDTC1 and BDTC3–BDTC7. Lower binding energy values in all studied compounds enabled higher exciton dissociation rates, which resulted in higher charge transference rates that were further supported by TDM and DOS analyses. Moreover, the Voc values were also determined relative to HOMOPBDBT–LUMOAcceptor and interesting data were found in the order: BDTC1 (3.27 V)> BDTC5 (3.25 V) > BDTC2 (3.19 V) > BDTC4 (3.02 V) > BDTC3 (3.00 V) > BDTC6 (2.31 V) > BDTC7 (1.97 V). Band gaps of 4.546–3.583 eV were found in these synthesized compounds, and a narrow band gap (3.583 eV) was found in BDTC7. Additionally, the highest λmax value (448.990 nm), along with a great charge density transfer from HOMO to LUMO, was also observed in BDTC7. With powerful electron-withdrawing acceptor units, the calculated photovoltaic characteristics such as a reduced band gap (3.583 eV), the largest λmax (448.990 nm), moderate reorganization energy (λe = 0.00, λh = −0.025 eV) and Voc (1.97 V) support the candidacy of BDTC7 as a potential OSC material.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Dr Muhammad Khalid gratefully acknowledges the financial support of HEC Pakistan (project no. 20-14703/NRPU/R&D/HEC/2021). Moreover, the authors extend their appreciation to the Researcher Supporting Project number (RSP2023R431), King Saud University, Riyadh, Saudi Arabia, for funding this research work.
References
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding and R. Xia, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- D. Sharma, R. Mehra and B. Raj, Superlattices Microstruct., 2021, 153, 106861 CrossRef CAS.
- S. A. Siddique, M. B. A. Siddique, R. Hussain, X. Liu, M. Y. Mehboob, Z. Irshad and M. Adnan, Comput. Theor. Chem., 2020, 1191, 113045 CrossRef CAS.
- J. Tan, Y. Zhao, G. Li, S. Yang, C. Huang and H. Yu, Adv. Funct. Mater., 2022, 2209094 CrossRef CAS.
- M. R. Busireddy, M. Chakali, G. R. Reddy, N. R. Chereddy, B. Shanigaram, B. Kotamarthi, G. D. Sharma and V. J. Rao, Sol. Energy, 2019, 186, 84–93 CrossRef CAS.
- C. Hou and H. Yu, Chem. Eng. J., 2021, 407, 127192 CrossRef CAS.
- V. Dusastre, Materials for sustainable energy: a collection of peer-reviewed research and review articles from Nature Publishing Group, World Scientific, 2010 Search PubMed.
- S. Sabir, R. A. Khera, S. Jabeen, Z. Shafiq, A. Musawwir and J. Iqbal, J. Theor. Comput. Chem., 2020, 19, 2050003 CrossRef CAS.
- A. Mahmood, S. U.-D. Khan and U. A. Rana, J. Comput. Electron., 2014, 13, 1033–1041 CrossRef CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- T. Saleem, S. Khan, M. Yaqub, M. Khalid, M. Islam, M. Y. ur Rehman, M. Rashid, I. Shafiq, A. A. Braga and A. Syed, New J. Chem., 2022, 46, 18233–18243 RSC.
- M. U. Khan, M. Khalid, I. Shafiq, R. A. Khera, Z. Shafiq, R. Jawaria, M. Shafiq, M. M. Alam, A. A. C. Braga and M. Imran, J. Saudi Chem. Soc., 2021, 25, 101339 CrossRef CAS.
- A. Mahmood, S. U.-D. Khan and U. A. Rana, J. Comput. Electron., 2014, 13, 1033–1041 CrossRef CAS.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- S. Sharma, K. K. Jain and A. Sharma, Mater. Sci. Appl., 2015, 6, 1145 CAS.
- Q. Yue, W. Liu and X. Zhu, J. Am. Chem. Soc., 2020, 142, 11613–11628 CrossRef CAS PubMed.
- L. Zhan, S. Li, T.-K. Lau, Y. Cui, X. Lu, M. Shi, C.-Z. Li, H. Li, J. Hou and H. Chen, Energy Environ. Sci., 2020, 13, 635–645 RSC.
- M. Khalid, R. Ahmed, M. Arshad, M. A. Asghar, K. S. Munawar, M. Imran and A. A. Braga, Sci. Rep., 2022, 12, 1–15 CrossRef PubMed.
- M. Khalid, R. Ahmed, M. Arshad, M. A. Asghar, K. S. Munawar, M. Imran and A. A. Braga, Sci. Rep., 2022, 12, 1–15 CrossRef PubMed.
- T. Albes, L. Xu, J. Wang, J. W. Hsu and A. Gagliardi, J. Phys. Chem. C, 2018, 122, 15140–15148 CrossRef CAS.
- J. Kalowekamo and E. Baker, Sol. Energy, 2009, 83, 1224–1231 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma and C. An, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- Y. Lin, Y. Firdaus, F. H. Isikgor, M. I. Nugraha, E. Yengel, G. T. Harrison, R. Hallani, A. El-Labban, H. Faber and C. Ma, ACS Energy Lett., 2020, 5, 2935–2944 CrossRef CAS.
- T. Wang, J. Qin, Z. Xiao, J. Zhang, Z. Chen, L. Zhang, M. Cheng, Z. Jin, Y. Yuan and W.-Q. Wu, Nano Energy, 2020, 77, 105161 CrossRef CAS.
- C. Huang and H. Yu, Nano Energy, 2022, 103, 107750 CrossRef CAS.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang and J. Zhu, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed.
- A. Mahmood, J. Cluster Sci., 2019, 30, 1123–1130 CrossRef CAS.
- A. Mahmood, J. Yang, J. Hu, X. Wang, A. Tang, Y. Geng, Q. Zeng and E. Zhou, J. Phys. Chem. C, 2018, 122, 29122–29128 CrossRef CAS.
- Z. Xiao, X. Jia and L. Ding, Sci. Bull., 2017, 62, 1562–1564 CrossRef CAS PubMed.
- Y. Zhang, Z. Liu, T. Shan, Y. Wang, L. Zhu, T. Li, F. Liu and H. Zhong, Mater. Chem. Front., 2020, 4, 2462–2471 RSC.
- A. Mahmood, M. HussainTahir, A. Irfan, B. Khalid and A. G. Al-Sehemi, Bull. Korean Chem. Soc., 2015, 36, 2615–2620 CrossRef CAS.
- A. Mahmood, A. Irfan, F. Ahmad and M. R. S. A. Janjua, Comput. Theor. Chem., 2021, 1204, 113387 CrossRef CAS.
- S. A. Siddique, S. Altaf, E. Ahmed, S. Naveed, M. B. A. Siddique, R. Hussain, X. Liu, A. Rauf and M. Arshad, Int. J. Energy Res., 2022, 46, 13393–13408 CrossRef CAS.
- M. Ans, J. Iqbal, B. Eliasson and K. Ayub, Comput. Mater. Sci., 2019, 159, 150–159 CrossRef CAS.
- A. Khalid, R. A. Khera, A. Saeed, M. Khalid, S. Iqbal and J. Iqbal, Optik, 2021, 228, 166138 CrossRef CAS.
- R. A. Shehzad, J. Iqbal, M. U. Khan, R. Hussain, H. M. A. Javed, A. ur Rehman, M. U. Alvi and M. Khalid, Comput. Theor. Chem., 2020, 1181, 112833 CrossRef CAS.
- L. S. C. Martins, F. E. Jorge, M. L. Franco and I. B. Ferreira, J. Chem. Phys., 2016, 145, 244113 CrossRef CAS PubMed.
- R. D. Dennington, T. A. Keith, J. M. Millam, GaussView 5.0, Gaussian. Inc., Wallingford, 2008 Search PubMed.
- M. D. Hanwell, et al., J. Cheminf., 2012, 4(1), 17 CAS.
- G. A. Zhurko and D. A. Zhurko, ChemCraf, version 1.6, http://www.chemcrafprog.com, 2009 Search PubMed.
- N. M. O’boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29(5), 839–845 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- N. M. O’boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- Y. Wang, P. Verma, X. Jin, D. G. Truhlar and X. He, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10257–10262 CrossRef CAS PubMed.
- Y.-S. Park, J.-S. Park, S. Lee, S.-H. Jung, S.-K. Kim and C.-M. Ryu, Comput. Struct. Biotechnol. J., 2021, 19, 2084–2096 CrossRef CAS PubMed.
- J. E. Del Bene, W. B. Person and K. Szczepaniak, J. Phys. Chem., 1995, 99, 10705–10707 CrossRef CAS.
- R. Hussain, M. U. Khan, M. Y. Mehboob, M. Khalid, J. Iqbal, K. Ayub, M. Adnan, M. Ahmed, K. Atiq and K. Mahmood, ChemistrySelect, 2020, 5, 5022–5034 CrossRef CAS.
- M. U. Khan, J. Iqbal, M. Khalid, R. Hussain, A. A. C. Braga, M. Hussain and S. Muhammad, RSC Adv., 2019, 9, 26402–26418 RSC.
- M. Y. Mehboob, R. Hussain, M. U. Khan, M. Adnan, A. Umar, M. U. Alvi, M. Ahmed, M. Khalid, J. Iqbal and M. N. Akhtar, Comput. Theor. Chem., 2020, 1186, 112908 CrossRef CAS.
- M. Y. Mehboob, M. U. Khan, R. Hussain, R. Fatima, Z. Irshad and M. Adnan, J. Theor. Comput. Chem., 2020, 19, 2050034 CrossRef CAS.
- M. U. Khan, M. Khalid, M. Ibrahim, A. A. C. Braga, M. Safdar, A. A. Al-Saadi and M. R. S. A. Janjua, J. Phys. Chem. C, 2018, 122, 4009–4018 CrossRef CAS.
- H. Layaida, A. Hellal, N. Chafai, I. Haddadi, K. Imene, B. Anis, E. Mouna, C. Bensouici, W. Sobhi, A. Attoui and A. Lilia, J. Biomol. Struct. Dyn., 2022, 1–16 CrossRef PubMed.
- M. R. S. A. Janjua, M. U. Khan, B. Bashir, M. A. Iqbal, Y. Song, S. A. R. Naqvi and Z. A. Khan, Comput. Theor. Chem., 2012, 994, 34–40 CrossRef CAS.
- M. U. Khan, M. Ibrahim, M. Khalid, M. S. Qureshi, T. Gulzar, K. M. Zia, A. A. Al-Saadi and M. R. S. A. Janjua, Chem. Phys. Lett., 2019, 715, 222–230 CrossRef CAS.
- M. U. Khan, M. Ibrahim, M. Khalid, A. A. C. Braga, S. Ahmed and A. Sultan, J. Cluster Sci., 2019, 30, 415–430 CrossRef CAS.
- M. U. Khan, M. Ibrahim, M. Khalid, S. Jamil, A. A. Al-Saadi and M. R. S. A. Janjua, Chem. Phys. Lett., 2019, 719, 59–66 CrossRef CAS.
- M. R. S. A. Janjua, J. Cluster Sci., 2021, 32, 243–253 CrossRef CAS.
- M. U. Khan, R. Hussain, M. Y. Mehboob, M. Khalid, M. A. Ehsan, A. Rehman and M. R. S. A. Janjua, Spectrochim. Acta, Part A, 2021, 245, 118938 CrossRef CAS PubMed.
- M. Ashfaq, A. Ali, M. N. Tahir, M. Khalid, M. A. Assiri, M. Imran, K. S. Munawar and U. Habiba, Chem. Phys. Lett., 2022, 803, 139843 CrossRef CAS.
- M. Haroon, T. Akhtar, M. Khalid, S. S. Zahra, I. Haq, M. A. Assiri, M. Imran and A. A. Braga, J. Mol. Struct., 2022, 1270, 133923 CrossRef CAS.
- A. Mahmood, S. U.-D. Khan, U. A. Rana, M. R. S. A. Janjua, M. H. Tahir, M. F. Nazar and Y. Song, J. Phys. Org. Chem., 2015, 28, 418–422 CrossRef CAS.
- A. Mahmood, S. U.-D. Khan, U. A. Rana and M. H. Tahir, Arabian J. Chem., 2019, 12, 1447–1453 CrossRef CAS.
- M. U. Khan, M. Y. Mehboob, R. Hussain, Z. Afzal, M. Khalid and M. Adnan, Int. J. Quantum Chem., 2020, 120, e26377 CrossRef CAS.
- M. U. Khan, M. Y. Mehboob, R. Hussain, R. Fatima, M. S. Tahir, M. Khalid and A. A. C. Braga, J. Phys. Org. Chem., 2021, 34, e4119 CAS.
- P. Goszczycki, K. Stadnicka, M. Z. Brela, J. Grolik and K. Ostrowska, J. Mol. Struct., 2017, 1146, 337–346 CrossRef CAS.
- S. A. Siddique, M. Arshad, S. Naveed, M. Y. Mehboob, M. Adnan, R. Hussain, B. Ali, M. B. A. Siddique and X. Liu, RSC Adv., 2021, 11, 27570–27582 RSC.
- W. Rahmalia, J.-F. Fabre, T. Usman and Z. Mouloungui, Spectrochim. Acta, Part A, 2014, 131, 455–460 CrossRef CAS PubMed.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef CAS.
- M. D. Adeoye, A. I. Adeogun, S. Adewuyi, S. A. Ahmed, N. W. Odozi and N. O. Obi-Egbeedi, Sci. Res. Essays, 2009, 4, 107–111 Search PubMed.
- X. Qian, L. Lu, Y.-Z. Zhu, H.-H. Gao and J.-Y. Zheng, Dyes Pigm., 2015, 113, 737–742 CrossRef CAS.
- R. Hussain, M. Y. Mehboob, M. U. Khan, M. Khalid, Z. Irshad, R. Fatima, A. Anwar, S. Nawab and M. Adnan, J. Mater. Sci., 2021, 56, 5113–5131 CrossRef CAS.
- M. Y. Mehboob, M. U. Khan, R. Hussain, K. Ayub, A. Sattar, M. K. Ahmad, Z. Irshad and M. Adnan, Spectrochim. Acta, Part A, 2021, 244, 118873 CrossRef CAS PubMed.
- M. Y. Mehboob, R. Hussain, Z. Irshad and M. Adnan, J. Phys. Org. Chem., 2021, 34, e4210 CAS.
- Z. Afzal, R. Hussain, M. U. Khan, M. Khalid, J. Iqbal, M. U. Alvi, M. Adnan, M. Ahmed, M. Y. Mehboob and M. Hussain, J. Mol. Model., 2020, 26, 1–17 CrossRef PubMed.
- M. Khalid, I. Shafiq, M. Zhu, M. U. Khan, Z. Shafiq, J. Iqbal, M. M. Alam, A. A. C. Braga and M. Imran, J. Saudi Chem. Soc., 2021, 25, 101305 CrossRef CAS.
- M. R. S. A. Janjua, Synth. Met., 2021, 279, 116865 CrossRef CAS.
- S. Kraner, G. Prampolini and G. Cuniberti, J. Phys. Chem. C, 2017, 121, 17088–17095 CrossRef CAS.
- M. Ans, J. Iqbal, Z. Ahmad, S. Muhammad, R. Hussain, B. Eliasson and K. Ayub, ChemistrySelect, 2018, 3, 12797–12804 CrossRef CAS.
- M. U. Khan, M. Khalid, R. Hussain, A. Umar, M. Y. Mehboob, Z. Shafiq, M. Imran and A. Irfan, Energy Fuels, 2021, 35, 12436–12450 CrossRef CAS.
- A. Dkhissi, Synth. Met., 2011, 161, 1441–1443 CrossRef CAS.
- M. Naeem, S. Jabeen, R. A. Khera, U. Mubashar and J. Iqbal, J. Theor. Comput. Chem., 2019, 18, 1950036 CrossRef CAS.
- M. N. Arshad, I. Shafiq, M. Khalid and A. M. Asiri, ACS Omega, 2022, 7, 11606–11617 CrossRef CAS PubMed.
- Z. Zheng, H. Yao, L. Ye, Y. Xu, S. Zhang and J. Hou, Mater. Today, 2020, 35, 115–130 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Muhammad Khalid
*a,
Muhammad Khalid